

Abstract
Human butyrylcholinesterase (BChE) is recognized as the most promising bioscavenger for organophosphorus (OP) warfare nerve agents. The G117H mutant of human BChE has been identified as a potential catalytic bioscavenger with a remarkably improved activity against OP nerve agents such as sarin, but it still does not satisfy the clinical use. For further design of the higher-activity mutants against OP nerve agents, it is essential to understand how the G117H mutation improves the activity. The reaction mechanisms and the free energy profiles for spontaneous reactivation of the wild-type BChE and its G117H mutant phosphorylated by sarin have been explored, in this study, by performing first-principles quantum mechanical/molecular mechanical free energy (QM/MM-FE) calculations, and the remarkable role of the G117H mutation on the activity has been elucidated. For both the wild-type and G117H mutant enzymes, H438 acts as a general base to initiate the spontaneous reactivation which consists of two reaction steps: the nucleophilic attack at the phosphorus by a water molecule and decomposition of the pentacoordinated phosphorus intermediate. The calculated overall free energy barriers, i.e. 30.2 and 23.9 kcal/mol for the wild-type and G117H mutant, respectively, are in good agreement with available experimental kinetic data. Based on the calculated results, the mutated residue (H117 in the G117H mutant) cannot initiate the spontaneous reactivation as a general base. Instead, it skews the oxyanion hole and makes the phosphorus more open to the nucleophilic water molecule, resulting in remarkable change of the rate-determining step and significantly improved catalytic activity of human BChE.
Introduction
Organophosphorus (OP) chemical warfare nerve agents, e.g. sarin, soman, tabun, VX, and VR, are highly neurotoxic compounds. They inhibit cholinesterases, including both acetylcholinesterase (AChE, E.C. 3.1.1.7) and butyrylcholinesterase (BChE, E.C. 3.1.1.8), by phosphorylation of the catalytic serine residue. Phosphorylation of AChE at neuronal and neuromuscular synapses is the main cause of symptoms of the OP nerve agent poisoning.(1–3) The production, stockpiling and use of OP chemical warfare nerve agents are now prohibited under the terms of the Chemical Weapons Convention (CWC) of the United Nations and, thus, the threat of military use has been diminished. However, there is still perceived to be a serious threat of use by terrorists. In the 1980s, sarin was used against Kurdish communities in Iraq.(4) In the 1990s, sarin was released by Japanese terrorists in Matsumoto, Nagano, and the Tokyo subway.(5–8) Moreover, VX was also used in an assassination in Japan by the same group in 1994.(9)
Cholinesterases phosphorylated by OP chemical warfare nerve agents can undergo three possible post-inhibitory reactions: (1) spontaneous reactivation by a water molecule to restore the enzyme activity; (2) nucleophile-induced reactivation to restore the enzyme activity: and (3) aging reaction by dealkylation of the phosphorylated cholinesterase resulting in the truly irreversible inhibition. Based on these reactions, many strategies, e.g. protective inhibitor, reactivator, and scavenger approaches, have been developed for the pretreatment and post-treatment against the effects of the chemical warfare nerve agents in the past 50 years.(10) The protective inhibitor, such as carbamate, may be able to block AChE against the reactions with the chemical warfare nerve agents. However, after an exposure, there will always be a time during which acetylcholine builds up pending reactivation of the enzyme, causing a period of incapacitation. The reactivator, such as oxime, must reach the phosphorylated enzyme and restore the enzyme activity in time to protect the life if it is to be effective. In the mean time, the developed stronger anticholinergics and superior anticonvulsants must be delivered in a timely fashion, otherwise the reactivator strategy is useless. Therefore, the best way to completely and efficiently protect humans from the OP nerve agents is to design and discover an effective scavenger to degrade the OP nerve agents in plasma. A desirable scavenger for this purpose is capable of intercepting and destroying an OP nerve agent before it reaches a critical target without causing any incapacitation or behavioral deficits. There are several approaches to achieve this goal. One approach is to isolate and characterize an organophosphorus acid anhydride hydrolase (OPAAH), for example, carboxylesterase, paraoxonase, and a bacterial OPAAH from Pseudomonas diminuta. The bacterial OPAAH is also known as phosphotriesterase (PTE) in literature.(11–15) Unfortunately, none of them can defend the effects of the chemical warfare nerve agents without measurable side effects. A more efficient approach is to construct a highly active OPAAH against the nerve agents by using protein engineering techniques. The progress was achieved by using human BChE as a scavenger for all of the OP chemical warfare nerve agents.(16–17) Human BChE was chosen as the scavenger for several reasons. (1) The OP-inhibited human BChE can restore the enzyme activity and generate a nontoxic product via spontaneous reactivation reaction. (2) Human BChE is present in human serum and could be used as a therapeutic agent injected into blood. (3) Human BChE has a history of use in the clinic and no adverse effects were reported.(18) (4) So far, about 65 different naturally occurring mutants of human BChE have been identified and none of them is antigenic.
The rationale for the initial attempt involving site-directed mutagenesis of human BChE was based on a hypothesis proposed by Jarv(19) that organophosphates are hemisubstrates for the cholinesterases. In the hypothesis of hemisubstrate, the first stage, i.e. phosphorylation, is similar to the acylation stage of acetylcholine hydrolysis, but the second stage is significantly different to the deacylation stage of acetylcholine hydrolysis. In the deacylation stage, a histidine-bound water molecule displaces the acyl group from the catalytic serine, whereas this histidine-bound water molecule is prevented sterically from attacking the appropriate face of the tetrahedral phosphorus atom in the corresponding stage of the OP hydrolysis, and thus prevents the reactivation of the phosphorylated enzyme by water. Therefore, introducing a new histidine residue into the active site of the enzyme might generate another nucleophilic center which activates a water molecule to the opposite side of the phosphorous-serine bond and thereby initiates the reactivation reaction. Given this scheme, G115, G117, E119, G121, L286, and V288 in human BChE were mutated to histidine and only the G117H mutant was reported to have the ability to catalyze the hydrolysis of OP compounds.(20–21) The experimental data indicated that the catalytic rate constant of G117H mutant of human BChE against sarin has been increased by at least ~174-fold than that of wild-type human BChE.(21–22) However, the significantly improved catalytic activity of the G117H mutant against sarin, where the catalytic rate constant is 8.7×10−5 s−1,(21–22) is still too low for clinical use. The search for new mutants of human BChE with a further improved catalytic activity continues.
The rationale for the G117H mutation was to introduce a new nucleophilic center where H117 may act as a general base in the hydrolysis of OP. An empirical valence bond (EVB) simulation on a cluster model of the G117H mutant predicted the conformation of the introduced H117 residue and, based on the EVB results, the authors suggested that H117 may act as a general base to hydrolyze OP.(23) If this suggestion was correct, then the spontaneous reactivation reaction should show bell-shaped pH-dependence curve with the rates(21), as similar to those in the deacylation and dephosphylation reactions catalyzed by wild-type cholinesterases.(19) However, neither the recent X-ray crystal structure of the G117H mutant of human BChE phosphorylated by VX(24) nor the relationship between the reactivation rate and pH value(21) supports this suggestion. Understanding why and how the G117H mutation increases the activity against OP is therefore becoming very interesting. There have been several efforts to understand the mechanism for the reaction of the G117H mutant. On the basis of molecular mechanics calculations, a hypothesis proposed by Fortier et al.(25) involves a phosphoryl-histidine intermediate which is subsequently hydrolyzed by a water or hydroxide ion. However, the formation of the phosphoryl-histidine intermediate is prevented by the steric effect of OP and, hence, this process should be very slow, as pointed by Broomfield et al.(10, 22) This suggests that the spontaneous reactivation reaction of the G117H mutant of human BChE is not really initiated by the introduced H117 residue. Hence, it is possible that the mechanism for the spontaneous reactivation of the G117H mutant might be similar to that proposed for the spontaneous reactivation of wild-type human BChE,(26–27) implying that H438 residue still acts as a general base to activate a water molecule which initiates the nucleophilic attack on the phosphorus atom. If this is true, the introduced H117 residue might play an indirect role to accelerate the reaction process. One hypothesis attributes the improved rate constant to the presence of a positive charge at H117 residue (as it was assumed that H117 is protonated).(22) However, the G117K mutation, which also increases a positive charge, makes the dephorsphorylation much slower than that of the G117H mutant, meaning that the hypothesis is not correct.(22) Another hypothesis(20) is that the G117H mutation causes a couple of structural changes. The first one is to distort the oxyanion hole, resulting in that the P=O bond sits askew and H438 no longer blocks attack by water. The other one is to destabilize the covalent bond between sarin and the enzyme.(20) The nature of the G117H mutation improving the catalytic activity for the OP hydrolysis, which is very important to the design of high-activity mutants, still remains uncovered. Understanding the role of G117H mutation is therefore crucial.
In the present study, we have performed first-principles quantum mechanical/molecular mechanical-free energy perturbation (QM/MM-FE) calculations(28–31) to uncover the role of G117H mutation by exploring the detailed reaction mechanisms for spontaneous reactivation of both the wild-type human BChE and the G117H mutant that are phosphorylated by chemical warfare nerve agent sarin. Sarin was chosen here because it is a typical representative of the G-type nerve agents and its experimental kinetic data were well-established. In the QM/MM-FE method, the first-principles QM/MM reaction-coordinate calculations were followed by free-energy perturbation (FEP) calculations in order to more reasonably account for the dynamic effects of the protein environment on the free energy barriers for the enzymatic reactions. Our QM/MM simulations are based on the pseudobond first-principles QM/MM approach,(29–32) which has been demonstrated to be a powerful tool in simulating a variety of enzymes,(28, 33–50) and some theoretical predictions(39, 49, 51) were subsequently confirmed by the experimental studies.(35, 49, 52–53) The computational results clearly reveal the reaction mechanisms and free energy profiles of the reactivation reactions of sarin-phosphorylated wild-type human BChE and the G117H mutant. The rate-determining steps are identified and the roles of the essential residues, including those forming the catalytic triad and oxyanion hole, are discussed on the basis of the determined free energy profiles and the QM/MM-optimized geometries. By comparing the detailed reactivation reaction mechanisms of sarin-phosphorylated wild-type enzyme and G117H mutant of human BChE, we are able to understand the role and the remarkable effects of the G117H mutation.
Computational Methods
Structure preparation
The initial model of BChE was constructed on the basis of the X-ray crystal structure of human BChE (PDB code 1P0M).(54) All small molecules and the carbohydrate in the crystal structure were removed except the crystal water molecules. The missing residues (D2, D3, D378, D379, Q455, and Q486) in the X-ray crystal structure were rebuilt by using the automated homology modeling tool Modeler/InsightII software (Accelrys, Inc.).(55–56) It should be noted that residues #17, #455, #481, and #486 are all asparagines (glycosylation sites) in the wild-type BChE. One had to mutate these asparagines residues into glutamines through site-directed mutagenesis in order to obtain a single crystal for structural analysis by X-ray crystallography. So, the artificially engineered residues Q17, Q455, Q481, and Q486 were modified back to N17, N455, N481, and N486, respectively, in order to obtain the wild-type BChE structure. In addition, G117 was mutated to be histidine, arginine, or valine for the G117H, G117K, or G117V mutant examined in this study, by using the LEaP module in Amber 9 package.(57–58) The missing hydrogen atoms were also added by using the LEaP module in Amber 9 package.(57–58) Then, sarin-phosphorylated wild-type human BChE and G117H mutant conjugates were built by referring to the orientation of the X-ray crystal structure of sarin-inhibited mouse AChE (non-aged enzyme, PDB code: 2JGG).(59) Each system was solvated in an orthorhombic box of TIP3P water molecules(60) with a minimum solute-wall distance of 10 Å.
The MD simulations in the current study were carried out by using the Sander module of Amber 9 package.(57–58) The prepared systems were fully energy-minimized, followed by the equilibration through gradually increasing the temperature from 10 to 298.15 K. Then, the production MD simulations at 298.15 K were kept running for ~2 ns. In the MD simulation, the time step of the simulation was 2 fs with a cutoff of 10 Å for nonbonded interactions. The Shake procedure(61–62) was employed to constrain all bonds involving hydrogen atoms. The partial atomic charges for the atoms in the non-standard residue (sarin-phosphorylated S198) were calculated by using the restrained electrostatic potential (RESP) protocol implemented in the Antechamber module of Amber 9 package.(57–58)
QM/MM-FE simulation
The initial structures used for the QM/MM calculations were chosen from snapshots of the MD simulations on sarin-phosphorylated wild-type human BChE and the G117H mutant, and the chosen structures were close to the corresponding average structures simulated. All of the QM/MM calculations were performed by using a pseudobond QM/MM method(29–30) implemented recently in a revised version(49) of the Gaussian03(63) and AMBER8(64) programs. The QM-MM interface was treated by a pseudobond approach, where a seven-valence-electron atom with an effective core potential is constructed to replace the boundary atom of the environment part and to form a pseudobond with the boundary atom of the active part. In the QM/MM calculation, the attacking water molecule and the side chains of the sarin-phosphorylated S198, H438, and E325 (that form the catalytic triad) were considered as the QM atoms, whereas the other atoms were regarded as MM atoms (Figure 1). The QM/MM calculations were performed using an iterative energy minimization procedure(31) at the B3LYP/6-31*:AMBER level, i.e., the QM calculations were carried out at the B3LYP/6-31G* level, whereas the MM calculations were carried out by using the AMBER force field implemented in the AMBER8 program. For the QM subsystem, the convergence criterion for the geometry optimizations follows the original Gaussian03 defaults; for the MM subsystem, the geometry optimization convergence criterion is the root-mean-square deviation (RMSD) of the energy gradient being less than 0.1 kcal·mol−1·Å−1. An iterative restrained optimization procedure was then repeatedly applied to different points along the reaction coordinate, resulting in a minimum-energy path. Full QM/MM geometry optimizations were carried out at the B3LYP/6- 31G*:AMBER level, followed by vibrational frequency analyses at the same level to characterize the reactants, intermediates, and transition states. The contribution of the QM subsystem fluctuation to the free energy change was then calculated with the obtained vibrational frequencies using the harmonic approximation. In addition, single-point energy calculations were carried out at the QM/MM(MP2/6-31+G*:AMBER) level for each geometry along the minimum-energy path.
Figure 1.

Division of the QM/MM systems for simulating spontaneous reactivation reactions of sarin-phosphorylated wild-type human BChE and the G117H mutant. Atoms in blue are treated by QM method. Three boundary carbon atoms in red are treated with the improved pseudobond parameters.(29) All other atoms belong to the MM subsystem.
The free energy changes associated with the QM-MM interaction were then determined by using the free energy perturbation (FEP) method(31) using a revised version(40) of the AMBER8 program. The FEP calculations enabled us to more reasonably determine the relative free energy changes due to the QM-MM interaction. In the FEP calculations, sampling of the MM subsystem was carried out with the QM subsystem frozen at different states along the reaction path.(31) Technically, the final (relative) free energy determined by the QM/MM-FE calculations is the QM part of the QM/MM energy (excluding the Columbic interaction energy between the point charges of the MM atoms and the ESP charges of the QM atoms) plus the relative free energy change determined by the FEP calculations. In FEP calculations, the time step used was 2 fs, and bond lengths involving hydrogen atoms were constrained. In sampling of the MM subsystem by MD simulations, the temperature was maintained at 298.15 K. Each FEP calculation (for each geometry of the QM subsystem along the minimum-energy path) consisted of 50 ps of equilibration and 300 ps of sampling.
The MD simulations and QM/MM-FE calculations were performed on a supercomputer (e.g. IBM X-series Cluster with 340 nodes or 1,360 processors) at University of Kentucky Computer Center. The other less-time-consuming modeling and computations were carried out on SGI Fuel workstations and a 34-processor IBM x335 Linux cluster in our own lab.
Results and Discussion
Determination of the nucleophilic attacking water molecule
A water molecule is involved and acts as a nucleophile in the spontaneous reactivation of sarin-phosphorylated wild-type BChE. Such a water molecule needs to be close to the phosphorus atom of sarin in order to perform the nucleophilic attack. From the stereochemical prospective, this water molecule was proposed to be located on the opposite side of the enzyme-phosphorus bond since the enzyme molecule (specifically the oxygen atom of S198 side chain) is the leaving group of the reactivation reaction.(19) As shown in Figure 2, a water molecule (denoted by WAT1) was found at such location in three crystal structures, i.e., the diethylphosphorylated BChE (PDB code 1XLW),(65) and the non-aged forms of human BChE inhibited by tabun (PDB code 3DJY)(66) and by tabun analogue TA5 (PDB code 2WIJ)(67). However, WAT1 was too far away from the phosphorus atom to initiate the nucleophilic attack. The respective distances between the oxygen atom of WAT1 and the phosphorus atom of the OP were 5.78 Å, 5.22 Å, and 5.54 Å in these crystal structures. Considering the significant effects of the protein environment, the true nucleophilic water molecule may not necessarily sit well on the opposite side of the enzyme-phosphorus bond to initiate a linear nucleophilic attack on the phosphorous atom. Another water molecule close to the phosphorous atom might be more appropriate to perform a non-linear nucleophilic attack on the phosphorous atom. By carefully checking the surrounding environment of the phosphorus atom, we noted that another water molecule (denoted by WAT2) was located on the opposite side of the ethoxy group toward the acyl pocket in the crystal structures of the above-mentioned diethylphosphorylated BChE. The distance of the phosphorus atom with the oxygen of WAT2 was 3.65 Å, which was 2.13 Å shorter than the corresponding distance with the oxygen of WAT1. In addition, there were two hydrogen bonds between WAT2 and the side chains of two essential residues, E197 and H438. The distances of the oxygen atom of WAT2 with the oxygen atom of E197 side chain and the nitrogen (epsilon-N) atom of H438 side chain were 2.82 and 2.90 Å, respectively, indicating that WAT2 was strongly stabilized by these two hydrogen bonds. Clearly, a water molecule at the location of WAT2 is favored to serve as the nucleophile to initiate the nucleophilic attack on the phosphorous atom.
Figure 2.
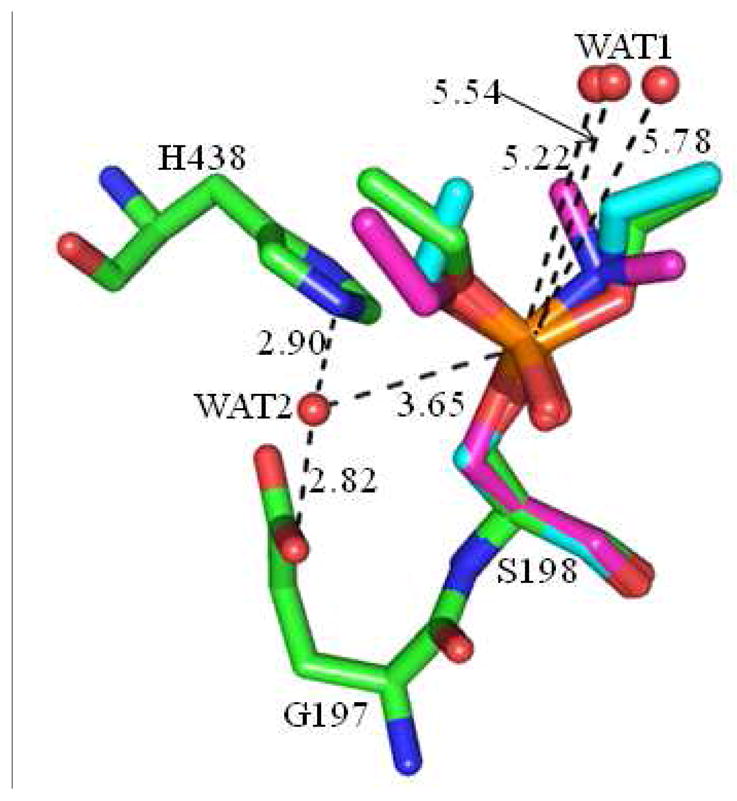
Superimposed conformations of two different water locations in the active sites of the three crystal structures of human BChE phosphorylated by different organophosphorus compounds with PDB codes of 1XLW, 3DJY, and 2WIJ, and the carbon atoms of organophosphorus compounds are colored in green, magenta, and cyan, respectively. The distances are given in Å.
To further identify which water molecule, WAT1 or WAT2, should be the nucleophile, a ~2 ns MD simulation was performed on sarin-phosphorylated wild-type human BChE. At the end of the MD simulation, due to the dynamic distribution of water molecules, another water molecule (denoted by WAT532) was observed to locate on the original position of WAT2 and WAT532 formed hydrogen bonds with the carboxylate group of E197 side chain and the nitrogen (epsilon- N) atom of H438 side chain. The traces of simulated key distances associated with WAT532 are given in Figure 3B, including the distance (D1) between the oxygen atom of WAT532 and the phosphorus atom as well as distances D2 and D3 reflecting the hydrogen bonds of WAT532 with H438 and E197, respectively. The average value of D1 is 3.67 ± 0.28 Å, suggesting that WAT532 is appropriate to attack the phosphorus atom. The average values of D2 and D3 are 2.00 ± 0.19 Å and 1.72 ± 0.12 Å, respectively, indicating that WAT532 has strong hydrogen- bonding interactions with E197 and H438. In addition, through the hydrogen bond between WAT532 and H438, it is possible that H438 accepts a proton from WAT532 and serves as a general base to facilitate the nucleophilic attack on the phosphorus atom by WAT532. Based on the results and discussion above, WAT532 is determined to be the nucleophile to initiate the spontaneous reactivation reaction of sarin-phosphorylated wild-type human BChE.
Figure 3.

Key internuclear distances (A) (D1 to D3) vs the simulation time in the molecular dynamics simulation on sarin-phosphorylated (B) wild-type enzyme and (C) G117H mutant of human BChE.
Spontaneous reactivation reactions of sarin-phosphorylated wild-type enzyme and G117H mutant of human BChE
Reaction pathway for wild-type human BChE
The QM/MM reaction-coordinate calculations at the B3LYP/6-31G*:AMBER level revealed that the spontaneous reactivation reaction consists of two reaction steps. The first step is the nucleophilic attack on the phosphorus atom by the oxygen atom of WAT532 and the second step is the decomposition of the pentacoordinate intermediate, resulting in leaving of the oxygen of S198 side chain and the generation of a nontoxic product, i.e. O-isopropyl methylphosphonic acid (IMPA). The schematic representation of the pathway is shown in Figure 4. The optimized geometries of the reactant, transition states, intermediate, and product are depicted in Figure 5. Below, we discuss each step in detail.
Figure 4.

Schematic representation of the pathway for spontaneous reactivation reaction of sarin-phosphorylated wild-type human BChE. The dashed lines in the transition states represent the covalent bonds that form or break during the reaction steps.
Figure 5.

The optimized geometries (A–E) and free energy profile (F) of spontaneous reactivation reaction of sarin-phosphorylated wild-type human BChE. The QM atoms are represented as balls and sticks. Carbon, oxygen, nitrogen, phosphorus, and hydrogen atoms are colored in green, red, blue, orange, and white, respectively. The key distances are given in Å. The figures below are represented using the same method.
In the nucleophilic attack process, the oxygen atom (Oω) of WAT532 gradually approaches and attacks the phosphorus atom (P), while a hydrogen atom (Hω) of WAT532 gradually transfers to the nitrogen atom (Nε) of the H438 side chain. Since this reaction step involves the formation of both the P–Oω and Nε–Hω bonds and the breaking of the Oω–Hω bond, the distances between the P and Oω atoms (RP–Oω), between the Nε and Hω atoms (RNε– Hω), and between the Oω and Hω atoms (ROω–Hω) reflect the nature of the current chemical reaction step and, thus, the reaction coordinate was set as ROω–Hω – RP–Oω – RNε– Hω.
As shown in Figure 5, ERWT has a tetrahedrally coordinated phosphorus conformation. The distances between the P and Oω atoms is 3.49 Å in ERWT and 1.87 Å in INT1WT, indicating the formation of P–Oω bond during the reaction process via transition state TS1WT. The distance between the Nε and Hω atoms is 1.95 Å in ERWT and 1.07 Å in INT1WT, along with the distance between the Oω and Hω atoms being elongated to be 1.64 Å in INT1WT, illustrating the completion of the proton (Hω) transfer from WAT532 to H438. INT1WT is a pentacoordinated phosphorus intermediate and it has a trigonal bipyramidal conformation with the Oω atom and methyl group of sarin in the axial orientation. As a general base, H438 accepts a proton (Hω) from WAT532 to increase the nucleophilicity of WAT532 and facilitates the nucleophilic attack on the phosphorus atom, resulting in the protonated H438 in INT1WT. Regarding the role of E325, which is one of the three members of the catalytic triad in human BChE, the optimized distances between the Hδ and Oδ atoms in all states indicate that no proton transfer takes place between H438 and E325. However, a strong Nδ–Hδ·Oδ hydrogen bond is formed between the side chains of these two residues and this hydrogen-bond distance is shortened to be 1.62 Å in INT1WT from 1.73 Å in ERWT, suggesting that the hydrogen bonding interaction becomes stronger during the reaction process. The stronger hydrogen bonding interaction is useful to stabilize the developed positive charge of H438 in TS1WT and INT1WT.
During the decomposition process of the pentacoordinate intermediate, the P–Oγ bond length gradually increases while the proton (Hω) in the protonated H438 gradually transfers to the Oγ atom of S198. Since this reaction step consists of the cleavages of the P–Oγ and Nε–Hω bonds and the bond formation between the Oγ and Hω atoms, the distances between the P and Oγ atoms (RP– Oγ), between the Nε and Hω atoms (RNε– Hω), and between the Oγ and Hω atoms (ROγ –Hω) reflect the nature of the current chemical reaction step and, thus, the reaction coordinate was set as RP–Oγ + RNε– Hω − ROγ –Hω.
As shown in Figure 5, the P–Oγ bond length is 1.75 Å in INT1WT and 2.60 Å in EPWT, and the P atom is tetrahedrally coordinated in EPWT, indicating the cleavage of the P–Oγ bond and the generation of the nontoxic IMPA. The Nε–Hω bond length is 1.07 Å in INT1WT and 1.53 Å in EPWT, and the distance between Oγ and Hω atoms is 2.46 Å in INT1WT and 1.02 Å in EPWT, showing that the proton (Hω) has been transferred from the nitrogen atom of H438 side chain to the Oγ atom of S198 side chain. Therefore, the enzyme activity is restored. Similar to the role of E325 in the first reaction step, E325 is also not involved in formation/breaking of any covalent bond during the decomposition reaction (second reaction step) as the Nδ–Hδ bond length is almost unchanged, but the Oδ atom of E325 side chain forms a strong Nδ–Hδ·Oδ hydrogen bond with H438. The distance of this hydrogen bond in EPWT is shorter than that in INT1WT, and this is probably caused by the rotation of H438 such that it facilitates the proton transfer from H438 to S198.
As well known, the oxyanion hole consists of the backbone NH groups of G116, G117, and A199. It is interesting to know the catalytic role of the oxyanion hole during the reaction process. Based on the geometries shown in Figure 5, the phosphoryl oxygen (Oα) atom forms strong N–H·O hydrogen bonds with the backbone NH groups of G116, G117, and A199 during the whole reaction process and these three hydrogen bonds help to stabilize the developed negative charge of the Oα atom of the sarin-phosphorylated S198 in TS1WT and INT1WT.
On the basis of the QM/MM-optimized geometries at the B3LYP/6-31G*:AMBER level, the QM/MM single-point energy calculations were performed at the MP2/6-31+G*:AMBER level for each geometry along the minimum-energy path for spontaneous reactivation reaction of sarin-phosphorylated human BChE. For each geometry along the minimum-energy path, the ESP charges determined in the QM subsystem of the QM/MM single-point energy calculation were used in further FEP calculations for estimating the free energy changes along the reaction path. The zero-point and thermal corrections for the QM subsystem are included in the free energy profile determined by the QM/MM-FE calculations and shown in Figure 5F. As seen in Figure 5F, the first reaction step, i.e. the nucleophilic attack on the phosphorus atom by the oxygen of WAT532, is the rate-determining step with the free energy barrier being 30.2 kcal/mol. The calculated free energy barrier of 30.2 kcal/mol is consistent with the available experimental kinetic data(22) that revealed no significant reaction or kcat < 5 × 10−5 min−1 (within the experimental error). So, it has been known that kcat < 5 × 10−5 min−1 although the exact kcat value remains to be determined. According to the conventional transition-state theory,(68) when kcat < 5 × 10−5 min−1, the activation free energy should be at least higher than 25.7 kcal/mol.
Reaction pathway for the G117H mutant of human BChE
After the G117H mutant was constructed by using the LEaP module in Amber 9 package, a ~2 ns MD simulation was performed to identify the conformation of sarin-phosphorylated G117H mutant of human BChE. Regarding the possibility that H117 acts as a general base, there is no water molecule observed close to H117 and sarin-phosphorylated S198. The H117 side chain is very close to the O-isopropyl group of sarin and there is clearly not enough space between the sarin-phosphorylated S198 and H117 to accommodate a water molecule, showing that H117 cannot serve as a general base for the reactivation reaction. Similar to the reaction pathway for wild-type human BChE, a water molecule (denoted by WAT532) locates on the position of WAT2 and forms two hydrogen bonds with the nitrogen (epsilon-N) atom of the H438 side chain and the carboxyl group of E197 in the sarin-phosphorylated G117H mutant. The traces of simulated key distances associated with WAT532, including the distance (D1) between the oxygen atom of WAT532 and the phosphorus atom of sarin as well as the hydrogen bonding distances (D2 and D3) of WAT532 with H438 and E197 are shown in Figure 3C. The average value of D1 is 3.68 ± 0.20 Å, suggesting that WAT532 is in a position appropriate to attack the phosphorus atom. The average values of D2 and D3 are 2.00 ± 0.16 Å and 1.78 ± 0.14 Å, respectively, indicating strong hydrogen bonding interactions with both H438 and E197. Through the hydrogen bond between WAT532 and H438, it is very possible that H438 serves as a general base to accept a proton from WAT532 and facilitate the nucleophilic attack on the phosphorus atom by the oxygen of WAT532.
The reaction pathway for the spontaneous reactivation of sarin-phosphorylated G117H mutant of human BChE has been investigated by the QM/MM reaction-coordinate calculations at the B3LYP/6-31G*:AMBER level. The results indicate that it follows a similar reaction pathway as that for the spontaneous reactivation of sarin-phosphorylated wild-type human BChE shown in Figure 4. The whole reaction process consists of two steps, i.e. the nucleophilic attack and the decomposition of the pentacoordinated phosphorus intermediate. For each reaction step, the same reaction coordinate used to represent the reaction for the wild-type BChE was used to represent the reaction for the G117H mutant. The optimized geometries of the reactant, transition states, intermediate, and product are depicted in Figure 6.
Figure 6.

The optimized geometries (A–E) and free energy profile (F) of spontaneous reactivation reaction of sarin-phosphorylated G117H mutant of human BChE.
In the first reaction step, the oxygen atom (Oω) of WAT532 gradually approaches and attacks the phosphorus atom (P) of sarin, while the hydrogen atom (Hω) of WAT532 gradually transfers to the nitrogen atom (Nε) of the H438 side chain. As seen in Figure 6, the distance between the P and Oω atoms is 3.21 Å in ERG117H and 1.89 Å in INT1G117H, indicating the formation of the P– Oω bond during the reaction process via transition state TS1G117H. The distance between the Nε and Hω atoms is 1.90 Å in ERG117H and 1.09 Å in INT1G117H, along with the distance between the Oω and Hω atoms being elongated to 1.52 Å in INT1G117H, revealing the proton (Hω) transfer from WAT532 to H438. During the reaction process, the distance between the Hδ and Oδ atoms is almost unchanged, indicating that there is no proton transfer between H438 and E325, but the strong Nδ–Hδ·Oδ hydrogen bond between H438 and E325 helps to stabilize the positively charged H438 in TS1G117H and INT1G117H.
In the second reaction step, the P–Oγ bond length gradually increases while the proton (Hω) in the protonated H438 gradually transfers to the Oγ atom of S198. As seen in Figure 6, in INT1G117H, the distance between the Oγ and Hω atoms is 2.59 Å, indicating a very weak Nε– Hω·Oγ hydrogen bond. This distance is 0.13 Å longer than that in INT1WT, suggesting that it is not as favorable as that in the wild-type human BChE for the porton (Hω) transfer from H438 to S198. During the reaction process via TS2G117H, the P–Oγ bond is elongated and the imidazole group of the protonated H438 residue is rotated to make the Hω atom closer to the Oγ atom. The distance between the Oγ and Hω atoms is shortened to 1.69 Å in TS2G117H. This change may facilitate the proton (Hω) transfer from the protonated H438 to S198. At the same time, an important angle between the Oγ, P, and Oω atoms (∠OγPOω) increases to 132.2° in TS2G117H from 110.7° in INT1G117H. This spatial reorganization results in the Oγ atom being in the axial orientation which is favored for the breaking of the P–Oγ bond. Finally, in EPG117H, the P–Oγ bond is broken with the distance between the P and Oγ atoms being 2.67 Å. The phosphorus atom is tetrahedrally coordinated, indicating that the nontoxic product, i.e. IMPA, is released. The angle ∠OγPOω equals to 160.1° in EPG117H, meaning that the Oγ and Oω are located in the axial orientation before the complete breaking of the P–Oγ bond. The distances between the Nε and Hω atoms and between the Oγ and Hω atoms are 1.49 Å and 1.05 Å, respectively, showing that the proton (Hω) transfer from the H438 side chain to the S198 side chain has been completed. During the reaction process, the strong Nδ–Hδ·Oδ hydrogen bond remains between H438 and E325 and there is no proton transfer between them.
It should be pointed out that the spatial reorganization observed for the G117H mutant has not been observed in the corresponding step of the reaction for the wild-type human BChE, which reveals a remarkable difference between the reactivation reactions of the sarin-phosphorylated wild-type enzyme and G117H mutant of human BChE.
Another significant difference can be noted in the hydrogen-bonding interaction between the phosphoryl oxygen (Oα) and the oxyanion hole during the reaction process. There are three N– H·O hydrogen bonds between the Oα atom and the NH groups of the three residues (G116, H117, and A199) forming the oxyanion hole during the reaction process for the G117H mutant, which is similar to the reaction process for the wild-type human BChE. However, there are relatively larger changes in the H·Oα distances of these hydrogen bonds, particularly those with G116 and A199, in the reaction of the G117H mutant compared to that of the wild-type human BChE. For example, the H·Oα distance with the NH group of G116 backbone increases from 2.22 Å in INT1G117H to 2.67 Å in TS2G117H, while the H·Oα distance with the NH group of A199 backbone decreases from 2.25 Å in INT1G117H to 2.09 Å in TS2G117H. In comparison, there are little changes (within 0.02 Å) in the corresponding distances in the reaction of the wild-type human BChE from INT1WT to TS2WT.
Further, the free energy profile depicted in Figure 6F reveals that the second reaction step is the rate-determining step for the G117H mutant and the overall free energy barrier is the free energy change from ERG117H to TS2G117H. The calculated overall free energy barrier is 23.9 kcal/mol. The calculated free energy barrier of 23.9 kcal/mol is good agreement with the available experimental kinetic data (kcat = 8.7 × 10−5 s−1).(21–22) According to the conventional transition-state theory,(68) when kcat = 8.7 × 10−5 s−1, the activation free energy should be 23.0 kcal/mol which is close to our computational value of 23.9 kcal/mol.
Effects of the G117H mutation
On the basis of the results discussed above, it is evident that the spontaneous reactivation reactions of the sarin-phosphorylated wild-type enzyme and G117H mutant of human BChE are all catalyzed by H438 which acts as a general base. The G117H mutation cannot introduce a second nucleophilic center into the active site of human BChE which was supposed to activate a water molecule to initiate the spontaneous reactivation reaction. Our results do not support the aforementioned hypothesis(19) that the H117 residue in the G117H mutant acts as a general base to catalyze the OP hydrolysis. A careful comparison between the uncovered spontaneous reactivation mechanisms of sarin-phosphorylated wild-type enzyme and G117H mutant of human BChE reveals some remarkable differences caused by the G117H mutation, as discussed below.
The G117H mutation-caused change in the oxyanion hole interacting with the phosphoryl oxygen
As discussed above, the oxyanion hole consists of the backbone NH groups of G116, G117 (or H117 in the G117H mutant), and A199. As seen in Figure 5, the oxyanion hole in wild-type human BChE forms three strong hydrogen bonds with the phosphoryl oxygen and the average values of the H·Oα distances of these three hydrogen bonds with G116, G117, and A199 in all of the optimized geometries (including ERWT, TS1WT, INT1WT, TS2WT, and EPWT) in Figure 5 are 1.77 ± 0.03 Å, 1.90 ± 0.07 Å, and 1.98 ± 0.05 Å, respectively, indicating that these hydrogen bonds are rather strong and stable during the reaction process. Similarly, for the G117H mutant of human BChE shown in Figure 6, the oxyanion hole also forms three hydrogen bonds with the phosphoryl oxygen. However, the average values of the H·Oα distances of these three hydrogen bonds with G116, H117, and A199 in all of the optimized geometries in Figure 6 are 2.44 ± 0.21 Å, 1.82 ± 0.02 Å, and 2.19 ± 0.09 Å, respectively. Clearly, the G117H mutation makes the hydrogen bond of the phosphoryl oxygen with H117 in the G117H mutant stronger and more stable than that with G117 in the wild-type human BChE, while making the other two hydrogen bonds (with G117 and A199) in the G117H mutant weaker and less stable than those in the wild-type human BChE. When G117 is replaced by H117, the local environment of the oxyanion hole is changed by the larger side chain of H117 and the three NH groups of the oxyanion hole are redistributed, resulting in the changes of the hydrogen bonds between the oxyanion hole and the phosphoryl oxygen. Thus, the oxyanion hole is skewed by the G117H mutation.
The G117H mutation-caused change in the conformation of the sarin-phosphorylated S198
The skewed oxyanion hole makes some change in the conformation of the sarinphosphorylated S198. Depicted in Figure 7 are the superimposed conformations of ERWT and ERG117H, showing that the O–isopropyl group of sarin in the G117H mutant of human BChE rotates away from the side chain of H117. This crucial structural difference caused by the G117H mutation makes the phosphorus atom in the G117H mutant more open to the nucleophilic water molecule and, thus, decreases the steric hindrance for the nuclephilic attack. According to the structures of ERWT and ERG117H depicted in Figures 5 and 6, the distances between the P and Oω atoms are 3.49 Å and 3.21 Å, respectively, in ERWT and ERG117H, illustrating that the water molecule in the G117H mutant is closer to the phosphorus atom compared to the wild-type. Hence, this conformational change caused by the G117H mutation facilitates the nucleophilic attack on the phosphorus atom of sarin by the water molecule.
Figure 7.
The superimposed conformations of ERWT and ERG117H. The wild-type and G117H mutant of human BChE are colored in green and orange, respectively.
The G117H mutation-caused change in the free energy profile
Based on the free energy profile for the spontaneous reactivation reaction of sarin-phosphorylated wild-type human BChE given in Figure 5F, the relative free energy of TS1WT is 30.2 kcal/mol which is 0.9 kcal/mol higher than that of TS2WT, meaning that the first reaction step is the rate-determining step and the overall free energy barrier is 30.2 kcal/mol for the reaction of the wild-type enzyme. However, after the G117H mutation, the relative free energy of TS1G117H, shown in Figure 6F, becomes 22.0 kcal/mol which is 1.9 kcal/mol lower than that of TS2G117H, meaning that the second reaction step is the rate-determining step and that the overall free energy barrier is 23.9 kcal/mol for the reaction of the G117H mutant. Apparently, the G117H mutation remarkably changes the rate-determining step from the first reaction step to the second reaction step and effectively decreases the overall free energy barrier of the spontaneous reactivation reaction of sarin-phosphorylated human BChE.
All of the geometric and energetic results discussed above indicate that the role of the G117H mutation is to skew the oxyanion hole and to make the phosphorus atom more open to the nucleophilic attack by a water molecule so that the overall free energy barrier for the spontaneous reactivation reaction of sarin-phosphorylated human BChE can be decreased effectively. For comparison, we also examined the G117V and G117K mutants for their ER structures (ERG117V and ERG117K) by using the same computational protocol, i.e. the MD simulation for 2 ns followed by the QM/MM geometry optimization, for each of the mutants. It turned out that in the QM/MM-optimized ERG117V and ERG117K structures (see Figures 8), the distances between the P and Oω atoms are 3.84 Å and 4.32 Å, respectively. The P·Oω distances in both the ERG117V (3.84 Å) and ERG117K (4.32 Å) structures are significantly longer than that in the ERWT structure (3.49 Å). So, whereas the G117H mutation made the P·Oω distance shorter, the G117V and G117K mutations made the P·Oω distance longer compared to that of 3.21 Å in ERWT. Based on these computational data, one may reasonably expect that the G117V and G117K mutations would increase the free energy barrier, whereas the G117H mutation decreased the free energy barrier for the spontaneous reactivation reaction of sarin-phosphorylated human BChE. In fact, the G117K mutant was examined in a previously reported experimental study,(22) and no spontaneous reactivation reaction was observed for the G117K mutant. The experimental observation for the G117K mutant was qualitatively consistent with our computational insights.
Figure 8.

The QM/MM-optimized geometries of ERG117V and ERG117K.
Conclusion
Spontaneous reactivation reactions of the sarin-phosphorylated wild-type enzyme and G117H mutant of human BChE have been studied by using the first-principles QM/MM-FE calculations. For wild-type human BChE, the attacking water molecule is located on the opposite side of methyl group toward the acyl pocket of human BChE. The water molecule is stabilized in this location by E197 and H438 through strong hydrogen bonds. The reaction pathway consists of two steps. The first step is the nucleophilic attack on the phosphorus atom of sarin by the oxygen atom of water molecule, along with a proton transfer from the water molecule to the H438 side chain. The second reaction step is the decomposition of the pentacoordinate intermediate, resulting in the active form of the human BChE. According to the calculated free energy profile, the first reaction step is the rate-determining step of the reaction with the calculated free energy barrier being 30.2 kcal/mol, which is consistent with the reported experimental observation that the rate constant (kcat) should be at least smaller than 5×10−5 min−1 (or the activation free energy should be at least higher than 25.7 kcal/mol).
For the G117H mutant of human BChE, the attacking water molecule is located on the same location as that of wild-type human BChE, and no water molecule can be close to H117, demonstrating that H117 cannot act as a general base in the spontaneous reactivation. The reaction of the G117H mutant also consists of two reaction steps. The first step is similar to that in spontaneous reactivation reaction of sarin-phosphorylated wild-type of human BChE. However, the second step is remarkably different. A spatial reorganization, which is not involved in the second step of the reaction of wild-type human BChE, is involved in the second step of the reaction of the G117H mutant. The spatial reorganization results in the oxygen atom of Ser198 side chain sitting in the axial orientation and being close to the transferring proton in the protonated H438, thus facilitating the leaving of the oxygen of S198 side chain from the phosphorus atom. The second reaction step is the rate-determining step of the entire reaction of the G117H mutant. The overall free energy barrier is the free energy change (23.9 kcal/mol) from the initial reactant ERG117H to transition state TS2G117H. The calculated overall free energy barrier of 23.9 kcal/mol is in good agreement with the experimentally derived activation free energy of 23.0 kcal/mol (based on the reported experimental rate constant of 8.7×10−5 s−1).
For both the wild-type human BChE and the G117H mutant, E325 serves as one of the three members of the catalytic triad to stabilize the developed positive charge on H438, but there is no proton transfer between H438 and E325. The oxyanion hole forms three hydrogen bonds with the phosphoryl oxygen to stabilize the developed negative charge on the phosphoryl oxygen during the reactivation reaction.
Further, concerning how the G117H mutation accelerates the reactivation reaction process, the obtained structural and energetic data reveal the specific role of the G117H mutation. It has been demonstrated that the G117H mutation does not introduce a second nucleophilic center into the active site of human BChE which was supposed to activate the water molecule to initiate spontaneous reactivation reaction. Instead, the G117H mutation skews the oxyanion hole and makes the phosphorus atom more open to the nucleophilic water molecule, resulting in the dramatically decreased overall free energy barrier of the reactivation reaction. In comparison, the G117V and G117K mutations make the phosphorus atom farther from the nucleophilic water molecule.
The obtained mechanistic insights provide a valuable guide to the rational design of new mutants of human BChE with an improved catalytic activity against sarin. Particularly, the future work should be focused on computational identification of the new mutants that can more effectively stabilize the transition state associated with the second reaction step.
In general, this computational study demonstrates how a single amino-acid mutation on an enzyme can dramatically change the rate-determining step of the enzymatic reaction from one reaction step to another and considerably improve the activity against a substrate of interest. The obtained novel mechanistic insights should be interesting to researchers in a variety of fields of chemistry and related disciplines, such as biochemistry, neurochemistry, phosphorus chemistry, agricultural chemistry, computational chemistry, chemical defense, human health sciences, catalyst design, drug design and discovery, and protein engineering design.
Acknowledgments
Funding. This work was supported in part by the NIH (grants R01 DA013930, R01 DA032910, and R01 DA025100), NSF (grant CHE-1111761), and NSFC (grant No.21102050).
The entire research was performed at the University of Kentucky. The authors acknowledge the Center for Computational Sciences (CCS) at University of Kentucky for supercomputing time on an IBM X-series Cluster with 340 nodes or 1,360 processors and a Dell X-series Cluster with 384 nodes or 4,768 processors.
References
- 1.MacPhee-Quigley K, Taylor P, Taylor S. Primary structures of the catalytic subunits from two molecular forms of acetylcholinesterase. A comparison of NH2-terminal and active center sequences. J Biol Chem. 1985;260:12185–12189. [PubMed] [Google Scholar]
- 2.Marrs TC. Organophosphate poisoning. Pharmacol Ther. 1993;58:51–66. doi: 10.1016/0163-7258(93)90066-m. [DOI] [PubMed] [Google Scholar]
- 3.Taylor P, Radic Z, Hosea NA, Camp S, Marchot P, Berman HA. Structural bases for the specificity of cholinesterase catalysis and inhibition. Toxicol Lett. 1995;82–83:453–458. doi: 10.1016/0378-4274(95)03575-3. [DOI] [PubMed] [Google Scholar]
- 4.Black RM, Clarke RJ, Read RW, Reid MT. Application of gas chromatography-mass spectrometry and gas chromatography-tandem mass spectrometry to the analysis of chemical warfare samples, found to contain residues of the nerve agent sarin, sulphur mustard and their degradation products. J Chromatogr A. 1994;662:301–321. doi: 10.1016/0021-9673(94)80518-0. [DOI] [PubMed] [Google Scholar]
- 5.Fidder A, Hulst AG, Noort D, de Ruiter R, van der Schans MJ, Benschop HP, Langenberg JP. Retrospective Detection of Exposure to Organophosphorus Anti-Cholinesterases: Mass Spectrometric Analysis of Phosphylated Human Butyrylcholinesterase. Chem Res Toxicol. 2002;15:582–590. doi: 10.1021/tx0101806. [DOI] [PubMed] [Google Scholar]
- 6.Nagao M, Takatori T, Matsuda Y, Nakajima M, Iwase H, Iwadate K. Definitive evidence for the acute sarin poisoning diagnosis in the Tokyo subway. Toxicol Appl Pharmacol. 1997;144:198–203. doi: 10.1006/taap.1997.8110. [DOI] [PubMed] [Google Scholar]
- 7.Noort D, Hulst AG, Platenburg DH, Polhuijs M, Benschop HP. Quantitative analysis of O-isopropyl methylphosphonic acid in serum samples of Japanese citizens allegedly exposed to sarin: estimation of internal dosage. Arch Toxicol. 1998;72:671–675. doi: 10.1007/s002040050559. [DOI] [PubMed] [Google Scholar]
- 8.Polhuijs M, Langenberg JP, Benschop HP. New method for retrospective detection of exposure to organophosphorus anticholinesterases: Application to alleged sarin victims of Japanese terrorists. Toxicol Appl Pharm. 1997;146:156–161. doi: 10.1006/taap.1997.8243. [DOI] [PubMed] [Google Scholar]
- 9.Tsuchihashi H, Katagi M, Nishikawa M, Tatsuno M. Identification of metabolites of nerve agent VX in serum collected from a victim. J Anal Toxicol. 1998;22:383–388. doi: 10.1093/jat/22.5.383. [DOI] [PubMed] [Google Scholar]
- 10.Broomfield CA, Kirby SD. Progress on the road to new nerve agent treatments. J Appl Toxicol. 2001;21(Suppl 1):S43–46. doi: 10.1002/jat.804. [DOI] [PubMed] [Google Scholar]
- 11.Koca J, Zhan CG, Rittenhouse RC, Ornstein RL. Coordination number of zinc ions in the phosphotriesterase active site by molecular dynamics and quantum mechanics. J Comput Chem. 2003;24:368–378. doi: 10.1002/jcc.10217. [DOI] [PubMed] [Google Scholar]
- 12.Zheng F, Zhan CG, Ornstein RL. Theoretical studies of reaction pathways and energy barriers for alkaline hydrolysis of phosphotriesterase substrates paraoxon and related toxic phosphofluoridate nerve agents. J Chem Soc-Perkin Transact. 2001;2:2355–2363. [Google Scholar]
- 13.Koca J, Zhan CG, Rittenhouse RC, Ornstein RL. Mobility of the active site bound paraoxon and sarin in zinc-phosphotriesterase by molecular dynamics simulation and quantum chemical calculation. J Am Chem Soc. 2001;123:817–826. doi: 10.1021/ja000439r. [DOI] [PubMed] [Google Scholar]
- 14.Zhan CG, de Souza ON, Rittenhouse R, Ornstein RL. Determination of two structural forms of catalytic bridging ligand in zinc-phosphotriesterase by molecular dynamics simulation and quantum chemical calculation. J Am Chem Soc. 1999;121:7279–7282. doi: 10.1021/ja000439r. [DOI] [PubMed] [Google Scholar]
- 15.Zheng F, Zhan CG, Ornstein RL. Theoretical Determination of Two Structural Forms of the Active Site in Cadmium-Containing Phosphotriesterases. J Phys Chem B. 2001;106:717–722. [Google Scholar]
- 16.Broomfield CA, Maxwell DM, Solana RP, Castro CA, Finger AV, Lenz DE. Protection by butyrylcholinesterase against organophosphorus poisoning in nonhuman primates. J Pharmacol Exp Ther. 1991;259:633–638. [PubMed] [Google Scholar]
- 17.Raveh L, Grauer E, Grunwald J, Cohen E, Ashani Y. The stoichiometry of protection against soman and VX toxicity in monkeys pretreated with human butyrylcholinesterase. Toxicol Appl Pharmacol. 1997;145:43–53. doi: 10.1006/taap.1997.8160. [DOI] [PubMed] [Google Scholar]
- 18.Gorelick DA. Enhancing cocaine metabolism with butyrylcholinesterase as a treatment strategy. Drug Alcohol Depend. 1997;48:159–165. doi: 10.1016/s0376-8716(97)00119-1. [DOI] [PubMed] [Google Scholar]
- 19.Jarv J. Stereochemical Aspects of Cholinesterase Catalysis. Bioorg Chem. 1984;12:259–278. [Google Scholar]
- 20.Lockridge O, Blong RM, Masson P, Froment MT, Millard CB, Broomfield CA. A single amino acid substitution, Gly117His, confers phosphotriesterase (organophosphorus acid anhydride hydrolase) activity on human butyrylcholinesterase. Biochemistry. 1997;36:786–795. doi: 10.1021/bi961412g. [DOI] [PubMed] [Google Scholar]
- 21.Millard CB, Lockridge O, Broomfield CA. Design and expression of organophosphorus acid anhydride hydrolase activity in human butyrylcholinesterase. Biochemistry. 1995;34:15925–15933. doi: 10.1021/bi00049a007. [DOI] [PubMed] [Google Scholar]
- 22.Broomfield CA, Lockridge O, Millard CB. Protein engineering of a human enzyme that hydrolyzes V and G nerve agents: design, construction and characterization. Chem Biol Interact 119–. 1999;120:413–418. doi: 10.1016/s0009-2797(99)00053-8. [DOI] [PubMed] [Google Scholar]
- 23.Amitay M, Shurki A. The structure of G117H mutant of butyrylcholinesterase: nerve agents scavenger. Proteins. 2009;77:370–377. doi: 10.1002/prot.22442. [DOI] [PubMed] [Google Scholar]
- 24.Nachon F, Carletti E, Wandhammer M, Nicolet Y, Schopfer LM, Masson P, Lockridge O. X-ray crystallographic snapshots of reaction intermediates in the G117H mutant of human butyrylcholinesterase, a nerve agent target engineered into a catalytic bioscavenger. Biochem J. 2011;434:73–82. doi: 10.1042/BJ20101648. [DOI] [PubMed] [Google Scholar]
- 25.Albaret C, Masson P, Broomfield CA, EI Kaim L, Fortier PL. Mechanical aspects of the phosphotriesterase activity of human butyrylcholinesterase G117H mutant. In: BP, editor. Structure and Function of Cholinesterases and Related Proteins. Plenum Press; New York: 1998. pp. 399–405. [Google Scholar]
- 26.Kovach IM. Structure and dynamics of serine hydrolase-organophosphate adducts. J Enzyme Inhib. 1988;2:199–208. doi: 10.3109/14756368809040726. [DOI] [PubMed] [Google Scholar]
- 27.Qian N, Kovach IM. Key active site residues in the inhibition of acetylcholinesterases by soman. FEBS Lett. 1993;336:263–266. doi: 10.1016/0014-5793(93)80816-d. [DOI] [PubMed] [Google Scholar]
- 28.Hu P, Zhang YK. Catalytic mechanism and product specificity of the histone lysine methyltransferase SET7/9: An ab initio QM/MM-FE study with multiple initial structures. J Am Chem Soc. 2006;128:1272–1278. doi: 10.1021/ja056153+. [DOI] [PubMed] [Google Scholar]
- 29.Zhang YK. Improved pseudobonds for combined ab initio quantum mechanical/molecular mechanical methods. J Chem Phys. 2005;122:024114. doi: 10.1063/1.1834899. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YK, Lee TS, Yang WT. A pseudobond approach to combining quantum mechanical and molecular mechanical methods. J Chem Phys. 1999;110:46–54. [Google Scholar]
- 31.Zhang YK, Liu HY, Yang WT. Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio QM/MM potential energy surface. J Chem Phys. 2000;112:3483–3492. [Google Scholar]
- 32.Zhang YK. Pseudobond ab initio QM/MM approach and its applications to enzyme reactions. Theor Chem Acc. 2006;116:43–50. [Google Scholar]
- 33.Cheng Y, Zhang Y, McCammon JA. How Does the cAMP-Dependent Protein Kinase Catalyze the Phosphorylation Reaction: An ab Initio QM/MM Study. J Am Chem Soc. 2005;127:1553–1562. doi: 10.1021/ja0464084. [DOI] [PubMed] [Google Scholar]
- 34.Cheng YH, Zhang YK, McCammon JA. How does activation loop phosphorylation modulate catalytic activity in the cAMP-dependent protein kinase: A theoretical study. Protein Sci. 2006;15:672–683. doi: 10.1110/ps.051852306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cisneros GA, Wang M, Silinski P, Fitzgerald MC, Yang W. The protein backbone makes important contributions to 4-oxalocrotonate tautomerase enzyme catalysis: understanding from theory and experiment. Biochemistry. 2004;43:6885–6892. doi: 10.1021/bi049943p. [DOI] [PubMed] [Google Scholar]
- 36.Corminboeuf C, Hu P, Tuckerman ME, Zhang Y. Unexpected Deacetylation Mechanism Suggested by a Density Functional Theory QM/MM Study of Histone-Deacetylase-Like Protein. J Am Chem Soc. 2006;128:4530–4531. doi: 10.1021/ja0600882. [DOI] [PubMed] [Google Scholar]
- 37.Hu P, Wang S, Zhang Y. How Do SET-Domain Protein Lysine Methyltransferases Achieve the Methylation State Specificity? Revisited by Ab Initio QM/MM Molecular Dynamics Simulations. J Am Chem Soc. 2008;130:3806–3813. doi: 10.1021/ja075896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ke Z, Wang S, Xie D, Zhang Y. Born–Oppenheimer ab Initio QM/MM Molecular Dynamics Simulations of the Hydrolysis Reaction Catalyzed by Protein Arginine Deiminase 4. J Phys Chem B. 2009;113:16705–16710. doi: 10.1021/jp9080614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu H, Zhang Y, Yang W. How Is the Active Site of Enolase Organized To Catalyze Two Different Reaction Steps? J Am Chem Soc. 2000;122:6560–6570. [Google Scholar]
- 40.Liu J, Hamza A, Zhan CG. Fundamental Reaction Mechanism and Free Energy Profile for (−)-Cocaine Hydrolysis Catalyzed by Cocaine Esterase. J Am Chem Soc. 2009;131:11964–11975. doi: 10.1021/ja903990p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Zhang Y, Zhan CG. Reaction Pathway and Free-Energy Barrier for Reactivation of Dimethylphosphoryl-Inhibited Human Acetylcholinesterase. J Phys Chem B. 2009;113:16226–16236. doi: 10.1021/jp9055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Yu X, Hu P, Broyde S, Zhang Y. A Water-Mediated and Substrate-Assisted Catalytic Mechanism for Sulfolobus solfataricus DNA Polymerase IV. J Am Chem Soc. 2007;129:4731–4737. doi: 10.1021/ja068821c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Hu P, Zhang Y. Ab Initio Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulation of Enzyme Catalysis: The Case of Histone Lysine Methyltransferase SET7/9. J Phys Chem B. 2007;111:3758–3764. doi: 10.1021/jp067147i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu R, Hu P, Wang S, Cao Z, Zhang Y. Flexibility of Catalytic Zinc Coordination in Thermolysin and HDAC8: A Born–Oppenheimer ab Initio QM/MM Molecular Dynamics Study. J Chem Theory Comput. 2009;6:337–343. doi: 10.1021/ct9005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu R, Wang S, Zhou N, Cao Z, Zhang Y. A Proton-Shuttle Reaction Mechanism for Histone Deacetylase 8 and the Catalytic Role of Metal Ions. J Am Chem Soc. 2010;132:9471–9479. doi: 10.1021/ja103932d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao C, Zhang Y. Catalytic Mechanism and Metal Specificity of Bacterial Peptide Deformylase: A Density Functional Theory QM/MM Study. J Phys Chem B. 2007;111:6229–6235. doi: 10.1021/jp068657f. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Kua J, McCammon JA. Role of the Catalytic Triad and Oxyanion Hole in Acetylcholinesterase Catalysis: An ab initio QM/MM Study. J Am Chem Soc. 2002;124:10572–10577. doi: 10.1021/ja020243m. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Kua J, McCammon JA. Influence of Structural Fluctuation on Enzyme Reaction Energy Barriers in Combined Quantum Mechanical/Molecular Mechanical Studies. J Phys Chem B. 2003;107:4459–4463. [Google Scholar]
- 49.Zheng F, Yang W, Ko MC, Liu J, Cho H, Gao D, Tong M, Tai HH, Woods JH, Zhan CG. Most efficient cocaine hydrolase designed by virtual screening of transition states. J Am Chem Soc. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y, Wang S, Zhang Y. Catalytic Reaction Mechanism of Acetylcholinesterase Determined by Born–Oppenheimer Ab Initio QM/MM Molecular Dynamics Simulations. J Phys Chem B. 2010;114:8817–8825. doi: 10.1021/jp104258d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cisneros GA, Liu H, Zhang Y, Yang W. Ab Initio QM/MM Study Shows There Is No General Acid in the Reaction Catalyzed by 4-Oxalocrotonate Tautomerase. J Am Chem Soc. 2003;125:10384–10393. doi: 10.1021/ja029672a. [DOI] [PubMed] [Google Scholar]
- 52.Metanis N, Brik A, Dawson PE, Keinan E. Electrostatic Interactions Dominate the Catalytic Contribution of Arg39 in 4-Oxalocrotonate Tautomerase. J Am Chem Soc. 2004;126:12726–12727. doi: 10.1021/ja0463841. [DOI] [PubMed] [Google Scholar]
- 53.Poyner RR, Larsen TM, Wong SW, Reed GH. Functional and structural changes due to a serine to alanine mutation in the active-site flap of enolase. Arch Biochem Biophys. 2002;401:155–163. doi: 10.1016/S0003-9861(02)00024-3. [DOI] [PubMed] [Google Scholar]
- 54.Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J Biol Chem. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 55.Sali A, Blundell TL. Definition of general topological equivalence in protein structures. A procedure involving comparison of properties and relationships through simulated annealing and dynamic programming. J Mol Biol. 1990;212:403–428. doi: 10.1016/0022-2836(90)90134-8. [DOI] [PubMed] [Google Scholar]
- 56.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 57.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Case DADTA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. Amber 9. University of California; San Francisco: 2006. [Google Scholar]
- 59.Hörnberg A, Tunemalm A-K, Ekström F. Crystal Structures of Acetylcholinesterase in Complex with Organophosphorus Compounds Suggest that the Acyl Pocket Modulates the Aging Reaction by Precluding the Formation of the Trigonal Bipyramidal Transition State. Biochemistry. 2007;46:4815–4825. doi: 10.1021/bi0621361. [DOI] [PubMed] [Google Scholar]
- 60.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 61.Miyamoto S, Kollman PA. Settle - an Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 62.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of NAlkanes. J Chem Phys. 1977;23:327–341. [Google Scholar]
- 63.Frisch MJTGW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 64.Case DADTATE, Cheatham I, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER 8. University of California; San Francisco: 2004. [Google Scholar]
- 65.Nachon F, Asojo OA, Borgstahl GE, Masson P, Lockridge O. Role of water in aging of human butyrylcholinesterase inhibited by echothiophate: the crystal structure suggests two alternative mechanisms of aging. Biochemistry. 2005;44:1154–1162. doi: 10.1021/bi048238d. [DOI] [PubMed] [Google Scholar]
- 66.Carletti E, Li H, Li B, Ekstrom F, Nicolet Y, Loiodice M, Gillon E, Froment MT, Lockridge O, Schopfer LM, Masson P, Nachon F. Aging of cholinesterases phosphylated by tabun proceeds through O-dealkylation. J Am Chem Soc. 2008;130:16011–16020. doi: 10.1021/ja804941z. [DOI] [PubMed] [Google Scholar]
- 67.Carletti E, Aurbek N, Gillon E, Loiodice M, Nicolet Y, Fontecilla-Camps JC, Masson P, Thiermann H, Nachon F, Worek F. Structure-activity analysis of aging and reactivation of human butyrylcholinesterase inhibited by analogues of tabun. Biochem J. 2009;421:97–106. doi: 10.1042/BJ20090091. [DOI] [PubMed] [Google Scholar]
- 68.Truhlar DG, Garrett BC. Variational Transition State Theory. Ann Rev Phys Chem. 1984;35:159–189. [Google Scholar]