

Abstract
Hepatic iron overload has been associated classically with the genetic disorder hereditary hemochromatosis. More recently, it has become apparent that mild-to-moderate degrees of elevated hepatic iron stores observed in other liver diseases also have clinical relevance. The goal was to use a mouse model of dietary hepatic iron overload and isobaric tag for relative and absolute quantitation proteomics to identify, at a global level, differentially expressed proteins in livers from mice fed a control or 3,5,5-trimethyl-hexanoyl-ferrocene (TMHF) supplemented diet for 4 weeks. The expression of 74 proteins was altered by ≥ ±1.5-fold, showing that the effects of iron on the liver proteome were extensive. The top canonical pathway altered by TMHF treatment was the NF-E2–related factor 2 (NRF2–)–mediated oxidative stress response. Because of the long-standing association of elevated hepatic iron with oxidative stress, the remainder of the study was focused on NRF2. TMHF treatment upregulated 25 phase I/II and antioxidant proteins previously categorized as NRF2 target gene products. Immunoblot analyses showed that TMHF treatment increased the levels of glutathione S-transferase (GST) M1, GSTM4, glutamate-cysteine ligase (GCL) catalytic subunit, GCL modifier subunit, glutathione synthetase, glutathione reductase, heme oxygenase 1, epoxide hydrolase 1, and NAD(P)H dehydrogenase quinone 1. Immunofluorescence, carried out to determine the cellular localization of NRF2, showed that NRF2 was detected in the nucleus of hepatocytes from TMHF-treated mice and not from control mice. We conclude that elevated hepatic iron in a mouse model activates NRF2, a key regulator of the cellular response to oxidative stress.
Key Words: NRF2-mediated oxidative stress response, antioxidant proteins, phase I/II metabolism, glutathione metabolism.
Hepatic iron overload has been associated historically with genetic disorders, viral hepatitis, unusual dietary habits, and combinations of these conditions (Isom et al., 2009). The genetic disorder hereditary hemochromatosis (HH) classically associated with hepatic iron overload is characterized by increased intestinal iron absorption, progressive parenchymal iron overload, and liver injury. However, more recently it has become apparent that mild-to-moderate degrees of elevated hepatic iron stores observed in other liver diseases also have clinical relevance. These include chronic hepatitis C, chronic hepatitis B, alcoholic liver disease, and nonalcoholic steatohepatitis (Sumida et al., 2009). Although iron is essential for life as a crucial component of cytochromes, oxygen binding molecules, and a variety of enzymes, the reactive properties that make iron useful also lead to free iron being toxic. As such, mammalian species have evolved systems for iron homeostasis that include meticulous control of intestinal absorption of iron and regulation of iron trafficking. Under normal conditions, almost all iron in the body is sequestered in proteins. Although the liver is capable of iron uptake and export and the major depot for iron storage, hepatocytes are particularly susceptible to damage by iron overload.
Reactive oxygen species (ROS) are required for normal cell function; however, excessive ROS production is detrimental. Evidence exists supporting a role for ROS in normal cellular signal transduction; however, elevated ROS also have been associated with aging and human diseases including carcinogenesis, radiation and reperfusion injury, cardiovascular disorders, and neurodegenerative diseases (Valko et al., 2007). ROS can be produced from both endogenous and exogenous sources. One exogenous source with the potential to be detrimental is iron. Free or chelatable iron, which is injurious to liver cells, is present not only in the cytosol but also in nuclei, lysosomes, and mitochondria of hepatocytes (Petrat et al., 2001).
The methods by which patients accumulate iron in the liver have been well studied (Isom et al., 2009). Also, it has been repeatedly demonstrated that elevated hepatic iron levels induce oxidative stress and the production of ROS. Iron functions as a catalyst in the Fenton reaction. Hydroxyl radicals produced by the Fenton reaction nonselectively attack proteins, nucleic acids, polysaccharides, and lipids. In rat models of hepatic iron overload, lipid peroxidation has been found in whole liver and also in isolated cellular fractions including mitochondria, microsomes, and lysosomes (Britton et al., 1987). Malondialdehyde and 4-hydroxynonenal-lysine adducts have been found in albumin and other plasma proteins and in the liver (Houglum et al., 1990). The highly reactive aldehydes 4-hydroxynonenal and malondialdehyde can also bind to DNA. DNA strand breaks or DNA containing 8-OH-dG has been demonstrated in rodents subjected to chronic dietary iron overload (Faux et al., 1992). Hydroxyl radicals have also been reported in iron-overloaded human patients. Increased amounts of malondialdehyde-protein and 4-hydroxynonenal protein adducts have been detected in liver biopsies and increased levels of thiobarbituric acid reactants in serum of patients with HH (Young et al., 1994).
Although it is clear that hepatic iron overload results in oxidative stress–mediated damage to proteins, cellular organelles, and nucleic acids, little is known about the effects of iron overload on protein expression and signaling. Because it is becoming increasingly apparent that hepatic iron indices are elevated in patients with numerous different liver diseases, it is important to further understand the effects of hepatic iron overload at the molecular level. Obtaining this information is critical not only to further understand the role of iron in liver toxicity but also the effect of iron on cellular signaling and drug metabolism.
In the present study, we used a mouse model of dietary hepatic iron overload, with 3,5,5-trimethyl-hexanoyl-ferrocene (TMHF) as the iron donor (Moon et al., 2011), and used isobaric tag for relative and absolute quantitation (iTRAQ) proteomics to identify, at a global level, proteins whose expression is changed by iron overload. In addition, we pursued a targeted analysis of one of the top canonical pathways altered by iron, specifically the transcription factor NF-E2–related factor 2 (NRF2) and proteins induced by NRF2 activation. Evidence has accumulated over the last decade pointing to NRF2 as a key regulator of the cellular response to oxidative stress in multiple cell types and tissues, including the liver (Klaassen and Reisman, 2010). Numerous studies using NRF2-null mice have shown that NRF2 facilitates hepatoprotection from a variety of chemical- and oxidative stress–induced pathologies (Aleksunes and Manautou, 2007). In this study, we demonstrate that expression of multiple different proteins encoded by NRF2 target genes is increased in the livers of mice fed a TMHF-supplemented diet compared with mice fed a control diet. We also show, using immunofluorescence, that TMHF treatment results not only in increased NRF2 levels in the cytoplasm but also in detection of NRF2 in the nucleus of hepatocytes in livers from mice fed a TMHF-supplemented diet compared with mice fed a control diet.
MATERIALS AND METHODS
Animals. Eight-week-old male C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). The animals received humane care in accordance with the guidelines established by the National Research Council as outlined in the Guide for the Care and Use of Laboratory Animals. At 12 weeks of age, mice were fed with either control diet for 2 or 4 weeks or TMHF diet for 1, 2, or 4 weeks. The control diet was modified from AIN-93M diet (ICN, Irvine, CA), whereas the TMHF diet was achieved by supplementing control diet with 0.5% TMHF (prepared by the Organic Synthesis Core, Penn State Hershey Cancer Institute, Hershey, PA). At the indicated time points, mice were sacrificed and the livers were extracted and processed for protein and liver sections.
Perls’ Prussian blue staining. Formalin-fixed paraffin-embedded tissue samples were prepared and stained for hemosiderin as previously described (Moon et al., 2011).
iTRAQ analysis. Samples were prepared according to the Pennsylvania State University College of Medicine Mass Spectrometry (MS) and Proteomics Core Facility standard protocol (http://med.psu.edu/web/core/proteinsmassspectometry/protocols/itraq), adapted from the manufacturer’s instructions (Applied Biosystems, Carlsbad, CA). Briefly, liver protein (100 μg) from four control and four TMHF-fed mice were subjected to acetone precipitation according to the manufacturer’s instructions (Applied Biosystems). Samples were then centrifuged at 6000 × g for 10min, and pellets were dissolved in 20 μl of 0.5M triethylammonium bicarbonate (pH 8.5). Samples were then denatured and reduced by adding 1 μl of 0.2% SDS and 1 μl of 110mM tris-(2-carboxyethyl) phosphine and subsequently incubated at 60°C for 1h. One microliter of freshly prepared 84mM iodoacetamide was added, and samples were incubated for 30min at room temperature in the dark. Samples were then digested by adding 10 μl (10 μg) of Sequencing Grade trypsin (Promega) and incubating overnight at 48°C. 8-plex iTRAQ reagents were dissolved in 50 μl of isopropanol and separately mixed with each of the eight tryptic digests, incubated for 2h at room temperature, quenched by addition of 100 μl of water, and pooled. The pooled sample was then dried in vacuo and resuspended thrice with 100 μl of water. The final resuspension was with 500 μl of strong cation exchange (SCX) loading buffer (10mM potassium phosphate, 25% acetonitrile (pH 2.5–3.0).
The pooled sample was analyzed by two-dimensional liquid chromatography (LC) separation and matrix-assisted laser desorption and ionization (MALDI) time-of-flight (TOF) tandem mass spectrometry (MS/MS) as described previously (Bortner et al., 2011; Fogle et al., 2011). The samples were first separated on a passivated Waters 600E HPLC system using a 4.6 × 250mm PolySULFOETHYL Aspartamide column (PolyLC, Columbia, MD). Fifteen SCX fractions obtained from that first separation were then separated again on a Chromolith CapRod column (150 × 0.1mm, Merck) on an LC-Tempo (ABSciex, Foster City, CA) MALDI spotting system, resulting in a total of 370 spots per SCX fraction. MALDI target plates were analyzed in a data-dependent manner on either ABI 4800 or 5800 MALDI TOF-TOFs, with MS/MS spectra obtained from collision-induced dissociation of selected peaks.
Protein identification and data processing. Peptide identification, protein grouping, and subsequent protein quantitation were done using the Paragon and ProGroup Algorithms (Shilov et al., 2007) in the ProteinPilot 4.0 Software package (ABSciex). Database searched for protein identification is a species-specific NCBInr database of 5 January 2010, containing 256,336 mouse protein sequences plus a list of 156 common contaminants, concatenated with a decoy database containing the reversed sequences of the original, normal database. This search of a concatenated normal and reversed database allowed estimation of the false discovery rate using the Proteomics System Performance Evaluation Pipeline algorithm (Tang et al., 2008). Identifications were only accepted with an estimated local false discovery rate of < 5% and an unused score > 1.3 (> 95% confidence interval). The total score for a protein match is the sum of all the peptide scores for peptide sequences belonging to that protein; however, in some cases, the same peptide sequence may also be contained in different protein sequences identified in the whole data set. When shared peptide sequences exist, the “unused” score for a protein is the sum of all the individual peptide scores for all the peptides which are not shared with higher ranking proteins, and as such, are “unused” by higher ranking proteins. Thus, the highest ranking protein containing shared peptide sequences will have the same unused score as its total score, whereas lower ranking proteins with shared evidence will have a lower unused score than total score.
For iTRAQ quantitation of changes in protein levels between control and TMHF-treated mice, several criteria were applied. First, proteins were only accepted if they were significantly altered based on all of the eight denominators as generated using the two-sample t-test. The level of stringency was further increased by applying two additional criteria: proteins with a minimum of two peptides and an error factor < 4. The error factor is a statistic for reporting errors/variability in ratios, such as iTraq ratio data, and is analogous to variance/SD in nonratio data. Unlike variance/SD, errors for ratios are not a symmetrical “±” error; instead they are better reported as a factor. If one uses a “±” error for a ratio, the uncertainty range depends upon whether the ratio is defined as Condition 1:Condition 2 or Condition 2:Condition 1. Using the error factor to report ratios gives the same results, irrespective of how the ratio is defined. Proteins that were differentially expressed by ≥ ±1.5-fold were then analyzed through the use of Ingenuity Pathway Analysis (IPA) software (version 9.0, Ingenuity Systems, Redwood City, CA, www.ingenuity.com).
Immunoblot analyses. Liver tissues were homogenized, lysates were prepared, protein concentrations were determined, and gel electrophoresis was carried out as previously described (Moon et al., 2011). Blots were blocked in 5% milk in tris-buffered saline-tween for 1h and incubated in primary antibodies, followed by incubation with a horseradish peroxidase–conjugated secondary antibody. Bound antibodies were visualized by enhanced chemiluminescence (PerkinElmer Inc., Waltham, MA). Primary antibodies used for immunoblot are as follows: ferritin (Abcam, Cambridge, MA); cyclin D1 (BD Pharmingen, San Diego, CA); proliferating cell nuclear antigen (PCNA, Santa Cruz Biotechnology Inc., Santa Cruz, CA); glutathione S-transferase mu 1 (GSTM1, Santa Cruz Biotechnology); GSTM4 (Abcam), glutamate-cysteine ligase, catalytic subunit (GCLC, Abcam); glutamate-cysteine ligase, modifier subunit (GCLM, Santa Cruz Biotechnology); glutathione synthetase (GSS, Abcam), glutathione reductase (GSR, Abcam); heme oxygenase 1 (HO-1, Abcam); epoxide hydrolase 1 (EPHX1, Santa Cruz Biotechnology); NAD(P)H dehydrogenase quinone 1 (NQO1, Santa Cruz Biotechnology); β-actin (Cell Signaling Technology, Beverly, MA).
Immunohistochemistry. Formalin-fixed, paraffin-embedded tissue sections were prepared and were stained as previously described (Moon et al., 2011). Anti-PCNA antibody (Santa Cruz Biotechnology) was used for incubating the sections. For quantification of PCNA positive cells, a minimum of nine fields (high power, ×400) was counted for each experimental condition. The percentage of PCNA positive cells for each field was calculated and the average represented graphically.
Immunofluorescence. Liver tissue cryosections (5 μm) were air-dried for 5min and then fixed in 4% paraformaldehyde in PBS for 5min. The staining procedures were performed as previously described (Aleksunes et al., 2008). Anti-NRF2 (H-300) (Santa Cruz Biotechnology) primary antibody was used for incubating the sections. Images were acquired on a Leica SP2 AOBS confocal microscope (Leica Microsystems Inc., Exton, PA). Negative control was prepared by incubating a section in the absence of primary antibody.
Statistical analysis. One-way ANOVA model was performed with SigmaStat 3.5 (Aspire Software International Inc., Ashburn, VA) using the Shapiro-Wilk test for multiple comparisons as appropriate. Data are expressed as mean ± SE. A p value of < 0.05 was considered statistically significant (Figs. 1B and C).
RESULTS
Characterization of Livers From Mice Fed TMHF
C57BL/6 mice were administered a TMHF-supplemented diet for 1, 2, or 4 weeks or a control diet for 2 or 4 weeks. At the indicated time points, tissue was harvested and subjected to hematoxylin and eosin staining to examine hepatic architecture. Sections from both control and iron-loaded mice exhibited typical hepatic architecture, including the presence of mono- and binucleated hepatocytes with circular-shaped nuclei and prominent nucleoli. No histological evidence of cytotoxicity was observed (data not shown) in agreement with our previous data for mice fed a TMHF diet for 4 weeks (Moon et al., 2011). To determine the time course and cellular distribution of TMHF-induced hepatic iron deposition, Perls’ Prussian blue staining was performed (Fig. 1A). In liver sections from mice fed a TMHF-supplemented diet for 1 or 2 weeks, Perls’ staining was restricted to the hepatocytes, whereas in sections from mice fed TMHF for 4 weeks, Perls’ positivity was detected both in the hepatocytes and nonparenchymal cells. Initial loading of iron into hepatocytes is consistent with the primary iron overload observed in patients with HH (Batts, 2007). Tissue sections from control mice were negative for Perls’ Prussian blue staining.
Fig. 1.

Detection of iron deposition, liver/body weight ratio, and alterations in cell proliferation associated proteins in control and TMHF-treated mouse livers. C57BL/6 mice were administered a TMHF-supplemented diet for 1, 2, or 4 weeks or a control diet for 2 or 4 weeks. (A) Detection of iron deposition. Whole livers from TMHF and control mice were fixed, sectioned, and subjected to Perl’s Prussian blue staining. Representative images of PCNA stained sections from mice fed a TMHF-supplemented diet for 1, 2, or 4 weeks or a control diet for 4 weeks are shown. (B) Liver/body weight ratio. The liver-to-body weight ratios were averaged and graphically represented. Data shown are mean ± SE for each group (n = 4). *p < 0.05, **p < 0.001. All changes for mice treated with TMHF were significant in comparison with either 2- or 4-week control. (C, D) Proliferation measured by PCNA positive nuclei. (C) The percentage of PCNA positive cells, as measured by immunohistochemistry, in tissue sections from mice administered a TMHF-supplemented diet for 1, 2, or 4 weeks or a control diet for 2 or 4 weeks was calculated and represented graphically. Data shown are mean ± SE for each group (n = 4). *p < 0.05, **p < 0.001. All changes for mice treated with TMHF were significant in comparison with either 2- or 4-week control. (D) Representative images of PCNA stained sections from mice fed a TMHF-supplemented diet for 1, 2, or 4 weeks or a control diet for 4 weeks are shown. Arrows indicate PCNA positive nuclei in hepatocytes in liver sections from TMHF-treated mice. (E) Immunoblot analysis for ferritin, PCNA, and cyclin D1. Protein was extracted from whole livers of mice fed a TMHF diet for 1, 2, or 4 weeks or control diet for 2 or 4 weeks and subjected to immunoblot analysis for ferritin, PCNA, cyclin D1, or β-actin. Stained tissue sections (panels A and D) were photographed by light microscopy (Original magnification: ×400).
Patients with HH (Nichols and Bacon, 1989) and rodents fed a carbonyl iron–supplemented diet (Brown et al., 2006; Troadec et al., 2006) develop hepatomegaly. It has also been shown that hepatocyte proliferation as measured by PCNA labeling index is significantly higher in mice fed an iron-supplemented diet compared with mice fed a control diet (Furutani et al., 2006). Similarly, increases in cyclin D1 have been observed in mice and rats with iron overload (Brown et al., 2006; Troadec et al., 2006). The ability of elevated hepatic iron levels to induce cyclin D1 is particularly important in light of the findings that patients with HH have an increased risk for hepatocellular carcinoma and that iron overload appears to function as a cofactor in the development of hepatocellular carcinoma in some mouse models (Carthew et al., 1997; Furutani et al., 2006; Smith et al., 1990). Transient overexpression of cyclin D1 in mouse liver was shown to induce hepatocyte replication and to increase liver mass indicating that overexpression of cyclin D1 is sufficient to induce hepatocyte proliferation (Nelsen et al., 2001). In addition, chronic overexpression of cyclin D1 targeted to the liver in transgenic mice has been shown to lead to a high rate of hepatocellular carcinoma development (Deane et al., 2001). Therefore, the ability of iron overload to chronically induce cyclin D1 may play an important role in hepatocarcinogenesis. For these reasons, these parameters were evaluated in the dietary TMHF mouse model. A significant increase in liver-to-body weight ratios was detected by 1 week of feeding a TMHF-supplemented diet and the ratios continued to rise through 4 weeks of treatment (Fig. 1B). To determine whether increased liver-to-body weight ratios were accompanied by an increase in PCNA levels, liver sections were subjected to immunohistochemistry for PCNA. TMHF treatment caused a significant increase in PCNA labeling index that rose with time (Figs. 1C and D), paralleling the results obtained for liver-to-body weight ratios. Immunoblotting for PCNA and cyclin D1 was also carried out (Fig. 1E). PCNA and cyclin D1 levels were elevated above control levels by 1 week of TMHF treatment and the increase progressed as TMHF treatment continued for 4 weeks.
The iron storage protein ferritin is rapidly induced by iron and is used as an indicator of cellular response to iron. As expected, treatment with TMHF resulted in a marked increase in total ferritin by 1 week that remained high through 4 weeks (Fig. 1E). The 4-week time point of TMHF treatment was selected for proteomic analysis.
iTRAQ Profiling of Liver Protein From Mice Fed a TMHF Compared With Mice Fed a Control Diet
To define global alterations in the liver proteome caused by hepatic iron overload, we used iTRAQ proteomics methodology. Total liver protein (100 μg) from each of four control and four TMHF-fed mice were concentrated, digested, labeled with distinct iTRAQ probes, and then pooled. After two-dimensional LC fractionation of the pooled peptide sample, peptides were identified by mass spectrometry (MALDI TOF-TOF). Mass spectra were taken from 5500 MALDI spots, and MS/MS spectra were acquired from up to 20 peaks per spot. A ProteinPilot search of the MS/MS data against an NCBInr Mouse database identified 1008 proteins with an Unused Score > 1.3 (> 95% confidence interval). Search of a concatenated normal and reversed database using the Proteomics System Performance Evaluation Pipeline algorithm and allowing a false discovery rate of < 5% reduced the list of confidently identified proteins to 868 proteins. For quantitation of changes in protein levels between control and TMHF-treated mice, several criteria were applied. Data analysis using the two-sample t-test identified 159 out of 868 proteins as significantly altered based on all of the eight denominators. When the additional criteria of including only significantly altered proteins with a minimum of two peptides and an error factor < 4 were applied, 119 proteins were accepted (Supplementary Table 1).
Applying cutoff thresholds of above or below ± 1.5-fold changes, a total of 74 proteins were significantly altered, with 46 upregulated and 28 downregulated. The 15 most upregulated and 15 most downregulated proteins are listed in Table 1. The top 5 upregulated proteins were increased by more than sevenfold. The top 5 most downregulated proteins were decreased by more than threefold. The top four canonical pathways altered by TMHF treatment, which were NRF2-mediated oxidative stress response, metabolism of xenobiotics by cytochrome P450, glutathione metabolism, and xenobiotic metabolism signaling (Table 2), had overlap with regard to specific proteins in particular the GSTs. Because of the long-standing association of elevated hepatic iron with oxidative stress and the evidence that the transcription factor NRF2 is a key regulator of the cellular response to oxidative stress, we focused the remainder of the study on the NRF2-mediated oxidative stress response pathway.
TABLE 1 .
List of Top 15 Proteins Differentially Expressed in TMHF-Fed Mice
| Symbol | Description | Accession no.a | p value | Fold changeb |
| Increased | ||||
| GSTM5 | Glutathione S-transferase mu 5 | gi|6754084 | 1.58E-06 | 18.82 |
| FTH1 | Ferritin, heavy polypeptide 1 | gi|74225483 | 3.19E-04 | 15.41 |
| GSTM3 | Glutathione S-transferase, mu 3 | gi|33468899 | 7.80E-05 | 14.11 |
| CYP2A6* | Cytochrome P450, family 2, subfamily A, polypeptide 6 | gi|75832129 | 1.19E-05 | 11.82 |
| UGDH | UDP-glucose 6-dehydrogenase | gi|74214298 | 4.74E-05 | 7.74 |
| GSTA4 | Glutathione S-transferase, alpha 4 | gi|20141353 | 1.50E-05 | 6.13 |
| CES1G | Carboxylesterase 1G | gi|74203588 | 1.26E-04 | 5.69 |
| EPHX1 | Epoxide hydrolase 1, microsomal (xenobiotic) | gi|6753762 | 6.59E-05 | 5.55 |
| BLVRB | Biliverdin reductase B (flavin reductase (NADPH)) | gi|85541765 | 6.83E-04 | 4.68 |
| UGT1A8* | UDP glucuronosyltransferase 1 family, polypeptide A8 | gi|436187 | 4.56E-04 | 4.41 |
| EHHADH | Enoyl-CoA, hydratase/3-hydroxyacyl CoA dehydrogenase | gi|31541815 | 6.03E-04 | 4.06 |
| ACOX2 | Acyl-CoA oxidase 2, branched chain | gi|259458401 | 7.18E-04 | 3.82 |
| ALDH1A7 | Aldehyde dehydrogenase family 1, subfamily A7 | gi|81886650 | 6.23E-04 | 3.75 |
| DECR2 | 2,4-dienoyl CoA reductase 2, peroxisomal | gi|74188033 | 2.38E-04 | 3.27 |
| UGT2B17 | UDP glucuronosyltransferase 2 family, polypeptide B17 | gi|74201996 | 3.12E-03 | 3.10 |
| Decreased | ||||
| HPD | 4-hydroxyphenylpyruvate dioxygenase | gi|849053 | 2.98E-03 | −2.04 |
| HSPA5 | Heat shock 70kDa protein 5 (glucose-regulated protein, 78kDa) | gi|74207401 | 1.53E-03 | −2.05 |
| GULO | Gulonolactone (L-) oxidase | gi|74195453 | 2.81E-04 | −2.14 |
| GLUD1 | Glutamate dehydrogenase 1 | gi|6680027 | 1.04E-05 | −2.30 |
| ACAA2 | Acetyl-CoA acyltransferase 2 | gi|74198556 | 4.68E-04 | −2.32 |
| PPIB | Peptidylprolyl isomerase B (cyclophilin B) | gi|83406529 | 3.65E-05 | −2.36 |
| TF | Transferrin | gi|74223225 | 4.59E-03 | −2.39 |
| CALR | Calreticulin | gi|6680836 | 8.23E-03 | −2.61 |
| LDHA | Lactate dehydrogenase A | gi|74223193 | 5.79E-04 | −2.64 |
| GLUL | Glutamate-ammonia ligase | gi|74214272 | 2.95E-03 | −2.76 |
| CPS1 | Carbamoyl-phosphate synthase 1, mitochondrial | gi|73918911 | 6.12E-04 | −3.52 |
| ASS1 | Argininosuccinate synthase 1 | gi|74146278 | 1.24E-03 | −3.76 |
| OAT | Ornithine aminotransferase | gi|8393866 | 8.14E-04 | −4.05 |
| SCP2 | Sterol carrier protein 2 | gi|45476581 | 1.30E-04 | −4.09 |
| SELENBP2 | Selenium binding protein 2 | gi|9507079 | 6.08E-05 | −18.70 |
Note. Listed proteins are differentially expressed (fold change ≥ ±1.5) in TMHF-fed mice compared with control mice.
aNational Center for Biotechnology Information (NCBI).
bRelative to control diet fed mice.
*Protein includes other isoforms.
TABLE 2 .
Top Canonical Pathways Obtained Through Ingenuity in TMHF-Fed Mice
| Pathway | p value | Ratio | Protein list |
| NRF2-mediated oxidative stress response | 1.50E-17 | 16/192 (0.083) | AKR1A1, CAT, CBR1, EPHX1, FTH1, GCLC, GCLM, GSTA5, GSTM1, GSTM3, GSTM4, GSTM5, GSTO1, PPIB, PRDX1, TXN |
| Metabolism of xenobiotics by cytochrome P450 | 2.10E-17 | 14/197 (0.071) | AKR1A1, CYP2A6*, GSTA4, GSTA5, GSTM1, GSTM3, GSTM4, GSTM5, GSTO1, GSTT3, GSTZ1, UGT1A8*, UGT2B15, UGT2B17 |
| Glutathione metabolism | 7.53E-16 | 11/92 (0.120) | GCLC, GCLM, GSTA4, GSTA5, GSTM1, GSTM3, GSTM4, GSTM5, GSTO1, GSTT3, GSTZ1 |
| Xenobiotic metabolism signaling | 6.17E-12 | 14/296 (0.047) | ALDH1A1, CAT, CES1G, CES2A, GCLC, GSTA5, GSTM1, GSTM3, GSTM4, GSTM5, GSTO1, UGT1A8*, UGT2B15, UGT2B17 |
Note. Listed proteins are differentially expressed (fold change ≥ ±1.5) in TMHF-fed mice compared with control mice.
Boldface type indicates upregulated protein; regular type indicates downregulated protein.
*Protein includes other isoforms.
Proteins Encoded by NRF2 Target Genes Identified by iTRAQ Profiling as Upregulated in Liver Protein From TMHF-Treated Mice
NRF2 was named for its homology to nuclear factor erythroid 2 p45, a protein important in erythroid cell-specific gene transcription (Moi et al., 1994). The generation of NRF2-null mice, which developed normally and were not anemic, disproved the concept that NRF2 was important in erythropoiesis (Chan et al., 1996). Four different groupings of agents, conditions that induce oxidative stress and ROS and that activate NRF2, and pathways downstream of NRF2 are shown in the IPA diagram (upper portion, Fig. 2). Proteins downstream of NRF2 are grouped using IPA in specific categories (bottom portion of Fig. 2). Further analysis focused only on proteins downstream of NRF2 that were upregulated in response to TMHF treatment. These proteins fell into two categories in the IPA canonical pathway, phase I/II proteins and antioxidant proteins (Fig. 2).
Fig. 2.

Alterations in the NRF2 canonical pathway in livers of mice treated with TMHF. Liver proteins from mice fed a control or TMHF diet mice for 4 weeks (four mice in each group) were labeled and analyzed by iTRAQ. The data from iTRAQ were then analyzed using IPA. A modified schematic of the NRF2 signaling pathway obtained from IPA is shown. The diagram was reduced in magnitude to include only those categories containing proteins that were identified as upregulated (shades of red) or downregulated (green) by TMHF by more than ± 1.5-fold of the control in this iTRAQ profile. Proteins in white circles were not among the 74 identified proteins.
Numerous phase I/II proteins and antioxidant proteins have been linked with NRF2 activation. Proteins in these categories upregulated in response to TMHF treatment to more than 1.5-fold of the control are listed in Table 3. With regard to phase I proteins, iTRAQ profiling identified greater than 1.5-fold upregulation of carbonyl reductase 1, two members of the carboxylesterase family (CES1G and CES2A), and cytochrome P450 (CYP) 2A6. Among phase II proteins, eight GSTs and five UDP glucuronosyltransferases (UGTs) were upregulated more than 1.5-fold of the control. With regard to glutathione (GSH) synthesis, both GCLC and GCLM were upregulated more than 1.5-fold of the control. This is particularly important because GCL is the rate-limiting step in GSH synthesis. GSH, a tripeptide mainly synthesized by the liver, is the predominant cellular thiol resource and protects against oxidative and electrophilic stress. Eight antioxidant proteins were also upregulated: aldo-keto reductase 1A1 (AKR1A1), aldehyde dehydrogenase (ALDH) 1A1 and ALDH1A7, catalase (CAT), EPHX1, ferritin, heavy polypeptide 1 (FTH1), peroxiredoxin 1 (PRDX1), and thioredoxin (TXN).
TABLE 3 .
List of Proteins Upregulated by NRF2 in TMHF-Fed Mice
| Category | Symbol | Description | Accession no.a | Fold changeb |
| Phase I | ||||
| CBR1 | Carbonyl reductase 1 | gi|187957220 | 2.66 | |
| CES1G | Carboxylesterase 1G | gi|74203588 | 5.69 | |
| CES2A | Carboxylesterase 2A | gi|26347655 | 2.16 | |
| CYP2A6* | Cytochrome P450, family 2, subfamily A, polypeptide 6 | gi|75832129 | 11.82 | |
| Phase II | ||||
| GSTs | GSTA4 | Glutathione S-transferase alpha 4 | gi|20141353 | 6.13 |
| GSTA5 | Glutathione S-transferase alpha 5 | gi|50263046 | 1.66 | |
| GSTM1 | Glutathione S-transferase mu 1 | gi|6680121 | 2.48 | |
| GSTM3 | Glutathione S-transferase, mu 3 | gi|33468899 | 14.11 | |
| GSTM4 | Glutathione S-transferase mu 4 | gi|28076911 | 2.85 | |
| GSTM5 | Glutathione S-transferase mu 5 | gi|6754084 | 18.82 | |
| GSTO1 | Glutathione S-transferase omega 1 | gi|74212315 | 1.75 | |
| GSTT3 | Glutathione S-transferase, theta 3 | gi|74216166 | 2.94 | |
| UGTs | UGT1A1 | UDP glucuronosyltransferase 1 family, polypeptide A1 | gi|62533164 | 1.56 |
| UGT1A8* | UDP glucuronosyltransferase 1 family, polypeptide A8 | gi|436187 | 4.41 | |
| UGT2B15 | UDP glucuronosyltransferase 2 family, polypeptide B15 | gi|74146299 | 2.08 | |
| UGT2B15 | UDP glucuronosyltransferase 2 family, polypeptide B15 | gi|74143635 | 1.75 | |
| UGT2B17 | UDP glucuronosyltransferase 2 family, polypeptide B17 | gi|74201996 | 3.10 | |
| Glutathione synthesis | ||||
| GCLC | Glutamate-cysteine ligase, catalytic subunit | gi|74188391 | 3.02 | |
| GCLM | Glutamate-cysteine ligase, modifier subunit | gi|71059841 | 2.88 | |
| Antioxidants | ||||
| AKR1A1 | Aldo-keto reductase family 1, member A1 | gi|7677318 | 1.73 | |
| ALDH1A1 | Aldehyde dehydrogenase 1 family, member A1 | gi|85861182 | 2.04 | |
| ALDH1A7 | Aldehyde dehydrogenase family 1, subfamily A7 | gi|81886650 | 3.75 | |
| CAT | Catalase | gi|74204790 | 2.19 | |
| EPHX1 | Epoxide hydrolase 1, microsomal (xenobiotic) | gi|6753762 | 5.55 | |
| FTH1 | Ferritin, heavy polypeptide 1 | gi|74225483 | 15.41 | |
| PRDX1 | Peroxiredoxin 1 | gi|74203142 | 1.90 | |
| TXN | Thioredoxin | gi|6755911 | 1.64 | |
Note. Listed proteins are differentially expressed (fold change ≥ ±1.5) in TMHF-fed mice compared with control mice.
aNational Center for Biotechnology Information (NCBI).
bRelative to control diet fed mice.
*Protein includes other isoforms.
Proteins Encoded by NRF2 Target Genes Identified by Immunoblot Analyses as Upregulated in Liver Protein From TMHF-Treated Mice
Immunoblot analyses of selected NRF2 target proteins were carried out to validate the results obtained from the proteomic screen and to expand the analyses to other NRF2 target gene products (Fig. 3). The nine proteins selected included (1) proteins identified by proteomics as upregulated more than 1.5-fold of the control, (2) proteins identified as upregulated but by less than 1.5-fold of the control, and (3) proteins previously identified as encoded by NRF2 target genes but not included among 119 proteins identified with a high level of confidence. An additional goal was to choose proteins for validation that fell into the four different categories listed in Fig. 3A. With regard to phase I/II drug metabolism, the data from immunoblot analyses clearly demonstrate that GSTM1 and GSTM4 are markedly upregulated in protein from all four TMHF-treated mice compared with the control mice. With regard to GSH synthesis, immunoblot analyses validated TMHF-induced increases in GCLC, GCLM, and GSS. The latter protein GSS, indicated as being increased 1.39-fold by iTRAQ quantitation, was clearly shown to be upregulated using immunoblotting. Three antioxidant proteins, GSR, HO-1, and EPHX1, were all shown to be upregulated by TMHF. Although iTRAQ ratios showed that GSR only upregulated 1.32-fold by the screen, immunoblotting clearly showed induction of this protein by TMHF treatment. HO-1 and NQO1, a prototypical NRF2 target gene, were not identified by iTRAQ profiling; however, immunoblot data clearly show that HO-1 and NQO1 were markedly upregulated in TMHF-treated mice.
Fig. 3.

Effect of iron overload on NRF2 downstream proteins in TMHF-fed mice. (A) Table showing NRF2-regulated enzymes that fell into the four categories. Upon binding to ARE, NRF2 transactivates genes associated with phase I/II drug metabolism, GSH synthesis, antioxidants, and NADPH regeneration. Numbers in parenthesis are fold increase from iTRAQ profiling. (B–E) Liver protein from mice fed a control or TMHF diet for 4 weeks was subjected to immunoblotting to verify and extend identification of NRF2-regulated proteins associated with (B) phase I/II drug metabolism, (C) GSH synthesis, (D) antioxidants, and (E) NADPH regeneration and increased in level of expression by TMHF treatment.
Nuclear Localization of NRF2 in Tissue Sections From TMHF-Treated Mice
Both iTRAQ proteomic profiling and immunoblotting of liver protein clearly showed that TMHF treatment induced many proteins previously identified as induced by NRF2 activation. To more directly address the question of NRF2 activation, immunofluorescence for NRF2 was carried out (Fig. 4). Positive staining for NRF2 was difficult to detect in the cytoplasm or nucleus of hepatocytes from control mice. However, a readily apparent signal for NRF2 was observed in the nuclei of hepatocytes from TMHF-treated mice. Faint positive staining for NRF2 was also detectable in the cytoplasm of hepatocytes from TMHF-treated mice. These results indicate that TMHF treatment increases NRF2 levels and results in NRF2 translocation to the nucleus.
Fig. 4.
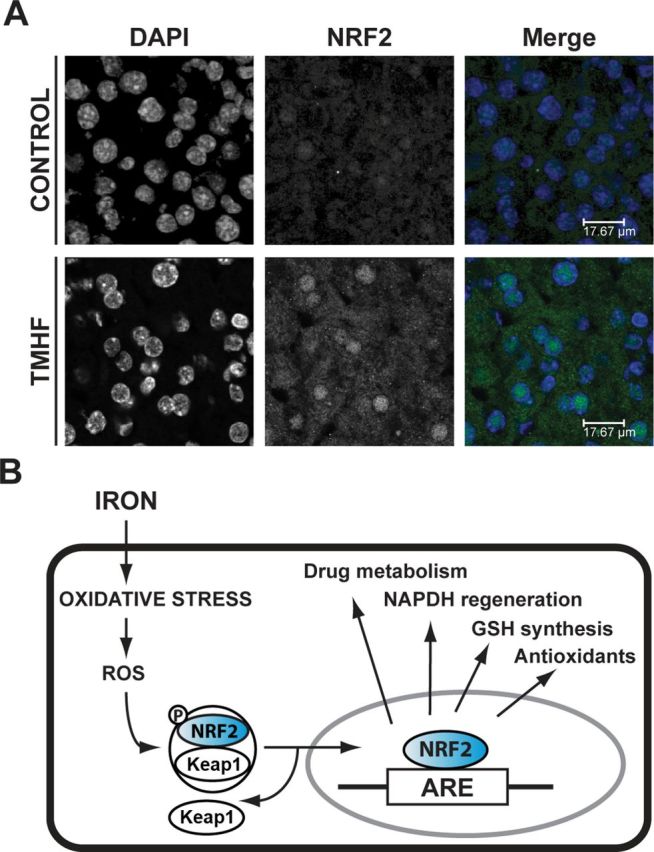
Translocation of NRF2 to the nucleus in livers from TMHF-treated mice. (A) Liver cryosections from control and TMHF-fed mice were fixed and subjected to indirect immunofluorescence to detect NRF2 (green). Sections were mounted in Prolong Gold containing 4′,6-diamidino-2-phenylindole for nuclear staining (blue). Representative images of each stain alone and merged are shown. (B) Proposed schematic of iron-induced activation of NRF2. Oxidative stress produced by overload iron causes generation of ROS, which triggers release of NRF2 from Keap1. NRF2 levels are increased. NRF2 translocates to the nucleus and binds to the ARE of NRF2 target genes activating phase I/II and antioxidant gene transcription.
DISCUSSION
We used iTRAQ proteomics to survey proteins differentially expressed in liver from mice fed a TMHF-supplemented diet compared with mice fed a control diet. The expression of 74 proteins, out of a total of 119 proteins identified with a high level of confidence, was altered by more than ± 1.5-fold showing that the effects of iron on the liver proteome were extensive. In addition, both up- and downregulation of proteins in multiple different metabolic and signaling pathways were observed. It is well known that hepatic iron overload induces oxidative stress leading to direct damage to proteins, cellular organelles, and nucleic acids. Indeed, in patients, continued assault by iron overload can result in the development of fibrosis, cirrhosis, and even hepatocellular carcinoma. Data obtained in this study indicate that iron, by inducing oxidative stress, also signals the liver to upregulate a battery of cytoprotective proteins and drug processing proteins.
The effect of dietary iron overload, using carbonyl iron as the iron donor, on gene expression in the liver at the level of the transcriptome was previously reported (Rodriguez et al., 2009). Interestingly, many of the 218 genes identified were genes that encode the proteins identified by iTRAQ in our study. Both studies identified pronounced changes in expression of genes responsive to oxidative stress. Our iTRAQ quantitative proteomics approach identified at a very high level of confidence proteins altered by hepatic iron overload. Many of these proteins contribute to biological processes not previously reported as altered by dietary hepatic iron overload. Postanalysis of the data was carried out using IPA. Forty-two different IPA canonical pathways were identified as altered at a confidence level of p < 0.05. The top IPA canonical pathway was NRF2-mediated oxidative stress response. The remainder of the study focused on this pathway.
Activation of NRF2 requires at least four components, NRF2, Keap1 (Kelch-like ECH-associated protein), a group of small musculoaponeurotic fibrosarcoma (Maf) proteins and a cis-acting enhancer called antioxidant response element (ARE) (Dinkova-Kostova et al., 2005). NRF2, a basic leucine zipper transcription factor featuring a Cap “n” collar, is rapidly degraded in a ubiquitin-dependent manner. Keap1 is a potent cytosolic repressor of NRF2. Under normal cellular conditions, only a minimal basal level of NRF2-directed gene expression is observed because NRF2 is held inactive in the cytoplasm by Keap1 (Kang et al., 2005). Gene knockout of Keap1 results in constitutively hyperactive NRF2 signaling. Once in the nucleus, NRF2 forms heterodimers with small Maf proteins and NRF2/Maf bind to ARE.
NRF2 is an important transcriptional regulator of antioxidative cellular protection. Upon cellular oxidative/electrophilic insult, NRF2 initiates a response by upregulating a battery of cytoprotective genes, such as NQO1, GCLC and GCLM, HO-1, and drug processing genes, such as GSTs, UGTs (Reisman et al., 2009). We report here that TMHF treatment resulted in upregulation of 25 phase I/II and antioxidant proteins previously categorized as being encoded by NRF2 target genes. Upregulation of nine of these proteins was verified by immunoblot analyses. Previous studies have shown that TMHF is an efficient hepatic iron loading compound in rats in vivo that results in hepatic changes consistent with many of the clinical features observed in patients with HH (Nielsen and Heinrich, 1993; Nielsen et al., 1993). We show here that mice fed a TMHF-supplemented diet demonstrate many of the characteristics previously reported for other mouse models of dietary hepatic iron overload and for patients with HH, including positive Perls’ Prussian blue staining, hepatomegaly, increased PCNA labeling index, increased levels of PCNA and cyclin D1 proteins, and increased ferritin, thereby validating the use of the TMHF mouse model. In addition, the data reported here also indicate the temporal nature of these iron-induced changes. All the effects of iron were progressive with continued TMHF feeding and none appeared to represent a threshold effect. Even at 4 weeks of TMHF treatment, liver tissue from mice fed a TMHF diet had no gross histological evidence of liver damage. Therefore, the multiple iron-mediated changes in the liver proteome identified by iTRAQ analysis appear to precede any gross histological evidence of liver damage.
The NRF2 molecule possesses multiple nuclear export signals and nuclear localization motifs (Li and Kong, 2009). Under basal conditions, the combined nuclear exporting forces counteract the combined nuclear importing forces, and NRF2 exhibits a predominantly whole-cell distribution. When NRF2 was overexpressed in HeLa cells using an enhanced green fluorescence protein tagged NRF2, nearly 60% of the cells showed whole-cell distribution, 30% of the cells showed nuclear distribution, and 12% of the cells showed cytosolic distribution (Li et al., 2006). Under normal conditions, an equilibrium exists between NRF2 synthesis and degradation. Two pools of NRF2 proteins have been proposed to exist in the cytoplasm (Li and Kong, 2009). One is free-floating NRF2. The other is Keap1-bound NRF2, which is destined for ubiquitination and proteasomal degradation. When cells are exposed to oxidative stress, NRF2 ubiquitination is impeded, Keap1 self-ubiquitinates, and the NRF2-binding capacity of Keap1 is saturated thereby enlarging the free-floating NRF2 pool. When NRF2 is challenged with oxidative stress, one NRF2 nuclear export motif is disabled with no effect on the nuclear localization motifs thereby triggering NRF2 nuclear translocation. The result is an increase in total NRF2 and an increase in the amount of NRF2 translocated to the nucleus. In response to TMHF treatment, we observed both an increase in NRF2 in the cytoplasm and translocation of NRF2 to the nucleus, two features previously seen in antioxidant responses induced by a variety of insults.
We previously used the TMHF mouse model to examine the effect of 4 weeks of TMHF feeding on acetaminophen (APAP) toxicity (Moon et al., 2011). A nonlethal APAP dose was used because our expectation was that TMHF-induced hepatic iron overload would exacerbate APAP toxicity. Contrary to expectations, TMHF treatment protected the mice from moderate transient APAP-induced hepatotoxicity, thereby acting as a hepatoprotectant. Protection occurred prior to the formation of APAP adducts, and TMHF treatment had no significant effect on CYP2E1 or CYP1A2 levels.
APAP is metabolically activated by the cytochrome P450 system to the highly reactive metabolite N-acetyl- p-benzoquinoneimine, which is then detoxified by GSH to form a nontoxic APAP-GSH conjugate. When depletion of GSH occurs at a rate that exceeds its replenishment, N-acetyl-p-benzoquinoneimine covalently binds to cysteine residues on cellular proteins forming APAP protein adducts. TMHF treatment markedly decreased the ability of APAP to deplete GSH (Moon et al., 2011), suggesting that one contributing factor to protection afforded by excess iron is partial blockage of APAP-induced depletion of GSH. The finding that THMF treatment increased expression of NRF2 target genes involved in GSH synthesis may partially explain how iron functions as a protectant for APAP. GSH synthesis involves two steps. The first and rate-limiting step, which results in the synthesis of gamma-glutamylcysteine from L-glutamate and cysteine, is catalyzed by GCL (made up of two subunits GCLC and GCLM). In the second step, glycine is added to gamma-glutamylcysteine by the enzyme GSS. GSH can also be regenerated from glutathione disulfide by the enzyme GSR. In the iTRAQ proteomics profile of proteins altered by TMHF treatment, GSH synthesis was the IPA pathway identified as having the third highest confidence level. GCLC, GCLM, GSS, and GSR were among the 119 proteins identified with a high level of confidence. All four were upregulated with GCLC, and GCLM being upregulated by greater than 1.5-fold of the control. In addition, all four proteins were verified as upregulated by immunoblot analyses.
Previous findings with NRF2-altered mice also help to explain TMHF-mediated protection from moderate transient APAP-induced hepatotoxicity. Mice genetically engineered to overexpress NRF2, by having hepatocyte-specific disruption of the Keap1 gene, exhibit dramatic resistance to APAP hepatotoxicity (Okawa et al., 2006). TMHF treatment and hepatocyte-specific disruption of Keap1 each activates NRF2 and therefore protects against APAP. Conversely, treatment of NRF2 knockout mice with APAP resulted in enhanced liver injury and mortality compared with wild-type and heterozygous counterparts (Chan et al., 2001; Enomoto et al., 2001).
Our global proteome analysis of mice fed a TMHF- supplemented diet has led to new findings that warrant further investigation. For example, the primary site of drug metabolism is the liver. NRF2 has been described as having a pivotal role in drug metabolism and detoxification. We report that dietary hepatic iron overload in mice activates NRF2 inducing expression of NRF2 phase I/II proteins. It is possible that patients with an elevated hepatic iron index may respond differently to treatment with specific drugs than individuals without elevated hepatic iron levels. In addition, the results of this study, showing that iron alone alters expression of numerous proteins, taken together with reports that hepatic iron levels are elevated in patients with numerous viral and nonviral liver diseases raise the question of whether elevated hepatic iron may exacerbate or counterbalance changes in liver protein expression caused by an underlying liver disease. This question could begin to be addressed by measuring protein expression in mice that model a specific liver disease (e.g., hepatitis C virus core protein transgenic mice) and have been fed a TMHF-supplemented diet.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (DK54482 to H.C.I.); Pennsylvania Department of Health (Core Facility services and instruments).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the use of the Penn State College of Medicine Mass Spectrometry and Proteomics Facility for the iTRAQ analysis/interpretation and the Microscopy Imaging Core for the Confocal Imaging and the use of the Penn State Hershey Cancer Institute (PSHCI) Organic Synthesis for TMHF and the PSHCI Biostatistics Cores for statistical analysis. We also acknowledge Anne Stanley who did the 2D-LC separations and acquired all the mass spectra, Dr James Bortner who provided assistance with data analyses, and Colleen Kelley who provided assistance with manuscript preparation. The Department of Health disclaims responsibility for any analyses, interpretations, or conclusions.
REFERENCES
- Aleksunes L. M., Manautou J. E. (2007). Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol. Pathol. 35, 459––473 [DOI] [PubMed] [Google Scholar]
- Aleksunes L. M., Slitt A. L., Maher J. M., Augustine L. M., Goedken M. J., Chan J. Y., Cherrington N. J., Klaassen C. D., Manautou J. E. (2008). Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol. Appl. Pharmacol. 226, 74––83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batts K. P. (2007). Iron overload syndromes and the liver. Mod. Pathol. 20,(Suppl. 1), S31––S39 [DOI] [PubMed] [Google Scholar]
- Bortner J. D., Jr, Richie J. P., Jr, Das A., Liao J., Umstead T. M., Stanley A., Stanley B. A., Belani C. P., El-Bayoumy K. (2011). Proteomic profiling of human plasma by iTRAQ reveals down-regulation of ITI-HC3 and VDBP by cigarette smoking. J. Proteome. Res. 10, 1151––1159 [DOI] [PubMed] [Google Scholar]
- Britton R. S., Bacon B. R., Recknagel R. O. (1987). Lipid peroxidation and associated hepatic organelle dysfunction in iron overload. Chem. Phys. Lipids 45, 207––239 [DOI] [PubMed] [Google Scholar]
- Brown K. E., Mathahs M. M., Broadhurst K. A., Weydert J. (2006). Chronic iron overload stimulates hepatocyte proliferation and cyclin D1 expression in rodent liver. Transl. Res. 148, 55––62 [DOI] [PubMed] [Google Scholar]
- Carthew P., Nolan B. M., Smith A. G., Edwards R. E. (1997). Iron promotes DEN initiated GST-P foci in rat liver. Carcinogenesis 18, 599––603 [DOI] [PubMed] [Google Scholar]
- Chan K., Han X. D., Kan Y. W. (2001). An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 98, 4611––4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K., Lu R., Chang J. C., Kan Y. W. (1996). NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U.S.A. 93, 13943––13948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane N. G., Parker M. A., Aramandla R., Diehl L., Lee W. J., Washington M. K., Nanney L. B., Shyr Y., Beauchamp R. D. (2001). Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 61, 5389––5395 [PubMed] [Google Scholar]
- Dinkova-Kostova A. T., Holtzclaw W. D., Kensler T. W. (2005). The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 18, 1779––1791 [DOI] [PubMed] [Google Scholar]
- Enomoto A., Itoh K., Nagayoshi E., Haruta J., Kimura T., O’Connor T., Harada T., Yamamoto M. (2001). High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 59, 169––177 [DOI] [PubMed] [Google Scholar]
- Faux S. P., Francis J. E., Smith A. G., Chipman J. K. (1992). Induction of 8-hydroxydeoxyguanosine in Ah-responsive mouse liver by iron and Aroclor 1254. Carcinogenesis 13, 247––250 [DOI] [PubMed] [Google Scholar]
- Fogle R. L., Hollenbeak C. S., Stanley B. A., Vary T. C., Kimball S. R., Lynch C. J. (2011). Functional proteomic analysis reveals sex-dependent differences in structural and energy-producing myocardial proteins in rat model of alcoholic cardiomyopathy. Physiol. Genomics 43, 346––356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furutani T., Hino K., Okuda M., Gondo T., Nishina S., Kitase A., Korenaga M., Xiao S. Y., Weinman S. A., Lemon S. M., et al. (2006). Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 130, 2087––2098 [DOI] [PubMed] [Google Scholar]
- Houglum K., Filip M., Witztum J. L., Chojkier M. (1990). Malondialdehyde and 4-hydroxynonenal protein adducts in plasma and liver of rats with iron overload. J. Clin. Invest. 86, 1991––1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom H. C., McDevitt E. I., Moon M. S. (2009). Elevated hepatic iron: A confounding factor in chronic hepatitis C. Biochim. Biophys. Acta 1790, 650––662 [DOI] [PubMed] [Google Scholar]
- Kang K. W., Lee S. J., Kim S. G. (2005). Molecular mechanism of nrf2 activation by oxidative stress. Antioxid. Redox Signal 7, 1664––1673 [DOI] [PubMed] [Google Scholar]
- Klaassen C. D., Reisman S. A. (2010). Nrf2 the rescue: Effects of the antioxidative/electrophilic response on the liver. Toxicol. Appl. Pharmacol. 244, 57––65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Kong A. N. (2009). Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 48, 91––104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Yu S. W., Kong A. N. (2006). Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J. Biol. Chem. 281, 27251––27263 [DOI] [PubMed] [Google Scholar]
- Moi P., Chan K., Asunis I., Cao A., Kan Y. W. (1994). Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U.S.A. 91, 9926––9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon M. S., Kang B. H., Krzeminski J., Amin S., Aliaga C., Zhu J., McDevitt E. I., Kocher S., Richie J. P., Isom H. C. (2011). 3,5,5-trimethyl-hexanoyl-ferrocene diet protects mice from moderate transient acetaminophen-induced hepatotoxicity. Toxicol. Sci. 124, 348––358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelsen C. J., Rickheim D. G., Timchenko N. A., Stanley M. W., Albrecht J. H. (2001). Transient expression of cyclin D1 is sufficient to promote hepatocyte replication and liver growth in vivo. Cancer Res. 61, 8564––8568 [PubMed] [Google Scholar]
- Nichols G. M., Bacon B. R. (1989). Hereditary hemochromatosis: Pathogenesis and clinical features of a common disease. Am. J. Gastroenterol. 84, 851––862 [PubMed] [Google Scholar]
- Nielsen P., Heinrich H. C. (1993). Metabolism of iron from (3,5,5-trimethylhexanoyl)ferrocene in rats. A dietary model for severe iron overload. Biochem. Pharmacol. 45, 385––391 [DOI] [PubMed] [Google Scholar]
- Nielsen P., Heinelt S., Dullmann J. (1993). Chronic feeding of carbonyl-iron and TMH-ferrocene in rats. Comparison of two iron-overload models with different iron absorption. Comp. Biochem. Physiol. C. 106, 429––436 [DOI] [PubMed] [Google Scholar]
- Okawa H., Motohashi H., Kobayashi A., Aburatani H., Kensler T. W., Yamamoto M. (2006). Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 339, 79––88 [DOI] [PubMed] [Google Scholar]
- Petrat F., de Groot H., Rauen U. (2001). Subcellular distribution of chelatable iron: A laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem. J. 356, 61––69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman S. A., Yeager R. L., Yamamoto M., Klaassen C. D. (2009). Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol. Sci. 108, 35––47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A., Luukkaala T., Fleming R. E., Britton R. S., Bacon B. R., Parkkila S. Global transcriptional response to Hfe deficiency and dietary iron overload in mouse liver and duodenum. PLoS One. (2009);4,:e7212. doi: 10.1371/journal.pone.0007212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007). The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell Proteomics 6, 1638––1655 [DOI] [PubMed] [Google Scholar]
- Smith A. G., Francis J. E., Carthew P. (1990). Iron as a synergist for hepatocellular carcinoma induced by polychlorinated biphenyls in Ah-responsive C57BL/10ScSn mice. Carcinogenesis 11, 437––444 [DOI] [PubMed] [Google Scholar]
- Sumida Y., Yoshikawa T., Okanoue T. (2009). Role of hepatic iron in non-alcoholic steatohepatitis. Hepatol. Res. 39, 213––222 [DOI] [PubMed] [Google Scholar]
- Tang W. H., Shilov I. V., Seymour S. L. (2008). Nonlinear fitting method for determining local false discovery rates from decoy database searches. J. Proteome. Res. 7, 3661––3667 [DOI] [PubMed] [Google Scholar]
- Troadec M. B., Courselaud B., Detivaud L., Haziza-Pigeon C., Leroyer P., Brissot P., Loreal O. (2006). Iron overload promotes Cyclin D1 expression and alters cell cycle in mouse hepatocytes. J. Hepatol. 44, 391––399 [DOI] [PubMed] [Google Scholar]
- Valko M., Leibfritz D., Moncol J., Cronin M. T., Mazur M., Telser J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44––84 [DOI] [PubMed] [Google Scholar]
- Young I. S., Trouton T. G., Torney J. J., McMaster D., Callender M. E., Trimble E. R. (1994). Antioxidant status and lipid peroxidation in hereditary haemochromatosis. Free Radic. Biol. Med. 16, 393––397 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.