

Abstract
The HIV-1 nucleocapsid (NC) is a RNA/DNA binding protein encoded within the Gag polyprotein, which is critical for the selection and chaperoning of viral genomic RNA during virion assembly. RNA/DNA binding occurs through a highly conserved zinc-knuckle motif present in NC. Given the necessity of NC-viral RNA/DNA interaction for viral replication, identification of compounds that disrupt the NC-RNA/DNA interaction may have value as an anti-viral strategy. To identify small molecules that disrupt NC-viral RNA/DNA binding a high-throughput fluorescence polarization assay was developed and a library of 14,400 diverse, drug-like compounds was screened. Compounds that disrupted NC binding to a fluorescence-labeled DNA tracer were next evaluated by differential scanning fluorimetry to identify compounds that must bind to NC or Gag to impart their effects. Two compounds were identified that inhibited NC-DNA interaction, specifically bound NC with nM affinity, and showed modest anti-HIV-1 activity in ex vivo cell assays.
INTRODUCTION
The HIV-1 nucleocapsid (NC) protein is a small basic 7 kDa protein encoded within the Gag polyprotein of the infected cell and is cleaved from Gag by the viral protease during maturation (For review, please see Muriaux and Darlix 2010)1. NC provides viral genomic binding specificity allowing viral RNA selection from amongst the cellular host RNAs, as well as the annealing of the tRNAlys to the primer-binding site.2–5 The NC protein is also required for proper dimer formation of genomic RNA in the virus nucleocapsid and in later stages is critical for facilitating reverse transcription.6, 7 Moreover, it has been shown that altered NC processing from the precursor Gag polyprotein by the viral protease during maturation affects stability of the RNA dimer and thus impairing infectivity of the virus.8
NC interacts with a 120 nt RNA segment via hydrophobic and hydrophilic interactions, termed the ψ-site, which is located between the viral 5’ Long Terminal Repeat (LTR) and the gag start codon in the genomic RNA.9 The structure of NC bound to stem-loop 3 (SL-3), one of four stem-loops present in the ψ-site, was solved and shown to be composed of two highly conserved CCHC-type zinc knuckles (a Cys-Xaa2-Cys-Xaa4-His-Xaa2-Cys motif) which directly interact with RNA/DNA.10, 11 Although the NC protein binds RNA with high affinity, the NC protein has been shown to bind a broad variety of nucleotide sequences and synthetic oligonucleotides.1
Given the required role of NC in genomic RNA selection and interaction during viral encapsidation, as well as early and late post-entry events, it is considered a potential anti-viral target. Moreover, the highly conserved NC zinc-knuckle motif, found in all HIV-1 subtypes, is required for viral RNA interaction and presumably has a low tolerance for drug-resistance mutations.1, 12 These biological properties of NC make it ideal for small molecule targeting. Screening efforts to identify compounds that directly disrupted NC-RNA interaction were undertaken in the early 1990s, resulting in inhibitors that promoted zinc ejection. Although these identified compounds demonstrated anti-viral effects, they had a number of shortcomings, including NC specificity and cellular toxicity.12–15 Newer reported compounds that disrupt NC-RNA/DNA interaction include Trp-containing peptides, nucleomimetics, RNA aptamers, and gallein-related compounds.16–21 Interference of the NC-RNA/DNA binding mechanism by targeting the SL-3 stem-loop, rather than the NC protein, has also led to inhibitors that have not yet been tested in anti-viral assays.12, 22 Recently, Shvadhak and colleagues have focused on the inherent NC chaperone activity for RNA and have identified five compounds that disrupt NC-cTAR DNA interaction, a stem–loop sequence complementary to the transactivation response element, without coordinating through the zinc-knuckle / finger motifs. 23 However, these compounds have yet to be evaluated for anti-HIV-1 activity in cellular assays.23 Chemotypes of N-substituted S-acyl-2-mercaptobenzamides (SAMTs) have been identified which disrupt NC zinc coordination resulting in NC protein aggregation and diminished protease processing.24–26 Thus, NC inhibitors can have a number of distinct modes of action.12, 25 Despite the significant screening efforts for identification of NC inhibitors, the lack of compound specificity as well as cellular toxicity of compounds identified has hampered the progression of NC anti-virals to the clinic.
To identify compounds that disrupt NC-RNA/DNA interaction, we developed a high-throughput screening (HTS) platform utilizing fluorescence polarization (FP) as a primary screen and differential scanning fluorimetry (DSF) as a secondary screen. The combination and order of the assays allowed us to first identify compounds that disrupted NC-DNA interaction and then in the secondary screen determine which of the compounds required NC binding for the disruption effect. A total of 14,400 drug-like compounds were systematically screened, 5 compounds were selected based on their NC-DNA disruption and NC binding affinity profiles. Of the 5 compounds selected, 2 demonstrated modest anti-HIV-1 activity in tissue culture infection assays. These compounds may provide a starting point for developing molecules with potential anti- HIV-1 effects.
RESULTS
Establishment and validation of screens for compound that disrupt nucleocapsid/DNA interaction
A fluorescence polarization (FP)-based competitive equilibrium-binding assay was utilized as the primary, high-throughput screen (HTS) to identify small molecules (Hits) from the HitFinder™ library that disrupt HIV-1 nucleocapsid (NC) protein - DNA interaction. The small molecule library collection from Maybridge contains 14,400 selected compounds with drug-like diversity, which complies with Lipinski’s Rule of Five. The FP-based HTS employed a simple one-step “mix-and measure” FP design, which was ideal for miniaturization and HTS automation. A 6- carboxyfluorescein-labeled SL-2 DNA (FSL-2), termed the tracer, was developed to interact with the zinc-knuckle motifs present in a GST-p2-NC fusion protein (NC’), termed the receptor (Figures 1A, B). The DNA bases selected for FSL-2 corresponds to the 12 RNA bases of the viral SL-2 RNA, which provides for contact with the NC zincknuckle motifs.27 Given that several studies have suggested that the spacer peptide 1 (SP1 or p2) influences RNA packaging and dimerization, the spacer was included in the GST-p2- NC fusion protein reporter.28–30
Figure 1.

Methodology for identifying compounds that disrupt nucleocapsid-RNA/DNA interaction. (A) Fluorescence polarization (FP) assay components. Shown are the N-terminal GST-tagged Gag (Gag’) fusion protein and N-terminal GST-tagged receptor nucleocapsid (NC’) fusion protein, containing, the spacer peptide 1 (p2) and nucleocapsid domains. NC and Gag are from HIV-1NL4-3. NC contains two zinc-knuckle motifs that interact with viral DNA or RNA. The 3'-6-carboxyfluorescein-labeled SL-2 DNA (FSL-2) is a tracer molecule that interacts with the NC’ zinc-knuckle motifs. (B) Electrophoresis mobility shift assay confirms interaction of FSL-2 with NC’ or full-length Gag’. Note that GST alone does not shift FSl-2. (C) A fluorescence polarization binding study of FSL-2 and NC’ interaction revealed a Kd of 324 ± 6 nM (mean ± s.e.m.). The data shown represents the mean ± s.e.m. of three independent experiments performed in duplicate. (D) Thermal protein denaturation of Gag’ or maturation inhibitor Bevirimat-Gag’ complex using differential scanning fluorometry (DSF). The maturation inhibitor Bevirimat increased the thermal stability of the Gag’ protein. The black line indicates a nonlinear curve fit based to the Boltzmann equation. The result shown is one representative measurement from three independent experiments.
SL-2 binding to NC’ and the Gag polyprotein (Gag’) was first validated with an electrophoresis mobility shift assay (EMSA) (Figure 1B), as well by Native PAGE utilizing a fluorescent protein fusion variation of NC’ (Supplementary Figure 1C). Importantly, FSL-2 binding to NC’ had a stabilizing effect resulting in a homogenous protein form, rather than an oligomerized form, an often-reported problem when using the disordered apo-form of zinc-knuckle / finger proteins (Figure 1B and Supplementary Figure 1C).1 The robust interaction of FSL-2 with NC’ revealed an 8-fold change of fluorescence polarization which provided an excellent signal to noise (S/N) ratio, high to low (H/L) signal ratio, and a Z’ factor of 0.84 ± 0.02 (mean ± s.d.) (Supplementary Figures 1D, E).31 The NC’-FSL-2 dissociation constant was determined to be 324 ± 6 nM (Figure 1C).
To ensure that we identified compounds that bound to NC in the context of full-length Gag to disrupt FSL-2 interaction, thereby potentially increasing in situ specificity, a differential scanning fluorimetry (DSF) screen was established. The rational for the inclusion of this screen was to allow a rapid throughput and determination if binding of the compound to NC increased or decreased the melting temperature, thus indicating that the Gag - compound complex was more thermally stable than Gag alone. The feasibility of this methodology was validated by denaturing the Gag’ protein in the presence and absence of the HIV-1 maturation inhibitor, Bevirimat (PA-457), which is reported to function in blocking maturation via Gag interaction.32 As seen in Figure 1D Gag’ in complex with Bevirimat has a higher melting temperature (Tm) than Gag’ alone, consistent with the notion that Bevirimat interacts with Gag and increases its thermal stability. This finding supports the use of the DSF assay for identifying compounds (Selected Hits) that bind to and increase or decrease NC’ and Gag’ stability. To place the screening and Hit validation strategy in a temporal perspective a flowchart is shown in Figure 2A, with Figure 2B providing an enumeration of the successful Hits identified during screening and the cellular validation process.
Figure 2. Overview of the strategy for identifying compounds that disrupt HIV-1 nucleocapsid/RNA interaction.

(A) Outline of the experimental procedure. (B) Enumeration of Hits during library screening. Utilizing a fluorescence polarization assay to detect small molecule displacement of 3'-6-carboxyfluorescein-labeled SL-2 DNA (FSL-2) from binding to the nucleocapsid (NC’) protein, 14,400 compounds were screened and 101 compounds (Hits) were identified as displacing FSL-2. The 101 Hits were retested for NC’-FSL-2 displacement and 65 did not show activity or demonstrated < 60 % FSL-2 displacement activity and were eliminated from further study. Of the remaining 36 compounds (Selected Hits), 18 failed to show a significant shift in thermal stability when the compound-Gag complexes were evaluated by differential scanning fluorometry assay, 8 displayed promiscuous β-galactosidase inhibition, and 5 were excluded due to low affinity binding to NC’. Of the 5 remaining Selected Hits, 3 Hits showed cellular toxicity at higher compound concentrations, whereas, 2 compounds demonstrated both low cellular toxicity and inhibition of HIV-1 replication in CD4+ T cells.
Compounds that disrupt nucleocapsid/DNA interaction
Use of the FP HTS for interrogating the 14,400 compound HitFinder™ library, resulted in 101 Hits that disrupted NC’- FSL-2 interaction (Figure 2B). Hit identification was based on two quantitative criteria: firstly, disruption of NC’-FSL-2 interaction was required to be > 50 % at 10 µM, and secondly, intrinsic compound fluorescence was negated by comparing FP to total fluorescence (Supplementary Figure 1D).33 Next, we determined whether the 101 Hits identified were functioning to disrupt NC’-FSL-2 by binding to NC’ in the context of the polyprotein Gag and forming a stable complex. Of the 101 compounds, 36 displayed ΔTm > 2 °C when analyzed by DSF, the criterium established for selection, but only 18 of the 36 compounds also demonstrated inhibition of NC’-FSL-2 interaction by more than 60 % (Supplementary Figure 2A). These 18 compounds were selected for further analyses (Figures 2A, B).
To determine if the 18 Selected Hits were promiscuous inhibitors two enzymes, beta-galactosidase and chymotrypsin commonly used to uncover compounds that bind promiscuously and inhibit enzymatic function, were used for evaluation.34 Of the 18 Selected Hits, 8 were eliminated since they demonstrated > 10 % inhibition of these enzymes at 10 µM (Supplementary Figure 2B). The remaining 10 compounds were selected for further evaluation (Figures 2A, B).
Biochemical profiling: Affinity and specificity evaluation of the 10 selected compounds
In order to quantify the inhibitor activity relationship of the 10 compounds the Kis were determined using the FP assay (see the Experimental Section for details) 35. It should be noted that newly synthesized compounds from Maybridge (see the Experimental Section for details) were utilized to avoid storage-associated effects. All 10 compounds were found to have Ki values in the nM range (Figures 3A, B). Moreover, the order of addition of compounds, NC', or FSL-2 did not alter compound effects or Ki (data not shown). To determine potential promiscuous activity, the 10 selected compounds were evaluated at higher concentrations utilizing DSF as well as the galactosidase and chymotrypsin enzymatic activity assays (data not shown). Five of the 10 compounds, 2, 3, 4, 6, and 7, displayed weaker affinity (Ki ≥ 100 nM), thermal stability (ΔTm ≤ 2 °C), and non-specific effects at higher concentrations (Figure 3), thus eliminating these compounds from further studies.
Figure 3.

Biochemical characterization of 10 compounds selected by fluorescence polarization and differential scanning fluorometry assays. (A) Correlation of DSF-based thermal stability versus FP-based Ki values of re-synthesized compounds. Only compounds with a Δ Tm ≥ 2 °C and a Ki ≤ 100 nM were selected for further evaluation. (B) Binding curve of compound 1 with NC’ in FP-based displacement experiment reveals a high affinity (Ki = 18 ± 14 nM). Shown is the mean ± s.d. of a representative experiment from 3 experiments. (C) DSF-based stability analysis of the compound 1 – Gag’ complex. Compound 1 enhances the stability of Gag’ by Δ Tm = 5.7 ± 1.0 °C (grey circles = control; black open circles = Gag’ + compound 1; nonlinear fit = black line).
The remaining 5 compounds, 1, 5, 8, 9, and 10, were next evaluated by EMSA for their ability to disrupt the NC’-FSL-2 complex. The pre-formed NC’-FSL-2 complex was incubated with the compounds, separated by Native PAGE, and the FSL-2 intensity was evaluated by fluorescence scanning. All compounds displaced FSL-2, confirming their disruption of the NC’-FSL-2 interaction (Figure 4A). Furthermore, studies indicated that the order of NC’, FSL-2, or compounds to the reaction mixture did alter the capacity of any of the 5 compounds to disrupt the NC’-FSL-2 complex (data not shown). This suggests that the compounds do not require prebinding to NC’ to impart their SL-2 inhibitory affect.
Figure 4.

Biochemical characterization of 5 selected compounds. (A) EMSA-based analyses of the binding of compounds 1, 5, 8, 9, or 10 to NC’ showing the displacement of FSL-2 from the NC’- FSL-2 complex or the internal control utilizing 10 µM unlabeled SL-2 DNA. (B) Resonant waveguide grating-based analyses of the binding of compounds 1, 5, 8, 9, or 10 to immobilized NC’. Real-time kinetics of compound binding was monitored by peak wavelength value shifts over time.
To determine NC’-compound stoichiometry, all 5 compounds were evaluated by a resonant waveguide grating-based assay employing SRU BIND™ technology.36 Resonant waveguide grating-based assessment has been useful for the evaluation of protein-drug interaction, protein-DNA inhibitors, as well as the identification of promiscuous binders to proteins.36 To evaluate the 5 compounds we immobilized NC’ on an optical biosensor, then monitored compound binding by measuring the peak wavelength value shift over time. The plateauing of NC’-compound binding kinetics implies a specific binding mode for compounds 1, 5, 8, and 10, with the NC’-compound stoichiometry determined to be approximately 1:1 (Figure 4B). Although compound 9 displayed a plateauing of NC’-compound binding kinetics, the stoichiometry was determined to be approximately 1:2. This finding is consistent with two molecules of compound 9 binding to NC. In summary, these findings indicate that the 5 selected compounds had Ki ≤ 100 nM, disrupted FSL-2 binding to NC’, recognized the NC domain in the Gag polyprotein, and 4 of the 5 compounds demonstrated 1:1 stoichiometry for NC’-compound interaction. Table 1 summarizes the chemical and biochemical findings for the 5 compounds.
Table 1.
Summary of the Biochemical and Biological Characterization of Compounds 1, 5, 8, 9, and 10
| Sample ID | MB ID | Name | PubChem ID |
MW (KDa) |
Ki (FP) nm (mean ± s.e.m.) |
DSF (°C) Δ Tm | optical biosensor (SRU bind) |
% inhibition (p24 assay) at 200 nM |
|---|---|---|---|---|---|---|---|---|
| CMPD-1 | 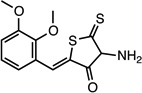 |
(Z)-4-amino-2-(2,3- dimethoxybenzylidene)-5- thioxodihydrothiophen-3 (2H)-one |
26532231 | 295.38 | 18 ± 14 | 4.8 ± 0.4 | n = 1:1; npa | 43 ± 8 |
| CMPD-5 | 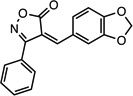 |
(Z)-4-(benzo[d][1,3] dioxol-5-ylmethylene)-3- phenylisoxazol-5(4H)-one |
26533388 | 293.27 | 20 ± 5 | 6.3 ± 0.1 | n = 1:1; np | 33 ± 10 |
| CMPD-8 | 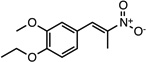 |
(E)-1-ethoxy-2- methoxy-4-(2-nitroprop-1- en-1-yl)benzene |
26541579 | 237.25 | 14 ± 7 | 2.2 ± 0.6 | n = 1:1; np | 50 ± 12 |
| CMPD-9 | 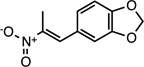 |
(E)-5-(2-nitroprop-1-en-1- yl)benzo[d][1,3]dioxole |
26529289 | 207.18 | 22 ± 9 | 3.4 ± 0.7 | n = 1:1.7; np | 18 ± 16 |
| CMPD-10 | 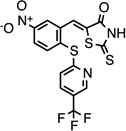 |
(Z)-5-(5-nitro-2-((5- (trifluoromethyl)pyridin-2- yl)thio)benzylidene)-2- thioxothiazolidin-4-one |
26540561 | 443.44 | 73 ± 50 | 2.4 ± 0.6 | n = 1:1; np | 0 ± 0 |
np = non-promiscuous binder
Biological profiling: Cellular toxicity and anti-HIV-1 activity of compounds 1, 5, 8, 9, and 10
The cellular toxicity of compounds 1, 5, 8, 9, and 10 were evaluated at concentrations of 0.1 and 10 µM at 3 and 7 days after addition to SupT1 T cells, a CD4+ T cell line (Figures 5A, B). At the 10 µM concentration, compounds 5, 8, 9, and 10 exhibited > 90 % cytotoxicity at 7 days (Figure 5A), at 30 µM compound 1 demonstrated > 70 % toxicity within 6 days (not shown). At the lower concentration of 0.1 µM compounds 1, 5, 8, 9, and 10 demonstrated 0 – 38 % cell toxicity within 7 days (Figure A), whereas at 1 µM compounds 5, 9, and 10 compound demonstrated > 70 % cell toxicity (not shown). In contrast to compounds 5, 9, and 10, compounds 1 and 8 evaluated at 0.1 or 1 µM exhibited < 3 % cell toxicity within 7 days (Figure 7A). Similar cellular toxicity patterns were seen with the HeLa cell line, as well as with activated, primary CD4+ T cells (not shown).
Figure 5.

Cellular toxicity and anti-HIV-1 evaluation of 5 selected compounds. (A) Cellular toxicity evaluation of compounds 1, 5, 8, 9, or 10. Cellular viability over time of SupT1 T-cells was evaluated in the presence of 0.1 or 10 µM of each of the 5 compounds using a fluorescence-based cytotoxicity assay. No compound control cultures contained 0.2 % DMSO, the diluent for the compounds, for comparison. (B) Inhibition of HIVLAI replication in primary CD4+ T cells. Cells were cultured with 0.2 µM compounds 1, 5, 8, 9, or 10 for 3 days before exposure to 100 ng p24 of HIVLAI. 9 days after HIV-1 exposure, cells were evaluated for infection by flow cytometry to determine cellular viral p24 and results were plotted as mean ± s.d. of % inhibition. 0.2 µM Amprenavir, a protease inhibitor, was used at 5-fold over EC50 as a known inhibitor of viral replication and was found to inhibit viral replication 98 % (data not shown). All assays were in triplicate and shown is 1 of 2 replicate experiments. (C) Compounds 1 and 8 disrupts HIV-1 production. The EC50s of compounds 1 and 8 were determined utilizing a HeLa cell line transiently transfected with plasmid HIVNL4-3 DNA in the absence or presence of increasing concentrations of each compound. HeLa cell culture controls contained 1% DMSO (v/v), the diluent used for the compounds. Viral production was determined by measuring p24 production. Results are shown as relative viral packaging in the presence of compounds as compared to DMSO controls. All points are the result of three independent transient transfections and results are shown as the mean ± s.d. of relative viral packaging. EC50s values were determined using non-linear regression analysis in Prism 5.0d to fit a three-parameter dose-response curve to normalized p24 values.
We next evaluated anti-HIV-1 activity of the 5 selected compounds on activated, primary CD4+ T cells. A concentration of 0.2 µM of the compounds was evaluated based on compound-NC’ affinity, Figure 3A, and the range of cellular toxicity, Figures 5A. Activated, primary CD4+ T cells were cultured for 3 days in the presence of compounds 1, 5, 8, 9, and 10, and then infected with 100 ng p24 of HIVLAI. After 9 days the level of HIV-1 infection in CD4+ T cells was enumerated through detection of infected, intracellular p24 positive T cells present in the culture (Figure 5B). Relative to the control cultures, compound 10 did not exhibit a viral inhibitory affect. In contrast, compounds 1, 5, 8, and 9 modestly reduced T cell infections from 18 – 50 %, with compounds 1 (43 ± 8% viral inhibition), 5 (33 ± 10 % viral inhibition), and 8 (50 ± 12 % viral inhibition) showing the greatest anti-HIV affect (Figure 5B and Table 1).
Compounds 1 and 8 were selected for further anti-HIV-1 evaluation given their slightly better anti-viral activity than compounds 5, 9, and 10. Moreover, the toxicity of 5, 9, and 10 precluded additional cell-based studies. The viral spreading assay used for the studies shown in Figure 5B allows all steps of viral replication to be targeted by an inhibitory compound. To determine inhibitory EC50s for compounds 1 and 8 we utilized a HIV DNA transient transfection assay to produce virus from HeLa cells in the presence or absence of each compound. This procedure bypasses the early stages of the viral replication cycle and provides information on compound activity in later stages of viral replication (ie, post-integration). Viral production was quantified by determining p24 amounts. When this assay was utilized the EC50 for compound 1 was determined to be 3.5 µM, whereas for compound 8 the EC50 was 0.32 µM (Figure 5C).
DISCUSSION
The primary role of NC during viral replication and its inability to tolerate mutations makes it an ideal target for small molecule interaction to disrupt HIV-1 replication.37 Moreover, a 20–30 % reduction in Gag processing results in a 3 log decrease in viral production.38 We have identified two compounds that interfered with NC’- and Gag’- DNA interaction in biochemical assays and viral replication in primary CD4+ T cells. A simple series of assays were developed that required compounds to disrupt NC’-DNA interaction for positive selection and a secondary screen to identify compounds that bound to NC’ to impart their disruptive affect. This second screen ensured that the mode of action of the compound requires NC’ binding and is not simply the result of compound interacting with DNA. Moreover, the use of the Gag polyprotein for later compound assessment takes into account the knowledge that NC domain and viral RNA interaction takes place in the context of the Gag polyprotein in host cells. The use of the 14,400 drug-like, small molecule HitFinder™ library for screening may have increased the probability for identifying compounds with favorable biological and chemical activities, as well as possibly reducing cellular toxicity and promiscuous inhibitory activities. Compounds 1, 5, 8, 9, and 10 formed stable complexes with NC’ (Figure 3A and Table 1), and except for compound 9, demonstrated NC’-compound binding stoichiometries of 1:1 (Table 1).
Of the 5 compounds selected for biological evaluation, compounds 1 and 8, which demonstrated modest anti-HIV affect in primary CD4+ T cells at 200 nM, 1, 43 ± 8 % viral inhibition and 8, 50 ± 12 % viral inhibition, respectively, had the lowest FP Ki, values, with no overt promiscuous activity (Figure 5 and Table 1). The inhibitory evaluation of compounds 1 and 8, the least toxic of the 5 compounds, in the viral packaging assay yielded EC50s of 3.5 µM for compound 1 and 32 nM for compound 8. Interestingly, although compound 8 showed comparable inhibitory activity in both the viral spreading assay (Figure 5B) and the viral production assay (Figure 5C), compound 1 did not show comparable inhibitory activity in the two assays. Whether the differences in the inhibitory activity of compound 1 reflect targeting of earlier stages of viral replication or cell-based mechanisms supporting viral replication, in addition to the later stages of viral replication, is unclear at the current time and is the subject of investigation. Nonetheless both compounds exhibited modest anti-viral activity.
SMAT-based NCp7 inhibitors have been shown to disrupt protease processing of Gag as the result of compound-NC interaction, followed by inhibitor-mediated NC aggregation resulting in decreased HIV-1 replication in cell culture.25 To evaluate whether the binding of compounds 1 and 8 to NC interfered with Gag polyprotein processing we monitored p2/NC and NC/p1 cleavage in the presence and absence of both compounds utilizing the Cleavage Enzyme-Cytometric Bead Array, a Gag-specific cleavage assay (data not shown).39 Our results revealed that compounds 1 and 8 did not interfere with HIV protease enzymatic function or block Gag cleavage. Thus, the inhibitory mechanism of compounds 1 and 8 in cell culture was most likely not the result of either compound interfering with Gag cleavage. Furthermore, and in contrast to the gallein-based inhibitors, compounds 1 and 8 inhibited NC’-DNA interaction independent of the order of addition of the compounds or DNA in the FP assay. The gallein-based NC inhibitors require binding to free NC’ before the addition of DNA to impart their inhibitory affect.21 Based on the differing NC binding requirements for gallein-based and compounds 1 and 8 for inhibitory function, our findings might suggest that compounds 1 and 8 bind to different NC locations than the gallein-based inhibitors to impart their anti-HIV effects.
Evident in all compounds obtained are similar chemical motifs, an electron-rich aromatic ring structure in conjugation with an electron-withdrawing nitro-substituent or an electron-poor heterocycle (Table 1). A closer evaluation of the 5 compounds revealed nitroalkenes in compounds 8 and 9, rhodanine in compound 10, and isoxazolone in compound 5, features suggestive of pan assay interference compounds, which can correlate with undesirable chemical properties.40 However, the behavior of the 5 compounds in the resonant waveguide-grating assessment was inconsistent with either reactivity or aggregation (Figure 4B). Moreover, mass spectrometry analysis did not reveal covalent binding between NC’ and any of the compounds (Supplementary Table 1). We next searched the PubChem Compound Database to determine if screening information was available on any of the 5 compounds identified.41 The search indicated that compounds 1, 5, and 8 were not active in 7 or more assays against various protein targets, whereas compound 10 were found to be active in 1 out of 7 assays (Supplementary Table 2). Compound 9 demonstrated promiscuous behavior in some assays, showing activity in 13 of 78 assays, albeit mostly from a related group of anti-cancer screens. Although the perusal of the PubChem Compound Database was suggestive of limited protein reactivity, compounds 5, 9, and 10 were cell toxic at moderate compound concentrations. Given the potentially undesirable chemical properties of compounds 5, 9, and 10, as discussed, further assays are required to fully evaluate potential off-target effects.
Interestingly, compounds 1, 5, 8, and 9 have some chemical similarities to E03 and H04, 2 compounds identified by Shvadchak and colleagues as NC polyprotein binders capable of disrupting DNA binding (Supplementary Figure 3).23 However, anti-HIV activities of those 2 compounds were not evaluated. Both compounds display similar structural features as compounds 8 and 9 regarding the benzene ring structure, but have branching oxygen and/or hydroxyl groups in contrast to the pendent nitro groups present on compounds 8 and 9 (Supplementary Figure 3). In the case of the compounds identified by Shvadchak and colleagues, the oxygen and/or hydroxyl groups likely have a role in NC binding. Moreover, their studies indicated that activity was not coordinated through interaction or zinc ejection, but possibly through competing with DNA binding to NC.23 Given similar chemical structural features of these compounds, and the compounds identified in our screen, it is tempting to speculate that our compounds may function to compete with DNA binding to NC.
In conclusion, development and implementation of our screen has resulted in the identification of 5 compounds, which are capable of disrupting p2-NC and Gag polyprotein interaction with SL-2 DNA. Of the 5 compounds, 2 were not overtly toxic to primary CD4+ T cells and both had modest effects on disrupting HIV-1 spread in tissue culture. Although NC binding is required for the inhibitory effect of the compounds, whether the compounds’ activity was due to DNA binding competition resulting from direct inhibition of the zinc knuckle motifs, or resulting from disruption of DNA binding caused by allosteric changes due to a distal NC binding site, is currently unknown. Efforts are planned to structurally define the NC binding sites of compounds 1 and 8 to provide rational insights for improving new compounds to enhance anti-HIV-1 efficacy. Lastly, given the high-throughput capacity of the FP-based NC' binding assay and the relative ease of use, it will be informative to screen a larger and more diverse chemical library to identify compounds that may have anti-viral activity.
EXPERIMENTAL SECTION
Cloning and recombinant protein synthesis
The HIV-1 NL4-3 p2-nc gene was cloned into the pGex4T2 vector (GE Healthcare) vector using BamHI and XhoI restriction to generate GST-p2-NC (NC’), transformed into BL21DE3(pLysS) (Invitrogen), inoculated into 1 – 6 liters (depending on need) of LB medium with ampicillin (100 µg/ml) and maintained at 28 °C for 16 hrs. At 0.8 OD600nm, expression was induced with 0.5 mM isopropyl-beta-D-thiogalactospyranosid. After 16 hrs, the cells were harvested by centrifugation (6,000 g, 15 min) and washed with buffer A (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 0.5 mM DTT, and 0.5 % NP40). Cells were then lysed by the addition of lysozyme and the use of a French press. To avoid bacterial protease activity, a protease inhibitor mix (Complete, Mini Protease inhibitor Tablets, Roche) and pepstatin A (Sigma-Aldrich) were added. DNA was removed from the lysate by adding 0.04 mg/ml DNase I for 1 hr. After separation by centrifugation (30,000 g, 45 min), the supernatant was loaded onto a GSH column (GE Healthcare) and washed with 20 CV buffer A. The protein was eluted by addition of 20 mM GSH in buffer A and the purity verified by SDS-PAGE. The protein was concentrated using Amicon Ultra-15 filter devices (Millipore). The protein size was confirmed by MALDI MS. All purification steps were conducted at 4 °C. GST-Gag (termed Gag’) was produced as reported previously.39
Electrophoretic mobility shift assay (EMSA)
15 µM NC’ or Gag’ was incubated with 0.5 µM FSL-2 in PBS with 10 nM ZnCl2. For the electrophoretic analysis, 15 µl samples were loaded on a NativePAGE Novex Bis-Tris Gel (Invitogen), run, and fluorescence measured by a Typhoon Trio fluorescence gel scanner (GE Healthcare).
Fluorescence polarization (FP)
The binding reaction was carried out in 384-well, low volume plates (Greiner) using 2.5 – 10,000 nM NC’ in PBS, 10 nM ZnCl2, 0.1 % pluronic F127 (SIGMA), and 10 nM 3’ fluorescein-labeled SL-2 DNA, GGGGCGACTGGTGAGTACGCCCC (Integrated DNA Technologies). The millipolarization (mP) units were calculated using:
S = fluorescence S-channel; P = fluorescence P-channel; G- = instrument G-factor The Kd was calculated using the least-squares fit to the equation:
where P0 is the total receptor concentration, L0 is total ligand concentration, Kd is the binding constant and FP is the fluorescence polarization.
IC50and Ki determination by FP
The Ki value was determined by a compound series dilution into 5 µM NC’ and 10 nM 6-carboxyfluorescein-labeled SL-2 DNA in PBS, 10 nM ZnCl2, and 0.1 % pluronic F127. All experiments were in triplicate. The NC’ concentration was chosen such that 90 % of tracer was bound. Since the concentration of free tracer was not equal to the concentration of total tracer, the Cheng-Prusoff equation could not be used. The IC50value was calculated using a four parameter logistic model, and the Ki value was determined by the as established by Kenakin 199335 and recommended by NIH Chemical Genomics Center: http://assay.nih.gov/assay/index.php/Section5:Practical_Use_of_Fluorescence_Polarization_in_Competitive_Receptor_Binding_Assays:
R0 = total receptor concentration; L0 = total tracer concentration; Lb = bound tracer fraction; Kd = dissociation constant
Small-compound library and high-throughput screening
The HitFinder™ library collection from Maybridge contains 14,400 compounds with drug-like diversity and the compounds conform to Lipinski’s Rule of Five with purity > 90 % based on mass spectrometry and NMR by the vendor (http://www.maybridge.com/portal/alias__Rainbow/lang__en/tabID__229/DesktopDefault.aspx). For single compounds used for biochemical and biological profiling a purity of > 95 % was confirmed by HPLC.
The binding reaction was carried out in 384-well low volume plates (Greiner) using 5 µM (tracer bound to receptor > 80%) NC’ in PBS, 10 nM ZnCl2, 0.1 % pluronic F127 (SIGMA), and 10 nM of the tracer 6-FAM-labeled SL-2 DNA. The compounds were dissolved in DMSO and used in a final concentration of 10 µM at room temperature. Dispensing and readout were executed using automated liquid handling systems (dispenser Aurora, liquid robot BiomekFX Beckman Coulter) and an EnVision 2104 (PerkinElmer) fluorescence plate reader. NC’ and compounds were pre-incubated at RT for 15 min in a volume of 10 µl before 10 nM of tracer was added to the solution. The mix was allowed to establish equilibrium, followed by FP readout 15 min later. The plate layout provided for 16 positive controls (tracer without NC’) and 16 negative controls (tracer with NC’) per plate. The Z’-factor was calculated using the equation established by Zhang and colleagues. 31
Differential Scanning Fluorimetry (DSF)
The melting temperatures of protein-compound complexes were determined using differential scanning fluorimetry. 5 µM Gag’ protein was incubated with 5X SYPRO Orange (Invitrogen) and 5 µM compound in 5 mM HEPES pH 7.5 with 150 mM NaCl. Fluorescence values were monitored in a LightCycler 480 II (Roche) instrument with temperature increased at 1°C/min, from 35–94 °C. The data were fitted to a Boltzmann sigmoid curve function and the Tm obtained as described by Vedadi et al., 2006 42 using GraphPad Prism. A shift of 2 °C corresponds to a 4-fold change of the standard deviation and was considered to be a hit.
Beta-Galactosidase Assay
To determine off-target hit inhibition, the conversion of 0.2 mM ONPG (Sigma) by 0.5 U/ml β-galactosidase (Sigma) was monitored in the presence or absence of 10 µM of the selected hit. The reactions were established in PBS with 1 % DMSO at 25 °C. The absorption at 420 nm was followed over time using an µQuant 800 microplate reader (Biotek). The rate was calculated from the initial linear phase of the enzyme reaction and the normalized inhibition was calculated relative to the rate of the DMSO control reaction.
Chymotrypsin Assay
0.5 U/ml chymotrypsin (Sigma) was mixed with 0.2 mM S-2586 (DiaPharma) and 10 µM of selected compounds in 20 mM Tris/HCl pH 8.3, 200 mM NaCl, 1 mM CaCl2, and 1 % DMSO, all at 25 °C. Absorption at 420 nm was followed over time using an JQuant 800 microplate reader (Biotek). The initial phase was fitted with a linear regression and the relative rate calculated for each compound.
Resonant Waveguide Grating-Based Assay
To evaluate label-free interaction between NC’ and selected compounds, the SRU BIND™ system (SRU Biosystems) was utilized. 5 µM of NC’ protein was immobilized on the wells of an aldehyde activated biosensor surface 384-well GA3 aldehyde plate (SRU Biosystems). Selected compounds at 25 µM in PBS with 1 % DMSO, were added to plated wells. The peak wavelength value (PWV) shift was monitored over the time at room temperature in a BIND® SCREENER reader (SRU Biosystems). The stoichiometry was calculated according to the vendor’s manual using the following equation:
n = stoichiometry; z = target activity in %
Cellular Toxicity Assay
The SupT1 T cell line or human CD4+ T cells were cultured in RPMI1640, containing 10 % fetal bovine serum (FBS), 2 mM L-glutamine, 0.1 mM non-essential amino acids (NEAA), 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 µg/ml streptomycin for all studies (see below for methodology for T cell isolation). Cells were maintained in a 37 humidified incubator containing 5 % CO2. Viability of the cells in presence of selected compounds was measured either by the conversion of the oxidation-reduction indicator resazurin (Alamar Blue) (Invitrogen) to resorufin43, 44 or enumerating dead vs. viable cells by Trypan Blue dye exclusion by cell counting utilizing a microscope. 15,000 SupT1 cells were cultured over 7-days with a series of selected compounds (0.01 – 10 µM) in 1 % DMSO and the viability was determined in the presence of Alamar Blue according to the manufacturer’s protocol. The 585 nm fluorescence value was normalized to a DMSO control. For primary T cells isolated from blood cells, and in some cases Sup T1 T cells cultured in the presence of selected compounds (0.01 – 10 µM) and viability determined at days 3 and 6 or 7 by Trypan Blue dye exclusion.
Determination of Compound Anti-HIV-1 Activity
After approval from The Scripps Research Institute (TSRI) Institutional Review Board (documentation is on file), La Jolla, CA, USA, human blood was obtained from healthy donors from the Normal Blood Donation Center at TSRI. PBMCs were isolated from blood via density-gradient centrifugation. Isolation of highly pure CD4+ T cells was achieved by depletion using Miltenyi products (Miltenyi Biotech). CD4+ T cell purity, > 95 %, was determined by flow cytometry with appropriate reagents. CD4+ T cells were maintained, in RPMI1640 medium, supplemented with 10 % FBS (HyClone), 100 U/ml IL-2, 2 mM L-glutamine, 0.1 mM NEAA, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 µg/ml streptomycin in a 37 °C humidified incubator containing 5 % CO2. After a 1-day stimulation with anti-CD3/CD28 beads (Dynabeads® Human T-Activator CD3/CD28, Invitrogen), 0.2 µM of selected compounds in 1 % (w/v/) DMSO were added for 3 days, then 1×105 CD4+ T cells were infected with 100 ng p24 HIV-1LAI virus. Cultures were maintained for 9 days and infection was determined by p24 flow cytometry analysis. Briefly, HIV-1 infected CD4+ T cells were permeabilized and fixed with Cytofix/CytopermTM buffer, followed by adding PE-conjugated HIV p24 antibody (Beckman Coulter) to a final 1:200 dilution. Data were acquired on Becton Dickinson FACScalibur using CELLQUEST software and analyzed with FlowJo (TreeStar) software. The FDA approved protease inhibitor amprenavir was used as a positive inhibitor of viral replication (data not shown). The significance of the difference between the DMSO control and a test compound was determined using a two-tailed t-test.
To determine the effective dose of compound to inhibit viral packaging in HeLa cells a viral packaging assay was utilized. Selected compounds were added to 2×105 HeLa cells in 1ml of Opti-MEM (Invitrogen) in 12-well plates. After 2 days, cells were transiently transfected with 1Jg of plasmid DNA of the pNL4-3 infectious molecular clone using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer’s instructions. Cell-free viral supernatants were collected 2 days post-transfection and p24 determined by CFAR at the University of California, San Diego, CA, utilizing a commercial p24 ELISA (http://cfar.ucsd.edu/core-facilities/translational-virology-core).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Drs. Hugh Rosen, Steven Brown and Jacqueline Lohse for providing valuable technical expertise with the high-throughput screening platform at The Scripps Research Molecular Screening Center, The Scripps Research Institute, La Jolla, CA. We thank Sheryll Espinola and Dr. Ingo H. Engels from the Genomics Institute of the Novartis Research Foundation, La Jolla, CA, for their expertise in SRU Bind™ assay technology. Karen Crain is thanked for helping with the cell culture experiments. M.G. Finn is gratefully thanked for his helpful discussions of chemical properties of the identified compounds. Drs. Christopher Aiken and Norman Chen, Vanderbilt University, Nashville, TN, gratefully provided Bevirimat. The studies were supported from the following NIH grants: 5 P01 GM083658 (BET), 5 R01 and HL091219 (BET), core support from CFAR, 5 P30 AI036214. JY is supported from 2T32AI007354 and S.B. is supported by CHRP-F09-SRI-205. This is publication MEM #21430 from The Scripps Research Institute.
ABBREVIATION LIST
- HIV
human immunodeficiency virus
- NC
nucleocapsid
- NC’
GST-p2-NC
- p2
spacer peptide 1
- Gag
group specific antigen
- SL
stem-loop
- Gag’
GST-Gag
- nt
nucleotides
- FSL-2
fluorescein-labeled SL-2 DNA
- DSF
differential scanning fluorimetry
- FP
fluorescence polarization
- HTS
high-throughput screening
- CMPD
compound
- GST
glutathione S-transferase
- s.d.
standard deviation
- s.e.m.
standard error of the mean
Footnotes
ASSOCIATED CONTENT
Supporting Information Available. Supporting information contains quality control and assay development of the FP-based HTS, positive and negative hit selection, analysis of NC’-compound complexes, structural comparison of selected small-molecule NC’ inhibitor compounds, summary of the mass spectrometry analysis of the NC’ – compounds, and PubChem information on the selected compounds. This material is free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Muriaux D, Darlix JL. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 2010;7:744–853. doi: 10.4161/rna.7.6.14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muriaux D, Mirro J, Harvin D, Rein A. RNA is a structural element in retrovirus particles. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5246–5251. doi: 10.1073/pnas.091000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mark-Danieli M, Laham N, Kenan-Eichler M, Castiel A, Melamed D, Landau M, Bouvier NM, Evans MJ, Bacharach E. Single point mutations in the zinc finger motifs of the human immunodeficiency virus type 1 nucleocapsid alter RNA binding specificities of the gag protein and enhance packaging and infectivity. J. Virol. 2005;79:7756–7767. doi: 10.1128/JVI.79.12.7756-7767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jouvenet N, Simon SM, Bieniasz PD. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA. 2009;106:19114–19119. doi: 10.1073/pnas.0907364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darlix JL, Garrido JL, Morellet N, Mely Y, de Rocquigny H. Properties, functions, and drug targeting of the multifunctional nucleocapsid protein of the human immunodeficiency virus. Adv. Pharmacol. 2007;55:299–346. doi: 10.1016/S1054-3589(07)55009-X. [DOI] [PubMed] [Google Scholar]
- 6.Feng YX, Campbell S, Harvin D, Ehresmann B, Ehresmann C, Rein A. The human immunodeficiency virus type 1 Gag polyprotein has nucleic acid chaperone activity: possible role in dimerization of genomic RNA and placement of tRNA on the primer binding site. J. Virol. 1999;73:4251–4256. doi: 10.1128/jvi.73.5.4251-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng YX, Copeland TD, Henderson LE, Gorelick RJ, Bosche WJ, Levin JG, Rein A. HIV-1 nucleocapsid protein induces "maturation" of dimeric retroviral RNA in vitro. Proc. Natl. Acad. Sci. USA. 1996;93:7577–7581. doi: 10.1073/pnas.93.15.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohishi M, Nakano T, Sakuragi S, Shioda T, Sano K, Sakuragi J. The relationship between HIV-1 genome RNA dimerization, virion maturation and infectivity. Nucleic Acids Res. 2011;39:3404–3417. doi: 10.1093/nar/gkq1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lever AM, Richardson JH, Harrison GP. Retroviral RNA packaging. Biochem. Soc. Trans. 1991;19:963–966. doi: 10.1042/bst0190963. [DOI] [PubMed] [Google Scholar]
- 10.De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF. Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science. 1998;279:384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 11.Amarasinghe GK, De Guzman RN, Turner RB, Summers MF. NMR structure of stem-loop SL2 of the HIV-1 psi RNA packaging signal reveals a novel A-U-A base-triple platform. J. Mol. Biol. 2000;299:145–156. doi: 10.1006/jmbi.2000.3710. [DOI] [PubMed] [Google Scholar]
- 12.de Rocquigny H, Shvadchak V, Avilov S, Dong CZ, Dietrich U, Darlix JL, Mely Y. Targeting the viral nucleocapsid protein in anti-HIV-1 therapy. Mini. Rev. Med. Chem. 2008;8:24–35. doi: 10.2174/138955708783331603. [DOI] [PubMed] [Google Scholar]
- 13.Rice WG, Supko JG, Malspeis L, Buckheit RW, Jr, Clanton D, Bu M, Graham L, Schaeffer CA, Turpin JA, Domagala J, Gogliotti R, Bader JP, Halliday SM, Coren L, Sowder RC, 2nd, Arthur LO, Henderson LE. Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science. 1995;270:1194–1197. doi: 10.1126/science.270.5239.1194. [DOI] [PubMed] [Google Scholar]
- 14.Arthur LO, Bess JW, Jr, Chertova EN, Rossio JL, Esser MT, Benveniste RE, Henderson LE, Lifson JD. Chemical inactivation of retroviral infectivity by targeting nucleocapsid protein zinc fingers: a candidate SIV vaccine. AIDS Res. Hum. Retroviruses. 1998;14(Suppl 3):S311–S319. [PubMed] [Google Scholar]
- 15.Chertova EN, Kane BP, McGrath C, Johnson DG, Sowder RC, 2nd, Arthur LO, Henderson LE. Probing the topography of HIV-1 nucleocapsid protein with the alkylating agent N-ethylmaleimide. Biochem. 1998;37:17890–17897. doi: 10.1021/bi980907y. [DOI] [PubMed] [Google Scholar]
- 16.Druillennec S, Dong CZ, Escaich S, Gresh N, Bousseau A, Roques BP, Fournie-Zaluski MC. A mimic of HIV-1 nucleocapsid protein impairs reverse transcription and displays antiviral activity. Proc. Natl. Acad. Sci. USA. 1999;96:4886–4891. doi: 10.1073/pnas.96.9.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Druillennec S, Meudal H, Roques BP, Fournie-Zaluski MC. Nucleomimetic strategy for the inhibition of HIV-1 nucleocapsid protein NCp7 activities. Bioorg. Med. Chem. Lett. 1999;9:627–632. doi: 10.1016/s0960-894x(99)00052-9. [DOI] [PubMed] [Google Scholar]
- 18.Raja C, Ferner J, Dietrich U, Avilov S, Ficheux D, Darlix JL, de Rocquigny H, Schwalbe H, Mely Y. A tryptophan-rich hexapeptide inhibits nucleic acid destabilization chaperoned by the HIV-1 nucleocapsid protein. Biochem. 2006;45:9254–9265. doi: 10.1021/bi052560m. [DOI] [PubMed] [Google Scholar]
- 19.Dietz J, Koch J, Kaur A, Raja C, Stein S, Grez M, Pustowka A, Mensch S, Ferner J, Moller L, Bannert N, Tampe R, Divita G, Mely Y, Schwalbe H, Dietrich U. Inhibition of HIV-1 by a peptide ligand of the genomic RNA packaging signal Psi. ChemMedChem. 2008;3:749–755. doi: 10.1002/cmdc.200700194. [DOI] [PubMed] [Google Scholar]
- 20.Cruceanu M, Stephen AG, Beuning PJ, Gorelick RJ, Fisher RJ, Williams MC. Single DNA molecule stretching measures the activity of chemicals that target the HIV-1 nucleocapsid protein. Anal. Biochem. 2006;358:159–170. doi: 10.1016/j.ab.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephen AG, Worthy KM, Towler E, Mikovits JA, Sei S, Roberts P, Yang QE, Akee RK, Klausmeyer P, McCloud TG, Henderson L, Rein A, Covell DG, Currens M, Shoemaker RH, Fisher RJ. Identification of HIV-1 nucleocapsid protein: nucleic acid antagonists with cellular anti-HIV activity. Biochem. Biophys. Res. Commun. 2002;296:1228–1237. doi: 10.1016/s0006-291x(02)02063-6. [DOI] [PubMed] [Google Scholar]
- 22.Warui DM, Baranger AM. Identification of specific small molecule ligands for stem loop 3 ribonucleic acid of the packaging signal Psi of human immunodeficiency virus-1. J. Med. Chem. 2009;52:5462–5473. doi: 10.1021/jm900599v. [DOI] [PubMed] [Google Scholar]
- 23.Shvadchak V, Sanglier S, Rocle S, Villa P, Haiech J, Hibert M, Van Dorsselaer A, Mely Y, de Rocquigny H. Identification by high throughput screening of small compounds inhibiting the nucleic acid destabilization activity of the HIV-1 nucleocapsid protein. Biochimie. 2009;91:916–923. doi: 10.1016/j.biochi.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 24.Jenkins LM, Byrd JC, Hara T, Srivastava P, Mazur SJ, Stahl SJ, Inman JK, Appella E, Omichinski JG, Legault P. Studies on the mechanism of inactivation of the HIV-1 nucleocapsid protein NCp7 with 2-mercaptobenzamide thioesters. J. Med. Chem. 2005;48:2847–2858. doi: 10.1021/jm0492195. [DOI] [PubMed] [Google Scholar]
- 25.Miller Jenkins LM, Ott DE, Hayashi R, Coren LV, Wang D, Xu Q, Schito ML, Inman JK, Appella DH, Appella E. Small-molecule inactivation of HIV-1 NCp7 by repetitive intracellular acyl transfer. Nat. Chem. Biol. 2010;6:887–889. doi: 10.1038/nchembio.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller Jenkins LM, Hara T, Durell SR, Hayashi R, Inman JK, Piquemal JP, Gresh N, Appella E. Specificity of acyl transfer from 2-mercaptobenzamide thioesters to the HIV-1 nucleocapsid protein. J. Am. Chem. Soc. 2007;129:11067–11078. doi: 10.1021/ja071254o. [DOI] [PubMed] [Google Scholar]
- 27.Amarasinghe GK, De Guzman RN, Turner RB, Chancellor KJ, Wu ZR, Summers MF. NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the psi-RNA packaging signal. Implications for genome recognition. J Mol Biol. 2000;301:491–511. doi: 10.1006/jmbi.2000.3979. [DOI] [PubMed] [Google Scholar]
- 28.Hill MK, Shehu-Xhilaga M, Crowe SM, Mak J. Proline residues within spacer peptide p1 are important for human immunodeficiency virus type 1 infectivity, protein processing, and genomic RNA dimer stability. J. Virol. 2002;76:11245–11253. doi: 10.1128/JVI.76.22.11245-11253.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. Characterization of a putative alpha-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 gag. J. Virol. 2002;76:11729–11737. doi: 10.1128/JVI.76.22.11729-11737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roldan A, Russell RS, Marchand B, Gotte M, Liang C, Wainberg MA. In vitro identification and characterization of an early complex linking HIV-1 genomic RNA recognition and Pr55Gag multimerization. J Biol Chem. 2004;279:39886–39894. doi: 10.1074/jbc.M405632200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. Journal of Biomolecular Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 32.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. USA. 2003;100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turconi S, Shea K, Ashman S, Fantom K, Earnshaw DL, Bingham RP, Haupts UM, Brown MJ, Pope AJ. Real experiences of uHTS: a prototypic 1536-well fluorescence anisotropy-based uHTS screen and application of well-level quality control procedures. J. Biomol. Screen. 2001;6:275–290. doi: 10.1177/108705710100600502. [DOI] [PubMed] [Google Scholar]
- 34.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 35.Kenakin T. Pharmacological Analysis of Drug-Receptor Interaction. Lippincott-Raven, Philadelphia. 1997 [Google Scholar]
- 36.Chan LL, Pineda M, Heeres JT, Hergenrother PJ, Cunningham BT. A general method for discovering inhibitors of protein-DNA interactions using photonic crystal biosensors. ACS Chem. Biol. 2008;3:437–448. doi: 10.1021/cb800057j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turpin JA. The next generation of HIV/AIDS drugs: novel and developmental antiHIV drugs and targets. Expert. Rev. Anti-Infect. Ther. 2003;1:97–128. doi: 10.1586/14787210.1.1.97. [DOI] [PubMed] [Google Scholar]
- 38.Kaplan AH, n JA, Knigge M, Paul DA, Kempf DJ, Norbeck DW, Swanstrom R. Partial inhibition of the human immunodeficiency virus type 1 protease results in aberrant virus assembly and the formation of noninfectious particles. J. Virol. 1993;67:4050–4055. doi: 10.1128/jvi.67.7.4050-4055.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breuer S, Sepulveda H, Chen Y, Trotter J, Torbett BE. A cleavage enzyme-cytometric bead array provides biochemical profiling of resistance mutations in HIV-1 Gag and protease. Biochem. 2011;50:4371–4381. doi: 10.1021/bi200031m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds PAINS. from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 41.Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: Integrated Platform of Small Molecules and Biological Activities. Ann. Reports Comp. Chem. 2008;4:217–241. [Google Scholar]
- 42.Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA. 2006;103:15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 44.Nociari MM, Shalev A, Benias P, Russo C. A novel one-step, highly sensitive fluorometric assay to evaluate cell-mediated cytotoxicity. J. Immunol. Meth. 1998;213:157–167. doi: 10.1016/s0022-1759(98)00028-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.