

Abstract
We have studied the recombination rate behaviour of a set of 140 genes which were investigated for their potential importance in inflammatory disease. Each gene was extensively sequenced in 24 individuals of African descent and 23 individuals of European descent, and the recombination process was studied separately in the two population samples. The results obtained from the two populations were highly correlated, suggesting that demographic bias does not affect our population genetic estimation procedure. We found evidence that levels of recombination correlate with levels of nucleotide diversity. High marker density allowed us to study recombination rate variation on a very fine spatial scale. We found that about 40 per cent of genes showed evidence of uniform recombination, while approximately 12 per cent of genes carried distinct signatures of recombination hotspots. On studying the locations of these hotspots, we found that they are not always confined to introns but can also stretch across exons. An investigation of the protein products of these genes suggested that recombination hotspots can sometimes separate exons belonging to different protein domains; however, this occurs much less frequently than might be expected based on evolutionary studies into the origins of recombination. This suggests that evolutionary analysis of the recombination process is greatly aided by considering nucleotide sequences and protein products jointly.
Keywords: molecular evolution, recombination hotspot, protein structure, protein domain
Introduction
The extent of intragenic recombination will be one of the factors determining the usefulness of case-control association studies [1]. Here, candidate genes are used in the search for genetic variants involved in clinical phenotypes such as complex diseases or variable drug response. There are also many interesting evolutionary questions related to intragenic recombination, which previously had to be treated using evolutionary models but without access to high quality data [2-6]. Recent advances in experimental technology, paired with new developments in population genetics theory, now allow us to infer details of the recombination process along the human genome from population genetic data [7-11]. Here, we will use a population genetic inferential procedure to study the recombination process in a set of 140 genes which were sequenced in two population samples of African (with 24 individuals) and European (with 23 individuals) descent.
Previously, it has often been assumed that recombination within genes can either be ignored or have a constant rate across the gene [12]. Recent studies, however, have shown that the assumption of a constant recombination rate is not justified in most genomic regions [13], irrespective of whether or not they contain genes. Here, we were interested in the local variation of the recombination rate and the physical position of recombination break-points.
The evolutionary consequences of intragenic recombination have also been studied previously [14], but only now are data of sufficient quality becoming available.
There has been a long-standing interest in recombination from an evolutionary point of view [4,6,15,16]. As originally pointed out by Muller, if mutations occur according to an infinite-sites (or infinite-allele) model, genetic loci which do not recombine will accumulate deleterious mutations over time; with each new deleterious mutation, the so-called Muller's ratchet is turned irreversibly by one notch. Thus, it has been argued that recombination is vital for purging deleterious alleles, as well as for the wider dissemination or redistribution of advantageous alleles. As far as intragenic recombination is concerned, there could be a potential tradeoff between such evolutionary advantages of recombination and the disruptive effects of crossover events.
For example, recombination between two epistatic genes may lead to a fitness decrease; however, some combinations may confer an evolutionary advantage under different conditions or at different times, as may be the case for genes involved in the immune response. To study this, it may be necessary to go beyond the sequence level and consider the corresponding protein products (see Figure 1 for a possible scenario).
Figure 1.
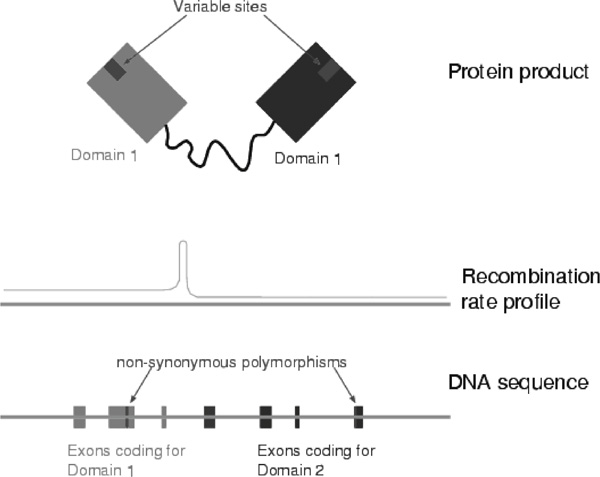
Potential explanation for why recombination at the DNA sequence level may be under evolutionary pressure resulting from selection at the protein level. If a protein has two variable sites in different domains, then, if the relative viabilities of the possible combinations change over time, recombination between exons carrying the different variants may be evolutionarily advantageous.
Recombination within genes (but also between genes) can therefore be intimately linked to the forces of natural selection [5,17,18], even in modern humans. Some of our results appear to shed some light on such evolutionary questions, although the data and uncertainties in the statistical estimator do not yet allow for a detailed and conclusive analysis. It should be noted, for example, that for some, but by no means all, genes, we find a spatial localisation of the recombination rate into (generally intronic) recombination hotspots which coincide with the sequence locations of protein domain boundaries. This offers a tantalising view into potential evolutionary reasons for why recombination might be localised in recombination hotspots along the genomic sequence. In order to understand the evolutionary causes and consequences of intragenic recombination, it appears necessary to consider levels of genetic organisation of genes into introns, exons and untranslated regions jointly with the structure of their protein products.
Below, after a brief discussion of the materials and methods used, we will discuss the properties of the average recombination behaviour across genes in the two populations, before turning to a more detailed investigation of the local recombination rate profiles across the 140 genes. We found strong evidence for recombination hotspots in several genes. The protein products of these genes were further analysed to see if there is a connection between recombination along the sequence and protein domain structure.
Materials and methods
Genetic data
We investigated 140 genes (see Table 1), which were extensively sequenced in 24 individuals of African descent and 23 individuals of European descent. The genes were studied as part of the UW-FHCRC Variation Discovery Resource (SeattleSNPs), which aims to discover and model associations between single nucleotide polymorphisms (SNPs) in genes and pathways underlying the inflammatory response. Because of SeattleSNPs resequencing, our analysis did not suffer from ascertainment bias and (apart froman overall shared genealogy) we were able to treat the African and European-derived population samples as independent. Genotypes, phased haplotypes, the proportion of coding DNA, missing data, repeats and other information are available at http://pga.gs.washington.edu/.
Table 1.
Heuristic assignment of genes to the seven classes of observed recombination properties outlined in the 'Materials and methods' section.
| Class | Gene name |
|---|---|
| I (47) | bf, ccr2, cebpb, crf, crp, csf3, csf3r, fga, fgb, fgg, fgl2, fgbp, f2rl1, f2rl3, f7, igf2, igf2as, il1a, il2, il3, il5, il6, il8, il9, il10, il13, il19, il22, il24, itga2, lta, ltb, mc1r, mmp9, pfc, plau, procr, thbd, tirap, tnfaip2, tnfaip1, vtn, proz1, rela, scya2, serpinc1, stat6 |
| II (18) | abo, f5, il10rb, itga8, jak3, klkb1, plaur, plg, pon1, pparg, ptgs1, sell, selp, sftpa1, sftpa2, tf, vcam1, vegf |
| III (17) | cd36, cyp4a11, f9, il1r1, il1r2, il1rn, il4r, il7r, il15ra, il21r, kng, plat, pon2, ppara, proz, selplg, serpina5 |
| IV (3) | agtrap, f11, f13a1 |
| V (2) | tnfrasf1b, hmox1 |
| VI (8) | bdkrb2, f2rl2, f10, il1b, il9r, il20, serpine1, tnfaip3 |
| VII (45) | ace2, apoh, cd9, csf2, c2, c3ar1, cyp4f2, dcn, ephb6, f2, f2r, f3, f12, gp1ba, icam1, ifng, il2rb, il4, il10ra, il11, il12a, il12b, il17, il17b, irak4, kel, klk1, map3k8, mmp3, nos3, proc, sele, sftpd, ptga2, sftpb, stfpc, smp1, stat4, tfpi, tgfb3, tnfaip2, tnfras1a, traf6, trpv5, trpv6 |
The number of genes in each class is given in brackets.
Recombination rate estimation
We used a composite likelihood estimator [8] to determine the population recombination rate ρ. This is proportional to the product of the molecular per-generation recombination rate r and the effective population size Ne;[19-21] it is defined as ρ = 4Ner. Composite likelihood estimators decompose a set of loci into distinct pairs of sites [8]. For each possible configuration of two loci, it is possible to calculate the corresponding likelihood for their genetic distance, which is given by the value of ρ. Averaging out the contributions from all distinct pairs of sites yields the composite likelihood estimator. Because twolocus configurations are easily enumerated, given the sample size, it is possible to calculate the likelihood for each possible two-locus haplotype resolution for a sample of n diploid individuals; a look-up table of likelihoods can therefore be calculated independently of the data. The approach can also readily be extended to deal with genotypic data by performing the weighted sum over all possible haplotype resolutions given the observed genotypes. We can thus avoid an intermediate haplotype inference step in our statistical procedure.
We used two different implementations of the composite likelihood estimator for estimation within a single locus. One was the standard form described by McVean et al., [9] which provides an average estimate across a region (see Tables 2 and 3). The second type, also described in McVean et al., [22] determines local estimates for ρ for each marker interval [22]. It uses a reversible jump Markov Chain Monte Carlo formalism on top of the composite likelihood estimator to run chains with one million steps, sampling from the chain after every thousand steps. Our block penalty is used only to capture changes in the recombination rate above a certain threshold. We chose the same parameter as McVean et al., [22] which satisfactorily reproduces the ρ profile (both in terms of location as well as relative intensity of the inferred hotspots) obtained by Jeffreys et al.[13] using sperm typing. Loosely speaking, low values of the 'block penalty'[22] would result in more structure in the profile. By contrast, high values of the block penalty would smooth out features in the recombination rate profiles. This second method is used in estimating hotspots (see later).
Table 2.
Statistical correlations of various summary statistics concerning the data between the two populations.
| Comparison | Spearman's ρ | Kendall's τ |
|---|---|---|
| Recombination distances | 0.59 | 0.75 |
| Average recombination rates | 0.50 | 0.66 |
| Heterozygosities | 0.19 | 0.28 |
| Nucleotide diversities | 0.55 | 0.71 |
| Tajima's D statistic | 0.20 | 0.29 |
| Number of non-synonymous polymorphisms | 0.66 | 0.75 |
| Number of synonymous polymorphisms | 0.67 | 0.74 |
All correlation coefficients are statistically significant. Note, in particular, that the average recombination rates across the genes correlate well between the two populations.
Table 3.
Inferred correlations between estimated average recombination rates and recombination distances (in brackets) with heterozygosities, nucleotide diversities, GC content, Tajima's D statistic and the numbers of non-synonymous and synonymous polymorphisms, in the two populations as measured using Spearman's ρ and Kendall's τ statistics.
| Test statistic | African-derived population sample | European-derived population sample | ||
|---|---|---|---|---|
| Spearman's ρ | Kendall's τ | Spearman's ρ | Kendall's τ | |
| Heterozygosity | - 0.11 (- 0.04) | - 0.08 (- 0.03) | 0.10 (0.09) | 0.07 (0.06) |
| Nucleotide diversity | 0.19 (0.32) | 0.12 (0.22) | 0.20 (0.22) | 0.13 (0.15) |
| GC-content | 0.37 (0.14) | 0.26 (0.10) | 0.26 (0.08) | 0.18 (0.06) |
| Tajima's D statistic | 0.04 (0.07) | 0.04 (0.07) | 0.09 (0.09) | 0.05 (0.06) |
| Number of non-synonymous polymorphisms |
0.13 (0.21) | 0.10 (0.16) | 0.17 (0.17) | 0.13 (0.13) |
| Number of synonymous polymorphisms |
0.01 (0.11) | 0.01 (0.08) | -0.12 (-0.05) | - 0.09 (- 0.04) |
Correlations which differ significantly from 0 (at the 5 per cent levels) are highlighted in bold.
From simulation studies (data not shown) in which we varied population parameters, we believe that bias resulting from mis-specification of the population model will usually be small compared with the intrinsic variability of the estimator (see also McVean et al's supplementary data [22]). Equally, comparisons with sperm-typing data suggest that the population genetic estimator does indeed capture the change in the recombination rate along a chromosomal region [11,22]. Moreover, for each population, we can compare recombination rates obtained from different genes in a meaningful way, as they have all undergone the same demography. Natural selection on some genes can, however, give rise to outliers. Comparisons of the recombination rate of the same gene in the two different populations are also possible. Strictly speaking, all results presented below apply to estimated, rather than to actual, recombination rates.
Analysis of recombination rate profiles
There have been recent reports suggesting temporal variability in recombination hotspot intensities and positions. Here, in order to safeguard against local biases in the recombination rate profiles in one population (for example, due to a selective sweep in one population), we scored hotspots only if there was a clearly localised and greater-than-fourfold increase in the recombination rate compared with the flanking regions.
We heuristically divided genes into different classes by considering their recombination rate profiles. The different classes are:
I: No evidence for non-uniform recombination from either population;
II: Clear indication of at least one hotspot shared between the two populations;
III: Evidence of a hotspot in one population and a ledge in the recombination rate profile in the second population;
IV: Increased recombination rate 5' from the genes in both populations;
V: Increased recombination rate 3' from the gene in both populations;
VI: Increased recombination rate over half the region considered in both populations;
VII: Other genes.
See Figure 2 for an illustration depicting the seven different classes used here.
Figure 2.
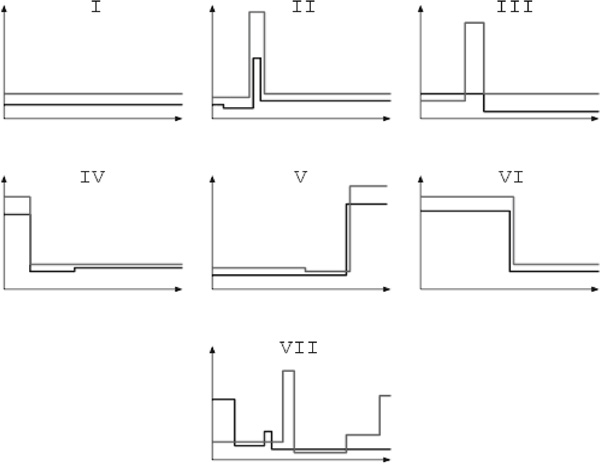
Illustrations of possible recombination rate profiles used in the heuristic analysis of intragenic recombination rate variation. Based on the observed behaviour, we divided the 140 genes under consideration into one of the seven different classes depicted here.
Class VII contains many genes in which we observe, for example, a hotspot-like feature in one population and 5' increases in the other or both populations. We found 45 genes for which the data and estimator did not allow unambiguous assignment to any of the other categories.
Results: Average recombination within genes
In Figure 3a, we have plotted the estimated recombination distance (genetic distance -- the product of recombination rate and gene size) corresponding to each gene (including 1 kilobase [kb] downstream and 2 kb upstream) obtained from the African-derived sample against the same value obtained from the European-derived sample. Note that there was a strong correlation between the two different values, which suggests that the estimator behaves consistently across regions in the two populations. The same was also observed for the average recombination rate across genes, which is shown in Figure 3b. Numerical values for the correlation coefficients of recombination rates and distances, as well as various summary statistics of the data, are given in Table 1. The high level of correlation suggests that inferred recombination rates from one population are statistically informative about the relative recombination rates in a different population.
Figure 3.
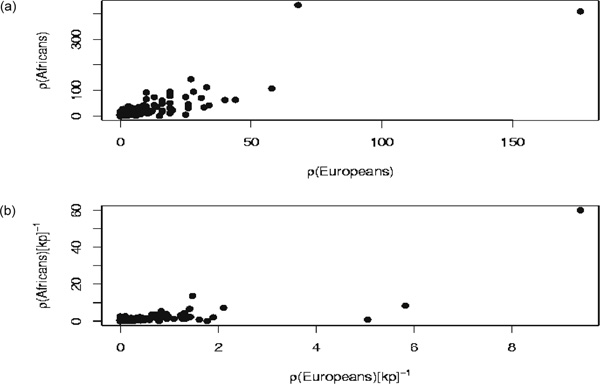
(a) Inferred recombination distances and (b) average rates across genes and their flanking regions in the African-derived sample compared with the European-derived sample. We found a strong correlation between the estimated recombination properties in the two populations [(a) Spearman's ρ = 0.75, Kendall's τ = 0.59; and (b) Spearman's ρ = 0.66, Kendall's τ = 0.50, with p values less than 10-10 in all cases]. Note that in (b), the correlation still holds with the removal of the righthandmost point.
If we assume that the molecular recombination rate is the same in both populations, and take the ratio of ρ (from pairs of loci) obtained from the two populations, then we have ρA/ρE = NeA/NeE, that is, the ratio of the effective population sizes. The results shown in Figures 3a and 3b thus allowed us to assess the relative effective population sizes. We found that the effective population size of the African-derived sample is approximately 2.5 times the value of the European-derived effective population size. This, incidentally, is in agreement with other estimates we have made (data not shown) for ρ for a set of 39 genomic regions first analysed by Gabriel et al.[23] This would suggest that: (i) ascertainment (the data of Gabriel et al.[23] was generated by genotyping combined with low levels of SNP discovery in predominantly European individuals) does not affect the estimator severely;[24] and (ii) selection on the genes does not appear to severely bias results compared with the genomic regions investigated by Gabriel et al. [23]
In order to assess what determines the population recombination rate, we studied the dependence of ρ on diversity, heterozygosity, Tajima's D[25] statistic, GC content [26], as well as the respective numbers of non-synonymous and synonymous polymorphisms (shown in Figure 4). The estimated correlation coefficients are given in Table 3, where we provide the correlation coefficients with the average recombination rate and the recombination distance or genetic distance (in brackets). We found that the nucleotide diversity correlated well with estimated recombination rates [22,27-29] and distances. The rates also correlated well with GC content, whereas the recombination distance correlated well with the number of non-synonymous polymorphisms; only in Europeans did we find a correlation between the rate and the number of non-synonymous polymorphisms. Thus, generally, the amount of adaptive (or non-synonymous) change correlates with the genetic distance (ie the product of recombination rate and physical distance) but not the rate itself. We note here that the sample size was relatively small and that many low-frequency polymorphisms will have been absent from the sample (for example, a polymorphism with a minor allele frequency of 10 per cent will be absent from the sample in 8.5 per cent of all cases). We also found a strong correlation between measures of haplotype diversity and estimated recombination rates, as well as with nucleotide diversity. Further, correcting for nucleotide diversity did not reduce the correlation between recombination rate and haplotype diversity measures.
Figure 4.
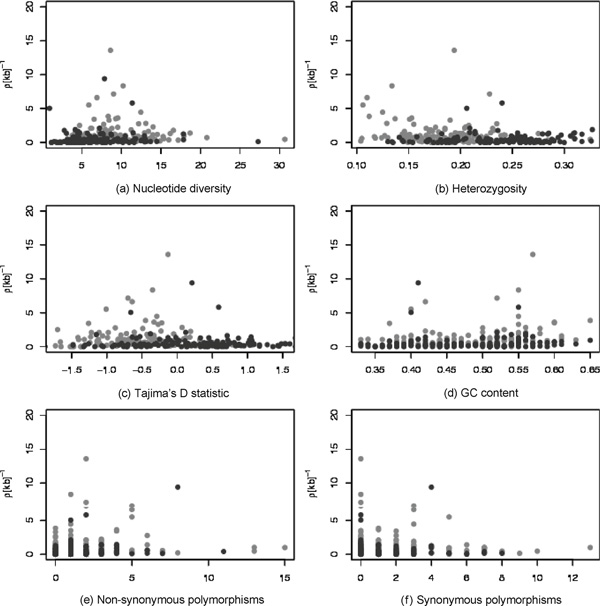
Average inferred recombination rates versus levels of: (a) nucleotide diversity, (b) heterozygosity, (c) values of Tajima's D statistic, (d) average GC content and (e) and (f) the numbers of non-synonymous and synonymous polymorphisms, respectively. The results obtained from the African-derived sample are shown in •, and those from the European-derived sample in •.
Previous studies have reported correlations between the recombination rate and nucleotide diversity and GC content [18,26,29,30]. These studies used estimates of ρ which were based on genetic maps; these afford a much lower resolution than is possible for the dense marker maps considered here with the population genetic estimator. It is thus encouraging that the fine and coarse scales appear to agree. We note that by considering only genes and their surrounding regions selection may crucially determine levels of diversity and linkage disequilibrium. Theoretical arguments suggest that both hitchhiking and background selection are expected to result in positive correlations between nucleotide diversity and recombination rates [18,31]. We may thus be comparing quantities that are very similar from the outset; that is, if both diversity and recombination rates within genes are evolutionarily constrained (compared with the neutral case) through selection, then we would expect only relatively weak correlations between them; the small sample size would exacerbate this problem. Nevertheless, we found statistically significant correlations. Interestingly, no correlation (at the 5 per cent level) was observed between GC content and recombination distance. Thus, while the amount of adaptive change also reflects the physical size of the gene, GC content appears to correlate more directly with the recombination activity in a gene.
For the most part, our results agree with those of Crawford et al., [32] who investigated some of the same genes and similarly found significant variation in recombination rate within genes, as well as a number of hotspot-like features.
The most tantalising result is probably the strong correlation found between the inferred recombination distances in genes and the number of non-synonymous--but not synonymous-- polymorphisms; for the rates, the correlation was statistically significant only in Europeans (Africans were just inside the 95th percentile). Although this does not, of course, prove a causal relationship between adaptability and recombination rate, it might suggest that there could be interplay between recombination at the nucleotide level and natural selection (which would act on the level of protein products).
Results: Intragenic recombination rate variation
The high marker density allows us to study the local variation in ρ across a gene and to determine regions in which we observe the highest levels of recombination activity. In Figure 5, we show four exemplary recombination rate profiles; Figure S1 (not included here, available at: http://www.bio.ic.ac.uk/research/stumpf/data.html), shows the inferred profiles for all 140 genes in the two populations. Table 1 summarises the inferred behaviour of the recombination rate profile; this should only be taken as indicative and is a largely heuristic assessment. In particular, however, assignment of genes to class II (genes with hotspots) was conservative, and we demanded that both populations should show an at least a fourfold increase in ρ in the same region (applying Anderson and Slatkin's criterion [33]); we allowed for slight differences in the position to account for the different marker sets in the two populations but we did not allow for 'moving' hotspots [34]. This possibility has recently been suggested and there is good experimental evidence that recombination hotspots may have only a limited lifetime under certain conditions. With the intrinsic variability of population genetic estimators, however, it becomes difficult to reliably detect such features ab initio. Comparisons with experimental data will reveal how reliable these methods really are. Where sperm-typing data are available, generally good agreement is found between the new classes of approximate population genetic estimators and the experimentally obtained results, both in terms of hotspot positions and relative intensities.
Figure 5.
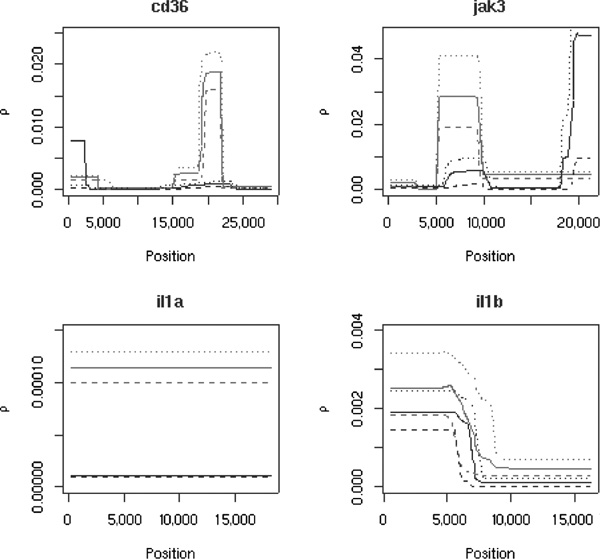
Recombination rate profiles for four different genes. The results obtained from the African-derived sample are shown in grey, those from the European-derived sample are in black. The solid lines indicate the mean values for the local values of ρ, while the dashed lines show the 2.5 and 97.5 percentiles obtained from the recombination rate estimator. In each case, the profiles of the quantiles are very similar to those of the average behaviour; all curves show comparable levels of recombination rate variation. Note that the recombination profiles in the two populations are vertically shifted, but areas of local change occur in similar positions across the genes.
A large fraction of the genes investigated here (47/140) showed no evidence for recombination rate variation; that is, the ρ profiles were flat in both populations and were assigned to class I. An equal fraction (45/140) was very difficult to assign (class VII). This is either because the profiles obtained for both populations were different or because several different features (such as hotspots, increases 3' or 5' from the gene, etc) were observed but not all were shared between the two samples. There were 18 genes for which we found localised increases in the ρ profiles from both populations (class II). Another 17 genes showed evidence for a clear hotspot in one of the populations and a ledge in the ρ profile from the other population (class III). Increases to the 5' and 3' ends of genes were observed in three and two genes, respectively (classes IV and V), and most of the unassignable genes also showed increases in ρ in the upstreamand downstream regions. Finally, we found eight genes which showed similar behaviour to that observed in IL1B, depicted in Figure 5 (class VI).
Does recombination at the sequence level affect properties at the protein level?
In order to investigate any potential relationship between intragenic recombination hotspots and exon shuffling, or domain boundaries in the protein structure, we selected the 18 genes which showed unambiguous evidence for recombination hotspots (see Table 1 and Figures 4 and 5) and one gene which belonged to our category III. We found that in some cases hotspots were intronic, while in others there was evidence that hotspot-like increases in the recombination rate extended well beyond several exons. As exons were typically rather short (eg compared with the introns), we may simply have lacked the resolution to localise recombination hotspots precisely.
From an evolutionary perspective, selection will act on the corresponding proteins. We therefore investigated whether the recombination structure at the DNA sequence level has any consequences at the protein level; for example, if recombination events occur between exons coding for different domains. Figure 6 shows the locations of the exons in a particular gene, vegf (encoding vascular endothelial growth factor). By entering the amino acid sequence corresponding to these exons into a package such as 3D-PSSM http://www.sbg.bio.ic.ac.uk/servers/3dpssm/, homology to known protein crystal structures can routinely be recognised with as little as 25 per cent sequence identity. In this work, the structurally characterised homologues identified by 3D-PSSM all had > 90 per cent sequence identity to the input exon sequence, and the 3D-PSSM models are thus expected to deviate by at most 2-3Å from the true structure [35].
Figure 6.
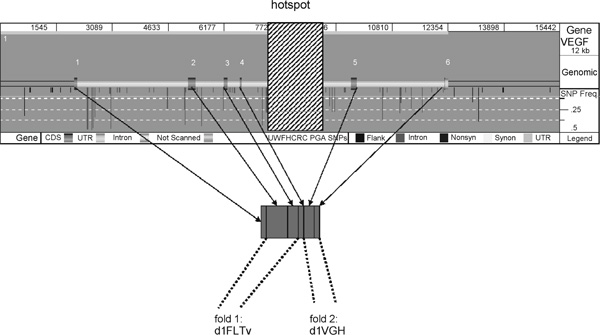
The sequence of the gene vegf, together with the single-nucleotide polymorphism (SNP) locations and their frequencies (grey vertical bars), taken from the website of the Seattle SNP project http://pga.gs.washington.edu, and the assignment of the exons to the inferred protein folds. The positions of exons along the gene are indicated in dark grey. The shaded region denotes the position of the inferred hotspot (in both populations). Exons 1 to 4 lie 5' of the hotspot, while exons 5 and 6 are on the 3' side. Below the sequence are the assignments of exonic DNA to the inferred protein folds. Exons 1 to 3 belong to the first fold (1FLTv), while DNA in exons 5 and 6 code for protein parts assigned to the second fold (1VGH). We note that in the case of vegf, no nonsynonymous polymorphism was detected in any of the exons.
Of the 18 genes, two (sftpa1 and sftpa2) have all their exons within the region covered by the hotspot, implying overall high recombination for these genes; two (abo and tf ) have a complex hotspot structure (ie several hotspots, some, but not all, of which are shared between populations) and one (itga8) has a narrow hotspot in between untranslated regions. Of the remaining genes, six (il10rb, jak3, klkb1, pon1, sell and selp) showed no evidence of a relationship between recombination hotspots and domain boundaries. In vegf, however, the hotspot appears to signpost the domain boundary (see below), that is, the region after which one fold begins and before which another fold ends.
Figure 6 shows the exon-intron boundaries, SNP locations and frequencies and the estimated recombination hotspot position in vegf. vegf is a mitogen (substance able to induce mitosis of certain eukaryotic cells), primarily for vascular endothelial cells. It is, however, structurally related to platelet-derived growth factor. In the case of vegf, 3D-PSSM identifies two folds, 1FLTv (PDB ID 1FLT, chain v) and 1VGH with 100 per cent and 91 per cent sequence identity, respectively. These are thus reliably recognised folds whose high identification implies that they have both been crystallised and structurally mapped. 1VGH is a heparin-binding domain. Interestingly, 1FLTv is built from the amino acid sequence of exon 2 and exon 3 (and also the first two amino acids in exon 4), while 1VGH is built from the amino acid sequence of exon 5 and exon 6. Given that the hotspot lies between exon 4 and exon 5, the identified folds are indicative of there being a relationship between the positions of the folds and the hotspot; perhaps the recombination hotspot influences the location of protein domain boundaries, or vice versa.
Further evidence for some sort of hotspot-fold relationship is seen in vcam1, where the hotspot appears to mark the end of a fold. The hotspot is positioned between exon 4 and exon 9, stretching across exons 5 to 8. 3D-PSSM recognises 1IJ9a, a cell adhesion fold with 100 per cent sequence identity, built from exon 2 and exon 3. Interestingly, exons 2 to 9 individually are immunoglobulin-like folds. Additionally, in both plaur and plg, the hotspot seems to mark the beginning of a fold. In pparg and ptgs1, the fold(s) seem(s) to cover the extent of the hotspot.
Finally, in f5 the hotspot covers exon 6 to exon 9 and there is 100 per cent sequence identity to the 1FV4h fold built from exon 1 to exon 13. A theoretical model of this fold was retrieved from the Protein Data Bank http://www.rcsb.org/pdb/ and is shown in Figure S2 (Not included here, available online at http://www.bio.ic.ac.uk/research/stumpf/data.html). An examination of the fold structure built-up from the residues corresponding to the recombination hotspot region (243-465) showed what appeared to be two domains connected by an interdomain bridge (shown in Figure S2). The interdomain bridge is made from residues 311-325 and has been shown [36] to contribute to the binding of f10 by f5. Kojima et al.[36] suggest that cleavage by a protein at arginine residue 306 breaks the joint between the two domains, disrupting the bridge structure (residues 311-325), and in so doing downregulating the binding of f10 to f5. It is not known at this time if the positioning of this interdomain bridge/cleavage site within the hotspot is coincidental, or is in some way related to the recombination process.
3D-PSSM was unable to identify any folds in one-third of the genes with flat recombination profiles (in genes with recombination hotspots, this figure was slightly more than one-third). In the remainder of the flat recombination profile genes, a range of different folds were identified with a range of sequence identity percentages, depending on the individual gene. It is not surprising that 3D-PSSM was able to identify domains in approximately the same fraction of genes with flat recombination profiles as those with recombination hotspots; here, we were only interested in the positioning of these folds (or the exons that make them) relative to the recombination hotspot.
Discussion
We have applied population genetic estimators to study the extent of recombination in genes and their immediate flanking regions. We were able to demonstrate that, in spite of the assumptions underlying the estimator [9,22], it is possible to obtain meaningful results for the level of recombination activity, both averaged across genes and within genes. Moreover, the results from two slightly different, although related, approaches were found to produce highly consistent pictures from the two populations in the majority of cases. Only for approximately one-third of the genes considered here was it not possible to characterise the recombination behaviour within the recombination profile classification scheme employed.
We found that nucleotide diversity, GC content and the number of non-synonymous polymorphisms (in the European sample) correlated with the inferred population recombination rates, while other measures of diversity--such as the average heterozygosity and Tajima's D statistic--did not. Moreover, we did not find a statistically significant correlation between ρ and the number of synonymous polymorphisms. Thus, the average recombination behaviour across the 140 genes appeared to correlate with measures of adaptive change. As outlined in the introduction and in the legend to Figure 1, such behaviour may be expected if combinations of polymorphisms within the same gene have a time-, environmentor context-dependent effect on the viability or Darwinian fitness. Larger sample sizes may, however, be necessary to ensure that lower frequency polymorphisms are adequately captured before a more detailed assessment of the interplay between evolutionary forces and recombination process can be established more conclusively.
We found, as expected from several previous studies, that estimates of ρ obtained from African population samples are higher than those derived from European-derived populations [11]. Within the scope of this analysis, in which we focused on recombination rate variation (and its potential role in exon shuffling), we did not investigate the extent to which recombination rate estimators can be used to detect the effects of natural selection [37]. We cannot rule out that some of the differences between populations observed in unclassified genes (class VII) are due to differences in selection pressures experienced by the two populations (or admixture effects).
We found considerable differences between the different genes but, generally, local profiles between the two populations were similar in terms of positions of rate changes, as well as relative intensities. The majority of genes considered here appeared to have a uniform recombination rate across the whole region. For 18 genes, however, we found persuasive evidence for recombination hotspots [38], with tentative evidence coming from a further 17. In unclassified genes (class VII), we often found increases towards the 5' and 3' flanking regions; three and two genes, respectively, also had constant recombination rates apart from their 5' and 3' regions, respectively.
A further analysis of genes with recombination hotspots (and one belonging to class III which has a hotspot in one population and a ledge in the other) were then analysed to assess the extent to which intragenic recombination can be understood evolutionarily. If recombination acts to shuffle advantageous genetic variants [2,39], and if these variants are confined to coding DNA (rather than, say, regulatory elements), then we may envisage that recombination hotspots between exons belonging to different domains are positively selected for in some instances. This would especially be the case if domain boundaries coincided with exon boundaries (see Figure 1). Unfortunately, we did not find conclusive evidence for such a scenario for the majority of cases and therefore cannot find evidence for a general rule. This may have been due either to limits imposed by marker density and/or sample size, or by lack of power of the estimator used here.
In some cases, however--and most clearly for the vegf gene--we found that the inferred recombination hotspot satisfactorily separated exons belonging to different protein domains. There are clearly a number of assumptions underlying the current approach, with respect to inferences drawn at the nucleotide and protein levels. We were, however, conservative in restricting our attention to genes for which the presence of a hotspot could be deduced with considerable certainty in both populations. Similarly, inferred folds for vegf were very reliable. The absence of high levels of concordance between recombination hotspots and protein properties can be due to a number of factors, including, but not limited to, failure of the estimator to capture fine-scale recombination rate variation, insufficient sample size (and hence marker density) and problems in correctly assigning protein structures and detecting domain boundaries. We hope we have demonstrated, however, that the joint consideration of DNA and protein levels holds great promise for further studies into the recombination process and properties of protein structures, and the evolutionary pressures which must have acted upon them.
If evolutionary pressures have been important in determining positions (and intensities) of recombination hotspots, this could explain why recombination hotspots might change over time [34] or be absent from other, closely related species [40]. The observation that ρ correlates with nucleotide diversity and (at least, in one of the population samples) the number of adaptive (non-synonymous) changes supports the notion that the two are linked and that some recombination hotspots may be species specific.
Acknowledgements
We thank the Wellcome Trust and the Royal Society for financial support for this work.
References
- Cardon LR, Bell JI. Association study designs for complex diseases. Nat Rev Genet. 2001;2:91–99. doi: 10.1038/35052543. [DOI] [PubMed] [Google Scholar]
- Felsenstein J, Yokoyama S. The evolutionary advantage of recombination, II. Individual selection for recombination. Genetics. 1976;83:845–859. doi: 10.1093/genetics/83.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A. Recombination and mammalian genome evolution. Proc R Soc Lond B Biol Sci. 1993;252:237–243. doi: 10.1098/rspb.1993.0071. [DOI] [PubMed] [Google Scholar]
- Barton NH. A general model for the evolution of recombination. Genet Res. 1995;65:123–145. doi: 10.1017/S0016672300033140. [DOI] [PubMed] [Google Scholar]
- Feldman MW, Otto SP, Christiansen FB. Population genetic perspectives on the evolution of recombination. Ann Rev Genet. 1996;30:261–295. doi: 10.1146/annurev.genet.30.1.261. [DOI] [PubMed] [Google Scholar]
- Barton NH, Charlesworth B. Why sex and recombination? Science. 1998;281:1986–1990. [PubMed] [Google Scholar]
- Fearnhead P, Donnelly P. Estimating recombination rates from population genetic data. Genetics. 2001;159:1299–1318. doi: 10.1093/genetics/159.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Two-locus sampling distributions and their application. Genetics. 2001;159:1805–1817. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA. A genealogical interpretation of linkage disequilibrium. Genetics. 2002;162:987–991. doi: 10.1093/genetics/162.2.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf MPH, McVean GAT. Estimating recombination rates from population-genetic data. Nat Rev Genet. 2003;4:959–968. doi: 10.1038/nrg1227. [DOI] [PubMed] [Google Scholar]
- Abbs S, Roberts RG, Mathew CG. et al. Accurate assessment of intragenic recombination frequency within the Duchenne muscular dystrophy gene. Genomics. 1990;7:602–606. doi: 10.1016/0888-7543(90)90205-9. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- Clarke CH, Johnston AW. Intragenic mutational spectra and hot spots. Mutat Res. 1976;36:147–164. doi: 10.1016/0027-5107(76)90003-8. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA, Holmes EC. Recombination in evolutionary genomics. Ann Rev Genet. 2002;36:75–97. doi: 10.1146/annurev.genet.36.040202.111115. [DOI] [PubMed] [Google Scholar]
- Awadalla P. The evolutionary genomics of pathogen recombination. Nat Rev Genet. 2003;4:50–60. doi: 10.1038/nrg964. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. Directional selection and the evolution of sex and recombination. Genet Res. 1993;61:205–224. doi: 10.1017/S0016672300031372. [DOI] [PubMed] [Google Scholar]
- Nachman MW. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 2001;17:481–485. doi: 10.1016/S0168-9525(01)02409-X. [DOI] [PubMed] [Google Scholar]
- Gillespie JH. Population Genetics: A Concise Guide. Johns Hopkins University Press, Baltimore, MD; 1998. [Google Scholar]
- Hartl DL, Clark AG. Principles of Population Genetics. Sinauer, Sunderland, MA; 1998. [Google Scholar]
- Donnelly P, Tavare S. Coalescents and genealogical structure under neutrality. Ann Rev Genet. 1995;29:401–421. doi: 10.1146/annurev.ge.29.120195.002153. [DOI] [PubMed] [Google Scholar]
- McVean GAT, Myers S, Hunt S. et al. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H. et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Signorovitch J. Correcting for ascertainment biases when analyzing SNP data: Applications to the estimation of linkage disequilibrium. Theor Popul Biol. 2003;63:245–255. doi: 10.1016/S0040-5809(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsell JA. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol Biol Evol. 2002;19:1181–1197. doi: 10.1093/oxfordjournals.molbev.a004176. [DOI] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J. et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 2002;18:337–340. doi: 10.1016/S0168-9525(02)02669-0. [DOI] [PubMed] [Google Scholar]
- Hellmann I, Ebersberger I, Ptak SE. et al. A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet. 2003;72:1527–1535. doi: 10.1086/375657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Bernardo-Carvalho A, Clark AG. Local rates of recombination are positively correlated with GC content in the human genome. Mol Biol Evol. 2001;18:1139–1142. doi: 10.1093/oxfordjournals.molbev.a003886. [DOI] [PubMed] [Google Scholar]
- Slatkin M. Balancing selection at closely linked, overdominant loci in a finite population. Genetics. 2000;154:1367–1378. doi: 10.1093/genetics/154.3.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Bhangale T, Li N. et al. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet. 2004;36:700–706. doi: 10.1038/ng1376. [DOI] [PubMed] [Google Scholar]
- Anderson EC, Slatkin M. Population-genetic basis of haplotype blocks in the 5q31 region. Am J Hum Genet. 2004;74:40–49. doi: 10.1086/381040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat Genet. 2002;31:267–271. doi: 10.1038/ng910. [DOI] [PubMed] [Google Scholar]
- Kelley LA, MacCallum RM, Sternberg MJ. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol. 2000;299:499–520. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- Kojima Y, Heeb MJ, Gale AJ. et al. Binding site for blood coagulation factor Xa involving residues 311-325 in factor Va. J Biol Chem. 1998;273:14900–14905. doi: 10.1074/jbc.273.24.14900. [DOI] [PubMed] [Google Scholar]
- Wall JD. Recombination and the power of statistical tests of neutrality. Genet Res. 1999;74:65–79. doi: 10.1017/S0016672399003870. [DOI] [Google Scholar]
- Arnheim N, Calabrese P, Nordborg M. Hot and cold spots of recombination in the human genome: The reason we should find them and how this can be achieved. Am J Hum Genet. 2003;73:5–16. doi: 10.1086/376419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt A. Perspective: sex, recombination, and the efficacy of selection--Was Weismann right? Evolution Int J Org Evolution. 2000;54:337–351. doi: 10.1111/j.0014-3820.2000.tb00038.x. [DOI] [PubMed] [Google Scholar]
- Wall JD, Frisse LA, Hudson RR, Di Rienzo A. Comparative linkage-disequilibrium analysis of the beta-globin hotspot in primates. Am J Hum Genet. 2003;73:1330–1340. doi: 10.1086/380311. [DOI] [PMC free article] [PubMed] [Google Scholar]