

Abstract
Enzyme-mediated disulfide bond formation is a highly conserved process affecting over one-third of all eukaryotic proteins. The enzymes primarily responsible for facilitating thiol-disulfide exchange are members of an expanding family of proteins known as protein disulfide isomerases (PDIs). These proteins are part of a larger superfamily of proteins known as the thioredoxin protein family (TRX). As members of the PDI family of proteins, all proteins contain a TRX-like structural domain and are predominantly expressed in the endoplasmic reticulum. Subcellular localization and the presence of a TRX domain, however, comprise the short list of distinguishing features required for gene family classification. To date, the PDI gene family contains 21 members, varying in domain composition, molecular weight, tissue expression, and cellular processing. Given their vital role in protein-folding, loss of PDI activity has been associated with the pathogenesis of numerous disease states, most commonly related to the unfolded protein response (UPR). Over the past decade, UPR has become a very attractive therapeutic target for multiple pathologies including Alzheimer disease, Parkinson disease, alcoholic and non-alcoholic liver disease, and type-2 diabetes. Understanding the mechanisms of protein-folding, specifically thiol-disulfide exchange, may lead to development of a novel class of therapeutics that would help alleviate a wide range of diseases by targeting the UPR.
Keywords: Disulfide bond, Thioredoxin, Calsequestrin, UPR, Unfolded protein response, ER stress
Introduction
Increasing evidence supports an important role for misfolded proteins in the pathogenesis of numerous diseases including diabetes, Alzheimer disease, Parkinson disease, and both alcoholic and non-alcoholic liver disease [1]. Accumulation of misfolded proteins (or erred protein load) is generally caused by either decreased disposal of erred protein or a decrease in the correct folding of proteins [1]. Disulfide bond formation represents a fundamentally important post-translational modification that is a critical step in the folding of nascent peptides in the endoplasmic reticulum (ER) [2]. These covalent linkages are formed between the side-chains of cysteine residues and represent a key rate-limiting step in protein maturation [3]. The enzymatic formation, breakage, and subsequent rearrangement of cysteine linkages are crucial to protein structure and function and primarily mediated by members of the protein disulfide isomerase (PDI) family [4]. All genes in the PDI family are part of a superfamily referred to as the thioredoxin (TRX) superfamily, which also includes the glutaredoxins, TRXs, ferroredoxins, and peroxidoxins [5].
The PDI gene family currently comprises 21 genes, varying in size, expression, localization, and enzymatic function. Although it is implied that all members of the PDI family possess the ability to rearrange disulfide bonds, only a subset is considered orthologous and able to carry out these reactions, with the other members being paralogous and linked to the family through evolution, not function [4]. While these proteins may be functionally different, the unifying feature of all PDI family members is the presence of a TRX-like domain [2]. These may be present as either a catalytically active a or a’ domain (the presence of a CXXC motif) or a catalytically inactive b or b’ domain (for a more detailed review on the precise role of these domains, see the work of Ellgaard et al.) [2,4]. Extensive research has assessed the roles of these domains and revealed the b’ domain to be the primary peptide- or protein-binding domain [4]. Previous literature has highlighted the features of a number of PDI family members; however, with an increasing amount of cDNA and EST sequence information deposited in the NCBI database, a composite review is required to further characterize and compare all 21 current members of the PDI gene family (Table 1).
Table 1.
Human PDI genes as listed in the Human Gene Nomenclature Committee (HGNC) database
| Gene name | Other aliases | Protein name | Chromosomal location | Amino acids |
|---|---|---|---|---|
|
AGR2 |
XAG-2, HAG-2, AG2, PDIA17 |
Anterior gradient protein 2 homolog |
7p21.3 |
175 |
|
AGR3 |
HAG3, hAG-3, BCMP11, PDIA18 |
Anterior gradient protein 3 homolog |
7p21.1 |
166 |
|
CASQ1 |
PDIB1 |
Calsequestrin-1 |
1q21 |
396 |
|
CASQ2 |
PDIB2 |
Calsequestrin-2 |
1p13.3-p11 |
399 |
|
DNAJC10 |
MTHr, ERdj5 |
DnaJ (Hsp40) homolog, subfamily C, member 10 |
2q32.1 |
793 |
|
ERP27 |
FLJ32115, ERp27, PDIA8 |
Endoplasmic reticulum resident protein 27 |
12p12.3 |
273 |
|
ERP29 |
ERp28, ERp31, ERp29, PDI-DB, PDIA9 |
Endoplasmic reticulum resident protein 29 |
12q24.13 |
261 |
|
ERP44 |
KIAA0573, PDIA10 |
Endoplasmic reticulum resident protein 44 |
9q22.33 |
406 |
|
P4HB |
DIA1, PROHB, DSI, GIT, PDI, PO4HB, P4Hb |
Protein disulfide-isomerase |
17q25 |
508 |
|
PDIA2 |
PDA2, PDIp |
Protein disulfide-isomerase A2 |
16p13.3 |
525 |
|
PDIA3 |
P58, ERp61, ERp57, ERp60, GRP57, PI-PLC, HsT17083 |
Protein disulfide-isomerase A3 |
15q15 |
505 |
|
PDIA4 |
ERP70, ERP72 |
Protein disulfide-isomerase A4 |
7q35 |
645 |
|
PDIA5 |
PDIR, FLJ30401 |
Protein disulfide-isomerase A5 |
3q21.1 |
519 |
|
PDIA6 |
P5, ERp5 |
Protein disulfide-isomerase A6 |
2p25.1 |
440 |
|
PDILT |
PDIA7, ERp65 |
Protein disulfide-isomerase-like protein of the testis |
16p12.3 |
584 |
|
TMX1 |
TMX, PDIA11 |
Thioredoxin-related transmembrane protein 1 |
14q22.1 |
280 |
|
TMX2 |
PDIA12 |
Thioredoxin-related transmembrane protein 2 |
11cen-q22.3 |
296 |
|
TMX3 |
FLJ20793, KIAA1830, PDIA13 |
Protein disulfide-isomerase TMX3 |
18q22 |
454 |
|
TMX4 |
DJ971N18.2, KIAA1162, PDIA14 |
Thioredoxin-related transmembrane protein 4 |
20p12 |
349 |
|
TXNDC5 |
MGC3178, FLJ21353, FLJ90810, EndoPDI, Hcc-2, ERp46, PDIA15 |
Thioredoxin domain-containing protein 5 |
6p24.3 |
432 |
| TXNDC12 | TLP19, ERP18, ERP19, hAG-1, AGR1, PDIA16 | Thioredoxin domain-containing protein 12 | 1p32.3 | 172 |
DNAJC10 has been included following identification of this gene in the PDI gene family. Synonyms of these genes have also been corrected.
Domain composition of the PDI family proteins
Proteins in the PDI family are largely expressed in the ER, although few family members have been detected in other subcellular compartments [6]. Due to their localization, the presence of a short NH2-terminal signal peptide exists in all members of the family. These peptides are typically 15–30 amino acids (a.a.) in length and are cleaved upon entry into the ER [7]; this has led to some sequence discrepancy among multiple PDI proteins. As indicated, the common thread between all members of the PDI proteins is the presence of at least one TRX-like domain, whether it is catalytically active (a) or inactive (b) [2]. The active site of the a-type domains also varies greatly, with the “classical” motif being comprised of Cys-Gly-His-Cys. The cysteine residues in these active sites are considered redox active, undergoing active shuffling of disulfide bonds [2]. The surrounding a.a. largely play a role in the regulation of the pKa of the cysteines, dictating the local redox potential and thus regulating the catalytic ability of these cysteines to actively oxidize or reduce disulfide bonds (for a more comprehensive review on the redox potential of PDI, see the work of Hatahet et al.) [3]. Extensive biochemical and biophysical experimentation has taken place analyzing TRX–like domain containing proteins; however, a complete crystal structure is currently not available for most family members. Another common characteristic of the PDI family of proteins is the presence of a COOH-terminal ER retention sequence comprised of a Lys-Asp-Glu-Leu-like sequence [2]. Whereas these sequences may differ greatly in a.a. composition, only four PDI proteins do not contain this sequence. Figure 1 shows the domain composition of the 21 proteins in the PDI family.
Figure 1.

Schematic representation of the domain compositions of the 21 proteins in the PDI gene family. All proteins contain a short NH2-terminal signal sequence designated in black. Catalytically active TRX-like domains (a or a’) are represented in red with active sites noted; inactive TRX domains in blue (b) and purple (b’); green represents the Asp/Glu rich Ca2+-binding domains; linker regions (gray); transmembrane domains (yellow); COOH-terminal ER-retention sequences in white with a.a. composition denoted. Although ERP29 does contain an a-like domain, this is based on homology and not catalytic efficiency. Sequences and domain compositions were acquired and verified from the National Center for Biotechnology Information (NCBI) database. Figure was adapted and modified from [4].
Evolutionary divergence of the PDI gene family
As mentioned, all genes encompassed in the PDI family belong to the TRX superfamily of genes [5]. The unifying theme between these proteins is the presence of at least one TRX-like domain, whether this be catalytically active (a or a’) or inactive (b or b’) [4]. These domains contain a TRX structural fold that has amino acids arranged in a conserved three-dimensional conformation [8]. While the enzymatic function of these domains is not conserved, the current theory proposes that all PDI family members evolved through domain duplications from an ancestral prokaryotic PDI which contained a single TRX domain [9]. Although all human PDIs possess a TRX-like domain, this remains one of the few similarities between these proteins, differing greatly in molecular mass and a.a. composition outside of the TRX fold. Phylogenetic analysis, presented as a dendrogram in Figure 2, outlines the evolutionary divergence of the human PDI proteins. Sequence analysis reveals a subset of genes within this family that are evolutionarily related, as shown by the calsequestrin (CASQ) and anterior gradient (AGR) branches (in red and blue, respectively). Supporting the hypothesis that these subsets of genes are phylogenetically related, a high degree of similarity was also observed with both domain architecture (Figure 1) and sequence identity (Table 2). Although similarities are evident, the overall homology between the proteins is quite minimal. This is supported by previous attempts to cluster large sets of both eukaryotic and prokaryotic PDIs where marginal resolution of PDI domains was also observed [9]. Given the broad spectrum of both enzymatic functions and domain compositions, it is not surprising that the proteins share little sequence homology with one another (Table 2).
Figure 2.
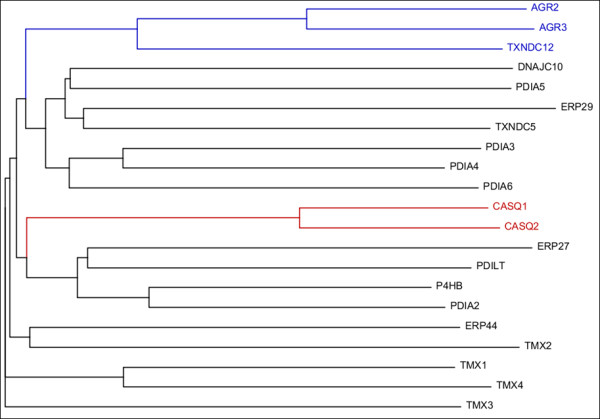
Clustering dendrogram of the human PDI gene family. Utilizing ClustalW alignment software of known protein sequences of the human PDI genes reveals divergence into AGR and CASQ subfamilies (red and blue, respectively).
Table 2.
Amino acid similarity between human PDI proteins, as reported using EMBOSS Water Pairwise Alignment Algorithm (http://www.ebi.ac.uk/Tools/psa/emboss_water/)
| AGR2 | AGR3 | CASQ1 | CASQ2 | DNAJC10 | ERP27 | ERP29 | ERP44 | P4HB | PDIA2 | PDIA3 | PDIA4 | PDIA5 | PDIA6 | PDILT | TMX1 | TMX2 | TMX3 | TMX4 | TXNDC5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGR2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| AGR3 |
65.0% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(81.4%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| CASQ1 |
18.8% |
17.4% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(34.8%) |
(40.2%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| CASQ2 |
15.3% |
17.40% |
68.4% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(37.4%) |
(34.0%) |
(86.1%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| DNAJC10 |
16.2% |
19.0% |
16.8% |
18.2% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(37.2%) |
(35.8%) |
(33.3%) |
(33.3%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ERP27 |
28.2% |
20.5% |
17.2% |
19.3% |
18.6% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(41.0%) |
(40.2%) |
(34.4%) |
(37.6%) |
(35.8%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ERP29 |
20.6% |
23.3% |
18.2% |
20.4% |
18.1% |
15.7% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(40.2%) |
(39.5%) |
(31.1%) |
(33.0%) |
(30.9%) |
(38.9%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ERP44 |
22.2% |
26.5% |
19.2% |
20.0% |
20.4% |
24.3% |
25.3% |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(39.4%) |
(47.0%) |
(42.9%) |
(40.8%) |
(30.6%) |
(40.5%) |
(42.0%) |
|
|
|
|
|
|
|
|
|
|
|
|
|
| P4HB |
20.1% |
21.7% |
21.6% |
22.9% |
20.2% |
32.5% |
24.50% |
23.5% |
|
|
|
|
|
|
|
|
|
|
|
|
| |
(34.3%) |
(29.2%) |
(39.3%) |
(38.3%) |
(36.0%) |
(54.1%) |
(39.9%) |
(42.9%) |
|
|
|
|
|
|
|
|
|
|
|
|
| PDIA2 |
23.5% |
20.8% |
21.2% |
20.6% |
20.8% |
28.6% |
23.3.% |
23.3% |
47.3% |
|
|
|
|
|
|
|
|
|
|
|
| |
(35.5%) |
(38.7%) |
(31.8%) |
(38.7%) |
(32.1%) |
(49.0%) |
(37.2%) |
(37.7%) |
(64.5%) |
|
|
|
|
|
|
|
|
|
|
|
| PDIA3 |
21.7% |
16.9% |
21.5% |
18.8% |
21.4% |
21.8% |
22.3% |
23.1% |
33.6% |
31.1% |
|
|
|
|
|
|
|
|
|
|
| |
(37.5%) |
(37.9%) |
(33.3%) |
(31.3%) |
(37.5%) |
(38.9%) |
(34.1%) |
(40.6%) |
(51.0%) |
(49.5%) |
|
|
|
|
|
|
|
|
|
|
| PDIA4 |
19.80% |
19.7% |
18.8% |
21.4% |
17.5% |
18.3% |
29.6% |
24.0% |
36.8% |
29.1% |
41.9% |
|
|
|
|
|
|
|
|
|
| |
(31.6%) |
(38.6%) |
(38.2%) |
(38.5%) |
(30.8%) |
(35.0)% |
(48.1%) |
(40.9%) |
(55.1%) |
(46.2%) |
(60.2%) |
|
|
|
|
|
|
|
|
|
| PDIA5 |
24.2% |
22.1% |
21.3% |
21.7% |
21.4% |
19.6% |
19.7% |
22.6% |
24.3% |
23.1% |
22.9% |
21.2% |
|
|
|
|
|
|
|
|
| |
(45.1%) |
(44.1%) |
(35.1%) |
(36.9%) |
(40.0%) |
(33.5%) |
(33.9%) |
(40.6%) |
(38.3%) |
(35.0%) |
(37.1%) |
(31.5%) |
|
|
|
|
|
|
|
|
| PDIA6 |
27.6% |
15.2% |
24.0% |
19.4% |
32.7% |
20.6% |
17.0% |
20.2% |
23.5% |
21.8% |
24.2% |
25.8% |
28.1% |
|
|
|
|
|
|
|
| |
(35.2%) |
(57.6%) |
(40.6%) |
(33.1%) |
(48.0%) |
(34.8)% |
(32.2%) |
(38.4%) |
(31.4%) |
(28.6%) |
(33.4%) |
(40.0%) |
(43.1%) |
|
|
|
|
|
|
|
| PDILT |
23.9% |
16.2% |
20.1% |
21.1% |
20.4% |
29.7% |
19.1% |
21.9% |
31.8% |
32.1% |
22.1% |
26.0% |
19.9% |
23.5% |
|
|
|
|
|
|
| |
(45.7%) |
(35.9%) |
(43.4%) |
(41.8%) |
(36.1%) |
(54.1%) |
(38.5%) |
(40.9%) |
(52.4%) |
(51.3%) |
(37.1%) |
(46.7%) |
(35.3%) |
(36.3%) |
|
|
|
|
|
|
| TMX1 |
18.6% |
24.8% |
23.7% |
17.2% |
21.1% |
23.9% |
15.5% |
25.8% |
29.9% |
25.7% |
30.0% |
25.1% |
35.0% |
36.2% |
27.0% |
|
|
|
|
|
| |
(36.1%) |
(36.0%) |
(37.8%) |
(29.9%) |
(38.2%) |
(39.4%) |
(32.3%) |
(49.0%) |
(46.9%) |
(40.0%) |
(50.0%) |
(41.3%) |
(56.4%) |
(59.0%) |
(39.3%) |
|
|
|
|
|
| TMX2 |
23.0% |
18.6% |
20.2% |
28.6% |
23.3% |
22.0% |
25.6% |
23.10% |
25.0% |
28.3% |
25.8% |
25.7% |
16.4% |
24.7% |
24.7% |
24.6% |
|
|
|
|
| |
(36.1%) |
(33.1%) |
(39.9%) |
(44.4%) |
(44.8%) |
(41.5%) |
(43.6%) |
(38.0%) |
(40.9) |
(44.6%) |
(36.9%) |
(43.7%) |
(34.9%) |
(38.9%) |
(42.9%) |
(43.1%) |
|
|
|
|
| TMX3 |
20.0% |
19.7% |
20.3% |
21.8% |
22.4% |
18.1% |
20.5% |
21.4% |
27.8% |
21.6% |
23.1% |
27.1% |
23.5% |
45.3% |
19.2% |
34.2% |
18.7% |
|
|
|
| |
(37.4%) |
(36.5%) |
(43.0%) |
(43.6%) |
(44.1%) |
(33.3%) |
(35.7%) |
(34.7%) |
(47.9%) |
(40.3%) |
(41.1%) |
(42.7%) |
(41.2%) |
(64.2%) |
(32.2%) |
(50.5%) |
(38.4%) |
|
|
|
| TMX4 |
23.5% |
25.7% |
27.0% |
23.1% |
23.7% |
28.6% |
19.5% |
20.1% |
21.8% |
24.9% |
24.3% |
20.5% |
27.2% |
24.0% |
25.0% |
43.6% |
24.4% |
22.4% |
|
|
| |
(39.5%) |
(36.6%) |
(62.2%) |
(47.3%) |
(39.4%) |
(42.9%) |
(36.0%) |
(37.0%) |
(34.4%) |
(37.0%) |
(45.7%) |
(32.1%) |
(41.6%) |
(37.1%) |
(42.5%) |
(65.0%) |
(42.2%) |
(34.2%) |
|
|
| TXNDC5 |
21.6% |
20.9% |
22.9% |
20.7% |
25.4% |
22.0% |
20.8% |
33.3% |
26.2% |
26.9% |
24.4% |
23.5% |
27.3% |
33.0% |
23.3% |
24.6% |
29.5% |
51.4% |
21.6% |
|
| |
(28.4%) |
(39.5%) |
(37.9%) |
(34.0%) |
(41.5%) |
(37.9%) |
(32.9%) |
(50.0%) |
(37.2%) |
(37.5%) |
(38.5%) |
(34.1%) |
(43.8%) |
(53.0%) |
(34.9%) |
(33.8%) |
(50.0%) |
(71.6%) |
(38.0%) |
|
| TXNDC12 |
34.2% |
38.3% |
20.0% |
41.2% |
18.5% |
20.2% |
27.5% |
21.1% |
26.2% |
26.9% |
24.1% |
22.9% |
22.7% |
26.9% |
23.1% |
27.7% |
21.6% |
31.9% |
18.7% |
20.1% |
| (50.6%) | (53.9%) | (30.8%) | (70.6%) | (36.3%) | (30.2%) | (40.0%) | (32.2%) | (37.2%) | (37.5%) | (39.7%) | (41.2%) | (43.8%) | (37.0%) | (38.2%) | (39.8%) | (37.4%) | (48.9%) | (32.1%) | (31.5%) |
Sequence identity and similarity (in parentheses) are noted.
The human PDI gene family
DNAJC10
The DNAJC10 gene is located at Chr 2q32.1 and encodes the 793-a.a. DNAJC10 protein (also commonly known as ERdj5 or MTHr) [10]. The DNAJC10 gene consists of 3483 bp; transcription of two splice variants has been identified due to a skipped exon, resulting in a 138-bp (46 a.a.) absence, present between nucleotides 1243 and 1244 [10]. To date, a total of four missense single-nucleotide polymorphisms (SNPs) have been reported for ERdj5, located at a.a. 76 (Asp → Asn), 347 (Leu → Ile), 414 (Tyr → Cys) and 646 (His → Gln); ERdj5 also contains one potential Asn-linked glycosylation site, present at a.a. 530. Like other PDI family members, the DNAJC10 promoter region contains a putative ER stress element (ERSE) box, yielding gene induction following ER stress [11]. Expression patterns of DNAJC10 revealed ubiquitous expression with varying intensities and high levels of expression in secretory tissues such as the pancreas and testis [10,11]. In addition to the PDI family, DNAJC10 is also a member of the ERdj family, being comprised of an unverified NH2-terminal signal peptide (32 a.a.), one DnaJ domain (which plays a major role in protein folding), five TRX domains (one b and four a type domains, active sites Cys-Ser-His-Cys, Cys-Pro-Pro-Cys, Cys-His-Pro-Cys, and Cys-Gly-Pro-Cys), and a COOH-terminal ER retention sequence (Lys-Asp-Glu-Leu) [10,12]. Despite the high number of TRX domains, DNAJC10 was found to possess roughly one-third the activity of P4HB and displayed no oxidase or isomerase activity [12]. These results were found to reflect an apparent redox equilibrium constant of 190 mM, roughly 100 times more reducing than that of the ER [12].
ERP27
Currently, little is known about the precise role and function of ERP27. ERP27 was discovered in 2006 following a database search for novel human PDI family members [13]. The ERP27 gene has been mapped to Chr 12p12.3 and encodes the 273-a.a protein. The ERP27 protein contains a cleavable NH2-terminal signal sequence, leaving the mature protein to be 248 a.a, beginning at Glu-26 [13]. ERP27 does not contain a redox active Cys-X-X-Cys TRX motif, however, the protein does contain one b and one b’ type domains and an ER retention sequence (Lys-Val-Glu-Leu) [13]. ERP27 was also found to directly interact with another PDI family member, PDIA3 [13]. Expression of ERP27 is fairly ubiquitous, with highest expression found in pancreas [13]. To date, no knockout studies have been conducted and future studies are needed to fully understand the function and role of ERP27.
ERP29
The ERP29 gene is located at Chr 12q24.13 and may represent a gene-duplication event with ERP27, given their close proximity. ERP29 encodes a protein of 261 a.a. termed ERP29 (also known as ERP28). In a comprehensive study of the genomic organization of ERP29, Sargsyan et al. studied a 5′-flanking region consisting of ~2 kb, 3 exons, 2 introns and a 3′-flanking region of 0.31 kb [14]. ERP29 contains an NH2-terminal signal peptide (32 a.a.), one a type TRX domain and a COOH-terminal ER-retention sequence (Lys-Glu-Glu-Leu) [15]. While the presence of one a type domain is present, ERP29 is unique in that it does not contain an active-site motif; assignment of the TRX domain is placed strictly on sequence homology with the a-type TRX domain, not on the activity of the domain. ERP29 contains two potential phosphorylation sites, both located on tyrosine residues, located at a.a. 64 and 66. ER localization of ERP29 was also confirmed using immunofluorescence and subcellular fractionation [15]. ERP29 is ubiquitously expressed with high levels being found in secretory tissues as well as the prostate, pancreas, and liver [14,16]. Although ERP29 lacks any identified ERSE in its promoter region, ERP29 is described as an ER stress-inducible protein and has been shown to co-localize with other ER stress-associated chaperones, glucose-regulated protein 78 (GRP78 or BiP) [16]. ERP29 has been postulated to play a role in the progression of tumorigenesis in mice; following implantation of both knockdown and over-expressed null ERP29 MCF-7 cells, a significant decrease in tumor size and altered morphogenesis was observed in mice [17]. Currently, no knockout mouse is available for Erp29.
ERP44
The ERP44 gene is located at Chr 9q22.33 and encodes the 406-a.a. ERP44 protein [18]. ERP44 was discovered following immunoprecipitation experiments with human endoplasmic reticulum oxidoreductase-1α (ERO1-Lα) and was originally identified as KIAA0573 [18]. The coding sequence of ERP44 contains 12 exons and there is an ERSE in the promoter region of the gene [18]. The human ERP44 protein contains an unverified NH2-terminal signal peptide (29 a.a.), three TRX-like domains (one being the catalytically active a domain with a Cys-Arg-Phe-Ser active site), and the ER-retention sequence (Arg-Asp-Glu-Leu) [18]. The precise physiological function of ERP44 has yet to be determined; however, oxidation of ERO1α has been observed, suggesting that ERP44 may control the function of the ERO1 proteins, thus controlling the redox state of the ER [18]. ERP44 is up-regulated during ER stress response and has also been proposed to play a role in adiponectin secretion which influences glucose regulation and fatty acid catabolism [18,19].
P4HB
P4HB is the first described member of the PDI gene family and was originally identified as the β-subunit of human prolyl-4-hydroxylase (P4H) [20,21]. The P4HB gene is located at Chr 17q25 and consists of 11 exons [20,22]. The promoter region of P4HB contains 11 protein binding sites, including an ERSE, underscoring the dynamic nature of P4HB transcriptional regulation [23]. Although the presence of an ERSE has been confirmed in the promoter region of the gene, P4HB is considered to be a weakly-induced ER stress protein, likely due to its high abundance. The promoter region also contains six CCAAT elements in the first 378 nt of the gene, and mutations introduced into any of these elements was found to reduce promoter activity by up to 50% [23].
The P4HB gene encodes a 508-a.a. protein containing a 17-a.a. signal peptide, four TRX domains with two a type (Cys-Gly-His-Cys, Cys-Gly-His-Cys), an Asp/Glu rich domain (a.a. 480 to 500), and a COOH-terminal ER retention sequence (Lys-Glu-Asp-Leu) [2]. P4HB is ubiquitously expressed in nearly all tissues and is very highly abundant; estimations predict P4HB to account for up to 0.8% of total cellular protein [24]. Currently, a crystal structure has yet to be resolved for the full, intact protein, although multiple domains have been solved. Despite this, P4HB remains the most widely studied and understood protein in the family. P4HB is effective at oxidizing, reducing, and isomerizing disulfide bonds both in vitro and in vivo and exists as a homodimer [25,26]. Although its role in disulfide bond generation remains the most widely studied enzymatic action, P4HB has also been shown to exhibit chaperone-like activity, demonstrating an additional role in maturation of nascent proteins regardless of the presence of disulfide bonds [27,28]. P4HB has also been shown to be an essential subunit for microsomal triglyceride transfer protein and P4H [21,29]. To date, no viable knockout mouse strain for P4hb has been reported, likely due to its critical role in disulfide bond generation [3].
PDIA2
PDIA2 was identified in 1996 as a pancreas-specific member of the PDI family, resulting in its common name, PDIp [30]. Located on Chr 16p13.3, the initial characterization of the PDIA2 gene revealed that it encodes a protein with an ORF of 511 a.a; these studies were unable to validate an in-frame stop codon located 5′ upstream of the ATG start site [30]. Due to this discrepancy, the PDIA2 sequence was verified in 2006 to have an NH2-terminal signal sequence, generating a protein of 525 a.a. in length. The PDIA2 gene also yields two splice variants encoding two isoforms of the mature PDIA2 protein, varying by three amino acids in length (isoform 2 does not contain a.a. 181 to 183). Multiple SNPs have been detected in the PDIA2 gene, resulting in mutations at a.a. 39 (Pro → Ser), 119 (Thr → Arg), 185 (Glu → Lys), 286 (Thr → Met), 382 (Pro → Ala), 388 (Arg → Gln), and 502 (Pro → Ser).
The PDIA2 protein contains two a type (Cys-Gly-His-Cys and Cys-Thr-His-Cys active sites) and one b and one b’ type domains; although redox active, PDIA2 was found to be less effective than P4HB in assays for both reduction and oxidation of disulfides [30]. In addition to its role as a folding catalyst, PDIA2 has been proposed to play a role in the production and secretion of digestive enzymes in vivo[31]. Evidence has also suggested a role for PDIA2 in the binding and regulation of intracellular 17β-estradiol levels, thus regulating estrogen synthesis [32]. PDIA2 contains three sites for Asn-linked glycosylation, located at residues 127, 284, and 516 [30]. To date, no knockout studies have been conducted with Pdia2.
PDIA3
Originally identified as phospholipase C alpha, the PDIA3gene, located at Chr 15q15, encodes the 505-a.a PDIA3 protein (also commonly known as ERp57, ERp60, P58) [33]. Like other PDI family members, PDIA3 contains a signal peptide, corresponding to the first 24 a.a., yielding a mature protein of 481 a.a. in length [34]. PDIA3 contains two a type (Cys-Gly-His-Cys and Cys-Gly-His-Cys active sites), one b and one b’ type domain and an ER retention sequence, Gln-Glu-Asp-Leu [35,36]. PDIA3 expression has been detected in liver, placenta, lung, pancreas, kidney, heart, skeletal muscle, brain, and spermatozoa [33,37]. Presently, only one missense SNP has been reported for PDIA3, resulting in a mutation of Lys → Arg at a.a. 415.
The precise physiological role of PDIA3 has come under much scrutiny. Following the initial characterization of the protein in vitro, PDIA3 (termed ERp60 or P58 at the time) was identified as a cysteine protease despite little sequence homology with other heavily studied cysteine proteases [35]. In 1995, however, PDIA3 was determined to be redox active, showing the ability to reduce insulin disulfides. Bourdi et al. were also able to definitively prove that PDIA3 possessed no protease activity [36]. PDIA3 has also been shown to play a role in the correct folding of glycoproteins when in a complex containing both calnexin and calreticulin [38]. The physiological role of PDIA3 has also been investigated in rodent models, utilizing Pdia3(−/−) knockout mice. Although ubiquitous deletion of Pdia3 was found to be embryolethal, heterozygous knockouts were generated, revealing multiple bone abnormalities, most notably in the femur [39]. Ablation of Pdia3 was found to abolish signaling induced by 1,25-dihydroxyvitamin D3, a crucial regulator of bone and cartilage development, by eliminating signaling through protein kinase C [39]. Additional knockout studies in murine B cells revealed a critical role for PDIA3 in the presentation of antigens by major histocompatibility complex I molecules [40]. Although additional work is needed, these studies suggest a wide array of physiological roles for PDIA3.
PDIA4
PDIA4 is located at Chr 7q35 and encodes the 645-a.a. PDIA4 protein (commonly known as ERp72). Although not much is known about the physiological role of PDIA4, studies indicate that this gene is induced following ER stress; these results have been found to be the result of a putative ERSE in the promoter region of the gene [41,42]. Like other PDI family members, PDIA4 contains an NH2-terminal signal peptide of 21 a.a., yielding a mature protein of 625 a.a. [43]. PDIA4 contains five TRX domains, three a type (with all three active sites being comprised of Cys-Gly-His-Cys), one b and one b’ type; PDIA4 also contains the ER retention sequence (Lys-Glu-Glu-Leu) [43,44]. Mutagenesis studies to the active-site cysteines revealed varying degrees of decreased enzymatic activity, whereas mutagenesis to multiple domains revealed a more pronounced reduction in enzymatic activity of the protein [44]. PDIA4 also contains a string of highly acidic residues near the NH2-terminus of the protein; while the precise role of these residues remains unknown, they have been proposed to play a role in regulation of Ca2+, yielding its rat homolog name calcium-binding protein-2 (CaBP2) [45]. PDIA4 is a fairly ubiquitously expressed protein, although less abundant than PDI, expression patterns are similar to those of PDIA3 [46]. One missense SNP has been reported in the PDIA4 gene, resulting in a mutation located at residue 173 (Thr → Met). Studies analyzing Pdia3 knockdown revealed partial functional restoration by the PDIA4 protein, although, to date, no Pdia4 knockout mouse has been generated [47].
PDIA5
Although discovered in 1995, little is known about the precise role of the PDIA5 gene. Located on Chr 3q21.1, PDIA5 encodes the 519-a.a. PDIA5 (or PDI-related protein). PDIA5 contains four TRX domains (three a and one b-like domain), made up of active sites Cys-Ser-Met-Cys, Cys-Gly-His-Cys and Cys-Pro-His-Cys, a COOH-terminal ER retention sequence (Lys-Glu-Glu-Leu), and an unverified signal sequence comprising the first 21 a.a. [48]. Despite an additional Cys-X-X-Cys motif, Horibe et al. revealed that PDIA5 has significantly less enzymatic activity than that of P4HB [49]. The contributions of each Cys-X-X-Cys active site were also investigated, revealing varying degrees of altered activity following mutations to each, or multiple, active sites [49]. It was concluded that the second active site (Cys-Gly-His-Cys) was the most critical for isomerase activity and that all three motifs are not required for activity [49]. Much like P4HB, PDIA5 was also shown to exhibit chaperone-like activity by refolding denatured rhodenase, which does not contain any disulfide bonds [49]. PDIA5 mRNA has been detected in liver, kidney, lung, and brain––with the highest level of secretion being noted in the liver [48]. Although to date no ERSE has been identified, PDIA5 has been shown to be moderately up-regulated following induction of the ER-stress response in cultured cells [48]. In a 2011 study by Carbone et al., a significant association was found between the SNP, rs11720822, and primary open-angle glaucoma in two separate populations [50]. No viable knockout mouse has been generated for Pdia5.
PDIA6
Much like PDIA5, little is known about the role of PDIA6 both in vitro and in vivo. The PDIA6 gene is located at Chr 2p25.1 and encodes the 440-a.a. PDIA6 protein (commonly reported as P5 or ERP5). PDIA6 contains an NH2-terminal signal sequence of 19 a.a., three TRX domains (two a type and one b) consisting of two Cys-Gly-His-Cys active sites, an Asp/Glu rich domain and a COOH-terminal Lys-Asp-Glu-Leu ER-retention sequence [51,52]. Recombinant PDIA6 demonstrates both isomerase and chaperone activities, although approximately 45% and 50% to 60% to that of P4HB, respectively [53]. Point mutations have also been conducted to the active-site cysteines, revealing that NH2-terminal cysteines in each active site exhibit the majority of isomerase activity [53]. PDIA6 contains an ERSE in its promoter region, which was recently validated in vitro utilizing over-expression of the ER-stress transcription factor, X-box protein-1 (XBP-1); PDIA6 was found to be significantly increased, demonstrating inducibility by the unfolded protein response [54,55]. A complete expression profile for PDIA6 has yet to be conducted; however, high levels of the protein were detected in platelets. Cell-surface expression of PDIA6 was found to be necessary for the proper development and function of platelets, whereas inhibition of the protein using anti- PDIA6 antibodies revealed inhibition of platelet aggregation [51]. PDIA6 has also been shown to directly interact with GRP78 (or BiP) suggesting a role for PDIA6 in the refolding of substrates that have been targeted to BiP [56]. Presently, one missense SNP has been identified (rs4807) resulting in a point mutation at a.a. 214 (Lys → Arg). At present, no viable Pdia6 knockout mouse is available.
PDILT
Expression of the PDILT gene has been reported to be exclusively limited to the testis. PDILT is located on Chr 16p12.3, encoding the 584-a.a. PDILT protein [57]. Despite the presence of two a type TRX domains (with non-classical Ser-Lys-Gln-Ser and Ser-Lys-Lys-Cys motifs), PDILT does not exhibit the ability to oxidize or reduce disulfide bonds, although evidence has supported PDILT to engage in disulfide-bonded complexes in vitro[57,58]. PDILT contains a predicted NH2-terminal signal peptide of 20 a.a. in length, a COOH-terminal ER-retention sequence (Lys-Glu-Glu-Leu) and is heavily glycosylated through nine potential Asn-linked glycosylation sites [57]. Much like P4HB, PDILT also interacts with the oxidoreductase ERo1α in cultured cells, suggesting a role in the shuffling of electrons in the ER lumen [57]. No knockout mouse is currently available and considerable research is needed to fully elucidate the precise role of PDILT.
TXNDC5
TXNDC5 is located at Chr 6p24.3 and encodes the 432-a.a. endothelial PDI, TXNDC5 (or EndoPDI) protein [59]. Despite its discovery in 2003, little research has been conducted on the role of TXNCD5 in vivo. TXNDC5 contains a signal peptide of 32 a.a., three a-type TRX domains all with Cys-Gly-His-Cys active sites, and a COOH-terminal ER retention sequence, KDEL [59]. TXNDC5 was originally identified in a screen for proteins highly expressed in endothelial cells, leading to its common name, EndoPDI. Subsequent studies revealed TXNDC5 expression in a number of tissues with the highest expression being found in lymph nodes, stomach, pancreatic islets, and heart [59,60]. TXNDC5 is induced under conditions of hypoxia, and loss of TXNDC5 leads to an increase in apoptotic cell death in microvascular endothelial cells during hypoxia, but not normoxia [59]. Preliminary studies have also implicated a role for TXNDC5 in diabetes, noting a decrease in the expression of the protein in pancreatic islets in animals with consistently elevated glucose levels [60]. In a 2010 study, Jeong et al. investigated the role of TXNDC5 in development of the skin disorder vitiligo [61]. A total of 230 Korean patients with non-segmental vitiligo were investigated for SNPs in the TXNDC5 gene; in total, seven SNPs were identified in the TXNDC5 gene, three of which (rs1043784, rs7764128, and rs8643) demonstrated an association with the vitiligo phenotype [61]. Although relevant in vivo studies have been conducted on the role of TXNDC5 no biochemical parameters have been evaluated, with regard to its role in disulfide bond oxidation and reduction. A viable knockout mouse has not been generated for the study of Txndc5.
The anterior gradient homolog genes
AGR2
The AGR2 gene is located at Chr 7p21.3 and encodes the 175-a.a. anterior gradient protein 2 homolog (AGR2) [62,63]. The AGR2 protein has an NH2-terminal signal sequence of 20 a.a., one TRX domain (with active site Cys-Pro-His-Ser), and a COOH-terminal ER-retention sequence (Lys-Thr-Glu-Leu) [63]. Human AGR2 was originally identified in estrogen receptor-positive MCF7 cells using suppression subtractive hybridization [64]. Expression of AGR2 transcripts has been detected in lung, pancreas, trachea, stomach, colon, prostate, and small intestine. AGR2 has also been investigated as a potential biomarker for hormone-responsive breast cancer in estrogen receptor-α-positive breast cancer cell lines [64].
Utilizing knockout studies in mice, the Agr2 gene was found to result in the inability to produce mucin, leading in an increased susceptibility to experimentally induced colitis and intestinal disease [65]. Due to its role in disulfide bond generation, it was hypothesized that AGR2 was responsible for the processing of MUC2, the major intestinal mucin. This protein contains >200 cysteine residues involved in various inter- and intra-protein disulfide bonds and has been found to directly associate with AGR2 [65]. Following this report, Zheng et al. investigated AGR2 and AGR3 as potential candidate genes for inflammatory bowel disease in humans [66]. A cohort of 2,540 patients having either ulcerative colitis or Chron’s disease was investigated for SNPs in AGR2 and AGR3; in total, 30 SNPs were identified, 25 were located in the AGR2 gene, while 5 were located in the AGR3 gene [66]. The promoter region of the AGR2 gene was also found to contain binding sites for hepatic nuclear factor-1, hepatocyte nuclear factor 3-α (FOXA1) and hepatocyte nuclear factor 3-β (FOXA2)––transcription factors that have been reported to play a role in the morphogenesis of goblet cell differentiation during formation. In summary, two total SNPs in the 5′ promoter region of the AGR2 gene were found to be associated with the risk haplotype of ulcerative colitis in two independent populations, providing further evidence for a role for AGR2 in disease pathogenesis [66].
AGR3
The AGR3 gene is located at Chr 7p21.1 and encodes the 166-a.a. anterior gradient protein-3 (AGR3) homolog [63]. AGR3 transcripts have been detected in lung and pancreas and, resembling AGR2, AGR3 has been reported to be co-expressed with estrogen receptor-α-containing breast cancer cell lines, suggesting it to be a marker for hormone-responsive breast cancer [63,64]. Unlike AGR2, however, little is known about the precise physiological role of AGR3. AGR3 protein was originally identified as breast cancer membrane protein 11 (BCMP11), following a proteomic screen of membrane proteins in breast cancer cell lines, and was later named AGR3 due to the high degree of sequence homology with AGR2 (see Table 2) [67]. Like AGR2, AGR3 contains one redox-active center, comprised of amino acids Cys-Gln-Tyr-Ser, an NH2-terminal signal peptide composed of 21 a.a., and a COOH-terminal ER-retention sequence (Gln-Ser-Glu-Leu) [63]. Recently, an increase in AGR3 expression was observed in serous border-line ovarian tumors and low-grade serous ovarian carcinoma [68]. Utilizing Kaplan-Meier survival curves, King et al. also established that patients with AGR3-expressing tumors survived significantly longer than those patients lacking AGR3-expressing tumors [68].
TXNDC12
TXNDC12, also known as AGR1TLP19, and ERP18/19, contains conserved intron positions with respect to amino acid sequence with the AGR2 and AGR3 genes [63]. Persson et al. also reported that several of the individual exon lengths are identical (or altered with one codon) to AGR2 and AGR3[63]. TXNDC12 has been mapped to Chr 1p32.3 and contains seven exons spanning more than 35 kb [69]. TXNDC12 is ubiquitously expressed in all tissues, with the highest expression being found in the liver and placenta. TXNDC12 in the placenta was found to express an additional transcript of 1.2 kb which is associated with two poly(A) addition signals in its 3′-UTR [69]. The TXNDC12 protein contains 172 a.a. with 149 a.a. comprising the mature form of the protein (Ser24 – Leu172); TXNDC12 has one active site comprised of Cys-Gly-Ala-Cys and an ER-retention sequence (Glu-Asp-Glu-Leu) [70]. Unlike the other members of the AGR subfamily, extensive work has been conducted on the biochemical and physiochemical actions of TXNDC12. The enzymatic activity of TXNDC12 has been found to be limited strictly to disulfide bond generation and not reduction; these studies were also confirmed with the use of point mutations to the active-site cysteines (Cys-Gly-Ala-Cys), after which no detectable activity was found [70]. Chemical denaturation curves were also found to favor greater protein stability in the reduced form over the oxidized form, a property consistent with other PDI family members [70].
The CASQ genes
The CASQ genes (1 and 2) are interesting members of the PDI family, possessing no cysteine containing redox-active sites and therefore playing no role in the formation or reduction of disulfide bonds. As indicated, many of the PDI family proteins bind Ca2+ with relatively high capacity and low affinity [71]. Following studies on CASQ, Shin et al. found that the COOH-terminal Asp-rich domain played a major role in storage of Ca2+ through interaction with ryanodine receptor (RYR), a protein involved in Ca2+ release from the sarcoplasmic reticulum (SR) [72]. These proteins, therefore, possess unique functions relating to the PDI family of proteins, despite limited sequence homology.
CASQ1
The CASQ1 gene is located on Chr 1q21 and encodes the 396-a.a. CASQ1 protein [73]. Expression of the mature CASQ1 protein is primarily limited to the SR of fast-twitch skeletal muscles [74]. Studies in rabbit have revealed CASQ1 to be a high-capacity (40 to 50 Ca2+ per molecule of CASQ1), moderate-affinity (Kd = 1 mM) Ca2+-binding protein that does not contain an ER-retention sequence [74-76]. Although experimentally unverified, the first 34 a.a. of CASQ1 encode the signal peptide, leaving the mature protein at 362 a.a. in length––which contains three TRX domains (two b and one b’) and a string of highly acidic residues from a.a. 353 – 396. Like other members of the PDI family, these Asp/Glu-rich stretches of a.a. are thought to be the primary binding regions for Ca2+; CASQ1 plays a major role in Ca2+ flux through the regulation of Ca2+ channel activity and interaction with Ca2+ directly [72]. Systemic knockout studies in mice revealed hypersensitivity to heat and volatile anesthetics, along with a phenotypic resemblance to malignant hyperthermia [77]; these effects were found to be due to increased Ca2+ following an increased exposure to heat [78]. CASQ1 also contains one potential Asn-linked glycosylation site, found at a.a. 350.
SNPs in the CASQ1 gene have been reported in cases of diabetes in both Old-Order Amish and Northern European Caucasians [79,80]. Out of 26 identified SNPs, SNP CASQ-1404 (rs1186694) in the 5′ flanking region was found to have a statistically significant association with type-2 diabetes in Northern European Caucasians [79]. In a similar study analyzing type-2 diabetes susceptibility in Old-Order Amish populations, SNPs rs2275703 and rs617698 were defined as the ‘at-risk alleles’ [80]. Although mechanistically these correlations have not been confirmed, previous work has identified a putative role for Ca2+ release from the SR into the cytosol in regulating glucose transporter-4 (GLUT4), a key enzyme in regulation of glucose transport by insulin [80,81].
CASQ2
The CASQ2 gene is located on Chr. 1p13.3-p11, and likely is the result of gene duplication with CASQ1––given their chromosomal location. CASQ2 encodes the 399-a.a. CASQ2, which shares 91% identity with its homologue, CASQ1 (see Table 2) [82,83] The CASQ2 protein is expressed exclusively in cardiac muscle and serves as the major Ca2+ reservoir in the SR of myocardium; CASQ2 also interacts with the RYR2 channel, regulating Ca2+ flux from the SR [76,84]. Much like CASQ1, CASQ2 contains an unverified NH2-terminal signal peptide (19 a.a.), three b-like TRX domains (two b and one b’), no ER-retention sequence, and a string of highly acidic residues (a.a. 356 to 399); CASQ2 does, however, contain one potential Asn-linked glycosylation site at a.a. 355.
The CASQ2 gene has become a heavily researched target for diseases associated with arrhythmic heartbeats. In 2001, Lahat et al. investigated missense mutations found in the coding region of the gene [83]. One SNP (G −> C) was found to result in an aspartic acid changed to a histidine at a.a. 307 of the mature protein, potentially altering the Ca2+-chelating function of that region [83]. This SNP was found to be associated with Bedouin families from Israel susceptible to catecholine-induced polymorphic ventricular tachycardia [83]. These studies were later confirmed in Casq2(−/−) knockout mice, revealing susceptibility to polymorphic ventricular tachycardia following exposure to catecholamines [85].
Thioredoxin-related transmembrane proteins
The thioredoxin-related transmembrane (TMX) genes are newly discovered members of the PDI gene family. To date, little is known about the precise function of these genes; however, all four members in the PDI family consist of one TRX domain, one transmembrane domain, and non-conventional ER-retention sequences.
TMX1
Discovered in 2001, TMX1 is located at Chr 14q22.1 and encodes the 280-a.a. TMX1 protein [86]. TMX1 contains an NH2-terminal signal sequence of 26 a.a., one a-type TRX domain with active site Cys-Pro-Ala-Cys, one transmembrane domain (a.a. 183 to 203), and lacks an ER-retention sequence [86]. Expression of TMX1 is fairly ubiquitous, with highest levels detected in liver, kidney, placenta, and lung [86]. Mature TMX1 possesses the ability to both oxidize and reduce disulfide bonds, although chaperone-like activity has yet to be investigated [86,87]. The TMX1 gene does not contain a putative ERSE in the promoter region, supporting evidence that TMX1 is not induced by numerous ER-stress-inducing agents; over-expression of the protein in cultured cells, however, has revealed amelioration of both Brefeldin A-induced apoptosis and tunicamycin-induced ER stress [86,88]. No knockout mouse has been generated for the study of Tmx1.
TMX2
Perhaps the least researched gene in the family, TMX2 was discovered in 2003 [89]. Located on Chr 11cen-q22.3, TMX2 encodes the 296-a.a. TMX2 protein [89]. Like TMX1, TMX2 contains a COOH-terminal signal peptide (48 a.a.), one a-type TRX domain (Ser-Asn-Asp-Cys active site), one transmembrane domain (a.a. 104 – 126), and an ER-retention sequence comprised of Lys-Lys-Glu-Ile [89]. Expression of TMX2 is fairly ubiquitous––with high levels detected in heart, brain, liver, kidney, and pancreas [89]. Although Meng et al. provided the initial characterization of the protein, sequence discrepancies have been found. The official NCBI sequence of the TMX2 protein reveals a protein of 296 a.a and a second isoform, lacking an in-frame exon in the central coding region, encoding a protein of 258 a.a (isoform 2 differs between a.a. 84 to 122). Future studies are required to fully elucidate the role of TMX2 in vivo. The availability of a TMX2 knockout mouse has not been reported.
TMX3
The human TMX3 gene is located at Chr 18q22 and encodes the 454-a.a. TMX3 protein [90]. Following cleavage of the 24-a.a. signal peptide, the 430-a.a. mature protein consists of one a-type TRX domain, with the active site being comprised of Cys-Gly-His-Cys, a transmembrane domain (located at a.a. 375 to 397), and the ER-retention sequence (Lys-Lys-Lys-Asp) [90]. Uncharacteristic to most PDI family members, TMX3 contains a luminal domain with weak sequence similar to that of the CASQ proteins [90]. Although no research has been conducted on the role of this domain in activity of the protein, it has been postulated to regulate Ca2+ in a manner similar to that of other CASQ proteins. NCBI reports two isoforms for TMX3 (one encoding a 195-a.a. protein), though experimentally this has not been validated [90]. TMX3 has been detected in brain, testis, lung, skin, kidney, uterus, bone, stomach, liver, prostate, placenta, eye, and muscle, with highest levels detected in heart and skeletal muscle [90]. TMX3 contains two sites of Asn-linked glycosylation (a.a. 258 and 313), which have been validated in vitro and is not induced under conditions of ER stress [90]. Although far less efficient than P4HB, TMX3 does display the ability to oxidize disulfide bonds; this is likely due to the presence of only one Cys-Gly-His-Cys active site [90].
Although no knockout mouse has been generated for TMX3, studies have been conducted in mice, targeting TMX3 transcripts using morpholinos [91]. Investigating the mechanisms behind microphthalmia in humans, a genetic disease associated with retarded growth of the eye, a 2.7-Mb deletion was found at Chr 18q22.1, leading to deletion of the TMX3 gene [91]. Studies in mice using a targeted approach to delete the TMX3 gene revealed a similar phenotype, which was rescued following injection of human TMX3 mRNA [91]. Sequencing of 162 patients with anopthalmia or microphthalmia revealed two missense mutations, leading to the missense SNPs, R39N, and D108N [91]. Future studies are required to fully elucidate the precise role of TMX3, although preliminary studies reveal exciting areas for research.
TMX4
The TMX4 gene is located at Chr 20p12 and encodes the 349-a.a. TMX4 protein [92]. TMX4 consists of an NH2-terminal signal sequence (23 a.a.), one a-type TRX domain (a.a. 39 – 136 with the active site comprised of Cys-Pro-Ser-Cys), a transmembrane domain (a.a. 188 to 210), a string of highly acidic a.a. (224 to 334), and (like the other TMX proteins) lacks an ER-retention sequence [93]. TMX4 is ubiquitously expressed, with highest levels detected in heart [92]. Preliminary studies show that TMX4 is not induced following conditions of ER stress and does not contain a putative ERSE in the promoter region of the gene [92]. TMX4 contains one site of Asn-linked glycosylation and two sites of Ser phosphorylation, which have all been experimentally validated [93]. Enzymatic activity was confirmed by observing reduction of insulin disulfides; a dominant-negative mutant, with the active-site cysteines mutated to serine, displayed no enzymatic activity [92]. No knockout mouse has been generated and substantial work will be required to understand the role of TMX4 in vivo.
Conclusions
The PDI family of proteins consists of 21 members varying greatly in enzymatic activity, domain architecture, and tissue specificity. Although the predominant role of the PDI proteins is the regulation of protein folding in vivo––through the oxidation, reduction and isomerization of disulfide bonds––these proteins have also been shown to regulate calcium homeostasis in the ER and induction of the unfolded protein response (UPR). Since its discovery over 40 years ago, PDI has become one of the most highly studied proteins and, despite these advances, extensive research is still needed to fully understand the role of PDI in vivo. The more recently characterized TMX genes have displayed promise in novel therapeutics, ranging from disorders of the eye to regulation of the ER-stress response. Many of the genes in the PDI family contain a putative ERSE sequence in the promoter region of the gene, suggesting a role in the UPR. Further research on the role of these proteins in the UPR is required before effective therapeutics can be generated for a plethora of disease states associated with the ER-stress response. The 21 members of the PDI family of proteins encompass many physiological responses, and these proteins will likely provide compelling avenues for future research.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JJG carried out the sequence alignment and drafted the manuscript. DRP designed and funded the study. Both authors read and approved the final manuscript.
Contributor Information
James J Galligan, Email: James.Galligan@UCDenver.edu.
Dennis R Petersen, Email: Dennis.Petersen@UCDenver.edu.
Acknowledgments
Studies were supported by the National Institutes of Health/National Institutes of Alcoholism and Alcohol Abuse under grant numbers R37AA09300 (DRP), R01DK074487-01 (DRP) and F31AA018606-01 (JJG).
References
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783:535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6:28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquot JP, Gelhaye E, Rouhier N, Corbier C, Didierjean C, Aubry A. Thioredoxins and related proteins in photosynthetic organisms: molecular basis for thiol dependent regulation. Biochem Pharmacol. 2002;64:1065–1069. doi: 10.1016/S0006-2952(02)01177-2. [DOI] [PubMed] [Google Scholar]
- Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- Rapoport TA. Protein transport across the endoplasmic reticulum membrane: facts, models, mysteries. FASEB J. 1991;5:2792–2798. doi: 10.1096/fasebj.5.13.1916103. [DOI] [PubMed] [Google Scholar]
- Houston NL, Fan C, Xiang JQ, Schulze JM, Jung R, Boston RS. Phylogenetic analyses identify 10 classes of the protein disulfide isomerase family in plants, including single-domain protein disulfide isomerase-related proteins. Plant Physiol. 2005;137:762–778. doi: 10.1104/pp.104.056507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AG, Knodler LA, Silberman JD, Davids BJ, Gillin F, Sogin ML. The evolutionary origins of eukaryotic protein disulfide isomerase domains: new evidence from the Amitochondriate protist Giardia lamblia. Mol Biol Evol. 2001;18:1455–1463. doi: 10.1093/oxfordjournals.molbev.a003931. [DOI] [PubMed] [Google Scholar]
- Gu SH, Chen JZ, Ying K, Wang S, Jin W, Qian J, Zhao EP, Xie Y, Mao YM. Cloning and identification of a novel cDNA which encodes a putative protein with a DnaJ domain and a thioredoxin active motif, human macrothioredoxin. Biochem Genet. 2003;41:245–253. doi: 10.1023/A:1025510502147. [DOI] [PubMed] [Google Scholar]
- Cunnea PM, Miranda-Vizuete A, Bertoli G, Simmen T, Damdimopoulos AE, Hermann S, Leinonen S, Huikko MP, Gustafsson JA, Sitia R, Spyrou G. ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress. J Biol Chem. 2003;278:1059–1066. doi: 10.1074/jbc.M206995200. [DOI] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- Alanen HI, Williamson RA, Howard MJ, Hatahet FS, Salo KE, Kauppila A, Kellokumpu S, Ruddock LW. ERp27, a new non-catalytic endoplasmic reticulum-located human protein disulfide isomerase family member, interacts with ERp57. J Biol Chem. 2006;281:33727–33738. doi: 10.1074/jbc.M604314200. [DOI] [PubMed] [Google Scholar]
- Sargsyan E, Baryshev M, Backlund M, Sharipo A, Mkrtchian S. Genomic organization and promoter characterization of the gene encoding a putative endoplasmic reticulum chaperone, ERp29. Gene. 2002;285:127–139. doi: 10.1016/S0378-1119(02)00417-1. [DOI] [PubMed] [Google Scholar]
- Ferrari DM, Van Nguyen P, Kratzin HD, Soling HD. ERp28, a human endoplasmic-reticulum-lumenal protein, is a member of the protein disulfide isomerase family but lacks a CXXC thioredoxin-box motif. Eur J Biochem. 1998;255:570–579. doi: 10.1046/j.1432-1327.1998.2550570.x. [DOI] [PubMed] [Google Scholar]
- Mkrtchian S, Fang C, Hellman U, Ingelman-Sundberg M. A stress-inducible rat liver endoplasmic reticulum protein, ERp29. Eur J Biochem. 1998;251:304–313. doi: 10.1046/j.1432-1327.1998.2510304.x. [DOI] [PubMed] [Google Scholar]
- Mkrtchian S, Baryshev M, Sargsyan E, Chatzistamou I, Volakaki AA, Chaviaras N, Pafiti A, Triantafyllou A, Kiaris H. ERp29, an endoplasmic reticulum secretion factor is involved in the growth of breast tumor xenografts. Mol Carcinog. 2008;47:886–892. doi: 10.1002/mc.20444. [DOI] [PubMed] [Google Scholar]
- Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A, Sitia R. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J. 2002;21:835–844. doi: 10.1093/emboj/21.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZV, Schraw TD, Kim JY, Khan T, Rajala MW, Follenzi A, Scherer PE. Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol Cell Biol. 2007;27:3716–3731. doi: 10.1128/MCB.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasanen K, Parkkonen T, Chow LT, Kivirikko KI, Pihlajaniemi T. Characterization of the human gene for a polypeptide that acts both as the beta subunit of prolyl 4-hydroxylase and as protein disulfide isomerase. J Biol Chem. 1988;263:16218–16224. [PubMed] [Google Scholar]
- Pihlajaniemi T, Helaakoski T, Tasanen K, Myllyla R, Huhtala ML, Koivu J, Kivirikko KI. Molecular cloning of the beta-subunit of human prolyl 4-hydroxylase. This subunit and protein disulphide isomerase are products of the same gene. EMBO J. 1987;6:643–649. doi: 10.1002/j.1460-2075.1987.tb04803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajunen L, Jones TA, Goddard A, Sheer D. Regional assignment of the human gene coding for a multifunctional polypeptide (P4HB) acting as the beta-subunit of prolyl 4-hydroxylase and the enzyme protein disulfide isomerase to 17q25. Cytogenet Cell Genet. 1991;56:165–168. doi: 10.1159/000133078. [DOI] [PubMed] [Google Scholar]
- Tasanen K, Oikarinen J, Kivirikko KI, Pihlajaniemi T. Promoter of the gene for the multifunctional protein disulfide isomerase polypeptide. Functional significance of the six CCAAT boxes and other promoter elements. J Biol Chem. 1992;267:11513–11519. [PubMed] [Google Scholar]
- Ferrari DM, Soling HD. The protein disulphide-isomerase family: unravelling a string of folds. Biochem J. 1999;339(Pt 1):1–10. [PMC free article] [PubMed] [Google Scholar]
- Freedman RB, Brockway BE, Lambert N. Protein disulphide-isomerase and the formation of native disulphide bonds. Biochem Soc Trans. 1984;12:929–932. doi: 10.1042/bst0120929. [DOI] [PubMed] [Google Scholar]
- Freedman RB, Hirst TR, Tuite MF. Protein disulphide isomerase: building bridges in protein folding. Trends Biochem Sci. 1994;19:331–336. doi: 10.1016/0968-0004(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Song JL, Wang CC. Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese. Eur J Biochem. 1995;231:312–316. doi: 10.1111/j.1432-1033.1995.tb20702.x. [DOI] [PubMed] [Google Scholar]
- Wang CC, Tsou CL. Protein disulfide isomerase is both an enzyme and a chaperone. FASEB J. 1993;7:1515–1517. doi: 10.1096/fasebj.7.15.7903263. [DOI] [PubMed] [Google Scholar]
- Wetterau JR, Combs KA, Spinner SN, Joiner BJ. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J Biol Chem. 1990;265:9800–9807. [PubMed] [Google Scholar]
- Desilva MG, Lu J, Donadel G, Modi WS, Xie H, Notkins AL, Lan MS. Characterization and chromosomal localization of a new protein disulfide isomerase, PDIp, highly expressed in human pancreas. DNA Cell Biol. 1996;15:9–16. doi: 10.1089/dna.1996.15.9. [DOI] [PubMed] [Google Scholar]
- Klappa P, Stromer T, Zimmermann R, Ruddock LW, Freedman RB. A pancreas-specific glycosylated protein disulphide-isomerase binds to misfolded proteins and peptides with an interaction inhibited by oestrogens. Eur J Biochem. 1998;254:63–69. doi: 10.1046/j.1432-1327.1998.2540063.x. [DOI] [PubMed] [Google Scholar]
- Fu XM, Zhu BT. Human pancreas-specific protein disulfide isomerase homolog (PDIp) is an intracellular estrogen-binding protein that modulates estrogen levels and actions in target cells. J Steroid Biochem Mol Biol. 2009;115:20–29. doi: 10.1016/j.jsbmb.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Horelli-Kuitunen N, Helaakoski T, Karvonen P, Jaakkola M, Palotie A, Kivirikko KI. Structures of the human gene for the protein disulfide isomerase-related polypeptide ERp60 and a processed gene and assignment of these genes to 15q15 and 1q21. Genomics. 1997;42:397–404. doi: 10.1006/geno.1997.4750. [DOI] [PubMed] [Google Scholar]
- Charnock-Jones DS, Day K, Smith SK. Cloning, expression and genomic organization of human placental protein disulfide isomerase (previously identified as phospholipase C alpha) Int J Biochem Cell Biol. 1996;28:81–89. doi: 10.1016/1357-2725(95)00120-4. [DOI] [PubMed] [Google Scholar]
- Urade R, Oda T, Ito H, Moriyama T, Utsumi S, Kito M. Functions of characteristic Cys-Gly-His-Cys (CGHC) and Gln-Glu-Asp-Leu (QEDL) motifs of microsomal ER-60 protease. J Biochem. 1997;122:834–842. doi: 10.1093/oxfordjournals.jbchem.a021830. [DOI] [PubMed] [Google Scholar]
- Bourdi M, Demady D, Martin JL, Jabbour SK, Martin BM, George JW, Pohl LR. cDNA cloning and baculovirus expression of the human liver endoplasmic reticulum P58: characterization as a protein disulfide isomerase isoform, but not as a protease or a carnitine acyltransferase. Arch Biochem Biophys. 1995;323:397–403. doi: 10.1006/abbi.1995.0060. [DOI] [PubMed] [Google Scholar]
- Kameshwari DB, Bhande S, Sundaram CS, Kota V, Siva AB, Shivaji S. Glucose-regulated protein precursor (GRP78) and tumor rejection antigen (GP96) are unique to hamster caput epididymal spermatozoa. Asian J Androl. 2010;12:344–355. doi: 10.1038/aja.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver JD, van der Wal FJ, Bulleid NJ, High S. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science. 1997;275:86–88. doi: 10.1126/science.275.5296.86. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen J, Lee CS, Nizkorodov A, Riemenschneider K, Martin D, Hyzy S, Schwartz Z, Boyan BD. Disruption of Pdia3 gene results in bone abnormality and affects 1alpha,25-dihydroxy-vitamin D3-induced rapid activation of PKC. J Steroid Biochem Mol Biol. 2010;121:257–260. doi: 10.1016/j.jsbmb.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Garbi N, Tanaka S, Momburg F, Hammerling GJ. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol. 2006;7:93–102. doi: 10.1038/ni1288. [DOI] [PubMed] [Google Scholar]
- Paschen W, Gissel C, Linden T, Doutheil J. Erp72 expression activated by transient cerebral ischemia or disturbance of neuronal endoplasmic reticulum calcium stores. Metab Brain Dis. 1998;13:55–68. doi: 10.1023/A:1020631029168. [DOI] [PubMed] [Google Scholar]
- Roy B, Lee AS. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999;27:1437–1443. doi: 10.1093/nar/27.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarella RA, Srinivasan M, Haugejorden SM, Green M. ERp72, an abundant luminal endoplasmic reticulum protein, contains three copies of the active site sequences of protein disulfide isomerase. J Biol Chem. 1990;265:1094–1101. [PubMed] [Google Scholar]
- Satoh M, Shimada A, Keino H, Kashiwai A, Nagai N, Saga S, Hosokawa M. Functional characterization of 3 thioredoxin homology domains of ERp72. Cell Stress Chaperones. 2005;10:278–284. doi: 10.1379/CSC-116R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam SK, Goldberg AL, Ho S, Rohde MF, Bush KT, Sherman MY. A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca(2+)-binding proteins and members of the thioredoxin superfamily. J Biol Chem. 1994;269:1744–1749. [PubMed] [Google Scholar]
- Marcus N, Shaffer D, Farrar P, Green M. Tissue distribution of three members of the murine protein disulfide isomerase (PDI) family. Biochim Biophys Acta. 1996;1309:253–260. doi: 10.1016/S0167-4781(96)00133-9. [DOI] [PubMed] [Google Scholar]
- Solda T, Garbi N, Hammerling GJ, Molinari M. Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J Biol Chem. 2006;281:6219–6226. doi: 10.1074/jbc.M513595200. [DOI] [PubMed] [Google Scholar]
- Hayano T, Kikuchi M. Molecular cloning of the cDNA encoding a novel protein disulfide isomerase-related protein (PDIR) FEBS Lett. 1995;372:210–214. doi: 10.1016/0014-5793(95)00996-M. [DOI] [PubMed] [Google Scholar]
- Horibe T, Gomi M, Iguchi D, Ito H, Kitamura Y, Masuoka T, Tsujimoto I, Kimura T, Kikuchi M. Different contributions of the three CXXC motifs of human protein-disulfide isomerase-related protein to isomerase activity and oxidative refolding. J Biol Chem. 2004;279:4604–4611. doi: 10.1074/jbc.M310922200. [DOI] [PubMed] [Google Scholar]
- Carbone MA, Chen Y, Hughes GA, Weinreb RN, Zabriskie NA, Zhang K, Anholt RRH. Genes of the unfolded protein response pathway harbor risk alleles for primary open angle glaucoma. PLoS One. 2011;6:e20649. doi: 10.1371/journal.pone.0020649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PA, Stevens JM, Hubbard GP, Barrett NE, Sage T, Authi KS, Gibbins JM. A role for the thiol isomerase protein ERP5 in platelet function. Blood. 2005;105:1500–1507. doi: 10.1182/blood-2004-02-0608. [DOI] [PubMed] [Google Scholar]
- Hayano T, Kikuchi M. Cloning and sequencing of the cDNA encoding human P5. Gene. 1995;164:377–378. doi: 10.1016/0378-1119(95)00474-K. [DOI] [PubMed] [Google Scholar]
- Kikuchi M, Doi E, Tsujimoto I, Horibe T, Tsujimoto Y. Functional analysis of human P5, a protein disulfide isomerase homologue. J Biochem. 2002;132:451–455. doi: 10.1093/oxfordjournals.jbchem.a003242. [DOI] [PubMed] [Google Scholar]
- Belmont PJ, Chen WJ, San Pedro MN, Thuerauf DJ, Gellings Lowe N, Gude N, Hilton B, Wolkowicz R, Sussman MA, Glembotski CC. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ Res. 2009;106:307–316. doi: 10.1161/CIRCRESAHA.109.203901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop CE, Watkins RH, Simmons JJ, Tasab M, Bulleid NJ. Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J Cell Sci. 2009;122:4287–4295. doi: 10.1242/jcs.059154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lith M, Hartigan N, Hatch J, Benham AM. PDILT, a divergent testis-specific protein disulfide isomerase with a non-classical SXXC motif that engages in disulfide-dependent interactions in the endoplasmic reticulum. J Biol Chem. 2005;280:1376–1383. doi: 10.1074/jbc.M408651200. [DOI] [PubMed] [Google Scholar]
- van Lith M, Karala AR, Bown D, Gatehouse JA, Ruddock LW, Saunders PT, Benham AM. A developmentally regulated chaperone complex for the endoplasmic reticulum of male haploid germ cells. Mol Biol Cell. 2007;18:2795–2804. doi: 10.1091/mbc.E07-02-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DC, Huminiecki L, Moore JW, Boyle JJ, Poulsom R, Creamer D, Barker J, Bicknell R. EndoPDI, a novel protein-disulfide isomerase-like protein that is preferentially expressed in endothelial cells acts as a stress survival factor. J Biol Chem. 2003;278:47079–47088. doi: 10.1074/jbc.M308124200. [DOI] [PubMed] [Google Scholar]
- Alberti A, Karamessinis P, Peroulis M, Kypreou K, Kavvadas P, Pagakis S, Politis PK, Charonis A. ERp46 is reduced by high glucose and regulates insulin content in pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2009;297:E812–E821. doi: 10.1152/ajpendo.00053.2009. [DOI] [PubMed] [Google Scholar]
- Jeong KH, Shin MK, Uhm YK, Kim HJ, Chung JH, Lee MH. Association of TXNDC5 gene polymorphisms and susceptibility to nonsegmental vitiligo in the Korean population. Br J Dermatol. 2010;162:759–764. doi: 10.1111/j.1365-2133.2009.09574.x. [DOI] [PubMed] [Google Scholar]
- Petek E, Windpassinger C, Egger H, Kroisel PM, Wagner K. Localization of the human anterior gradient-2 gene (AGR2) to chromosome band 7p21.3 by radiation hybrid mapping and fluorescencein situ hybridisation. Cytogenet Cell Genet. 2000;89:141–142. doi: 10.1159/000015594. [DOI] [PubMed] [Google Scholar]
- Persson S, Rosenquist M, Knoblach B, Khosravi-Far R, Sommarin M, Michalak M. Diversity of the protein disulfide isomerase family: identification of breast tumor induced Hag2 and Hag3 as novel members of the protein family. Mol Phylogenet Evol. 2005;36:734–740. doi: 10.1016/j.ympev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Thompson DA, Weigel RJ. hAG-2, the human homologue of the Xenopus laevis cement gland gene XAG-2, is coexpressed with estrogen receptor in breast cancer cell lines. Biochem Biophys Res Commun. 1998;251:111–116. doi: 10.1006/bbrc.1998.9440. [DOI] [PubMed] [Google Scholar]
- Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle D. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A. 2009;106:6950–6955. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Rosenstiel P, Huse K, Sina C, Valentonyte R, Mah N, Zeitlmann L, Grosse J, Ruf N, Nürnberg P, Costello CM, Onnie C, Mathew C, Platzer M, Schreiber S, Hampe J. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun. 2006;7:11–18. doi: 10.1038/sj.gene.6364263. [DOI] [PubMed] [Google Scholar]
- Adam PJ, Boyd R, Tyson KL, Fletcher GC, Stamps A, Hudson L, Poyser HR, Redpath N, Griffiths M, Steers G, Harris AL, Patel S, Berry J, Loader JA, Townsend RR, Daviet L, Legrain P, Parekh R, Terrett JA. Comprehensive proteomic analysis of breast cancer cell membranes reveals unique proteins with potential roles in clinical cancer. J Biol Chem. 2003;278:6482–6489. doi: 10.1074/jbc.M210184200. [DOI] [PubMed] [Google Scholar]
- King ER, Tung CS, Tsang YT, Zu Z, Lok GT, Deavers MT, Malpica A, Wolf JK, Lu KH, Birrer MJ, Mok SC, Gershenson DM, Wong KK. The anterior gradient homolog 3 (AGR3) gene is associated with differentiation and survival in ovarian cancer. Am J Surg Pathol. 2011;35:904–912. doi: 10.1097/PAS.0b013e318212ae22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Rong YP, Zeng LC, Zhang X, Han ZG. Isolation and characterization of a novel human thioredoxin-like gene hTLP19 encoding a secretory protein. Gene. 2003;315:71–78. doi: 10.1016/s0378-1119(03)00732-7. [DOI] [PubMed] [Google Scholar]
- Alanen HI, Williamson RA, Howard MJ, Lappi AK, Jäntti HP, Rautio SM, Kellokumpu S, Ruddock LW. Functional characterization of ERp18, a new endoplasmic reticulum-located thioredoxin superfamily member. J Biol Chem. 2003;278:28912–28920. doi: 10.1074/jbc.M304598200. [DOI] [PubMed] [Google Scholar]
- Coe H, Michalak M. Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys. 2009;28:F96–F103. Spec No Focus. [PubMed] [Google Scholar]
- Shin DW, Pan Z, Kim EK, Lee JM, Bhat MB, Parness J, Kim DH, Ma J. A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J Biol Chem. 2003;278:3286–3292. doi: 10.1074/jbc.M209045200. [DOI] [PubMed] [Google Scholar]
- Fujii J, Willard HF, MacLennan DH. Characterization and localization to human chromosome 1 of human fast-twitch skeletal muscle calsequestrin gene. Somat Cell Mol Genet. 1990;16:185–189. doi: 10.1007/BF01233048. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Wong PT. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1971;68:1231–1235. doi: 10.1073/pnas.68.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegel L, Ohnishi M, Carpenter MR, Khanna VK, Reithmeier RA, MacLennan DH. Amino acid sequence of rabbit fast-twitch skeletal muscle calsequestrin deduced from cDNA and peptide sequencing. Proc Natl Acad Sci U S A. 1987;84:1167–1171. doi: 10.1073/pnas.84.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem. 1994;135:61–70. doi: 10.1007/BF00925961. [DOI] [PubMed] [Google Scholar]
- Dainese M, Quarta M, Lyfenko AD, Paolini C, Canato M, Reggiani C, Dirksen RT, Protasi F. Anesthetic- and heat-induced sudden death in calsequestrin-1-knockout mice. FASEB J. 2009;23:1710–1720. doi: 10.1096/fj.08-121335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Min CK, Ko JK, Parness J, do Kim H, Weisleder N, Ma J. Increased store-operated Ca2+ entry in skeletal muscle with reduced calsequestrin-1 expression. Biophys J. 2010;99:1556–1564. doi: 10.1016/j.bpj.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Chu W, Zhang Z, Hasstedt SJ, Elbein SC. Calsquestrin 1 (CASQ1) gene polymorphisms under chromosome 1q21 linkage peak are associated with type 2 diabetes in Northern European Caucasians. Diabetes. 2004;53:3300–3306. doi: 10.2337/diabetes.53.12.3300. [DOI] [PubMed] [Google Scholar]
- Fu M, Damcott CM, Sabra M, Pollin TI, Ott SH, Wang J, Garant MJ, O’Connell JR, Mitchell BD, Shuldiner AR. Polymorphism in the calsequestrin 1 (CASQ1) gene on chromosome 1q21 is associated with type 2 diabetes in the old order Amish. Diabetes. 2004;53:3292–3299. doi: 10.2337/diabetes.53.12.3292. [DOI] [PubMed] [Google Scholar]
- Youn JH, Gulve EA, Holloszy JO. Calcium stimulates glucose transport in skeletal muscle by a pathway independent of contraction. Am J Physiol. 1991;260:C555–C561. doi: 10.1152/ajpcell.1991.260.3.C555. [DOI] [PubMed] [Google Scholar]
- Otsu K, Fujii J, Periasamy M, Difilippantonio M, Uppender M, Ward DC, MacLennan DH. Chromosome mapping of five human cardiac and skeletal muscle sarcoplasmic reticulum protein genes. Genomics. 1993;17:507–509. doi: 10.1006/geno.1993.1357. [DOI] [PubMed] [Google Scholar]
- Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Akiyama N, Nakamura H, Yodoi J, Noda M, Kizaka-Kondoh S. Identification of a novel thioredoxin-related transmembrane protein. J Biol Chem. 2001;276:10032–10038. doi: 10.1074/jbc.M011037200. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Nishinaka Y, Suzuki S, Kojima M, Kizaka-Kondoh S, Kondo N, Son A, Sakakura-Nishiyama J, Yamaguchi Y, Masutani H, Ishii Y, Yodoi J. TMX, a human transmembrane oxidoreductase of the thioredoxin family: the possible role in disulfide-linked protein folding in the endoplasmic reticulum. Arch Biochem Biophys. 2004;423:81–87. doi: 10.1016/j.abb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Masutani H, Son A, Kizaka-Kondoh S, Yodoi J. Physical and functional interaction of transmembrane thioredoxin-related protein with major histocompatibility complex class I heavy chain: redox-based protein quality control and its potential relevance to immune responses. Mol Biol Cell. 2009;20:4552–4562. doi: 10.1091/mbc.E09-05-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Zhang C, Chen J, Peng S, Cao Y, Ying K, Xie Y, Mao Y. Cloning and identification of a novel cDNA coding thioredoxin-related transmembrane protein 2. Biochem Genet. 2003;41:99–106. doi: 10.1023/A:1022073917044. [DOI] [PubMed] [Google Scholar]
- Haugstetter J, Blicher T, Ellgaard L. Identification and characterization of a novel thioredoxin-related transmembrane protein of the endoplasmic reticulum. J Biol Chem. 2005;280:8371–8380. doi: 10.1074/jbc.M413924200. [DOI] [PubMed] [Google Scholar]
- Chao R, Nevin L, Agarwal P, Riemer J, Bai X, Delaney A, Akana M, JimenezLopez N, Bardakjian T, Schneider A, Chassaing N, Schorderet DF, FitzPatrick D, Kwok P, Ellgaard L, Gould DB, Zhang Y, Malicki J, Baier H, Slavotinek A. A male with unilateral microphthalmia reveals a role for TMX3 in eye development. PLoS One. 2010;5:e10565. doi: 10.1371/journal.pone.0010565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Araki K, Iemura S, Natsume T, Jun Hoseki J, Nagata K. Novel thioredoxin-related transmembrane protein TMX4 has reductase activity. J Biol Chem. 2010;285:7135–7142. doi: 10.1074/jbc.M109.082545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth D, Lynes E, Riemer J, Hansen HG, Althaus N, Simmen T, Ellgaard L. A di-arginine motif contributes to the ER localization of the type I transmembrane ER oxidoreductase TMX4. Biochem J. 2010;425:195–205. doi: 10.1042/BJ20091064. [DOI] [PubMed] [Google Scholar]