

Abstract
Over the past few years, the number of known protein-protein interactions has increased substantially. To make this information more readily available, a number of publicly available databases have set out to collect and store protein-protein interaction data. Protein-protein interactions have been retrieved from six major databases, integrated and the results compared. The six databases (the Biological General Repository for Interaction Datasets [BioGRID], the Molecular INTeraction database [MINT], the Biomolecular Interaction Network Database [BIND], the Database of Interacting Proteins [DIP], the IntAct molecular interaction database [IntAct] and the Human Protein Reference Database [HPRD]) differ in scope and content; integration of all datasets is non-trivial owing to differences in data annotation. With respect to human protein-protein interaction data, HPRD seems to be the most comprehensive. To obtain a complete dataset, however, interactions from all six databases have to be combined. To overcome this limitation, meta-databases such as the Agile Protein Interaction Database (APID) offer access to integrated protein-protein interaction datasets, although these also currently have certain restrictions.
Keywords: protein-protein interactions, PPI, database, bioinformatics, IMEx, PSI-MI
The nature of protein-protein interaction data
Proteins do not act independently but in a network of complex molecular interactions. Therefore, it is important to identify physical interactions between proteins. Different experimental techniques have been developed to measure physical interactions between proteins; these methods vary considerably, not least in terms of the data they produce.
To give some examples, two widely used methods adapted for high-throughput approaches are the yeast two-hybrid (Y2H) system [1] and affinity purification followed by mass spectrometry (AP-MS) [2].
The Y2H system assays whether two proteins physically interact with each other (Figure 1). Genetically modified yeast strains are used to express a 'bait' and a 'prey' protein, which, if they interact, trigger the expression of a reporter gene. The method has been used for large-scale screening studies of a variety of model organisms, including yeast, fly and humans.
Figure 1.
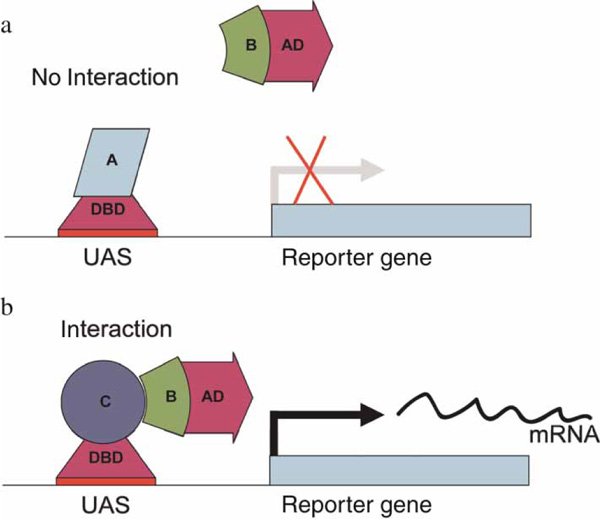
The yeast two-hybrid experiment. The yeast two-hybrid system utilises the DNA binding domain and the activation domain of a yeast transcription factor. The bait proteins A and C are fused to a DNA binding domain, allowing them to bind to a binding site in the promoter region of a reporter gene. The prey protein B is fused to an activation domain that can activate the expression of a gene. (a) Protein B does not bind to protein A, therefore, the reporter gene is not activated. (b) Protein B binds to protein C, thereby activating the reporter gene.
In an AP-MS experiment, a protein of interest is fused to a protein fragment (the 'tag'), which allows its purification (Figure 2). This modified or tagged protein is expressed and purified from the cell extract using the tag -- for example, by antibodies binding specifically to the tag. Proteins binding the tagged protein are co-purified and subsequently identified by MS. The most widely used variation of the AP-MS method is tandem affinity purification followed by mass spectrometry (TAP-MS). In TAP-MS, the protein of interest is attached to a larger protein tag, which allows two consecutive affinity purification steps [2]. Large-scale TAP-MS experiments have been performed for yeast and human proteins [3-5]. Currently, several variations of these two methods, as well as a number of other methods, are used to identify protein--protein interactions (PPIs) [6-8].
Figure 2.
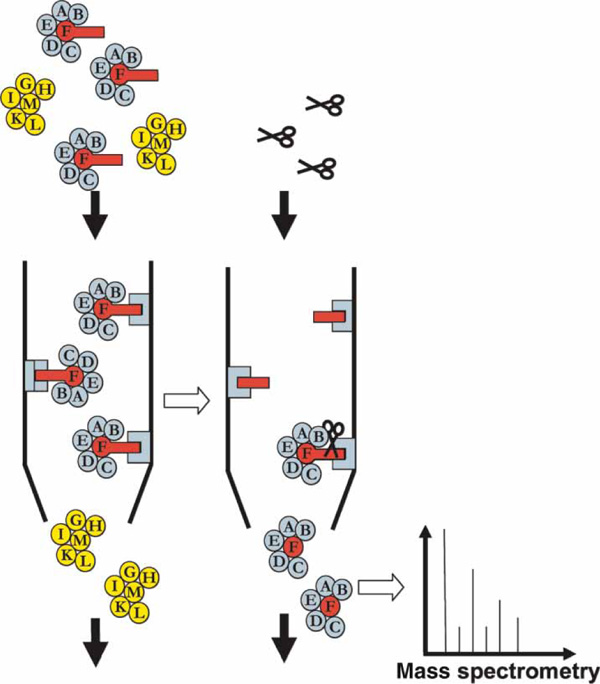
An affinity purification experiment followed by mass spectrometry. The protein of interest, F (red circle), is fused to a protein fragment -- the 'tag' (red rectangle). The tag allows this protein to be purified biochemically. Proteins binding to the tagged protein (blue) are co-purified, whereas proteins not binding to protein F (yellow) are discarded. The purified proteins can be released using enzymatic cleavage (scissors) or other methods, depending on the nature of the tag. These proteins are then identified by mass spectrometry.
PPI datasets are often visualised as graphs [9,10]. Proteins are represented as nodes, and interactions as connections between nodes. For example, if the interaction between two proteins is detected by a Y2H experiment, we represent this physical interaction by an undirected connection between the two nodes. In a more detailed representation, we could make a distinction between bait and prey proteins and use a directed connection to represent the interaction between two proteins, using an arrow pointing from bait to prey. The use of graphs to describe the experimental results of AP-MS protein interaction screens is not always as straightforward as for Y2H data. Due to the nature of an AP-MS experiment, which identifies a whole protein complex rather than pairwise interactions, its results can be represented as a graph, using either the matrix or the spokes model (Figure 3). The matrix model assumes that all proteins of a purified complex interact; therefore, in the graph each protein is connected to each other. The spokes model assumes no additional interactions between proteins in a complex other than between the tagged protein and each co-purified protein.
Figure 3.
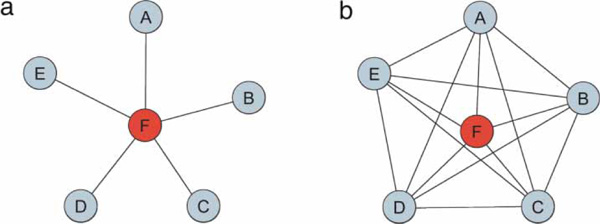
Spokes and matrix models. In order to represent the complexes identified by the AP-MS methods as binary pairs, the spokes model (a) or the matrix model (b) can be used. In the spokes model, it is assumed that only the tagged protein interacts with the other proteins in the complex. In the matrix model, it is assumed that all proteins in a complex interact with each other. While the spokes model probably leads to an underestimate of the real interactions, the matrix model leads to an overestimate.
Graph representation allows the data to be analysed using a graph-theoretical framework. Many graph analysis algorithms have been applied to PPI datasets; these approaches have been reviewed in detail elsewhere [11-16].
PPI databases
The primary resources for PPI data are individual scientific publications. Several public databases collect published PPI data and provide researchers access to their curated datasets. These usually reference the original publication and the experimental method that determined every individual interaction. Database designers choose to represent these data in different ways, and the wide spectrum of experimental methods makes it difficult to design a single data model to capture all necessary experimental detail. To overcome this problem, the International Molecular Exchange (IMEx; http://imex.sourceforge.net/) consortium was formed. IMEx aims to enable the exchange of data and to avoid the duplication of the curation effort. To that end, an XML-based proteomics standard, termed the proteomics standards initiative - molecular interaction (PSI-MI) has been developed [17]. At the time of writing, however, no data had yet been exchanged, and it was therefore necessary to combine PPI data from all available databases using the authors' own scripts to obtain as comprehensive a network as possible.
Here, the focus is on six databases: the Biological General Repository for Interaction Datasets (BioGRID) [18], the Molecular INTeraction database (MINT) [19], the Biomolecular Interaction Network Database (BIND) [20], the Database of Interacting Proteins (DIP) [21], the IntAct molecular interaction database (IntAct)[22] and the Human Protein Reference Database (HPRD)[23] (see Table 1). These databases report only experimentally verified interactions.
Table 1.
PPI databases
| Database | URL | Proteins | Interactions | Publications | Organisms |
|---|---|---|---|---|---|
| BioGRID | http://www.thebiogrid.org | 23,341 | 90,972 | 16,369 | 10 |
| MINT | http://mint.bio.uniroma2.it/mint | 27,306 | 80,039 | 3,047 | 144 |
| BIND | http://bond.unleashedinformatics.com | 23,643 | 43,050 | 6,364 | 80 |
| DIP | http://dip.doe-mbi.ucla.edu | 21,167 | 53,431 | 3,193 | 134 |
| IntAct | http://www.ebi.ac.uk/intact | 37,904 | 129,559 | 3,166 | 131 |
| HPRD | http://www.hprd.org | 9,182 | 36,169 | 18,777 | 1 |
DIP, IntAct and MINT are active members of the IMEx initiative; the curation accuracy of these three databases was assessed recently by Cusick et al. [24] HPRD focuses entirely on human proteins, providing not only information on protein interactions, but also a variety of protein-specific information, such as post-translational modifications, disease associations and enzyme-substrate relationships. One of the first interaction databases, BIND, initiated in 2001 by the University of Toronto and the University of British Columbia, is part of the Biomolecular Object Network Databank (BOND) and was subsequently acquired by the company Thomson Reuters.
The following comparison is based on complete sets of binary interactions that were downloaded from the individual databases in May 2008. IntAct and MINT derive binary interactions from protein complexes using the spokes model. No other database provided any information on which model is applied. Only 'physical interactions' are considered here, although most databases also provide 'genetic interactions' -- that is, two non-essential genes that lead to a non-viable phenotype if they are knocked out simultaneously. Furthermore, interactions were only accepted if a publication identifier was provided along with the interacting proteins.
Currently, the most comprehensive database in terms of individual interactions is IntAct, with almost 130,000 unique interactions from up to 131 different organisms. Despite these large numbers, it cites only about 3,000 different publications. Whereas IntAct seems to be concentrating on high-throughput studies, HPRD also takes into account small-scale publications. Although being restricted to human proteins, it reports over 36,000 unique interactions from more than 18,000 publications. Only BioGRID cites a similar number of publications (16,369); it is also the second largest database in terms of the number of unique interactions. It should be noted that the databases examine publications in different depth, and that higher numbers of publications do not necessarily involve a higher curation effort.
The majority of known protein interactions account for proteins from Saccharomyces cerevisiae and Homo sapiens. Individual high-throughput interaction screens were carried out for some other organisms; these high-throughput studies usually account for the majority of all known interactions in the corresponding organism. By contrast, known protein interactions for S. cerevisiae and H. sapiens are dispersed over numerous publications. For this reason, the number of interactions for humans and yeast can vary considerably between different databases, depending on their coverage of the literature.
Differences between the PPI databases
Ideally, every database would extract the same interactions from a given publication. Unfortunately, this is not the case. Of the 14,899 publications shared by at least two databases, 5,782 (39 per cent) were reported with a different number of interactions in different databases. For example, for the publication reporting the most interactions [25], a minimum of 18,877 (BIND) and a maximum of 20,800 interactions (DIP) were reported. According to the abstract, the number of interactions is 20,405, which, again, is different from the number reported by all five databases that cite this publication. In this case, the variation is presumably due to problems with identifier mapping. Many databases use different identifiers, which do not always map in a perfect one-to-one relationship to the originally published identifiers. BioGRID (20,220 interactions) uses the original gene identifiers, but still lacks 185 interactions.
As a second example, using a Y2H screen, Rual et al. detected 2,754 interactions between human proteins [26]. The authors compared their experimental findings with a literature-curated PPI network of 4,076 interactions. This resulted in a combined network of 6,438 interactions. HPRD (2,371 interactions), IntAct (2,671 interactions) and MINT (2,463 interactions) report only experimentally detected interactions for this reference. BioGRID reports 6,295 interactions for this study, of which 2,594 quote Y2H as the detection method. These also overlap with the interactions reported by the other databases for this reference. The remaining 3,895 interactions quote affinity capture as the detection method and possibly refer to the literature-curated interactions.
For a number of other publications, differences can be explained by different confidence sets or thresholds [27,28] or differences in the application of the matrix or spokes model. Often, no obvious reason for different numbers of interactions could be found.
Integration of PPI data
Integration of data from the different databases is not trivial. Although many databases provide their interactions in the proteomics standards initiative-molecular interactions (PSI-MI) format, its controlled vocabulary is often not used or is used incorrectly. Furthermore, a variety of different gene or protein identifiers are used, even within some of the databases. Although a gene can give rise to several different proteins (due to alternative splicing), we mapped all identifiers to Ensembl gene identifiers to avoid any ambiguities. This procedure is based on mapping tables obtained from UniProt [29]. Only interactions in which both proteins could be mapped to an Ensembl gene identifier were considered for further analysis.
After unifying all identifiers for eukaryotic organisms, the four model organisms Caenorhabditis elegans, Drosophila melanogaster, S. cerevisiae and H. sapiens showed the highest number of interactions (Table 2). The focus here has been on PPIs in eukaryotes, but the reader should note that high-throughput datasets also exist for a variety of pro-karyotes, including Escherichia coli, Campylobacter jejuni and Helicobacter pylori. Previous studies reported little overlap between individual PPI datasets [15]. Likewise, there is little redundancy in the combined set of interactions (Table 2). Between 1 per cent (D. melanogaster) and 18 per cent (H. sapiens) of all interactions are reported by more than one publication. Interestingly, the proportion of interactions that were reported by different methods reaches up to 25 per cent for yeast and 42 per cent for humans (Table 2). Although many small-scale publications apply more than one method to confirm an interaction, this number is most likely an overestimate, because databases use different nomenclature and spelling variations to describe experimental detection methods. Therefore, more interactions appear to be confirmed by several methods than really are.
Table 2.
Redundancy of PPIs.
| Species | Proteins | Interactions | >1 publication | >1 method | >1 database |
|---|---|---|---|---|---|
| C. elegans | 3,173 | 5,300 | 668 (13%) | 155 (3%) | 4,536 (86%) |
| D. melanogaster | 7,529 | 24,811 | 198 (1%) | 298 (1%) | 17,904 (72%) |
| H. sapiens | 10,397 | 51,308 | 9,358 (18%) | 21,036 (41%) | 26,263 (51%) |
| S. cerevisiae | 5,806 | 69,059 | 12,037 (17%) | 17,219 (25%) | 29,053 (42%) |
The total number of proteins and interactions (that could be mapped to Ensembl gene identifiers), as well as the number of interactions reported by more than one publication, more than one method or more than one database, is shown. Relative numbers were obtained through normalisation with the total number of interactions.
As mentioned above, databases focus their curation efforts on different publications. Consequently, only a subset of all protein interactions can be found in more than one database (Table 2). These range from 42 per cent of yeast interactions and 51 per cent of human interactions to 72 per cent of fly interactions and 86 per cent of worm interactions.
To assess these differences in more detail, the relative pairwise overlap of human protein interactions between databases was calculated (Table 3). All databases have their highest relative overlap when compared with HPRD, which reports the most interactions. High overlaps were also found between DIP and BioGRID (55 per cent) and between MINT and IntAct (59 per cent). Even the most abundant database (HPRD), however, covers only two-thirds of all reported human protein interactions.
Table 3.
Overlap of human PPIs between databases.
| PPIs total | BIND | DIP | BioGRID | HPRD | IntAct | MINT | |
|---|---|---|---|---|---|---|---|
| BIND | 5304 | 3% | 33% | 73% | 20% | 25% | |
| DIP | 737 | 22% | 55% | 73% | 34% | 33% | |
| BioGRID | 17645 | 10% | 2% | 75% | 17% | 8% | |
| HPRD | 34970 | 11% | 2% | 38% | 21% | 21% | |
| IntAct | 17746 | 6% | 1% | 17% | 42% | 37% | |
| MINT | 11185 | 12% | 2% | 3% | 66% | 59% | |
| PPI relative overlap: | 0-19% | 20-39% | 40-59% | >60% | |||
The pairwise relative overlap between databases was calculated for H. sapiens. Absolute numbers were normalised to the total number of PPIs for every row
Meta-databases
None of the existing PPI databases provides an exhaustive dataset. Therefore, some groups have set up meta-databases that provide protein interaction data extracted and integrated from other databases. Currently, one of the most comprehensive meta-database appears to be the Agile Protein Interaction Database (APID) [30]. APID extracts interactions from the six databases described above, mapping all proteins to UniProt identifiers [29]. Via a web interface, the user can query for proteins of interest. APID references the database from which an interaction is derived and provides the related information available in the original database, such as the detection method and the publication identifier. In addition, APID incorporates biological information from various other databases, such as the Gene Ontology [31] and Pfam databases [32]. Unfortunately, a download of the complete dataset is currently not possible due to licensing issues. APID is generally in good agreement with the results of the authors' data integration. For the time being, APID seems a good source of interactome data.
Several other meta-databases exist, but these usually focus on a single organism [33] or incorporate various other types of interactions, such as computationally predicted protein interactions and co-citation of proteins [34]. For a comprehensive list of available databases, the reader is referred to the Pathguide [35].
Conclusions
PPI databases not only report their data in different ways, using different ontologies, but their curators also report different PPIs when examining the same publication. In addition, all databases include different publications. It is therefore not surprising that every database reports different PPIs. The pairwise overlap among databases analysed here reaches up to 75 per cent, but always falls short of a perfect 100 per cent. Similar results were obtained in related studies [12,24]. Until a data exchange between databases is implemented, a comprehensive set of interactions can only be obtained through data integration of several databases. Meta-databases, such as APID, provide access to more comprehensive datasets, but do not always allow the download of their complete data. Furthermore, by their very nature, meta-databases will always be less up to date than the original databases.
PPI databases have improved greatly over the past couple of years, and important issues, such as data exchange, are being currently addressed by some of the databases described here. An important step towards increasing the number and quality of protein interaction data would be to introduce a submission requirement -- as, indeed, already exists for sequence and microarray data. These data have to be submitted to public databases prior to publication in a scientific journal, which ensures data availability and consistent annotation, and enables researchers to utilise the data with greatest efficiency.
Acknowledgements
The authors would like to thank all developers and curators of the protein--protein interaction databases. Without their effort, our life would be much harder. We thank Henning Hermjakob for helpful discussions. We are grateful for funding from the British Council/DAAD as part of the ARC programme (ARC1297).
References
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B. et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotech. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P. et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Bouwmeester T, Bauch A, Ruffner H. et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R. et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Berggård T, Linse S, James P. Methods for the detection and analysis of protein-protein interactions. Proteomics. 2007;7:2833–2842. doi: 10.1002/pmic.200700131. [DOI] [PubMed] [Google Scholar]
- Phizicky EM, Fields S. Protein-protein interactions: Methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part I. Experimental techniques and databases. PLoS Comput Biol. 2007;3:e42. doi: 10.1371/journal.pcbi.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suderman M, Hallett M. Tools for visually exploring biological networks. Bioinformatics. 2007;23:2651–2659. doi: 10.1093/bioinformatics/btm401. [DOI] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E. et al. Integration of biologi-cal networks and gene expression data using Cytoscape. Nat Protocols. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert R, Barabasi AL. Statistical mechanics of complex networks. Rev Mod Phys. 2002;74:47–97. doi: 10.1103/RevModPhys.74.47. [DOI] [Google Scholar]
- Futschik ME, Chaurasia G, Herzel H. Comparison of human protein protein interaction maps. Bioinformatics. 2007;23:605–611. doi: 10.1093/bioinformatics/btl683. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey V, Long L. et al. Graphs in molecular biology. BMC Bioinformatics. 2007;8:S8. doi: 10.1186/1471-2105-8-S6-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Krause R, Snel B. et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002;417:399–403. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- Schwikowski B, Uetz P, Fields S. A network of protein-protein interactions in yeast. Nat Biotechnol. 2000;18:1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- Kerrien S, Orchard S, Montecchi-Palazzi L. et al. Broadening the horizon - Level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biol. 2007;5:44. doi: 10.1186/1741-7007-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T. et al. BioGRID: A general repository for interaction datasets. Nucl Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanzoni A, Montecchi-Palazzi L, Quondam M. et al. MINT: A Molecular INTeraction database. FEBS Lett. 2002;513:135–140. doi: 10.1016/S0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- Bader GD, Donaldson I, Wolting C. et al. BIND - The Biomolecular Interaction Network Database. Nucl Acids Res. 2001;29:242–245. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenarios I, Rice DW, Salwinski L. et al. DIP: The Database of Interacting Proteins. Nucl Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermjakob H, Montecchi-Palazzi L, Lewington C. et al. IntAct: An open source molecular interaction database. Nucl Acids Res. 2004;32:D452–D455. doi: 10.1093/nar/gkh052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri S, Navarro JD, Amanchy R. et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003;13:2363–2371. doi: 10.1101/gr.1680803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick ME, Hu H, Smolyar A. et al. Literature-curated protein interaction datasets. Nat Meth. 2009;6:39–46. doi: 10.1038/nmeth.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, A protein interaction map of Drosophila melanogaster, Science. 2003. pp. 1727–1736. [DOI] [PubMed]
- Rual J-F, Venkatesan K, Hao T. et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- John PM, Russell SL, Asa BH. et al. Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci USA. 2005;102:12123–12128. doi: 10.1073/pnas.0505482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formstecher E, Aresta S, Collura V. et al. Protein interaction mapping: A Drosophila case study. Genome Res. 2005;15:376–384. doi: 10.1101/gr.2659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt C. The Universal Protein Resource (UniProt) Nucl Acids Res. 2008;36:D190–D195. doi: 10.1093/nar/gkn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto C, De Las Rivas J. APID: Agile Protein Interaction Data Analyzer. Nucl Acids Res. 2006;34:W298–W302. doi: 10.1093/nar/gkl128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA. et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J. et al. The Pfam protein families database. Nucl Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasia G, Iqbal Y, Hanig C. et al. UniHI: An entry gate to the human protein interactome. Nucl Acids Res. 2007;35:D590–D594. doi: 10.1093/nar/gkl817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M. et al. STRING 8 - A global view on proteins and their functional interactions in 630 organisms. Nucl Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Cary MP, Sander C. Pathguide: A pathway resource list. Nucl Acids Res. 2006;34:D504–D506. doi: 10.1093/nar/gkj126. [DOI] [PMC free article] [PubMed] [Google Scholar]