

Abstract
Although inflammatory bowel disease is associated with higher risk of colorectal cancer, the precise pathogenic mechanisms underlying this association are not completely understood. Prohibitin 1 (PHB), a protein implicated in the regulation of proliferation, apoptosis, and transcription, is decreased in intestinal inflammation. In this study, we have established a key function for PHB in mediating colitis-associated cancer. Wild-type and transgenic (Tg) mice specifically overexpressing PHB in intestinal epithelial cells were subjected to a classical two-stage protocol of colitis-associated carcinogenesis Additionally, wild-type and p53 null human cell models were used to assess PHB interaction with STAT3 and p53. Wild-type mice exhibited decreased mucosal PHB protein expression during colitis-associated carcinogenesis. Tg mice exhibited decreased susceptibility in a manner associated with increased apoptosis, p53, Bax and Bad expression plus decreased Bcl-xL and Bcl-2 expression. PHB overexpression in wild-type but not p53 null human cells increased expression of Bax, Bad and caspase-3 cleavage. In wild-type p53 cells, PHB overexpression decreased basal and IL-6-induced STAT3 activation and expression of the STAT3 responsive genes Bcl-xL and Bcl-2. PHB co-immunoprecipitated with phospho-STAT3 in addition to p53 in cultured cell lysates and colon mucosa. This is the first study to show interaction between PHB and STAT3 in vivo. In summary, our findings suggest that PHB protects against colitis-associated cancer by modulating p53- and STAT3-mediated apoptosis. Modulation of PHB expression in intestinal epithelial cells may offer a potential therapeutic approach to prevent colitis-associated carcinogenesis.
Keywords: colorectal cancer, inflammatory bowel disease, apoptosis
Introduction
Inflammatory bowel disease (IBD) includes two major chronic intestinal disorders, Crohn’s disease and ulcerative colitis (UC), which share related characteristics such as mucosal damage and diarrhea but have distinguishing clinical features. Evidence suggests that in addition to the widely accepted aberrant mucosal immune response, non-immune cells including epithelial cells play an emerging role in the pathogenesis of disease by modulating mucosal barrier integrity and homeostasis (1). An association between IBD and colorectal cancer (CRC) has been well established with a cumulative risk of developing CRC of 7% after 20 years for UC and 8% for Crohn’s disease (2). Previous studies have shown that production of proinflammatory cytokines in the lamina propria contribute to tumor growth and cancer development during intestinal inflammation (3, 4).
Prohibitin 1 (PHB) is an evolutionarily conserved, multifunctional 32 kDa protein implicated in cellular processes including the regulation of cell cycle progression, apoptosis, and transcription (5-7). Expression of PHB is decreased in mucosal biopsies from ulcerative colitis and Crohn’s disease afflicted patients and in the dextran sodium sulfate (DSS) and IL-10−/− mouse models of colitis (8, 9). Pro-inflammatory cytokines such as tumor necrosis factor α(TNFα) and reactive oxygen species decrease expression of intestinal epithelial PHB in vivo and in vitro (8-10). Restoration of colonic epithelial PHB expression using genetic manipulation (villin-PHB transgenic mice) or therapeutic delivery to the colon via nanoparticles or adenovirus protected mice from experimental colitis (11, 12).
The role of PHB in cancer cell proliferation and/or tumor suppression remains controversial especially since PHB expression is increased in many transformed cells and tumors (13-19). Consensus binding sites for the oncoprotein Myc are present in the PHB promoter and likely contribute to the increased PHB levels in many tumors (20). Although somatic mutations in the PHB gene were observed in a few sporadic breast cancers, none were identified in the ovary, hepatic, or lung cancers examined (16). Previous studies have shown that PHB has an anti-tumorigenic role in gastric, prostate, and liver cancers (21-23). Despite the increasing number of studies on PHB, its role in colorectal cancer or colitis-associated cancer (CAC) has not been explored. Here, we used mice specifically overexpressing PHB in intestinal epithelial cells (IECs) to assess the role and mechanism of PHB in CAC.
Materials and methods
Animal models
Wild-type (WT) and villin-prohibitin transgenic (Tg) mice specifically overexpressing prohibitin in intestinal epithelial cells (12) were 9 wk old at the beginning of the experimental protocol. All mice were grouped-housed in standard cages under a controlled temperature (25°C) and photoperiod (12-hour light /dark cycle) and were allowed standard chow and tap water ad libitum. All experiments were approved by the Baylor Research Institute Institutional Animal Care and Use Committee.
Induction of colitis-associated cancer in mice
Age-matched male and female C57BL/6 WT and Tg mice were i.p. injected with 7.6 mg/kg azoxymethane (AOM; Sigma-Aldrich, St. Louis, MO) on day 0. Colitis was induced 7 days after AOM injection by oral administration of 3% (wt/vol) dextran sodium sulfate (DSS; molecular weight, 50,000; MP Biomedicals, Solon, OH) in drinking water ad libitum for 7 days followed by 14 days of recovery of water alone. After recovery, a second cycle of DSS was repeated and mice were sacrificed after 3 weeks of recovery (day 56) as previously described (24). Three separate groups of mice were used as controls: one group of mice was maintained with regular water (n = 16 WT; n = 10 Tg), a second group of mice was injected with AOM and given water for the remainder of the experiment (n = 9 WT; n = 7 Tg), and the third group of mice was treated with DSS as described above without AOM injection (n = 20 WT; 8 Tg). AOM alone represents treatment with the alkylating agent in the absence of inflammation and is expected to result in no tumor formation in wild-type mice at the dose used (25). DSS alone represents repeated cycles of inflammation and was included to assess whether inflammation alone caused dysplasia in these mice. The study was powered in such a fashion that the control arms required less mice since death prior to the end of the study was not anticipated for these treatment groups. Body weight, stool consistency, and stool occult blood was monitored during the DSS treatment and recovery phases. Upon sacrifice, colon was excised from the ileocecal junction to anus, cut open longitudinally, and prepared for histological evaluation.
To assess mortality, the same protocol was followed with the exception of extending through 126 days or until mice developed rectal prolapse and/or >20% body weight loss. Colons were assessed macroscopically for polyps using a dissecting microscope.
Clinical score assessment
Assessment of body weight loss, stool consistency, and the presence of occult/gross blood by a guaiac test (Hemoccult Sensa; Beckman Coulter, Fullerton, CA) were determined daily during DSS administration to generate a clinical activity score as described previously (12). During recovery periods, body weight was measured every week for each group.
Endoscopic assessment of polyp formation and polyp count
Colonoscopy was done to assess polyp formation in mice by using the Coloview (Karl Storz Veterinary Endoscopy, Tuttlingen, Germany). Mice were euthanized with 1.5% - 2% isoflurane and ~3 cm of the colon proximal to the anus was visualized after inflation of the colon with air. Mice were then sacrificed by cervical dislocation after euthanizing with isoflurane. Colon was excised from ileocecal junction to anus, washed with 0.9% NaCl, cut open longitudinally and preserved in 10% neutral buffered formalin. The number and size of the polyps were determined using a Jenko dissecting microscope.
Histopathology scoring
Colons fixed in 10% neutral buffered formalin were Swiss-rolled, embedded in paraffin, sectioned at 5 μm, and stained with H&E for histopathological examination of polyps and adenocarcinoma (neoplasia). Scoring was performed in a blind fashion by two qualified pathologists. Score was given based on these criteria: normal (score 0), low grade dysplasia (score 1), high grade dysplasia (score 2), intramucosal adenocarcinoma (score 3), and invasive adenocarcinoma (score 4).
Immunohistochemistry
5-μm paraffin-embedded sections of colon were analyzed for Ki67 staining as a marker of cell proliferation as previously described (12). A minimum of 15 crypts with normal morphology were counted for Ki67-positive cells per section.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining
Immunofluorescent TUNEL staining was performed to measure apoptosis from paraffin-embedded sections using the In Situ Cell Death Detection kit as described by the manufacturer (Roche, Indianapolis, IN). Nuclei were stained with 4, 6′diamidino-2-phenylindole (DAPI) to count total cells per crypt. A minimum of 10 crypts with normal morphology were counted per section.
p53 mutational analyses
Tumor tissue was microdissected from paraffin-embedded sections (7 μm) of Swiss-rolled mouse colon. Genomic DNA was isolated from microdissected tissues using the QIAamp DNA FFPE Tissue purification kit (Qiagen, Valencia, CA). The PCR amplicons were generated and sequenced as previously described (26). See Supplementary Material and Methods section for primer sequences.
Human Tissue Samples
Matching normal and tumor tissues were obtained at the time of surgical resection from patients with one or more UC-associated colorectal neoplasms, consisting of adenocarcinomas or dysplasias. Normal control samples consisted of colonic normal mucosa adjacent to tumors or ileal mucosa. Inflamed UC tissues were obtained from patients without evident dysplasia. All tissues were grossly dissected free of normal surrounding tissue, and parallel sections were used for histological characterization. Tissue collection was approved by patients according to Institutional Review Board guidelines.
Total RNA extraction and quantitative real-time PCR
Total RNA was extracted for human tissues using the RNeasy kit (Qiagen). Quantitative real-time PCR was performed as described previously (11). See Supplementary Materials and Methods section for primer sequences.
Cell culture and transfection
Caco2-BBE cells were used to assess the interaction of PHB with STAT3. Since Caco2-BBE cells have mutated p53, wild-type (WT) HCT116 human colorectal cancer cells were utilized to assess PHB interaction with p53. All cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA). Cells were grown and transfected as previously described (27).
γ- Irradiation
γ-Irradiation was used to induce DNA damage in WT and p53−/− HCT116 cells (28) after 72 hrs of transfection with pEGFPN1 vector or pEGFPN1-PHB. Cells were irradiated by using 137Cs γ-irradiator at 6.5 cGy/sec for 32 seconds for a total of 209.4 cGy. Cells were harvested 24 hours after γ-irradiation for subsequent assays.
Protein extraction, Western blot analysis, and immunoprecipitation
Mucosal strippings from Tg and WT mice were obtained for Western blot analysis as described previously (12). Total protein was isolated from cultured cells as described previously (27). Antibodies used were mouse monoclonal PHB (Thermo Fisher, Fremont, CA), mouse monoclonal GFP, p53, Bcl-2, Bcl-xL and Bad and rabbit polyclonal BAX, p-Bad (ser 155), STAT3 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal PCNA antibody (Abcam, Cambridge, MA), rabbit polyclonal Caspase 3, p53, pSTAT3 (Cell Signaling Technology, Danvers, MA), rabbit polyclonal PUMA and mouse monoclonal anti-β-actin (Sigma-Aldrich).
PHB was immunoprecipitated from 0.6 mg total protein lysates from HCT116 or Caco2- BBE cells or 0.20 mg total protein lysates from mouse mucosa with 1 μg mouse anti-PHB, anti-p53, or anti-pSTAT3 antibody and 30 μl 50% protein A sepharose beads (GE Healthcare, Piscataway NJ). Blots were incubated with the rabbit p53 antibody or PHB antibody, respectively. Omission of primary antibody during the immunoprecipitation was performed as a negative control.
Statistical analysis
Values are expressed as mean ± SEM. Statistical analysis was performed using two-way analysis of variance and subsequent pair-wise comparisons using Bonferroni post-hoc tests. A P value less than 0.05 was considered statistically significant in all analyses.
Results
IEC-specific PHB overexpression decreases colonic tumorigenesis in a mouse model of CAC
Previous studies have shown that PHB has an anti-tumorigenic role in gastric, prostate, and liver cancers (21-23). We used the AOM DSS mouse model to study the role of PHB in CAC. Body weight was measured weekly as one parameter to assess the severity of disease. WT and Tg mice given water only throughout the experiment and mice injected with AOM followed by water alone showed similar body weight gain over the 8-week protocol (Supplemental Figure 1A) and did not develop polyps as previously reported for this dose of AOM (25). WT mice given 2 cycles of DSS without AOM injection lost more body weight following DSS administration and recovered less weight compared to Tg mice over the 8-week protocol (Supplemental Figure 1B), similar to our previous findings (12). Although both WT and Tg AOM DSS-treated mice lost weight after the first administration of DSS, Tg mice gained body weight more rapidly during the recovery phase and maintained body weight better than WT mice throughout the remainder of the study (Figure 1A).
Figure 1. IEC-specific PHB overexpression reduces colonic tumorigenesis in a mouse model of CAC.

Mice were intraperitoneally injected with azoxymethane (AOM) and 7 days later treated with two cycles of 3% DSS, each cycle consisting of 7 days DSS followed by 14 days of water recovery, and sacrificed on day 56 after AOM injection. (A) Percent change in body weight during each week of the CAC protocol. (B) Clinical score following DSS treatment. End 1st cycle DSS: n = 13 WT; n = 14 Tg. End 2nd cycle DSS: n = 12 WT; n = 14 Tg. (C) Percent survival. P = 0.146. (D) Number of polyps and size distribution of polyps as determined using a dissecting microscope. (E) Representative gross anatomy of the colon (upper panels) and representative colonoscopy images (lower panels) at the end of the CAC protocol. (F) Histopathology scores. *P < 0.05 vs. AOM DSS-treated WT.
Stool consistency, stool occult blood, and body weight loss were monitored following DSS administration to generate a clinical disease score. AOM DSS-treated Tg mice showed a decreased clinical score after the 1st administration of DSS compared to WT mice, but this did not reach statistical significance. Tg mice exhibited a significantly lower clinical score after the second administration of DSS (Figure 1B). Tg mice given DSS without AOM injection had lower clinical scores after DSS administration compared to WT mice (Supplemental Figure 1C), similar to our previous findings (12).
The overall survival rate for AOM DSS-treated Tg mice was 82% (14 of 17 mice) compared to 60% for WT (12 of 20 mice) (Figure 1C). Tg mice given DSS without AOM injection exhibited no mortality throughout the 8-week protocol (n = 8) compared to 60% survival rate for WT mice (12 of 20 mice; Supplemental Figure 1D). Upon sacrifice the entire colon from ileocecal junction to anus was excised and assessed for number and size of polyps. Tg mice showed considerably fewer polyps with the majority being smaller than 3 mm in diameter compared to WT mice (Figure 1D). Figure 1E shows representative photos of excised colons and endoscopic images of polyp formation in WT and Tg mice following AOM-DSS treatment. After polyps were counted and measured, colons were Swiss-rolled and assessed for histopathological score by a trained pathologist. WT mice had a higher occurrence of high grade dysplasia and adenocarcinoma compared to Tg mice which exhibited more low grade dysplasia and normal morphology (Figure 1F). Collectively, these results suggest that PHB Tg mice show less susceptibility to CAC than WT mice.
PHB Tg mice are less susceptible to CAC and mortality
To assess mortality and cancer development, the same protocol was followed with the exception of extending through 126 days or until mice developed rectal prolapse and/or >20% body weight loss. WT mice showed greater mortality than Tg mice throughout the study. The difference in mortality was especially pronounced after the formation of polyps which occurs at approximately 8 weeks (Supplemental Figure 2A). Tg mice showed significantly less total number of polyps, and although rare, a significantly less number of large polyps (>6 mm in diameter) compared to WT mice (Supplemental Figure 2B). Mice that survived from 8 through 18 weeks were assessed for histopathological score since animals that died prior to 8 weeks did not yet develop polyps. As shown in Supplemental Figure 2C, the majority of WT mice developed adenocarcinoma (invasive adenocarcinoma: n = 2, intramucosal adenocarcinoma: n = 7) compared to 5 Tg mice (intramucosal adenocarcinoma). The majority of Tg mice had low-grade or high-grade dysplasia. Supplemental Figure 2D shows representative gross anatomy of colon from three WT and Tg mice, which also reflects the number and size of polyps. These findings suggest that IEC-specific PHB overexpression improves mortality associated with CAC.
IEC-specific PHB overexpression does not affect cell proliferation during CAC
Alteration in cell proliferation can contribute to tumorigenesis (29). Ki67 immunohistochemical staining and proliferating cell nuclear antigen (PCNA) were assessed to examine the effect of PHB overexpression on cell proliferation in CAC. Both WT and Tg mice showed increased Ki67 staining following AOM-DSS treatment (Figure 3A, middle panels) compared to mice given water (Supplemental Figure 3A, upper panels and Supplemental Figure 3B). There was no significant difference in the percent Ki67-positive cells per crypt in normal epithelium (Supplemental Figure 3A, middle panels and Supplemental Figure 3B) or areas of dysplasia (Supplemental Figure 3A, bottom panels) between WT and Tg mice treated with AOM DSS. Similar results were corroborated by PCNA Western blot analysis. PCNA protein expression was increased in both WT and Tg mice following AOM DSS treatment compared to mice given water (Supplemental Figure 3, C and D). There was no significant difference in PCNA protein expression between WT and Tg mice treated with AOM DSS. These results suggest that WT and Tg mice exhibited increased cell proliferation after AOM DSS, and that IEC-specific PHB overexpression does not alter cell proliferation induced by AOM DSS treatment or in colonic tumors/polyps.
Figure 3. IEC-specific overexpression of PHB is associated with altered levels of p53, PUMA, Bax and activation of Bad.

Representative Western blots of total protein isolated from colonic mucosal strippings were probed with (A) anti-PHB, (B) anti-p53, (C) anti-PUMA, (D) anti-Bax, and (E) anti-phospho-Bad and anti-Bad. Anti-β-actin was used as a loading control. Histograms show cumulative mean band densitometry ± SEM, respectively. n ≥ 5 per treatment. *P < 0.05, **P < 0.01.
IEC-specific PHB overexpression is associated with increased apoptosis during CAC
Suppression of apoptosis plays an important role in tumorigenesis by allowing genetically compromised cells to proliferate (30). TUNEL staining and Western blot analysis of cleaved Caspase-3 levels were used to assess IEC apoptosis during CAC. WT and Tg mice exhibited increased number of TUNEL-positive cells per crypt after AOM DSS treatment (Figure 2, A and B). Tg mice exhibited a higher number of TUNEL-positive cells compared to WT mice after AOM DSS treatment in normal epithelium (Figure 2, A and B). Figure 2, C and D corroborate the TUNEL immunofluorescence staining by showing increased protein expression of cleaved Caspase-3 in Tg mice compared to WT mice with CAC. Thus, IEC-specific PHB overexpression may suppress tumor development by promoting IEC apoptosis.
Figure 2. IEC-specific PHB overexpression is associated with increased apoptosis during CAC.

(A) Fluorescent micrograph of TUNEL staining (green) of colonic sections from untreated (water) and AOM-DSS treated WT and PHB Tg mice. Sections were stained with DAPI to visualize nuclei (blue). Scale bars = 100 μm. (B) Number of TUNEL-positive cells per crypt in normal epithelium. *P < 0.05. n = 3 for water; n = 9 for AOM DSS. (C) Representative Western blots showing colonic mucosal cleaved caspase-3 levels and β-actin as a loading control. (D) Cumulative mean ± SEM relative to WT water. *P < 0.05. vs. WT AOM DSS. n ≥ 5 per treatment.
IEC-specific overexpression of PHB is associated with altered levels of p53, PUMA, Bax and p-Bad during CAC
Given the effect of PHB overexpression on apoptosis during CAC, we examined levels of p53, a tumor suppressor, and its downstream targets p53 upregulated modulator of apoptosis (PUMA) and Bax as well as the activation of the pro-apoptotic protein Bad. p53 regulates cell survival through induction of cell cycle arrest or apoptosis. Our previous study showed that PHB protein levels are decreased in mucosal biopsies of active inflammation from Crohn’s disease afflicted patients and in the DSS and IL-10−/− mouse models of colitis (9). To determine whether mucosal PHB levels are affected during CAC, Western blot analysis was performed on total protein isolated from mucosal stippings from WT mice. Figure 3A shows that mucosal PHB protein levels were decreased in WT mice after AOM DSS treatment compared to water control WT mice (water = 1.0 ± 0.09 vs. AOM DSS = 0.75 ± 0.06, P < 0.05). Tg mice exhibited higher expression of mucosal PHB protein levels compared to WT mice when given water or AOM DSS treatment (Figure 3A). Mucosal PHB protein expression in Tg mice was similar to our previous studies utilizing these animals (12). Although induction of CAC increased p53 protein expression in both WT and Tg mice compared to water control mice, p53 levels were significantly higher in Tg mice compared to WT mice after AOM DSS treatment (Figure 3B). Sequencing of exons 5-8 of p53 from DNA isolated from areas of dysplasia from 5 WT and 5 Tg mice found no mutations. PUMA, a central mediator of the p53 apoptotic response and suppressor of intestinal tumorigenesis in mice (31), was increased only in Tg mice during CAC (Figure 3C). Similarly, the pro-apoptotic protein Bax was also increased in Tg mice after AOM DSS treatment (Figure 3D). Phosphorylation of Bad at serine 155 inhibits the pro-apoptotic function of Bad by disrupting its binding to anti-apoptotic Bcl-2 (32). Western blot analysis showed that Tg mice exhibited less phosphorylation of Bad at serine 155 compared to WT mice during CAC (Figure 3E), suggesting that in Tg mice Bad retains its pro-apoptotic function. Together, these data suggest that CAC in PHB Tg mice is associated with increased expression of p53, PUMA, and Bax and decreased Bad phosphorylation.
PHB-induced altered expression of PUMA, Bax and Bad requires p53 signaling in HCT116 cells
HCT116 colorectal carcinoma cells were used as an in vitro model to understand the underlying role of PHB in modulating p53 activation and apoptosis. WT and p53−/− HCT116 cells were transiently transfected with GFP-tagged PHB (GFP PHB) or vector alone and downstream targets of p53 were measured by Western blot analysis. WT HCT116 cells overexpressing PHB (Figure 4A, lane 2) showed increased PUMA and Bax and decreased phospho-Bad protein expression compared to vector-transfected cells (lane 1). Cleaved Caspase-3, a marker of apoptosis, was also increased in PHB overexpressing cells (Figure 4A, lane 2 vs. lane 1). p53−/− HCT116 cells showed no change in PUMA, Bax, phospho-Bad, or cleaved Caspase-3 protein expression after PHB transfection (Figure 4A, lane 6) compared to vector-transfected cells (lane 5), suggesting that p53 is required for PHB-induced alteration of these proteins. WT and p53−/− HCT116 cells were exposed to γ-irradiation to induce DNA damage. Following γ-irradiation, PHB overexpressing WT HCT116 cells showed significantly increased PUMA, Bax, and cleaved Caspase-3 protein expression while phospho-Bad levels were decreased (Figure 4A, lane 4) compared to vector-transfected cells (lane 3). PHB overexpressing WT cells exposed to γ-irradiation exhibited a further increase in Bax and cleaved Caspase-3 and decrease in phospho-Bad compared to PHB overexpressing cells left untreated (Figure 4A, lane 4 versus lane 2). γ-irradiation did not alter expression of PUMA, Bax, phospho-Bad, or cleaved Caspase-3 in p53−/− HCT116 cells regardless of PHB transfection (Figure 4A, lanes 7 and 8).
Figure 4. PHB-induced altered expression of PUMA, Bax and Bad requires p53 signaling in HCT116 cells and PHB interacts with p53 in HCT116 cells and in colon mucosa.

(A) WT and p53−/− HCT116 cells were transiently transfected with pEGFPN1 vector or pEGFPN1-PHB (GFP-PHB) and subjected to γ-irradiation to induce DNA damage 72 hours post-transfection. Total protein was collected 24 hours after γ-irradiation. Western blots were probed with anti-PUMA, anti-Bax, anti-phospho-Bad, anti-Bad and anti-Caspase-3. Blots were subsequently probed with anti-GFP to ensure transfection efficiency and anti-p53 to ensure p53 knockout. Anti-β-actin was used as a loading control. Histograms show cumulative mean band densitometry ± SEM. n = 3 per treatment. *P < 0.05. PHB (B) or p53 (C) was immunoprecipitated (IP) from total HCT116 WT cell lysates overexpressing vector or GFP-PHB and subjected to γ-irradiation to induce DNA mutation 72 hours post-transfection. Total protein was collected 24 hours after γ-irradiation. Immunoprecipitates were separated by SDS-PAGE and membranes were immunoblotted (IB) for p53 or PHB expression. Omission of primary antibody during the immunoprecipitation was performed as a negative control. (D) PHB was immunoprecipitated from colon mucosal protein lysates from water- and AOM DSS-treated mice, followed by immunoblot for p53 expression.
PHB interacts with p53 in HCT116 cells and in colon mucosa
To determine whether PHB interacts with p53 in intestinal epithelial cells, PHB was immunoprecipitated from total cell lysates from WT HCT116 cells overexpressing vector or PHB and exposed to γ-irradiation to induce DNA damage. Following immunoprecipitation and SDS-PAGE, membranes were immunoblotted for p53 and subsequently PHB expression. p53 co-immunoprecipitated with PHB during basal conditions and after γ-irradiation (Figure 4B and 4C). To determine whether PHB interacts with p53 in vivo, we performed co-immunoprecipitation experiments for PHB and p53 in colon mucosal lysates from water- and AOM DSS-treated mice. p53 co-immunoprecipitated with PHB in lystates from both WT and Tg mice (Figure 4D), with increased levels in Tg mice (Figure 4D). Collectively, these results suggest that PHB interacts with p53 in cultured human colorectal cancer cells and in colon mucosa.
PHB interacts with phospho-STAT3 and modulates STAT3 responsive genes, Bcl-xL and Bcl-2 in vivo and in vitro
The IL-6/STAT3 signaling pathway is known to play a pathogenic role in intestinal inflammation and colonic cancer through the induction of the anti-apoptotic genes Bcl-xL and Bcl-2 (33). Caco2-BBE cells, which are responsive to IL-6, show decreased basal and IL-6- induced STAT3 activation, and decreased Bcl-xL and Bcl-2 protein expression when PHB is overexpressed (Figure 5A). Co-immunoprecipitation experiments using Caco2-BBE cells treated with IL-6 suggest that PHB interacts with phospho-STAT3 (Figure 5B and 5C). To determine whether PHB interacts with STAT3 in vivo, we performed co-immunoprecipitation experiments for PHB and phospho-STAT3 in colon mucosal lysates from water- and AOM DSS- treated mice. phospho-STAT3 co-immunoprecipitated with PHB in lystates from Tg mice (Figure 5D). Induction of CAC increased protein expression of STAT3 responsive genes Bcl-2 and Bcl-xL in both WT and Tg mice compared to water control mice. Bcl-2 and Bcl-xL levels were significantly decreased in Tg mice compared to WT mice after AOM DSS treatment (Figure 5E).
Figure 5. PHB interacts with phospho-STAT3 and modulates STAT3 responsive genes, Bcl-xL and Bcl-2 in vivo and in vitro.

(A) Representative Western blots of pSTAT3, Bcl-2, Bcl-xL protein expression in Caco2-BBE cells stably overexpressing pEGFPN1-PHB (GFP-PHB) or empty vector and treated with 100 ng/ml IL-6 basolaterally for 1 hour. Blots were subsequently probed with anti-GFP to ensure transfection efficiency. (B-C) Co-immunoprecipitation of PHB and pSTAT3 using cell lysates described in (A). (D) PHB was immunoprecipitated from colon mucosal protein lysates from water- and AOM DSS-treated mice, followed by immunoblot for pSTAT3 expression. (E) Representative Western blots of total protein isolated from colonic mucosal strippings were probed with anti-Bcl-2, anti-Bcl-xL, anti-β-actin (loading control). Histograms show mean band densitometry ± SEM. n ≥ 5 per treatment. *P < 0.05,
PHB mRNA expression is decreased in inflamed ulcerative colitis and ulcerative colitis-associated dysplasia and inversely correlates with STAT3 and p53 mRNA expression
Quantitative real-time PCR analysis of total RNA isolated from mucosal samples from patients with inflamed ulcerative colitis (UC) or UC-associated adenocarcinoma/dysplasia revealed that PHB expression is significantly decreased compared to matched normal samples (Figure 6). p53 expression was increased in inflamed UC and UC-associated dysplasia tissues, however this increase did not achieve statistical significance in the inflamed tissue samples. STAT3 expression was markedly increased in inflamed UC and UC-associated dysplasia but did not achieve statistical significance.
Figure 6. PHB mRNA expression is decreased in inflamed ulcerative colitis and ulcerative colitis-associated dysplasia and inversely correlates with STAT3 and p53 mRNA expression.
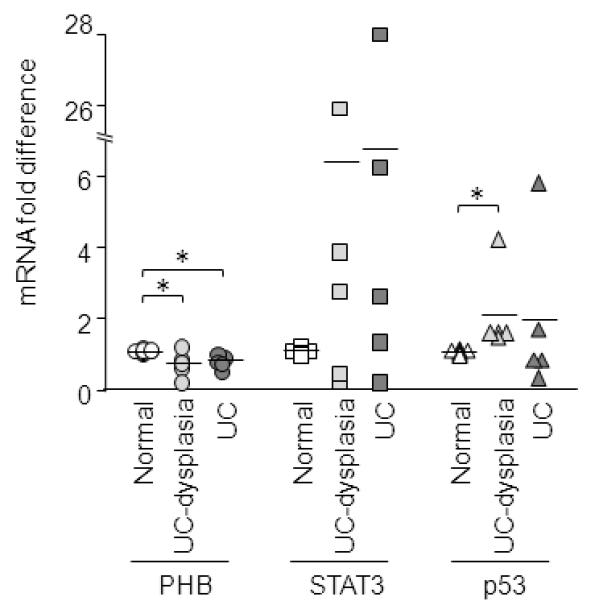
Quantitative real-time PCR analysis of PHB, STAT3, and p53 in total RNA isolated from patients with inflamed ulcerative colitis (UC) or UC-associated adenocarcinoma/dysplasia. n = 8 normal; n = 5 UC-dysplasia; n = 5 UC. *P < 0.05 vs. normal.
Discussion
The specific mechanism by which IBD leads to CRC is poorly understood, but it is clear chronic mucosal inflammation plays a causative role in the transition to adenocarcinoma. Our previous studies have shown that restoration of mucosal PHB can ameliorate experimental colitis (11, 12). For this reason, we chose to use the AOM DSS model to assess the role of PHB in CAC. Here, we show that in mice with CAC, IEC-specific PHB overexpression suppresses colonic neoplastic formation in association with increased rates of apoptosis. IEC-specific overexpression of PHB was associated with altered levels of p53, PUMA, Bax, p-Bad, Bcl-xL and Bcl-2 during CAC. We show, for the first time, that PHB interacts with phospho-STAT3 in addition to p53 in human colon cancer cells and colon mucosa, thereby modulating the apoptotic response (Figure 7).
Figure 7. Model of PHB modulation of p53 and STAT3 apoptotic responses.

PHB induces apoptosis in cells with DNA damage during inflammation through inhibition of STAT3-induced Bcl-xL and Bcl-2 and induction of p53 signaling via activation of Bax and PUMA. Reduced PHB expression in inflamed tissues may make genetically compromised intestinal epithelial cells more resistant to cell death, thereby promoting tumorigenesis.
IBD patients with longstanding colitis require surveillance colonoscopy after 8 years of disease duration due to the high risk of cancer development (34). Several features make colitis-associated cancer distinct from sporadic colon cancer. Colitis-associated cancer is often anaplastic, broadly infiltrating, rapidly growing, develops in flat dysplastic tissue, and occurs at a younger age. While genetic alterations are similar to those in sporadic colon cancer, the frequency and sequence of these events differ in colitis-associated cancer (35). Loss of p53 function is an early critical event in colitis-associated neoplasia in humans in the pathway from dysplasia to cancer (36). Furthermore, aberrent IL-6/STAT3 signaling is associated with inflammatory bowel disease and colitis-associated cancer (33). Activated STAT3 is enhanced in CRC patients and has been shown to induce the anti-apoptotic proteins Bcl-2 and Bcl-xL (37). Inhibition of STAT3 signaling induces apoptosis of colorectal cancer cells via the mitochondrial pathway by modulating Bcl-2 and Bcl-xL (38). Here, we show that in mice with CAC, PHB suppresses colonic tumor formation in association with increased rates of apoptosis. In human colon cancer cells, PHB interacts with p53 and phospho-STAT3 and modulates their downstream apoptotic proteins. These findings suggest that PHB protects against CAC by modulating p53 and STAT3 apoptotic responses.
The best-characterized function of PHB is as a chaperone involved in the stabilization of mitochondrial proteins thereby maintaining normal mitochondrial function and morphology (reviewed in (39)). In most cell types studied, including intestinal epithelial cells, PHB is predominantly localized to the mitochondria (9). However, it has been localized to the cell membrane, cytoplasm, or the nucleus in some cell types (6, 40, 41). Although PHB expression is increased in many transformed cells and tumors (13-19), in our study, colonic mucosal PHB protein expression was reduced after the development of colitis-associated tumors in WT mice. PHB expression was decreased in mucosal samples from human patients with inflamed UC or UC-associated adenocarcinoma/dysplasia. Pro-inflammatory cytokines such as TNFα and reactive oxygen species decrease expression of intestinal epithelial PHB in vivo and in vitro (8-10). Thus, it is likely that PHB levels are decreased in the colonic mucosa following AOM DSS treatment due to the inflammatory process. Tg mice with forced overexpression of PHB in intestinal epithelial cells exhibited no inflammation-induced loss of PHB expression and showed decreased susceptibility to CAC and mortality.
PHB has been reported to have an anti-tumorigenic role in prostate cancer (21), gastric cancer (23), and liver cancer (22). Mice with hepatocyte-specific deletion of PHB exhibited liver injury, fibrosis, oxidative stress and hepatocellular carcinoma developing by 8 months of age (22). Mitochondrial abnormalities in hepatocytes are evident by 3 weeks of age including no cristae formation (22). Knockdown of PHB expression by siRNA in Caco2-BBE intestinal epithelial cells caused mitochondrial depolarization and increased intracellular reactive oxygen species (27). Interestingly, mitochondrial dysfunction is a common feature of cancer cells and multiple studies suggest that mitochondrial chaperone proteins, including PHB, modulate tumorigenesis (reviewed in (42)).
A functional role for PHB as a regulator of transcription has become evident due to its interactions with p53, E2F and Rb (6, 7, 43). Here, we show that in addition to p53, PHB interacts with STAT3 in vitro and in vivo. Studies in breast cancer, prostate cancer, and retinal pigment epithelial cell lines indicate that PHB increases the transcriptional activity and prevents denaturation of p53, thereby regulating apoptosis (44-46). Our study showed that PHB suppresses colonic tumor formation in association with increased rates of apoptosis and p53 apoptotic responses. Sequencing of exons 5, 6, 7 and 8 of p53 from DNA isolated from areas of dysplasia from 5 WT and 5 Tg mice found no mutations. Unlike human UC-associated cancers, DSS-induced colon tumors in mice do not commonly harbor p53 mutations (26). Therefore, it is likely that the increased p53 in Tg mice is functional, unlike the increased, but mutant p53, in human IBD patients. PHB interaction with phospho-STAT3 was associated with decreased Bcl-xL and Bcl-2 expression in cultured intestinal epithelial cells and colon mucosa. The localization of PHB in the mitochondria and nucleus makes it an ideal candidate to modulate the mitochondrial apoptotic pathway and transcription activation. Further studies will assess the mechanism of PHB modulation of STAT3 signaling. PHB interacts with p53 and STAT3 in colon mucosa from mice treated with DSS alone without AOM injection (Supplemental Figure 1E), suggesting interaction is not dependent upon the presence of tumors. Furthermore, Tg mice treated with DSS alone without AOM injection exhibited altered mucosal p53, PUMA, Bcl-2 and Bcl-xL protein expression (Supplemental Figure 1F). Since the development of CAC is dependent on the severity of chronic inflammation, PHB transgene expression likely decreases the occurrence of CAC by decreasing the severity of DSS-induced inflammation and enhancing p53-mediated apoptosis and inhibiting STAT3 induction of anti-apoptotic genes during tumor formation. Early loss of PHB expression during inflammation may make genetically compromised intestinal epithelial cells more resistant to cell death, thereby promoting tumorigenesis.
In conclusion, PHB suppresses colonic tumor formation in association with increased rates of apoptosis in mice with colitis-associated cancer. PHB interacts with p53 and phospho-STAT3 in intestinal epithelial cells in vivo and in vitro and modulates downstream apoptotic responses. Reduced levels of PHB during chronic intestinal inflammation may be an underlying factor promoting tumorigenesis. Modulation of PHB expression in IECs represents a potential therapeutic approach to prevent colitis-associated tumorigenesis.
Supplementary Material
Acknowledgements
The authors thank the late Dr. Shanthi V. Sitaraman from Emory University, Atlanta, GA for her mentoring role with this project. We are grateful to Dr. Ajay Goel from Baylor Research Institute, Baylor University Medical Center, Dallas, TX for providing to us the HCT116 cell line as well as Bax, Bcl-2 and Bcl-xL antibodies. We thank Dr. Linda A. Feagins from Veterans Affairs North Texas Health Care System, University of Texas Southwestern Medical Center, Dallas, TX for helpful scientific discussions. The authors thank Dr. Shinichi Matsumoto, Ana Rahman, Jeff SoRelle, and Mazhar Kanak for their assistance with tissue sectioning through the Baylor Islet Cell Transplant Program, Baylor Research Institute. The authors also thank Dr. Steven Phillips from Baylor Research Institute for his assistance with the γ-irradiator.
Grant support This work was supported by National Institutes of Health grants K01-DK085222 (A.L.T.) and R01-CA133012 (S. J. M) and funds from the Baylor Research Institute (A.L.T).
Abbreviations
- AOM
azoxymethane
- CAC
colitis-associated cancer
- CRC
colorectal cancer
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- IEC
intestinal epithelial cell
- PHB
prohibitin
- PUMA
p53 upregulated modulator of apoptosis
- STAT3
signal transducer and activator of transcription 3
- Tg
transgenic
- UC
ulcerative colitis
- WT
wild-type
Footnotes
Disclosures: The authors have nothing to disclose. No conflicts of interest exist.
Author contributions: Study concept and design: ALT, RFS. Acquisition of data: ASK, WLN, RMG, JR, EH, YC, ALT. Analysis and interpretation of data: ASK, RFS, EH, ALT. Drafting the manuscript: AKS, ALT. Critical revision of the manuscript for important intellectual content: WLM, RMG, SJM, RFS.
References
- 1.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–78. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillen CD, Walmsley RS, Prior P, Andrews HA, Allan RN. Ulcerative colitis and Crohn’s disease: a comparison of the colorectal cancer risk in extensive colitis. Gut. 1994;35:1590–2. doi: 10.1136/gut.35.11.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClung JK, Jupe ER, Liu XT, Dell’Orco RT. Prohibitin: potential role in senescence, development, and tumor suppression. Exp Gerontol. 1995;30:99–124. doi: 10.1016/0531-5565(94)00069-7. [DOI] [PubMed] [Google Scholar]
- 6.Wang S, Fusaro G, Padmanabhan J, Chellappan SP. Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene. 2002;21:8388–96. doi: 10.1038/sj.onc.1205944. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, Zhang B, Faller DV. Prohibitin requires Brg-1 and Brm for the repression of E2F and cell growth. Embo J. 2002;21:3019–28. doi: 10.1093/emboj/cdf302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh SY, Shih TC, Yeh CY, Lin CJ, Chou YY, Lee YS. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics. 2006 doi: 10.1002/pmic.200500541. In Press. [DOI] [PubMed] [Google Scholar]
- 9.Theiss AL, Idell RD, Srinivasan S, Klapproth JM, Jones DP, Merlin D, et al. Prohibitin protects against oxidative stress in intestinal epithelial cells. Faseb J. 2007;21:197–206. doi: 10.1096/fj.06-6801com. [DOI] [PubMed] [Google Scholar]
- 10.Theiss AL, Jenkins AK, Okoro NI, Klapproth JM, Merlin D, Sitaraman SV. Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa B nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol Biol Cell. 2009;20:4412–23. doi: 10.1091/mbc.E09-05-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theiss AL, Laroui H, Obertone TS, Chowdhury I, Thompson WE, Merlin D, et al. Nanoparticle-based therapeutic delivery of prohibitin to the colonic epithelial cells ameliorates acute murine colitis. Inflamm Bowel Dis. 2011;17:1163–76. doi: 10.1002/ibd.21469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theiss AL, Vijay-Kumar M, Obertone TS, Jones DP, Hansen JM, Gewirtz AT, et al. Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology. 2009;137:199–208. e1–6. doi: 10.1053/j.gastro.2009.03.033. 08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai HW, Chow NH, Lin CP, Chan SH, Chou CY, Ho CL. The significance of prohibitin and c-Met/hepatocyte growth factor receptor in the progression of cervical adenocarcinoma. Hum Pathol. 2006;37:198–204. doi: 10.1016/j.humpath.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Ren HZ, Wang JS, Wang P, Pan GQ, Wen JF, Fu H, et al. Increased expression of prohibitin and its relationship with poor prognosis in esophageal squamous cell carcinoma. Pathol Oncol Res. 2010;16:515–22. doi: 10.1007/s12253-009-9242-1. [DOI] [PubMed] [Google Scholar]
- 15.Kang X, Zhang L, Sun J, Ni Z, Ma Y, Chen X, et al. Prohibitin: a potential biomarker for tissue-based detection of gastric cancer. J Gastroenterol. 2008;43:618–25. doi: 10.1007/s00535-008-2208-3. [DOI] [PubMed] [Google Scholar]
- 16.Sato T, Sakamoto T, Takita K, Saito H, Okui K, Nakamura Y. The human prohibitin (PHB) gene family and its somatic mutations in human tumors. Genomics. 1993;17:762–4. doi: 10.1006/geno.1993.1402. [DOI] [PubMed] [Google Scholar]
- 17.Nan Y, Yang S, Tian Y, Zhang W, Zhou B, Bu L, et al. Analysis of the expression protein profiles of lung squamous carcinoma cell using shot-gun proteomics strategy. Med Oncol. 2009;26:215–21. doi: 10.1007/s12032-008-9109-4. [DOI] [PubMed] [Google Scholar]
- 18.Wu TF, Wu H, Wang YW, Chang TY, Chan SH, Lin YP, et al. Prohibitin in the pathogenesis of transitional cell bladder cancer. Anticancer Res. 2007;27:895–900. [PubMed] [Google Scholar]
- 19.Gregory-Bass RC, Olatinwo M, Xu W, Matthews R, Stiles JK, Thomas K, et al. Prohibitin silencing reverses stabilization of mitochondrial integrity and chemoresistance in ovarian cancer cells by increasing their sensitivity to apoptosis. Int J Cancer. 2008;122:1923–30. doi: 10.1002/ijc.23351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coates PJ, Nenutil R, McGregor A, Picksley SM, Crouch DH, Hall PA, et al. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exp Cell Res. 2001;265:262–73. doi: 10.1006/excr.2001.5166. [DOI] [PubMed] [Google Scholar]
- 21.Dart DA, Spencer-Dene B, Gamble SC, Waxman J, Bevan CL. Manipulating prohibitin levels provides evidence for an in vivo role in androgen regulation of prostate tumours. Endocr Relat Cancer. 2009;16:1157–69. doi: 10.1677/ERC-09-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko KS, Tomasi ML, Iglesias-Ara A, French BA, French SW, Ramani K, et al. Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology. 2010 doi: 10.1002/hep.23919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu T, Tang H, Lang Y, Liu M, Li X. MicroRNA-27a functions as an oncogene in gastric adenocarcinoma by targeting prohibitin. Cancer Lett. 2009;273:233–42. doi: 10.1016/j.canlet.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–81. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007;28:1450–9. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 26.Chang WC, Coudry RA, Clapper ML, Zhang X, Williams KL, Spittle CS, et al. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis. 2007;28:2375–81. doi: 10.1093/carcin/bgm134. [DOI] [PubMed] [Google Scholar]
- 27.Kathiria AS, Butcher LD, Feagins LA, Souza RF, Boland CR, Theiss AL. Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS One. 2012 doi: 10.1371/journal.pone.0031231. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–9. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–14. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 30.Huerta S, Goulet EJ, Livingston EH. Colon cancer and apoptosis. Am J Surg. 2006;191:517–26. doi: 10.1016/j.amjsurg.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Qiu W, Carson-Walter EB, Kuan SF, Zhang L, Yu J. PUMA suppresses intestinal tumorigenesis in mice. Cancer Res. 2009;69:4999–5006. doi: 10.1158/0008-5472.CAN-09-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Downward J. How BAD phosphorylation is good for survival. Nat Cell Biol. 1999;1:E33–5. doi: 10.1038/10026. [DOI] [PubMed] [Google Scholar]
- 33.Atreya R, Neurath MF. Signaling molecules: the pathogenic role of the IL-6/STAT-3 trans signaling pathway in intestinal inflammation and in colonic cancer. Curr Drug Targets. 2008;9:369–74. doi: 10.2174/138945008784221116. [DOI] [PubMed] [Google Scholar]
- 34.Neumann H, Vieth M, Langner C, Neurath MF, Mudter J. Cancer risk in IBD: how to diagnose and how to manage DALM and ALM. World J Gastroenterol. 2011;17:3184–91. doi: 10.3748/wjg.v17.i27.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6:297–305. doi: 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- 36.Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology. 1994;107:369–78. doi: 10.1016/0016-5085(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 37.Lassmann S, Schuster I, Walch A, Gobel H, Jutting U, Makowiec F, et al. STAT3 mRNA and protein expression in colorectal cancer: effects on STAT3-inducible targets linked to cell survival and proliferation. J Clin Pathol. 2007;60:173–9. doi: 10.1136/jcp.2005.035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du W, Hong J, Wang YC, Zhang YJ, Wang P, Su WY, et al. Inhibition of JAK2/STAT3 signaling induces colorectal cancer cell apoptosis via mitochondrial pathway. J Cell Mol Med. doi: 10.1111/j.1582-4934.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Artal-Sanz M, Tavernarakis N. Prohibitin and mitochondrial biology. Trends Endocrinol Metab. 2009;20:394–401. doi: 10.1016/j.tem.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Rastogi S, Joshi B, Fusaro G, Chellappan S. Camptothecin induces nuclear export of prohibitin preferentially in transformed cells through a CRM-1-dependent mechanism. J Biol Chem. 2006;281:2951–9. doi: 10.1074/jbc.M508669200. [DOI] [PubMed] [Google Scholar]
- 41.Sharma A, Qadri A. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc Natl Acad Sci U S A. 2004;101:17492–7. doi: 10.1073/pnas.0407536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Czarnecka AM, Campanella C, Zummo G, Cappello F. Mitochondrial chaperones in cancer: from molecular biology to clinical diagnostics. Cancer Biol Ther. 2006;5:714–20. doi: 10.4161/cbt.5.7.2975. [DOI] [PubMed] [Google Scholar]
- 43.Joshi B, Rastogi S, Morris M, Carastro LM, DeCook C, Seto E, et al. Differential regulation of human YY1 and caspase 7 promoters by prohibitin through E2F1 and p53 binding sites. Biochem J. 2007;401:155–66. doi: 10.1042/BJ20060364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chander H, Halpern M, Resnick-Silverman L, Manfredi JJ, Germain D. Skp2B attenuates p53 function by inhibiting prohibitin. EMBO Rep. 2009;11:220–5. doi: 10.1038/embor.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem. 2003;278:47853–61. doi: 10.1074/jbc.M305171200. [DOI] [PubMed] [Google Scholar]
- 46.Chander H, Halpern M, Resnick-Silverman L, Manfredi JJ, Germain D. Skp2B overexpression alters a prohibitin-p53 axis and the transcription of PAPP-A, the protease of insulin-like growth factor binding protein 4. PLoS One. 2011;6:e22456. doi: 10.1371/journal.pone.0022456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.