

Abstract
Bacterial infections are becoming increasingly difficult to treat due to the development and spread of antibiotic resistance. Therefore, identifying novel antibacterial targets and new antibacterial agents capable of treating infections from drug-resistant bacteria is of vital importance. Structurally simple, yet potent fusaricidin or LI-F class of natural products represents a particularly attractive source of candidates for the development of new antibacterial agents. We have synthesized eighteen fusaricidin/LI-F analogs and investigated the effect of their structure modification on conformation, serum stability, antibacterial activity and human cell toxicity. Our findings show that substitution of an ester bond in depsipeptides with an amide bond may afford equally potent analogs with improved stability and greatly decreased cytotoxicity. Lower overall hydrophobicity/amphiphilicity of amide analogs in comparison to their parent depsipeptides, as indicated by the HPLC retention times, may explain dissociation of antibacterial activity and human cell cytotoxicity. These results indicate that amide analogs may have significant advantages over fusaricidin/LI-F natural products and their depsipeptide analogs as lead structures for the development of new antibacterial agents.
Keywords: antibiotics, drug resistance, ester-to-amide substitution, peptides, structure-activity relationship
Introduction
Antibiotic resistance is increasing at a faster rate than the development of new antibiotics,[1–4] and the prevalence of multi-drug resistant bacteria has become a global public health problem.[4] The majority of life-threatening infections worldwide are caused by the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species).[5–6] This group of bacteria is encountered in more than 40% of hospital-acquired infections, and is resistant to the majority of commonly used antibiotics. Therefore, identifying novel antibacterial targets and new antibacterial agents capable of treating infections from drug-resistant bacteria is of vital importance.
Naturally occurring cyclic lipodepsipeptides that contain one or more ester bonds in addition to the amide bonds have emerged as promising lead compounds for new antibiotic discovery.[7–10] The biosynthesis of these peptides proceeds nonribosomally and is catalyzed by a complex of multi-functional enzymes; termed non-ribosomal peptide synthases (NRPSs). NRPSs have unique modular structure in which each module contains the requisite domains for the recognition and activation of a single amino acid, generating huge structural and functional diversity of nonribosomal peptides.[11] Within this class of natural products, cyclic lipodepsipeptide daptomycin (Cubicin®, Cubist Pharmaceuticals, Inc.)[7, 12–13] is already approved in the USA, European Union and Canada for the treatment of infections caused by multidrug-resistant bacterial strains. Ramoplanin (Nanotherapeutics, Inc.)[7, 14–16] represents another example of cyclic lipodepsipeptide potentials for reverting multi-drug bacterial resistance. Ramoplanin is currently entering Phase III trials for the treatment of Clostridium difficile-associated diarrhea.
Despite the progress in development of new antibacterial agents, it is inevitable that resistant strains of bacteria will emerge in response to widespread use of a particular antibiotic and limit its usefulness. Structurally simple, yet potent fusaricidins or LI-F family of natural products, Figure 1, represent particularly attractive candidates for the development of new antibacterial agents capable of reverting infections caused by multi-drug resistant bacteria, Figure 1.[7, 17–20]
Figure 1.

Structures of the fusaricidin/LI-F family of compounds.
Fusaricidins/LI-Fs are cyclic lipodepsipeptide antifungal antibiotics isolated from Paenibacillus sp. Their common structural feature is the macrocyclic ring consisting of six amino acid residues, three of which, Thr1, D-allo-Thr4 (D-aThr4) and D-Ala6, are conserved throughout the family, as well as the 15-guanidino-3-hydroxypentadecanoic acid tail attached via an amide bond to the N-terminal Thr1. Fusaricidins/LI-Fs are cyclized by a lactone bridge between N-terminal Thr1 hydroxyl group and C-terminal D-Ala6. Among isolated fusaricidin/LI-F antibiotics, fusaricidin A or LI-F04a, Figure 1,[20] showed the most promising antimicrobial activity against a variety of fungi, including clinically important Candida albicans and Cryptococcus neoformans, and against Gram-positive bacteria such as S. aureus (MICs ranging from 0.78–3.12 μg/mL). Fusaricidins/LI-Fs did not, however, show activity against Gram-negative bacteria.[18–19] Fusaricidins/LI-Fs’ mode of action is still unknown. Total synthesis of fusaricidin A/LI-F04a natural product using a combination of solid-phase and solution synthetic approaches was recently reported by Cochrane et al.[21] Two groups, Jensen et al.[22] and Park et al.,[23] have reported identification and isolation of putative fusaricidin/LI-F synthetase gene, fusA, from Paenibacillus polymyxa, opening the possibility for the development of biosynthetic approaches toward this family of naturally occurring cyclic lipodepsipeptides and their analogs.
We have previously reported a complete Fmoc solid-phase synthesis of a fusaricidin A/LI-F04a analog containing 12-guanidino dodecanoic acid instead of naturally occurring 15-guanidino-3-hydroxypentadecanoic acid, Figure 1.[24] Total synthesis of fusaricidin/LI-F antifungal antibiotics and particularly unlimited access to their synthetic analogs represent important initial steps toward full exploitation of their antimicrobial potentials.
Herein, we have described our efforts to determine the structural requirements for the antimicrobial activity, improved serum stability and dissociation of antibacterial activity and low human cell toxicity of fusaricidin/LI-F peptides.
Results
Solid-phase synthesis
Amino acid sequences and lipid tails of synthesized fusaricidin A/LI-F04a analogs 1–18 are shown in Figure 2. Analogs 1–12 are depsipeptides containing an ester bond between Thr1 and D-Ala6 or Gly6 residues. In analog 13, naturally occurring amino acid residues Thr1 and D-Ala6 are replaced with Lys thus substituting the ester moiety, as well as two chiral centers while keeping the same number of the atoms in the ring. The remaining four cyclic analogs, 14–17, have an ester bond replaced with an amide bond by substituting Thr1 for Dap1. Analog 18 is a linear version of 6 and was prepared as a control. Synthesis of depsipeptide analogs 1–12 is shown in Scheme 1.[24] In the first step, the C-terminal amino acid Fmoc-D-Asp-OAllyl was attached to a PEG-PS based amide resin (TentaGel S RAM) via side chain using standard HBTU/HOBt/NMM synthetic protocol. Standard Fmoc SPPS strategy was used throughout. Ester bond was formed between Thr1 side chain hydroxyl group and Alloc-D-Ala6-OH or Alloc-Gly6-OH carboxyl group using DIC/DMAP coupling conditions in CH2Cl2. In order to avoid potential epimerization during Alloc-D-Ala6-OH coupling, a catalytic amount of DMAP (0.2 eq.) was used.[25–26] Under applied experimental conditions no epimerization was observed as indicated by analytical RP HPLC (data not shown). The lipid tail, Fmoc-aminododecanoic acid (Fmoc-ADA-OH), was incorporated into the linear peptide precursor prior to D-Ala6/Gly6 coupling via ester bond and on-resin cyclization in order to avoid an undesired O→N acyl shift known to occur under basic conditions required for Fmoc removal.[24, 27–28]
Figure 2.

Sequences of synthesized fusaricidin A/LI-F04a analogs. [a] Differences among the sequences of naturally occurring fusaricidin A/LI-F04a and synthetic analogs are highlighted in bold. [b] Rt=retention time obtained by analytical RP HPLC. Method: 2% B for 0.5 min followed by linear gradient 2–98% B over 30 min where A is 0.1% TFA in water and B is 0.08%TFA in CH3CN.
Scheme 1.

Synthesis of fusaricidin A/LI-F04a analogs 1–17. Reagents and conditions: a) Fmoc-D-Asp-OAllyl, and Fmoc-AA-OH, standard Fmoc-SPPS deprotection and coupling protocols; b) Ac-Thr-OH, standard Fmoc-SPPS deprotection and coupling protocols; c) Fmoc-Thr-OH, standard Fmoc-SPPS deprotection and coupling protocols; d) Fmoc-ADA-OH, standard Fmoc-SPPS deprotection and coupling protocols; e) Fmoc-Lys-OH, standard Fmoc-SPPS deprotection and coupling protocols; f) Fmoc-Dap(Mtt)-OH, standard Fmoc-SPPS deprotection and coupling protocols; g) Alloc-D-Ala-OH or Alloc-Gly-OH (4 eq.), DIC (4 eq.), DMAP (0.2 eq.), CH2Cl2, r.t., 18 h; h) 1% TFA/CH2Cl2 (v/v), r.t. 30 min.; i) Pd(Ph3P)4 (0.1 eq.), HN(CH3)2·BH3 (4 eq.), CH2Cl2, r.t. 2×10 min.; j) PyBOP/HOBt/DIEA (2/2/6 eq.), DMF, r.t. 18 h; k) 20% piperidine/DMF (v/v), r.t., 25 min.), N,N-bis(tert-butoxycarbonyl)thiourea (3 eq.), 2-chloro-1-methylpyridinium iodide (3 eq.), TEA (4 eq), DMF, r.t. 18 h; l) 20% piperidine/DMF (v/v), r.t., 25 min.); m) TFA/TIA/H2O=95:2.5:2.5 (v/v/v), r.t. 3 h.
After selective removal of Alloc and Allyl protective groups by treatment with Pd(Ph3P)4 and non-basic borane dimethylamine complex as scavenger,[13] the linear peptide was cyclized through an amide bond between D-Ala6 or Gly6 and D-Asn5 residues using PyBOP. The conversion of the lipid tail’s amino into the desired guanidino group was achieved by the removal of the Fmoc-protecting group using standard piperidine deprotection protocol and subsequent treatment of the peptidyl-resin with N,N-bis(tert-butoxycarbonyl)thiourea followed by Mukaiyama’s reagent, 2-chloro-1-methylpyridinium iodide.[14] Final deprotection and cleavage from the resins was carried out using a cleavage cocktail of TFA/TIA/water (95:2.5:2.5 v/v/v).
Amide analogs 13–17 were prepared by replacing Thr1 residue with Lys 13 or Dap, 14–17, Scheme 1. For this purpose, an identical Fmoc SPPS strategy was employed. In the case of analog 13, upon synthesis of linear precursor, Allyl and Alloc protecting groups were removed and the peptide was cyclized through Lys1 ε-amino group and D-Asn5 α-carboxyl group. In the case of analogs 14–17, selective removal of Mtt protecting group from Dap1 with 2% TFA in CH2Cl2, allowed coupling of Alloc-D-Ala-OH using standard coupling protocols. Final synthetic steps were performed as described above. Linear analog 18 was prepared on 2-chlorotrityl chloride resin (Cl-TrtCl)[29] using standard Fmoc chemistry, starting with attachment of Fmoc-D-Ala-OH via carboxylic group. Loading of the resin was determined to be 0.5 mmol/g.[30] Use of Cl-TrtCl resin in this case afforded a linear peptide with a C-terminus carboxylic group, identical to the hydrolysis product of 6. All analogs were purified using preparative RP HPLC, and purity (≥95%) was confirmed with analytical RP HPLC and MALDI-TOF MS.
Conformational study
The structural features of representative fusaricidin analogs 6 and 14, as well as the linear peptide 18 were monitored by circular dichroism (CD) spectroscopy, Figure 3. CD spectra were recorded in aqueous medium as well as in less polar trifluoroethanol (TFE), a membrane mimicking solvent system.[31] Besides membrane mimicking properties, TFE is also known to induce formation of the stable conformations in peptides which are otherwise unstructured in aqueous solutions.[32–33] In order to increase solubility of peptides in aqueous media and to inhibit potential peptide aggregation at the concentrations required for CD experiments, all analyzed analogs were dissolved in 0.5% aqueous AcOH.[34–35]
Figure 3.
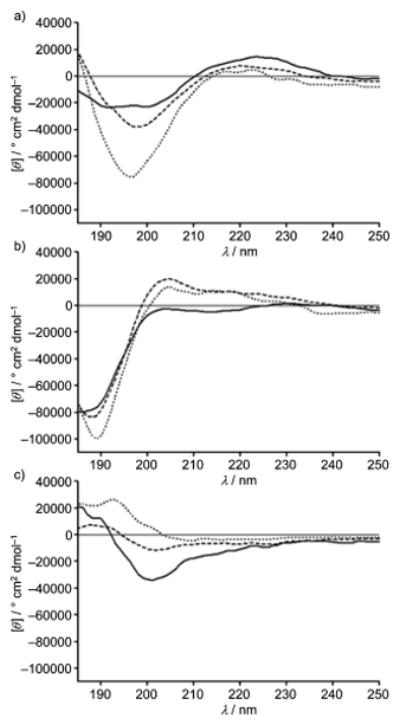
CD spectra of fusaricidin A/LI-F04 analogs: a) 6, b) 14 and c) 18.
Since small cyclic peptides still have considerable mobility of their backbones,[36–39] it is reasonable to expect that the ester-to-amide substitution in our case will also lead to significant conformational changes. Therefore, we should expect different CD spectra of depsipeptide 6 and amide analog 14 in water and less polar TFE, due to their different conformational flexibility and the ability of TFE to promote intramolecular H-bonds and stabilize preferential conformation.[33] It is important to note that D-amino acid residues predominate within the depsipeptide 6, amide 14 and linear peptide 18 sequences, Figure 3, causing inversion of their CD spectra, Figure 3. The CD spectrum of the depsipeptide 6 in 0.5% AcOH exhibits a minimum at approximately 197 nm and a weak maximum around 220 nm, reminiscent of an inverted antiparallel β-sheet structure, Figure 3a. Water replacement with less polar 50% TFE/water mixture and 100% TFE resulted in drastic change of depsipeptide 6 CD spectra. The CD spectrum in 100% TFE shows a double minimum at 192 nm and 201 nm and maximum at 225 nm. In addition to these changes, a marked decrease in the intensity of the CD spectrum in TFE was observed as well. All these spectral changes are characteristic of overall β-sheet/β-turn structures.[40–42] β-Sheet/β-turn containing small cyclic peptides are not unusual, and examples can be found in gramicidin S and its analogs.[41, 43–44] CD spectra of amide analog 14 are markedly different from the parent depsipeptide 6 in both aqueous and TFE solutions, indicating significant differences in structural flexibility and conformations induced by ester-to-amide substitution. The CD spectrum of amide analog 14 in 0.5% AcOH is characterized by a maximum at 205 nm and minimum at 188 nm, whereas in TFE CD maximum at 205 nm is completely lost and the intensity of the spectral minimum at 188 nm is decreased and slightly shifted toward shorter wavelengths, Figure 3b.
As expected, due to lack of the structural constraints, linear peptide 18 shows different CD spectra in aqueous medium and TFE. In 0.5% AcOH, the CD spectrum of peptide 18 has characteristics of an inverted unordered structure with a weak maximum at 195 nm, whereas in TFE the CD spectrum with a weak minimum at 200 nm indicates presence of an inverted type I β-turn, Figure 3c.[40, 42] Besides conformational changes, ester-to-amide substitution in cyclic peptides may also alter overall hydrophobicity and amphiphilicity, structural characteristics that are important for the biological activities of peptides.[46–50] Typically, peptides RP HPLC retention times (Rt) are used as indications of their overall hydrophobicity.[44–45] Rt values of synthesized fusaricidin A/LI-F04a analogs collected in Figure 2 show that amide analogs are less hydrophobic in comparison to their parent depsipeptides, which is expected due to the loss of a Thr1 methyl group. Hydrophobicity of synthetic analogs was further altered by replacing Val3 residue with Ala, Tyr, and Phe, based on the amino acid sequences found in fusaricidin/LI-F natural products. The observed change in overall hydrophobicity in both depsipeptide and amide analogs expressed by Rt is in the order Ala < Tyr < Val < Phe.
In order to get a better insight into conformational changes induced by ester-to-amide substitution in fusaricidin A/LI-F 04a analogs, we conducted molecular dynamics (MD) simulations. Low energy conformations were generated by conformational analysis in Molecular Operating Environment (MOE)[47] using a Monte Carlo search with the Generalized Born solvation model implemented in MOE.[48] The amphiphilic moment descriptor, which is an established measure of balance between hydrophilic and lipophilic moieties, was computed in MOE for the lowest energy conformations.[49]
As shown in Figure 4, MD studies are in qualitative agreement with the experimental CD data indicating that depsipeptide 6 can adopt different conformations in polar and nonpolar environments, whereas conformational differences of amide analog 14 under the same conditions are less pronounced suggesting more rigid peptide backbone structure. In addition, calculated amphiphilic moments for depsipeptide 6 and amide 14 are markedly different with depsipeptide being more amphiphilic in both solvent systems, Figure 4.
Figure 4.
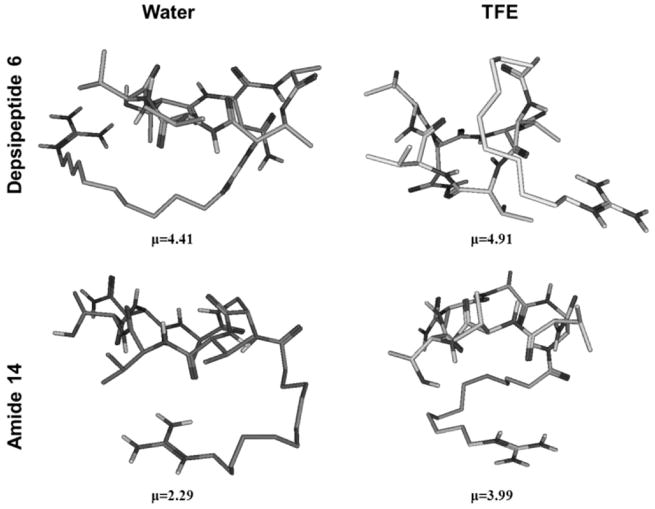
Representative structures of low energy conformers of depsipeptide 6 (upper panels) and amide 14 (lower panels) in water and TFE and their calculated amphiphilic moments, μ (see Experimental section).
Structure-activity study
To identify the structural determinants for the antibacterial activity and cytotoxicity, a total of eighteen fusaricidin A/LI-F04a analogs were prepared, Figure 2. The structural variations of the natural product include modification of the lipid tail, substitution of amino acid residues including alanine-scan analogs (Ala-scan), ester-to-amide substitution, and a linear control peptide for comparison. The antibacterial in vitro activity of synthesized fusaricidin A/LI-F04a analogs 1–18, expressed as the minimum inhibitory concentration (MIC, μg/mL), was determined for Gram-positive and Gram-negative bacteria using standard micro dilution broth method in 96-well plates, Table 1.[50–51] Similar to the natural products, fusaricidin/LI-F analogs are active against Gram-positive bacteria including antibiotic resistant strains, and did not show activity against Gram-negative bacteria (data not shown). To probe the role of the lipid tail in antibacterial activity, depsipeptides containing an acetyl group 1, 2, 12-ADA 3, 4, and 12-GDA 5–12 attached to Thr1 were synthesized. No antibacterial activity was observed for the analogs containing an acetyl group or 12-ADA, even at the highest concentration tested. On the other hand, depsipeptide analog containing 12-GDA 6 showed potent antibacterial activity (MIC 8 μg/mL) against Gram-positive bacteria, Table 1. Ala-scan and several other substitutions at key amino acid residues were performed to reveal the role of each individual amino acid in the antimicrobial activity of synthetic fusaricidin analogs. However, requirements of our solid-phase synthetic approach[24], Scheme 1, permitted replacement of four (D-Val2, Val3, D-aThr4, and D-Ala6) out of six amino acids. Obtained MIC values, Table 1, showed that the substitution of Val3 with Ala, analog 8, preserved the antibacterial activity of the parent depsipeptide 6, whereas substitutions of D-aThr4 and D-Val2 with D-Ala, analogs 7 and 9, respectively, resulted in significant reduction of antimicrobial activity. In contrast, replacement of either D-Ala6 with Gly, analog 10, or both Thr1 and D-Ala6 with εLys1, analog 13, led to a complete loss of antibacterial activity. No significant reduction in antimicrobial activity was observed in the case where D-aThr4 was replaced with D-Thr4, analog 5. Quite interestingly, despite significant changes in hydrophobicity, amphiphilicity and conformation caused by ester-to-amide substitution, antibacterial activities of amide analogs 14–17 parallel those of the parent depsipeptides 6, 8, 11 and 12. As mentioned earlier, fusaricidins/LI-F is a family of naturally occurring cyclic lipodepsipeptide antibiotics consisting of 12 compounds in total, with conserved Thr1, D-aThr4, D-Ala6 amino acid residues.[7] Aliphatic nonpolar amino acid residues are present at the position 2 (D-Val, D-Ile and D-aIle), and polar amino acids D-Asn and D-Gln are present at position 6 of the peptide sequence. Most diverse changes occur in position 3 where Tyr, Val, Ile, Phe, and D-Ile were found. Taking this into consideration, we have synthesized fusaricidin/LI-F analogs possessing Phe, analogs 11 and 16, and Tyr, analogs 12 and 17, residues in position 3 in the sequence. Replacement of Val3 with polar Tyr resulted in reduction of antimicrobial activity, whereas substitution with nonpolar hydrophobic Phe did not affect the antibacterial activity, Table 1. In addition, the MIC data obtained for depsipeptides 6, 8, 11 and 12 showed that an increase of overall hydrophobicity, as indicated by the HPLC retention times, Figure 2, did not correlate with their antibacterial activities. Analogs 6, 8 and 11 showed almost identical, within experimental error, antibacterial activities for tested bacterial strains while analog 12 showed reduced activity. In contrast, similar comparison of amide analogs 14–16 showed different results. Amide analogs possessing hydrophobic amino acids in position 3 of the peptide sequence such as Val and Phe (analogs 14 and 16) exhibited better antibacterial activity than analogs with more polar Ala and Tyr (analogs 15 and 17), indicating in this case correlation between the peptides’ overall hydrophobicity and antibacterial activity. Despite a much higher degree of conformational flexibility and complete sequence homology with biologically active depsipeptide 6, control linear peptide 18 did not show any biological activities in described assays.
Table 1.
Antimicrobial activity of fusaricidin A/LI-F04a analogs.
| MIC (μg/mL)
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
| S. aureus ATCC 29213 | ND[a] | ND | ND | ND | 16 | 8 | ND | 16 | ND | ND | 8 | >128 | ND | 8 | ND | 8 | ND | >128 |
| S. aureus ATCC 33591 | >128 | >128 | >128 | >128 | 16 | 8 | 64 | 16 | 32 | 64 | 8 | >128 | 64 | 16 | 64 | 8 | 32 | >128 |
| S. aureus ATCC 700699 | >128 | >128 | >128 | >128 | 16 | 16 | 64 | 16 | 64 | 64 | 16 | ND | 64 | 16 | ND | ND | ND | ND |
| S. epidermidis ATCC 27626 | ND | ND | ND | ND | 16 | 8 | 32 | 8 | 16 | 64 | 8 | 16 | 64 | 16 | 64 | 16 | 64 | >128 |
| S. pyogenes ATCC 19615 | ND | ND | ND | ND | 32 | 16 | 64 | 16 | 64 | 64 | 8 | 64 | 64 | 16 | >64 | 16 | 64 | >128 |
ND=not determined.
Cytotoxicity
Hemolytic activity was determined against human erythrocytes (0.5% in PBS buffer), as described in the Experimental Section. PBS and 0.5% Triton X-100 were used as a reference for 0 and 100% hemolysis, respectively. Peptides that were not active in the antimicrobial assays did not give noticeable hemolytic activity. None of the active depsipeptides 6, 8, 11 and their amide counterparts, peptides 14–16, showed hemolytic activity at MIC concentrations. However, at higher concentrations, depsipeptides 6, 8 and 11 showed considerable hemolysis relative to reference Triton X-100. The degree of hemolysis appears to correlate with the increase in hydrophobicity of depsipeptides. For example, at 64 μg/mL (8 x MIC) least hydrophobic depsipeptide 8 with Ala3 exhibited the lowest hemolytic activity (~30%) followed by analog 6 with Val3 causing ~45% hemolysis, whereas most hydrophobic analog 11 with Phe3 showed the highest level of hemolysis, ~65%. Interestingly, none of the amide analogs 14–16 exhibited appreciable hemolytic activity at this concentration. At much higher concentrations, 128 and 256 μg/mL (16 and 32 x MIC), depsipeptide analogs 6 and 11 showed hemolysis comparable to that of Triton X-100, whereas amide analogs 14 and 16 at 256 μg/mL reached 15 % and 50% hemolysis, respectively, Figure 5. Control peptide 18 did not exhibit appreciable hemolytic activity even at the highest tested concentration of 64 μg/ml.
Figure 5.
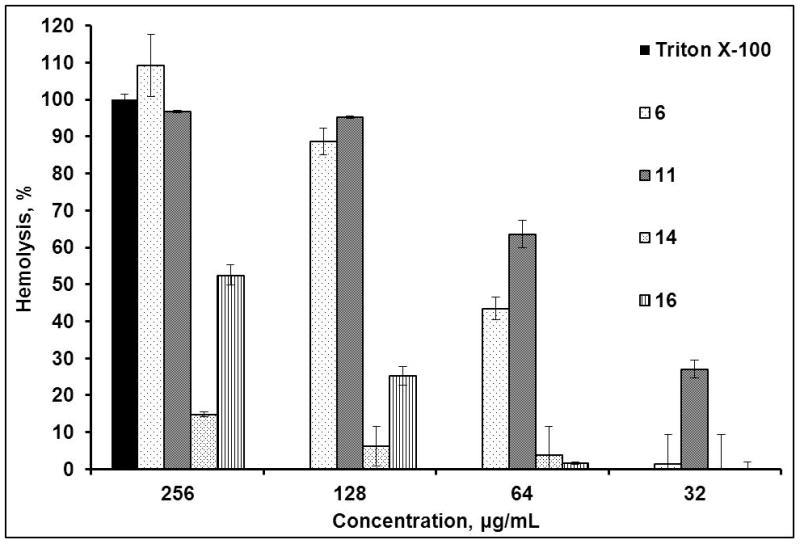
Hemolytic activity of fusaricidin A/LI-F04a analogs (see Supplementary materials).
It is worthwhile noticing that in this assay DMSO (up to 5% v/v final concentration) was added to increase tested fusaricidin A/LI-F04a analog solubility. Therefore, potential aggregation due to lower solubility of amide analogs at higher concentrations may contribute to the observed hemolytic activity.[52–54] Although DMSO is hemolytic,[55–56] our control experiments showed that in the conditions used DMSO did not cause hemolysis.
To further assess the therapeutic potential of synthetic fusaricidin A/LI-F04a analogs, we tested the in vitro toxicity of analogs 1, 4, 6 and 14 on the human liver embryonic cell line WRL 68 and the HepG2 cancer cell line. A known cytotoxic drug Adriamycin (Doxorubicin)[57] was used as a positive control in these assays. Depsipeptide analog 1, lacking a lipid tail and inactive in the antimicrobial assays, did not show appreciable cytotoxicity toward WRL 68 or HepG2 cells within the tested concentration range. Interestingly, lipidated depsipeptide analogs 4 and 6, regardless of their antibacterial or hemolytic activities, were cytotoxic at higher concentrations to both tested cell lines. Analog 4 was highly cytotoxic at 150 μg/mL, whereas more active depsipeptide analog 6 showed a somewhat similar trend in cytotoxicity to Adriamycin and was highly cytotoxic at 90 μg/mL concentration. In comparison, the cytotoxicity of less hydrophobic and amphiphylic amide analog 14 was much lower at the same concentrations (data not shown).
Serum stability
To investigate the effect of ester-to-amide substitution on cyclic peptide proteolytic stability, the disappearance of the intact peptide incubated in 50% human serum for 24 h at 37°C was followed by RP HPLC,[58] Figure 6. Depsipeptide 6 and amide 14 were used in this study as representative examples of each group of synthetic fusaricidin A/LI-F04a analogs. Approximately 35% change in the concentration of depsipeptide 6 was observed within an hour of incubation in 50% human serum. However, prolonged incubation of 6 did not result in complete depsipeptide degradation; and after 24 h, about 35% of 6 was still detected in the serum. Stability of 6 in EMEM buffer containing 10% FBS used for cytotoxicity assays was significantly better. In this case approximately 23% loss in concentration of 6 was observed after 24h (see Supplementary materials). Analysis of depsipeptide 6 degradation using RP HPLC and MALDI-TOF mass spectrometry revealed that the main degradation product is a result of this depsipeptide’s ring opening via ester bond hydrolysis (depsipeptide 6: Rt=15.8 min, [M+H]+ m/z=825.9709; hydrolysis product: Rt=15.04 min, found [M+H]+ m/z=843.9988,). Obtained identical MALDI-TOF m/z and RP HPLC Rt for the depsipeptide 6 degradation product and control peptide 18 (Rt=15.05 min, [M+H]+ m/z=844.1160, calculated [M+H]+ m/z=843.0228) additionally confirmed this finding. In contrast, no degradation was observed for amide analog 14 under the same experimental conditions, Figure 6.
Figure 6.
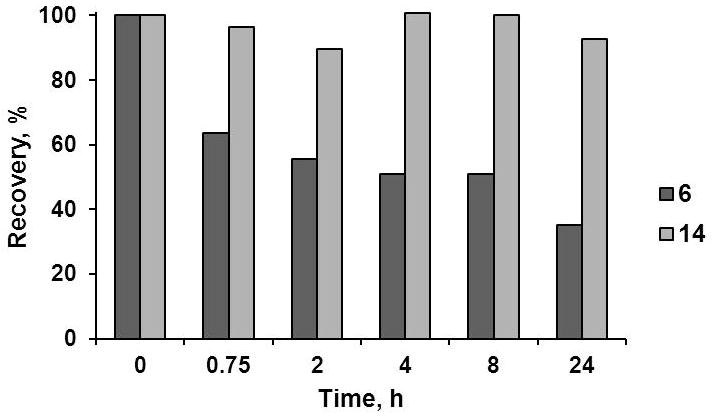
Serum stability of depsipeptide 6 and amide 14 in 50% human serum.
Discussion
Due to their structural diversity and high potency, naturally occurring cyclic depsipeptides have attracted a great deal of attention for discovery of new antibiotics with superior activity against multidrug-resistant bacteria.[7, 59] However, full exploitation of the antimicrobial potentials of this class of natural products strongly depends on the synthetic access to their structures, in particular their analogs, and understanding important details of their mode of action. It has been shown in the literature that the antibacterial and hemolytic activities of antimicrobial peptides depend on different structural components such as charge, size, secondary structure, hydrophobicity and amphipathicity.[43, 60–62] Among naturally occurring antibacterial peptides, the fusaricidins/LI-F family of cyclic lipodepsipeptides represents attractive lead compounds for the development of new antibiotics because of their unique structure in which peptide sequences are composed of six, mostly hydrophobic, amino acids and a positive charge located at the end of the lipid tail.[7] In addition, there is substantial evidence in the literature showing that substitution of the ester bond in depsipeptides with an amide bond may afford derivatives with comparable activities and markedly increased serum stabilities.[63–65] With this in mind, we synthesized eighteen fusaricidin A/LI-F04a analogs and investigated the effects of structural modification on their overall hydrophobicity/amphiphilicity, conformation, stability and biological activity. All analogs were synthesized on a solid support using Fmoc methodology. However, solid-phase synthesis of depsipeptides 1–13 turned out to be particularly challenging. Our strategy for the solid-phase synthesis of cyclic lipodepsipeptides involved attachment of D-Asp5 via side-chain, stepwise Fmoc-synthesis of a linear peptide precursor, and on-resin head-to-tail cyclization, Scheme 1. The key step in the synthesis of cyclic depsipeptides is the ring closure. Considering the greater reactivity of the amino group, and therefore minimal possibility of side reactions, we have chosen macrolactamization for depsipeptide ring closure, as opposed to macrolactonization. Although this synthetic strategy appears to be a better choice for the solid-phase depsipeptide synthesis, an undesired intramolecular O→N acyl shift may occur if basic conditions were to be used.[27, 66] Reversible intramolecular O→N or N→O acyl shifts are well described side reactions in peptide chemistry.[27, 66] Typically, peptides containing Ser or Thr residues undergo N→O acyl shift under acidic conditions, whereas the exposure of corresponding depsipeptides to basic conditions leads to opposite O→N acyl shift. Incorporation of the lipid tail or acetyl group into the linear peptide precursor prior to D-Ala6 coupling via ester bond and on-resin cyclization completely suppressed the O→N acyl shift resulting in the desired cyclic lipodepsipeptide, Scheme 1. In addition, we have found that the efficacy of the ester bond formation strongly depends on the type of solid support and the solvent. Optimal results were obtained using PEG-based resins, DIC/DMAP coupling method, and CH2Cl2.[24] Superiority of PEG-PS over PS-based resins in ester bond formation could be explained by a better solvation of peptide-PEG-PS resins, [67–68] rapid DIC activation of the carboxylic group,[69] and significant suppression of N-acylurea byproduct formation[70] in a non-polar solvent such as CH2Cl2. On the other hand, synthesis of amide analogs 13–17 using the same approach was straightforward, Scheme 1. The desired cyclic lipopeptides were obtained in satisfactory yields.
Several studies have pointed out the importance of amide-to-ester substitution on peptide conformation.[36, 71–73] In general, replacement of a hydrogen-donating NH-group by the oxygen atom in depsipeptides results in removal of the backbone H-bond, affecting therefore conformation of peptides. As long as the side chains are not altered, intramolecular hydrogen bonds affect the equilibrium distribution of peptide conformers. The effect of ester-to-amide substitution on the conformation of fusaridicin A/LI-F04a analogs was assessed by CD spectroscopy, Figure 3. Observed drastic differences in CD spectra of depsipeptide 6 in aqueous medium and less polar TFE, and absence of such spectral changes for amide analog 14, indicate a higher degree of flexibility of ester bond as opposed to the amide bond. These differences also suggest greater ability of synthesized depsipeptide analogs to alternate conformations in different cellular environments. However, complete interpretation of both depsipeptide 6 and amide 14 CD spectra in aqueous medium and TFE is rather difficult, due to the fact that obtained spectra cannot be attributed to a single conformation. MD were performed to further explore the conformational differences caused by ester-to-amide substitution in fusaricidin A/LI-F04a analogs and to assess the possibility of forming more organized structures under conditions that mimic the membrane environment. The results are shown in Figure 4. Our experimental CD data fully supports the MD simulations indicating higher conformational flexibility of depsipeptide analogs. MD calculations showed no significant conformational differences between depsipeptide 6 and amide analog 14 in water, whereas the differences become markedly more pronounced in a less polar environment such as TFE.
Besides conformational changes, ester-to-amide substitution altered the biochemical properties of the fusaricidin analogs. The assessments of synthesized fusaricidin A/LI-F-04a analogs’ stability in human serum and cytotoxicity are important secondary screening assays, mainly because these assays eliminate cytotoxic analogs and analogs with short half-lives.[74] Our experimental data suggest that low serum stability of depsipeptide 6 can be attributed mainly to the hydrolysis of its ester bond. This finding is in accordance with the literature data showing that cyclic depsipeptides can be degraded through competition between the protease action and esterase action. However, the depsipeptides were more easily subjected to hydrolysis by esterase-type enzymes.[75–77] Cyclic structure, the presence of D-amino acids, and greater stability of an amide bond all together contribute to the enhanced proteolytic stability of the amide analogs.[78–85] Quite importantly, ester-to-amide substitution did not affect the antibacterial potency of these peptides. A positive charge positioned at the end of the lipid tail, hydrophobicity and amphiphilicity appeared to be crucial for fusaricidin A/LI-F04a analogs’ antibacterial activities and separation of their antibacterial and human cell toxicity. Antibacterial assays showed that only analogs bearing a guanylated lipid tail are effective against Gram-positive bacteria, indicating the importance of the lipid tail and positively charged guanidino functionality for their antimicrobial activity, Table 1. Based on the role of the lipid tail in other lipopeptide antibiotics,[86–89] and our experimental data showing that nonlipidated analog 2 is not active against tested bacterial strains, we can assume that the lipid moiety helps target fusaricidin analogs to bacterial membrane. The antibacterial activity of peptides having a guanidilated lipid moiety could possibly be explained by the strong ability of the guanidinium group to bind the anionic phosphate of the bacterial phospholipid membrane through a combination of H-bonding and charge-charge interactions. In general, high pKa value, diffused charge density, and the geometry of the guanidinium group which allows better alignment of H-bonds, all contribute to better interactions with the phosphate anion compared to the ammonium group.[90]
The results of the antibacterial assays of the Ala-scan analogs revealed that residues D-Val2, D-aThr4 and D-Ala6 are crucial for antibacterial activity. Replacement of any of these amino acids with corresponding Ala, analogs 7, 9 and 10, resulted in complete loss of depsipeptide antibacterial activity, Table 1. On the other hand, position 3 in the depsipeptide sequence is more tolerable to changes. Depsipeptide analogs containing neutral Ala3, 8, and bulky hydrophobic Val3, 6, or Phe3, 11, exhibited almost identical antibacterial activities, whereas analog 12 with polar Tyr3 was not active. Equally potent in vitro antibacterial activity was determined for their amide counterparts, analogs 14 and 16 possessing hydrophobic Val3 and Phe3, respectively. However, differences in the antibacterial activity were observed between depsipeptide 8 and its amide counterpart 15, both containing Ala in position 3. Depsipeptide 8 exhibited antibacterial activity comparable to the most potent analogs, whereas the amide 15 showed significant decrease in activity, Table 1. Since the relative overall hydrophobicity order of Ala<Val<Phe is the same for both amide and depsipeptide analogs, greater change in amphiphilicity upon substitution of lipophylic Val3 with neutral Ala3 in structurally constrained amide 15 may explain the loss of its antibacterial activity.
As mentioned earlier, comparisons of the overall hydrophobicity of depsipeptides 6, 8, 11 and 12, Figure 2, show no correlation with their antibacterial activities. In contrast, an increase in depsipeptide hydrophobicity can be associated with the enhanced hemolytic activities, Figure 5. More hydrophobic depsipeptides 6 and 11 were also more hemolytic. Similarly, depsipeptides 6, 8, 11 and 12 exhibited higher hemolytic activities than their less hydrophobic amide counterparts 14–17. At concentrations 8 x MIC (64 μg/mL) none of the amide analogs 14–17 were hemolytic. The fact that linear peptide 18 is not hemolytic (see Supplementary materials), suggests that the hydrolysis of the ester bond which leads to opening of the depsipeptide ring is not a cause of hemolysis.
Liver toxicity is one of the most critical issues in drug development, often leading to the delay or failure of drug candidates.[91–93] Therefore, peptides that show no in vitro liver cell toxicity may prove to be better candidates for further preclinical or clinical studies. HepG2 and WRL 68 cell lines were used to assess the potential toxicity of fusaricidin A/LI-F04a analogs. In both cell lines, depsipeptide 6 exhibited much higher in vitro cytotoxicity than amide counterpart 14 (see Supplementary materials). However, absence of cytotoxicity to human cells observed for amide analog 14 cannot be solely explained by lower hydrophobicity. Conformational differences among these fusaricidin/LI-F analogs, as illustrated by the MD calculations and the differences in the CD spectra between analogs 6 and 14, Figures 3 and 4, should be taken into consideration as well. While hemolytic depsipeptide 6 has the ability to form more ordered structures in a membrane mimicking environment, non-hemolytic amide analog 14 fails to undergo any significant conformational changes regardless of the solvent system. Conformational changes and hence changes in the amphipathicity has been ascribed to the low hemolytic activity of some synthetic and naturally occurring cyclic cationic antimicrobial peptides.[43, 60, 62, 94] Typically, in these cyclic antimicrobial peptides, presence of cationic amino acids in the peptide sequence, defined secondary structure (β-sheet and hairpin loop), and amphiphilic character are common structural characteristics that determine their biological activities.[60, 94] In the case of fusaricidin A/LI-F04a and its analogs, a positive charge is positioned at the terminus of the 12-carbon-atom lipid tail, and peptide sequences are composed mostly of hydrophobic amino acids, Figure 2. Nevertheless, experimental CD data and molecular modeling studies showed that the peptide ring in both depsipeptide 6 and amide 14 analogs can adopt different amphiphilic conformations depending on the environment, with depsipeptides being more amphiphilic. Relatively lower amphiphilicity of the amide analogs may contribute to dissociation of antibacterial activity and human cell cytotoxicity.
Conclusion
Structure-activity relationship studies of fusaricidin A/LIF-04a depsipeptide analogs reported in this work reveal key structural requirements for antibacterial activity and decreasing cytotoxicity. The positively charged guanidinium group at the end of the 12-carbon-atom lipid tail, and the presence of hydrophobic amino acids in the depsipeptide sequence are crucial for antibacterial activity. Ala-scan results and comparison of the fusaricidin/LI-F natural product sequences suggest that position 3 in the depsipeptide sequence is more tolerable to amino acid change. By introduction of neutral and hydrophobic amino acids into this position, we were able to manipulate the depsipeptide’s overall hydrophobicity and amphiphilicity without losing antibacterial potency. However, structural changes leading to an increase in the depsipeptide’s overall hydrophobicity and amphiphilicity resulted in an increase of their cytotoxicity. On the other hand, substitution of an ester bond in depsipeptides with an amide bond gave more stable cyclic lipopeptide analogs with preserved in vitro antibacterial activities, yet greatly improved serum stabilities and minimized human cell toxicity. Lower over all hydrophobicity/amphiphilicity of amide analogs in comparison to their parent depsipeptides may explain dissociation of antibacterial activity and human cell cytotoxicity. More stable and less cytotoxic amide analogs may have significant advantages over naturally occurring fusaricidin A/LI-F04a and its depsipeptide analogs as lead structures for the development of new antibacterial agents. In addition, amide analogs are synthetically more accessible than the parent depsipeptides, allowing for further structural optimization using a combinatorial chemistry approach. Synthesis of a focused combinatorial library based on fusaricidin A/LI-F04a amide analogs, and elucidation of the mode of action of both the depsipeptide and amide analogs are currently underway.
Experimental Section
Chemicals and instrumentation
TentaGel S RAM resin was obtained from Advanced ChemTech (Louisville, KY). 2-chlorotrityl chloride was obtained from Novabiochem (Gibbstown, NJ). Fmoc-protected amino acids and coupling reagents (HOBt, HBTU, PyBOP) were purchased from Chem-Impex (Wood Dale, IL) or Novabiochem (Gibbstown, NJ). DIC was purchased from Acros Organics (Thermo Fisher Scientific, Waltham, MA). DMAP was purchased from Sigma Aldrich (St. Louis, MO). All solvents were purchased from Fisher Scientific (Atlanta, GA) or Sigma Aldrich (St. Louis, MO) and were analytical reagent grade or better. Linear peptidyl-resin precursors were synthesized on a PS3 automated peptide synthesizer (Protein Technologies Inc., Tucson, AZ). Mass spectrometry was performed on MALDI-TOF Voyager-DE™ STR (Applied Biosystems, Foster City, CA) in reflector mode using α-cyano-4-hydroxycinnamic acid as a matrix and positive mode. Analytical RP HPLC analyses and peptide purifications were performed on 1260 Infinity (Agilent Technologies, Santa Clara, CA) liquid chromatography systems equipped with a UV/Vis detector. For analytical RP HPLC analysis, a C18 monomeric column (Grace Vydac, 250 × 4.6 mm, 5 μm, 120 Å), 1 mL/min flow rate, and elution method with a linear gradient of 2–98% B over 30 min, where A was 0.1% TFA in H2O and B was 0.08% TFA in ACN was used. For peptide purification, a preparative C18 monomeric column (Grace Vydac, 250 × 22 mm, 10 μm, 120 Å) was used. Elution method was identical to the analytical method except for the flow rate, which was 19 mL/min. CD spectra were recorded on a JASCO 810 spectropolarimeter (Easton, MD) using a quartz cell of 0.1 mm optical path length. Spectra were measured over a wavelength range of 180–250 nm with an instrument scanning speed 200 nm/min and a response time of 1 s. The concentrations of peptides were 0.1–0.2 mM. Cytotoxicity assays were analyzed on a Synergy H4 microplate reader (BioTek, Winooski, VT). Microbial strains and human cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). Dehydrated culture media and agar, and polystyrene plates used for antimicrobial assays were purchased from BD (Franklin Lakes, NJ). Control antibiotics were purchased from Sigma Aldrich (St. Louis, MO). Antimicrobial activity assays were carried out in standard, sterile 96-well plates, and MIC values were determined by measuring turbidity at 600 nm using a Stat Fax 2100 Microplate reader (Awareness Technology Inc., Palm City, FL). Human red blood cells (hRBCs) were purchased from Innovative Research (Novi, MI). Human serum was purchased from Sigma Aldrich (St. Louis, MO).
General procedure for peptide synthesis and purification
Linear peptidyl-resin precursors for cyclic lipopeptides 1–17 were synthesized on amide TentaGel S RAM resin (substitution 0.26 mmol/g, 0.25 mmol scale) using automated peptide synthesizer. The solid-phase synthesis of cyclic peptides 1–17 started by attaching C-terminal Fmoc-D-Asp-OAllyl via side chain to the resin using HBTU/HOBt/NMM protocol. The same coupling protocol was used throughout, including coupling of the lipid tail (Fmoc-ADA-OH, 1.5 eq.). In the case of depsipeptide analogs 1–12, Alloc-D-Ala-OH (4 eq.) or Alloc-Gly (4 e.q) was coupled manually via ester bond to the hydroxyl group of Fmoc-Thr using DIC (4 eq.) and DMAP (1 eq.) coupling reagents in CH2Cl2. Amide analogs 13–17 were prepared by coupling Fmoc-Dap(Mtt) instead of Fmoc-Thr-OH using the same coupling protocol as above. Selective removal of Allyl and Alloc protecting groups was performed by treatment of peptidyl-resin precursors with borane dimethylamine complex (4 eq.), followed by addition of Pd(PPh3)4 (0.1 eq.) in CH2Cl2 under argon.[95] Mtt was selectively removed under mild acidic conditions (1% TFA in CH2Cl2, 30 min.). Solid-phase cyclization of linear precursors was carried out in a manual reaction vessel overnight using PyBOP/HOBt/DIEA (2/2/6 eq.) in DMF. The conversion of the lipid tail’s amino into the desired guanidino group was achieved by the removal of the Fmoc-protecting group using standard piperidine deprotection protocol and the treatment of the peptidyl-resin with N,N-bis(tert-butoxycarbonyl)thiourea (3 eq.) followed by Mukaiyama’s reagent 2-chloro-1-methylpyridinium iodide/TEA (3:4 eq.) in DMF.[96]
Control peptide 18 was synthesized on 2-chlorotrityl chloride resin (substitution 1.3 mmol/g). The synthesis started by attaching Fmoc-D-Ala-OH (4 eq.) to the resin using an equimolar amount of DIEA in CH2Cl2 followed by resin endcapping with MeOH,[65] and the chain elongation using standard Fmoc-chemistry. Quantitative Fmoc substitution of the resin (0.5 mmol/g) was determined by Fmoc cleavage and absorption measurement at 304 nm.[30] In all cases, the reaction progress was monitored by RP HPLC, MALDI-TOF MS, and where applicable ninhydrin colorimetric test.[97]
Peptides were removed from the resin using TFA/TIA/H2O (95:2.5:2.5 v/v/v) for 3 h. The crude peptides were precipitated with cold methyl tert-butyl ether, and purified using preparative RP HPLC. HPLC fractions were analyzed for purity, combined and lyophilized to give white powder. Final purity of synthesized peptides was confirmed by analytical RP HPLC, and was ≥ 95% in all cases.
Concentrations of peptides in all experiments were determined using RP HPLC and calibration curve based on analog 6. The peptide content of 6 was determined by quantitative amino acid analysis to be 62.32%.
Circular dichroism
All CD spectra were recorded on JASCO 810 spectropolarimeter at 25°C using a 0.1 cm path length cell. The spectra were acquired in the range 180–250 nm, 1 nm band width, 4 accumulations and 200 nm/min scanning speed. All spectra were obtained using 0.1–0.2 mM concentrations in 0.5% or 1% AcOH, 25–100% TFE/water (v/v) solution. Spectra at 0.5 and 1% AcOH were virtually identical for each peptide. Each experiment was repeated at least once and at various concentrations. No concentration dependent CD spectral changes were observed.
Molecular modeling
First low energy conformations were generated by conformational analysis in Molecular Operating Environment (MOE)[47] using a Monte Carlo search with the Generalized Born solvation model implemented in MOE.[48] The dielectric constant was set up to 26.14 or 80 to simulate the search in TFE or water, respectively. The search was conducted with the MMFF94x force field using default parameters. The lowest energy conformers found for each compound in the Monte Carlo search were the starting point of 40 ns of molecular dynamics simulation using the module MacroModel 9.9 from Maestro software.[98] In brief, the initial structures were equilibrated by 2 ps. The “Bonds to Hydrogens” option from the Shake procedure was selected in order to use 2 fs as time step. The simulation temperature was set to 300.0 K as default. The Optimized Potential for Liquid Simulation (OPLS)-2005 force field[99] was used to calculate the potential energy. Conformations were sampled every 20 ps and optimized using the same force field. The dielectric constant was set up to 26.14, to simulate the search in TFE. The GB/SA solvation model implemented in MacroModel[100] was used for water simulation.
The amphiphilic moment descriptor, μ which is an established measure of the balance between hydrophilic and lipophilic molecular properties,[26] was computed in MOE for the lowest energy conformations obtained in the dynamics within 5 kcal/mol of the lowest minimum.
Antibacterial activity
Total of six Gram-positive and two Gram-negative bacterial strains were used, including multiple-drug resistant bacterial strains; Staphylococcus aureus ATCC 29213, Staphylococcus aureus (MRSA) ATCC 33591, Staphylococcus aureus Mu50 (VRSA) ATCC 700699, Staphylococcus epidermidis (MRSE) ATCC 27626, Streptococcus pyogenes ATCC 19615, Escherichia coli K-12 ATCC 29181 and Klebsiella pneumoniae K6 ATCC 700603. Quantification of the antibacterial activity of synthesized analogs 1–18 was performed in sterile 96-well flat-bottomed polystyrene plates by the standard micro dilution broth method.[50–51] Tests were performed using Müller-Hinton broth (MHB) without dilution. Controls on each plate were media without bacteria, bacterial inoculum without antimicrobials added, and bacterial inoculum containing methicillin or vancomycin. Assay setup: Stocks of microorganisms maintained at −80°C in 30% glycerol were thawed and grown in media recommended by ATCC protocols for each particular microorganism. The following day, an aliquot of the bacterial suspension (100 μL) was transferred into 10 mL of fresh media and incubated for 4–5 h, until OD600 of the suspension was 0.3–0.4. Bacterial suspension (100 μL) was then transferred into sterile eppendorf tubes and centrifuged for 5 min at 4000 rpm. Supernatant was discarded and cell pellet resuspended in MHB. Upon measuring OD600 of the suspension, appropriate dilution was made so that the final OD of the suspension was approximately 0.001 (based on calculation). Concentrations of the analogs 1–18, as well as control antibiotics, were in the range 1–128 μg/mL. All samples were loaded in duplicate, and the average OD value was taken for calculating MIC. Each assay was repeated twice. Stock solutions of synthesized analogs 1–18, as well as control antibiotics were prepared in 5–10% of DMSO/water (v/v) solvent mixture, depending on the analog solubility. After dilutions with MHB medium, final concentration of DMSO in wells was 0.5–1%. Plates were loaded with 90 μL of bacterial suspension (with initial OD600 of 0.001) of the tested microorganism, and 10 μL aliquots of two-fold serial dilutions of the analogs 1–18 or control antibiotics. Plates were then incubated at 37°C overnight with gentle shaking. In the case of S. pyogenes, plates were incubated in 5% CO2 atmosphere. The inhibition of the bacterial growth was determined by measuring OD600; reduction in OD600 indicates inhibition of bacterial growth.
Hemolytic activity
Human red blood cells (hRBCs) were diluted with PBS to 1%. Depending on solubility, analogs 1–18 were dissolved in 5–10% DMSO/water (v/v) solvent mixture to concentrations of 16–512 μg/mL. Into each well of the clear, flat-bottom 96-well plate, 50 μL of the hRBCs were placed followed by addition of 50 μL of analog solution to final peptide concentrations of 8–256 μg/mL. Assays were performed in triplicate, and each experiment was repeated twice. To determine the potential effect of DMSO on hemolytic activity, controls containing 5–10% DMSO in water (v/v) were added to the assay setup. As a positive control, 50 μL of Triton X-100 in water was used at a final concentration of 0.5% (v/v). As a negative control, 50 μL of water and PBS was used. Plates were incubated for 1h at 37°C. To each well 100 μL of PBS was added, and the plates were centrifuged for 10 min at 1000 g. Supernatants (150 μL) were transferred into a new plate, and absorbance at 405 nm was measured. Within the tested concentration range, the effect of DMSO on hemolysis was minimal, and was deducted to obtain solely hemolytic activity of the peptides. The degree of hemolysis was expressed in percent relative to total hemolysis caused by Triton-X.
Cytotoxicity
Cytotoxicity of analogs 1–18 was determined using MTT colorimetric assay. Assay was set up in flat-bottom polystyrene 96-well plates with 10,000 cells/well grown in EMEM media containing 10% FBS, 5% penicillin/streptomycin and 5% of L-glutamine (v/v). After an overnight incubation at 37°C in humidified atmosphere with 5% CO2, media was removed, and fresh media containing analogs 1–18 in the concentration range of 1–250 μM was added. Plates were again incubated at 37°C in humidified atmosphere with 5% CO2. As a control, doxorubicin was used in the same concentration range. After 24 or 48 h incubation, media was removed, and 100 μL of MTT dissolved in serum free medium (1 mg/mL) was added to each well. Plates were again incubated for 3 h under the same conditions. Media containing MTT was removed and 100 μL of DMSO was added to each well. Plates were shaken for 5 min before reading absorbance at 540 nm.
Stability assays
Stabilities of selected fusaricidin A/LI-F04a analogs in 50% human serum, and as well as in EMEM medium (used in cell toxicity assays) were determined. For stability in 50% human serum, peptides 6 and 14 (0.5 mg each) were dissolved in 200 μL of DMSO to which 800 μL of water and 1 mL of human serum was added. The solution was incubated at 37°C. After 0 min, 45 min, 2 h, 4 h, 8 h and 24 h, three samples (3 × 100 μL) of each peptide were taken and precipitated by addition of 20 μL of 15 % aqueous TCA. Samples were quickly vortexed and then centrifuged at 10,000 rpm for 10 min. Supernatant was analyzed by analytical RP HPLC and MALDI-TOF MS. For stability of depsipeptide 6 in the EMEM medium containing 10% FBS, 0.25 mg of the peptide was dissolved in 10 μL of DMSO, followed by addition of 990 μL of media and incubation at 37°C. Peptide samples were treated and analyzed in the same way as described above.
Supplementary Material
Acknowledgments
We wish to acknowledge the support of cyclic lipopeptide work described in this article by the NIH (1S06-GM073621-01) and AHA (0630175N) Grant to P.C. We also would like to thank our colleague Dr. Laszlo Otvos, Jr. for the helpful comments, and Ms. Karen Gottwald for editing the text.
ABBREVIATIONS
- AcOH
acetic acid
- Alloc
allyloxycarbonyl
- 12-ADA
12-aminododecanoic acid
- CD
circular dichroism
- Dap
diaminopropionic acid
- DIC
diisopropylcarbodiimide
- DIEA
diisopropylethyl amine
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EMEM
Eagle’s minimal essential medium
- FBS
fetal bovine serum
- Fmoc
fluorenylmethyloxycarbonyl
- 12-GDA
12-guanidino dodecanoic acid
- HOBt
N-hydroxybenzotriazole
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- NMM
hRBCs, human red blood cells
- MALDI-TOF MS
matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy
- MHB
Müller-Hinton broth
- MIC
minimum inhibitory concentration
- MRSA
methicillin-resistant S. aureus
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Mtt
4-methyltrityl chloride
- NMM
N-methylmorpholine
- PBS
phosphate buffered saline
- PyBOP
benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
- RP HPLC
reversed-phase high performance liquid chromatography
- SPPS
solid-phase peptide synthesis
- TIA
thioanisole
- TCA
trichloroacetic acid
- TFA
trifluoroacetic acid
- TFE
trifluoroethanol
- VRSA
vancomycin resistant S. aureus
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
References
- 1.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 2.U.S. Food and Drug Administration. Antimicrob Agents Chemother. 2010;54:4033–4035. doi: 10.1128/AAC.00819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischbach MA, Walsh CT. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J. Clin Infect Dis. 2008;46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 5.Rice LB. J Infect Dis. 2008;197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 6.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Clin Infec Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 7.Bionda N, Cudic P. Croat Chem Acta. 2011;84:315–329. [Google Scholar]
- 8.Roongsawang N, Washio K, Morikawa M. Int J Molec Sci. 2010;12:141–172. doi: 10.3390/ijms12010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marahiel MA. J Pept Sci. 2009;15:799–807. doi: 10.1002/psc.1183. [DOI] [PubMed] [Google Scholar]
- 10.Sieber SA, Marahiel MA. J Bacteriol. 2003;185:7036–7043. doi: 10.1128/JB.185.24.7036-7043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grunewald J, Marahiel MA. Microbiol Mol Biol Rev. 2006;70:121–146. doi: 10.1128/MMBR.70.1.121-146.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodford N. Expert Opin Invest Drugs. 2003;12:117–137. doi: 10.1517/13543784.12.2.117. [DOI] [PubMed] [Google Scholar]
- 13.Kern WV. Int J Clin Pract. 2006;60:370–378. doi: 10.1111/j.1368-5031.2005.00885.x. [DOI] [PubMed] [Google Scholar]
- 14.McCafferty DG, Cudic P, Frankel BA, Barkallah S, Kruger RG, Li W. Biopolymers. 2002;66:261–284. doi: 10.1002/bip.10296. [DOI] [PubMed] [Google Scholar]
- 15.Shah D, Dang MD, Hasbun R, Koo HL, Jiang ZD, DuPont HL, Garey KW. Expert Rev Anti Infect Ther. 2010;8(510):555. doi: 10.1586/eri.10.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckert R. Future Microbiol. 2011;6:635–651. doi: 10.2217/fmb.11.27. [DOI] [PubMed] [Google Scholar]
- 17.Kurusu K, Ohba K. J Antibiot. 1987;40:1506–1514. doi: 10.7164/antibiotics.40.1506. [DOI] [PubMed] [Google Scholar]
- 18.Kajimura Y, Kaneda M. J Antibiot. 1996;49:129–135. doi: 10.7164/antibiotics.49.129. [DOI] [PubMed] [Google Scholar]
- 19.Kajimura Y, Kaneda M. J Antibiot. 1997;50:220–228. [PubMed] [Google Scholar]
- 20.Kuroda J, Fukai T, Konishi M, Uno J, Kurusu K, Nomura T. Heterocycles. 2000;53:1533–1549. [Google Scholar]
- 21.Cochrane JR, McErlean CSP, Jolliffe KA. Org Lett. 2010;12:3394–3397. doi: 10.1021/ol101254m. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Beatty PK, Shah S, Jensen SE. Appl Environ Microbiol. 2007;73:3480–3489. doi: 10.1128/AEM.02662-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi SK, Park SY, Kim R, Lee CH, Kim JF, Park SH. Biochem Bioph Res Co. 2008;365:89–95. doi: 10.1016/j.bbrc.2007.10.147. [DOI] [PubMed] [Google Scholar]
- 24.Stawikowski M, Cudic P. Tetrahedron Lett. 2006;47:8587–8590. doi: 10.1016/j.tetlet.2006.09.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bionda N, Cudic M, Barisic L, Stawikowski M, Stawikowska R, Binetti D, Cudic P. Amino Acids. 2012;42:285–293. doi: 10.1007/s00726-010-0806-x. [DOI] [PubMed] [Google Scholar]
- 26.Atherton E, Benoiton NL, Brown E, Sheppard RC, Williams BJ. J Chem Soc, Chem Commun. 1981:336–337. [Google Scholar]
- 27.Mouls L, Subra G, Enjalbal C, Martinez J, Aubagnac JL. Tetrahedron Lett. 2004;45:1173–1178. [Google Scholar]
- 28.Skwarczynski M, Kiso Y. Curr Med Chem. 2007;14:2813–2823. doi: 10.2174/092986707782360123. [DOI] [PubMed] [Google Scholar]
- 29.Barlos K. Tetrahedron Lett. 1989;30:3943–3946. [Google Scholar]
- 30.Chan WC, White PD. In: Fmoc Solid Phase Peptide Synthesis. Hames BD, editor. Vol. 222. Oxford University Press; New York: 2003. [Google Scholar]
- 31.Sonnichsen FD, Van Eyk JE, Hodges RS, Sykes BD. Biochemistry. 1992;31:8790–8798. doi: 10.1021/bi00152a015. [DOI] [PubMed] [Google Scholar]
- 32.Otvos L, Cudic M. In: Peptide Characterization and Application Protocols. Fields GB, editor. Humana Press; New Jersey: 2007. [Google Scholar]
- 33.Roccatano D, Colombo G, Fioroni M, Mark AE. Proc Natl Acad Sci USA. 2002;99:12179–12184. doi: 10.1073/pnas.182199699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fields C, Mickelson D, Drake S, McCarthy J, Fields G. J Biol Chem. 1993;268:14153–14160. [PubMed] [Google Scholar]
- 35.Sivaraman T, Kumar TKS, Huang CC, Yu C. Biochem Mol Biol Int. 1998;44:29–39. doi: 10.1080/15216549800201032. [DOI] [PubMed] [Google Scholar]
- 36.Cupido T, Spengler J, Ruiz-Rodriguez J, Adan J, Mitjans F, Piulats J, Albericio F. Angew Chem. 2010;122:2792–2797. doi: 10.1002/anie.200907274. [DOI] [PubMed] [Google Scholar]; Cupido T, Spengler J, Ruiz-Rodriguez J, Adan J, Mitjans F, Piulats J, Albericio F. Angew Chem Int Edit. 2010;49:2732–2737. doi: 10.1002/anie.200907274. [DOI] [PubMed] [Google Scholar]
- 37.Freidinger RM, Perlow DS, Randall WC, Saperstein R, Arison BH, Veber DF. Int J Pept Prot Res. 1984;23:142–150. doi: 10.1111/j.1399-3011.1984.tb02704.x. [DOI] [PubMed] [Google Scholar]
- 38.Haubner R, Gratias R, Diefenbach B, Goodman SL, Jonczyk A, Kessler H. J Am Chem Soc. 1996;118:7461–7472. [Google Scholar]
- 39.Kessler H, Gemmecker G, Haupt A, Klein M, Wagner K, Will M. Tetrahedron. 1988;44:745–759. [Google Scholar]
- 40.Otvos L. In: Use of circular dichroism to determine secondary structure of neuropeptides. Irvine GB, Williams CH, editors. Vol. 73. Clifton; 1997. pp. 153–161. [PubMed] [Google Scholar]
- 41.Jelokhani-Niaraki M, Kondejewski LH, Farmer SW, Hancock REW, Kay CM, Hodges RS. Biochem J. 2000;349:747–755. doi: 10.1042/bj3490747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woody RW. In: Circular dichroism. Goodman M, Felix A, Moroder L, Toniolo C, editors. E22b. Thieme; Stuttgart/New York: 2004. pp. 739–765. [Google Scholar]
- 43.Kondejewski LH, Jelokhani-Niaraki M, Farmer SW, Lix B, Kay CM, Sykes BD, Hancock REW, Hodges RS. J Biol Chem. 1999;274:13181–13192. doi: 10.1074/jbc.274.19.13181. [DOI] [PubMed] [Google Scholar]
- 44.Lee DL, Powers JP, Pflegerl K, Vasil ML, Hancock RE, Hodges RS. J Pept Res. 2004;63:69–84. doi: 10.1046/j.1399-3011.2003.00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS. Antimicrob Agents Chemother. 2007;51:1398–1406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mant CT, Kovacs JM, Kim HM, Pollock DD, Hodges RS. Biopolymers. 2009;92:573–595. doi: 10.1002/bip.21316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chemical Computing Group Inc. Molecular Operating Environment (MOE), version 2008, Vol. Montreal, Quebec, Canada: [accessed Sep. 15, 2011]. http://www.chemcomp.com. [Google Scholar]
- 48.Wojciechowski M, Lesyng B. J Phys Chem B. 2004;108:18368–18376. [Google Scholar]
- 49.Cruciani G, Crivori P, Carrupt PA, Testa B. J Mol Struc -THEOCHEM. 2000;503:17–30. [Google Scholar]
- 50.Otvos L, Cudic M. In: Broth microdilution antibacterial assay of peptides Vol. Fields GB, editor. Humana Press; Totowa: 2007. pp. 309–320. [DOI] [PubMed] [Google Scholar]
- 51.CLSI. Approved Standard M7-A8. Clinical and Laboratory Standards Institute; Wayne, PA: 2011. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. [Google Scholar]
- 52.Subbalakshmi C, Krishnakumari V, Nagaraj R, Sitaram N. FEBS Lett. 1996;395:48–52. doi: 10.1016/0014-5793(96)00996-9. [DOI] [PubMed] [Google Scholar]
- 53.John E, Jahning F. Biophys J. 1992;63:1536–1543. doi: 10.1016/S0006-3495(92)81737-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feder R, Dagan A, Mor A. J Biol Chem. 2000;275:4230–4238. doi: 10.1074/jbc.275.6.4230. [DOI] [PubMed] [Google Scholar]
- 55.Ansel HC, Cabre GE. J Phys Sci. 1970;59:478–481. doi: 10.1002/jps.2600590408. [DOI] [PubMed] [Google Scholar]
- 56.Santos NC, Figuera-Coelho J, Martins-Silva J, Saldanha C. Biochem Pharmacol. 2003;65:1035–1041. doi: 10.1016/s0006-2952(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 57.Weiss RB. Semin Oncol. 1992;19:670–686. [PubMed] [Google Scholar]
- 58.Jenssen H, Aspmo SI. Peptide-based drug design serum stability of peptides. Vol. 494. Humana Press; Totowa: 2008. pp. 177–186. [DOI] [PubMed] [Google Scholar]
- 59.Giuliani A, Pirri GP, Nicoletto SF. Cent Eur J Biol. 2007;2:1–33. [Google Scholar]
- 60.Kondejewski LH, Farmer SW, Wishart DS, Kay CM, Hancock REW, Hodges RS. J Biol Chem. 1996;271:25261–25268. doi: 10.1074/jbc.271.41.25261. [DOI] [PubMed] [Google Scholar]
- 61.McInnes C, Kondejewski LH, Hodges RS, Sykes BD. J Biol Chem. 2000;275:14287–14294. doi: 10.1074/jbc.275.19.14287. [DOI] [PubMed] [Google Scholar]
- 62.Qin C, Zhong X, Bu X, Ng NLJ, Guo Z. J Med Chem. 2003;46:4830–4833. doi: 10.1021/jm0341352. [DOI] [PubMed] [Google Scholar]
- 63.Nam J, Shin D, Rew Y, Boger DL. J Am Chem Soc. 2007;129:8747–8755. doi: 10.1021/ja068573k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rew Y, Shin D, Hwang I, Boger DL. J Am Chem Soc. 2004;126:1041–1043. doi: 10.1021/ja039671y. [DOI] [PubMed] [Google Scholar]
- 65.Izzo I, Acosta GA, Tulla-Puche J, Cupido T, Martin-Lopez MJ, Cuevas C, Albericio F. Eur J Org Chem. 2010;2010:2536–2543. [Google Scholar]
- 66.Mauger AB, Stuart OA. Int J Pept Prot Res. 1987;30:481–488. doi: 10.1111/j.1399-3011.1987.tb03356.x. [DOI] [PubMed] [Google Scholar]
- 67.Meldal M. In: Properties of solid supports. Fields GB, editor. Vol. 289. Academic Press, Inc; New York: 1997. pp. 83–104. [DOI] [PubMed] [Google Scholar]
- 68.Fields GB, Fields Cg. J Am Chem Soc. 1991;113:4202–4207. [Google Scholar]
- 69.Hudson D. J Org Chem. 1988;53:617–624. [Google Scholar]
- 70.Podlech J. In: 3.5 Carbodiimides. Goodman M, Felix A, Moroder L, Toniolo C, editors. Stuttgart/New York: 2004. pp. 517–533. [Google Scholar]
- 71.Powers ET, Deechongkit S, Kelly JW. In: Backbone-backbone H-bonds make context-dependent contributions to protein folding kinetics and thermodynamics: Lessons from amide-to-ester mutations. Robert LB, David B, editors. Vol. 72. Academic Press; 2005. pp. 39–78. [DOI] [PubMed] [Google Scholar]
- 72.Kato T, Mizuno H, Lee S, Aoyagi H, Kodama H, Go N, Kato T. Int J Pept Prot Res. 1992;39:485–492. [PubMed] [Google Scholar]
- 73.Scheike JA, Baldauf C, Spengler J, Albericio F, Pisabarro MT, Koksch B. Angew Chem. 2007;199:7912–7916. doi: 10.1002/anie.200702218. [DOI] [PubMed] [Google Scholar]; Scheike JA, Baldauf C, Spengler J, Albericio F, Pisabarro MT, Koksch B. Angew Chem Int Edit. 2007;46:7766–7769. doi: 10.1002/anie.200702218. [DOI] [PubMed] [Google Scholar]
- 74.Powell M, Stewart T, Otvos L, Urge L, Gaeta F, Sette A, Arrhenius T, Thomson D, Soda K, Colon S. Pharmaceut Res. 1993;10:1268–1273. doi: 10.1023/a:1018953309913. [DOI] [PubMed] [Google Scholar]
- 75.Shirahama H, Umemoto K, Yasuda H. J Biomat Sci - Polym Edit. 1999;10:621–639. doi: 10.1163/156856299x00847. [DOI] [PubMed] [Google Scholar]
- 76.Ouchi T, Nozaki T, Ishikawa A, Fujimoto I, Ohya Y. J Polym Sci Pol Chem. 1997;35:377–383. [Google Scholar]
- 77.Schaeffer A, Auld DS. Biochemistry. 1986;25:2476–2479. doi: 10.1021/bi00357a028. [DOI] [PubMed] [Google Scholar]
- 78.Sela M, Zisman E. FASEB J. 1997;11:449–456. doi: 10.1096/fasebj.11.6.9194525. [DOI] [PubMed] [Google Scholar]
- 79.Molhoek EM, van Dijk A, Veldhuizen EJA, Haagsman HP, Bikker FJ. Peptides. 2011;32:875–880. doi: 10.1016/j.peptides.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 80.Halai R, Callaghan B, Daly NL, Clark RJ, Adams DJ, Craik DJ. J Med Chem. 2011;54:6984–6992. doi: 10.1021/jm201060r. [DOI] [PubMed] [Google Scholar]
- 81.Davies JS. J Pept Sci. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 82.Li P, Roller PP. Curr Top Med Chem. 2002;2:325–341. doi: 10.2174/1568026023394209. [DOI] [PubMed] [Google Scholar]
- 83.Blackburn C, Kates SA. In: Solid-phase synthesis of cyclic homodetic peptides. Gregg BF, editor. Vol. 289. Academic Press; 1997. pp. 175–198. [DOI] [PubMed] [Google Scholar]
- 84.Kates SA, Sole NA, Albericio F, Barany G. In: Solid-phase synthesis of cyclic peptides. Basava C, Anantharamaiah GN, editors. Brikhauser; Boston: 1994. pp. 39–59. [Google Scholar]
- 85.Tugyi R, Uray K, Iván D, Fellinger E, Perkins A, Hudecz F. Proc Natl Acad Sci USA. 2005;102:413–418. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baltz RH, Miao V, Wrigley SK. Nat Prod Rep. 2005;22:717–741. doi: 10.1039/b416648p. [DOI] [PubMed] [Google Scholar]
- 87.Kopp F, Grünewald J, Mahlert C, Marahiel MA. Biochemistry. 2006;45:10474–10481. doi: 10.1021/bi0609422. [DOI] [PubMed] [Google Scholar]
- 88.Counter FT, Allen NE, Fukuda DS, Hobbs JN, Ott J, Ensminger PW, Mynderse JS, Preston DA, Wu CYE. J Antibiot. 1990;43:616–622. doi: 10.7164/antibiotics.43.616. [DOI] [PubMed] [Google Scholar]
- 89.Debono M, Abbot BJ, Molloy MR, Fukuda DS, Hunt AH, Daupert VM, Counter FT, Ott JL, Carrell CB, Howard LC, Boeck LVD, Hamill RL. J Antibiot. 1988;41:1093–1105. doi: 10.7164/antibiotics.41.1093. [DOI] [PubMed] [Google Scholar]
- 90.Houk RJT, Tobey SL, Anslyn EV. Top Curr Chem. 2005;255:199–229. [Google Scholar]
- 91.Fung M, Thornton A, Mybeck K, Wu JHH, Hornbuckle K, Muniz E. Drug Inf J. 2001;35:293–317. [Google Scholar]
- 92.Kaplowitz N. Drug Safety. 2001;24:483–490. doi: 10.2165/00002018-200124070-00001. [DOI] [PubMed] [Google Scholar]
- 93.Schuster D, Laggner C, Langer T. Curr Pharm Design. 2005;11:3545–3559. doi: 10.2174/138161205774414510. [DOI] [PubMed] [Google Scholar]
- 94.Frecer V, Ho B, Ding JL. Antimicrob Agents Chemother. 2004;48:3349–3357. doi: 10.1128/AAC.48.9.3349-3357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gomez-Martinez P, Dessolin M, Guibe F, Albericio F. J Chem Soc Perk T. 1999;1:2871–2874. [Google Scholar]
- 96.Yong YF, Kowalski JA, Lipton MA. J Org Chem. 1997;62:1540–1542. [Google Scholar]
- 97.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 98.Maestro, version 9.2, Schrödinger. LLC; New York, NY: 2011. [Google Scholar]
- 99.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 100.Still CW, Tempczyk A, Hawley RC, Hendrickson T. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.