

Abstract
Nitroxyl (HNO), the one-electron reduced and protonated congener of nitric oxide (NO), has received recent attention as a potential pharmacological agent for the treatment of heart failure and as a pre-conditioning agent to mitigate ischemia-reperfusion injury, among other possible uses. The interest in the pharmacology and biology of HNO has prompted examination (and/or re-examination) of the many of its chemical properties and has provided some clues as to how it may elicit its biological actions. In this review, a brief description of the biologically relevant chemistry of HNO is given, followed by a more in depth discussion of its pharmacology and potential toxicology.
Keywords: Nitroxyl, nitric oxide, calcitonin gene related peptide, thiols, ischemia-reperfusion, vasorelaxation
1. Introduction
The first reports of endogenous generation of nitric oxide (NO) in mammalian systems occurred almost 20 years ago [Furchgott, 1988; Ignarro et al., 1987; Palmer et al., 1987]. Since then, the physiology, pathophysiology, biochemistry and pharmacology of NO and related/derived nitrogen oxides has been the focus of intense research interest [for example, Ignarro, 2000]. Besides NO, other nitrogen oxides have garnered significant attention from the research and medical communities. Most of the research focus has been on nitrogen oxides that are oxidized relative to NO and generated in biological systems primarily via the reaction of NO with molecular oxygen or oxygen-derived species. For example, peroxynitrite (ONOO−), the reaction product of NO with superoxide (O2−), has been extensively examined as an oxidant potentially responsible for the toxicity associated with NO [for example, Pryor and Squadrito, 1995; Beckman and Koppenol, 1996]. Nitrogen dioxide (NO2) and dinitrogen trioxide (N2O3), derived from the aerobic decomposition of NO, have also been reported to be relevant to biological signaling and macromolecule modification [for example, Keshive et al., 1996; Ridnour et al., 2004; Wink and Mitchell, 1998]. More recently, nitrite (NO2−), also derived from the aerobic/biological decomposition of NO, has been postulated to be subject to reduction in mammalian systems and therefore may be a “storage” form of NO [Gladwin et al., 2005]. Interestingly, the physiology, pharmacology and biochemistry of nitrogen oxide species that are reduced relative to NO have been relatively ignored. The reasons for this are not entirely clear but likely due, in part, to the chemical predisposition of NO to react with O2 and O2−, to give oxidized species, and the preponderance of older chemical and toxicological literature that focuses on the oxidative properties of NO oxidation products. Several reports allude to the possibility that the more reduced nitrogen oxides, namely nitroxyl (HNO), hydroxylamine (NH2OH) and ammonia (NH3), among others, can be generated endogenously from NO-derived species, although the chemistry/biochemistry by which this may occur is not entirely established [for example, Singh et al., 1996; Wong et al., 1998; Spencer et al., 2003; Hausladen et al., 2001]. Over the past few years, there has been increased interest in HNO, the product of one-electron reduction and protonation of NO. Among all commonly studied nitrogen oxides (NO3−, NO2−, NO2, N2O3, ONOO−, NO, NH2OH, NH3), HNO is the most enigmatic and misunderstood [Fukuto et al, 2005a], a situation likely due to its ephemeral nature (vide infra). The current interest in HNO is due primarily to a series of recently published studies describing the extremely interesting and potentially important biological/pharmacological activity associated with HNO. Thus, this brief review focuses on the biology and pharmacology of HNO. However, prior to discussing the pharmacology of HNO, a short overview of the biologically important chemistry of HNO is given to provide some background and clues as to how HNO may elicit its biological activity. For more comprehensive treatments of HNO chemistry several other recent reviews are available [Farmer and Sulc, 2005; Miranda, 2005a; Fukuto et al., 2005a; Fukuto et al., 2005b].
2. The Chemistry of HNO
2.1 Nomenclature
The term “nitroxyl”, as used in this review, refers to the species with the molecular formula HNO. It has also been used to describe the deprotonated species, NO−. Unfortunately, this term is somewhat ambiguous as it is often used when referring to a paramagnetic species that possesses an unpaired electron associated with an N-O functionality (more commonly termed a nitroxide). In some of the chemical literature, HNO has been more appropriately called nitrosyl hydride and hydrogen oxonitrate. Regardless, the majority of the recent chemical and biological literature utilizes the term “nitroxyl” when referring to HNO.
2.2 General Properties
Although only three atoms, the chemistry of HNO is surprisingly complex. Even the simplest of all chemical reactions, deprotonation (Reaction 1), is not straightforward with HNO.
 |
(1) |
The earliest report of the acidity of HNO indicated that it was fairly good acid (pKa of 4.7) and therefore should exist primarily as the corresponding anion, NO−, at physiological pH [Gratzel et al., 1970]. However, more recent studies have shown that HNO is actually a very poor acid (pKa of 11.4) [Bartberger et al., 2002; Shafirovich and Lymar, 2002]. This means that at physiological pH (7.0) the equilibrium concentrations of HNO to NO− will be approximately 25,000 to 1, making HNO the near exclusive species present. A unique aspect of the acid-base equilibrium of HNO is that the acid and conjugate base forms have different electronic spin states. The protonated species, HNO, is an electronic singlet (all electrons are spin paired). However, the deprotonated form, NO−, has the same electronic configuration as molecular oxygen and, like O2, is a groundstate triplet (3NO−, two unpaired electrons in different orbitals and of parallel spin). The fact that the electronic groundstates for the acid and conjugate base forms of HNO are different means that protonation-deprotonation events to groundstate products require a spin state change. In other words, the deprotonation of HNO or the protonation of 3NO− is “spin-forbidden” (although other factors may also be important with regards to the forbidden nature of these processes [Shafirovich and Lymar, 2003]). Although this reaction is not strictly forbidden (as it can occur) it does mean that the rate of protonation and deprotonation are significantly slowed. This is an important aspect of HNO chemistry and indicates that even though the equilibrium ratio of HNO/3NO− is approximately 25,000/1 (at physiologically relevant pH 7), if HNO were generated in solution, deprotonation of HNO to give equilibrium concentrations of 3NO− will occur very slowly due to the spin restriction. This kinetic barrier to deprotonation of HNO may be important since it may prohibit the reactions of 3NO− in biological systems. That is, in situations where HNO is generated in solution, it would otherwise be possible to observe significant 3NO− chemistry in spite of its low concentration since 3NO− could be consumed by a fast reaction, driving the formation of more of the anion as the system tries to maintain equilibrium (a simple mass-action effect). However, with the slowed kinetics of HNO deprotonation, the likelihood of this mass-action effect is significantly diminished since HNO has a greater opportunity to react rather than deprotonate to 3NO−. Thus, HNO is the predominant species present at physiological pH (not 3NO−) and is slow to deprotonate to the anion limiting possible 3NO− chemistry (assuming 3NO− is generated from HNO deprotonation). As a corollary to this idea, if 3NO− were generated in a biological solution, it would not rapidly protonate, in spite of the thermodynamic driving force, and could have a significant lifetime.
The possible formation of HNO (or 3NO−) via one-electron reduction of NO (Reaction 2) is largely dependent on the one electron reduction potential for NO.
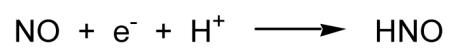 |
(2) |
It has been recently determined that the reduction potential for the proton coupled one electron reduction of NO to HNO is −0.5 to −0.6V (vs NHE) [Bartberger et al., 2002; Shafirovich and Lymar, 2002]. The reduction potential for the NO/3NO− couple is expectedly even more unfavorable (-0.8V vs NHE). Thus, the likelihood that HNO can be formed from direct one electron reduction of NO under biological conditions, even when proton coupled, seems low. However, it has been argued that at high relative concentrations of biological reductants, this may occur [Liochev and Fridovich, 2003].
One of the most prevalent and bothersome reactions of HNO is dimerization to give hyponitrous acid which further decomposes to give nitrous oxide (N2O) and water (Reaction 3) [Bazylinski and Hollocher, 1985a].
| (3) |
The existence of this reaction makes working with HNO problematic since it is unstable with respect to spontaneous dimerization and subsequent generation of N2O and H2O. Thus, if one were able to prepare a bottle of pure HNO, it would eventually turn into a bottle of N2O and water. Since there is no convenient way of working with pure HNO for extended periods of time, studies examining HNO biology are always performed using HNO-donors (vide infra).
2.3 HNO as an electrophile
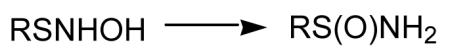
It appears that an important reactivity of HNO in biological systems is as an electrophile [Doyle et al., 1988; Bartberger et al., 2001]. However, it is somewhat selective with regards to the nucleophiles it reacts with. Theoretical examination of HNO reactivity predicts that it will not significantly hydrate (reaction with nucleophilic water) or react with alcohols [Bartberger et al., 2001]. However, it does appear to be very reactive towards thiols. The reaction with amines is predicted to be intermediate between water/alcohols and thiols. Reaction of HNO with a thiol leads to an initial formation of a putative N-hydroxysulfenamide via attack of a nucleophilic sulfur atom on the electrophilic nitrogen atom of HNO (Reaction 4). The N-hydroxysulfenamide can further react with excess thiol (or a vicinal thiol if on a protein) to give the corresponding disulfide and hydroxylamine (Reaction 5). Competing with this reaction is a process whereby the N-hydroxysulfenamide rearranges to a sulfinamide (Reaction 6) [Wong et al., 1998].
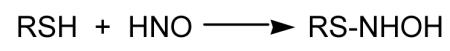 |
(4) |
| (5) |
 |
(6) |
Thus, reaction of a thiol with HNO can result in a disulfide (Reaction 5) that, under biological conditions, can be readily converted back to the starting thiol. A sulfinamide formed from the thiol-HNO reaction (Reaction 6) is much less likely to be reduced back to the thiol under biological conditions and may be considered to be an irreversible modification. However, recent studies indicate that certain protein thiols oxidized to an equivalent oxidation state, the sulfinic acid, can be reversed by specific ATP-dependent enzymes [Woo et al., 2003]. This raises the possibility that HNO-mediated sulfinamide formation (which can hydrolyze to the sulfinic acid) can also be biologically reversible with select proteins. In support of the ability of N-hydroxysulfenamides to rearrange to sulfinamides (Reaction 6), it has been reported that enzymatic reduction of S-nitroso glutathione by human aldehyde dehydrogenase generates the N-hydroxysulfenamide intermediate which then rearranges to the corresponding sulfinamide which was characterized by mass spectral analysis [Hedberg et al, 2003].
2.4 HNO Coordination chemistry
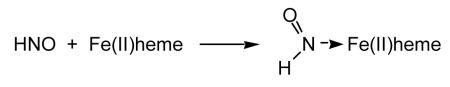
Besides reactions with thiols and thiol proteins, the other major biological target for HNO appears to be metalloproteins [for example, Farmer and Sulc, 2005]. The reaction of HNO with ferrichemes results in the generation of the ferrous-nitrosyl adduct (Reaction 7) [Doyle et al., 1988]. HNO can also react with a ferrousheme to generate the HNO-Fe(II) complex (Reaction 8) which appears to have unexpected stability (though it is reported to be unstable in the presence of O2) [Sulc et al., 2004]. HNO also can react with dioxygen-ferrousheme species giving the corresponding ferricheme, hydrogen peroxide and NO (Reaction 9) [Doyle et al., 1988].
| (7) |
 |
(8) |
| (9) |
2.5 HNO Kinetics
The biological targets for HNO will be dependent on the concentrations of the target species and the reaction rate constant for HNO and the target. Miranda and coworkers [2003] have approximated the rates of reaction of HNO with a variety of likely biological entities. They find that the rate constants for HNO reactions are as follows; oxymyoglobin (1 × 107 M−1s−1) > glutathione (GSH), horseradish peroxidase (2 × 106 M−1s−1) > N-acetyl cysteine, CuZn superoxide dismutase, Mn superoxide dismutase, metmyoglobin, catalase (3-10 × 105 M−1s−1) > tempol, ferricytochrome c (4-8 × 104 M−1s−1) > O2 (3 × 103 M−1s−1). More recently, Farmer’s group has shown that the reaction of ferrousheme proteins with HNO (Reaction 8) may also be fast (> 1.4 × 104 M−1s−1) [Sulc et al., 2004]. These kinetic studies indicate that the reaction of HNO with O2 is relatively slow and likely not of biological relevance. This is an important finding since numerous studies indicate that the HNO/O2 reaction can result in the production of a potent oxidant (vide infra). Considering the high concentrations of GSH in cells, it was postulated that scavenging of HNO by GSH may be its predominant fate in biological systems. In fact, Wink and coworkers [1998] demonstrated that high levels of HNO can deplete cells almost completely of GSH. However more recent work in yeast model systems shows that select thiol proteins can be substantially inhibited at very low HNO levels without altering GSH concentrations or leading to its oxidation [Lopez et al., 2005]. This indicates that certain thiol proteins may be significantly more reactive towards HNO and therefore can be selectively modified by HNO even in the presence of relatively high levels of GSH.
2.6 HNO oxidation chemistry
HNO, either directly or in concert with other reactive oxygen and nitrogen species, can affect the oxidation of diverse biomolecules. As mentioned above, HNO is capable of facile thiol oxidation and this oxidation is believed to take place without free radical intermediates. Organic targets of oxidation by nitroxyl may be limited to thiols and some nitrogenous moieties [Bartberger et al., 2001]. From a chemical perspective, the reaction of HNO with amines has been noted as a method to synthesize diazenes (RN=NH) [Lemal and Rave, 1965], which are unstable and liberate nitrogen gas to give a deaminated product. The reactivity of HNO with another amino acid, tryptophan, has recently been examined [Suzuki et al., 2004; Peyrot et al., 2006], but nitrosation was the ultimate outcome of that reaction. Purine DNA bases can also be oxidized by HNO under some circumstances (vide infra). Several studies have utilized common “antioxidants” to abrogate the experimental effects of nitroxyl [Ohshima et al., 1999; Fedeli et al., 2003]. We have recently completed studies detailing the kinetics of HNO reactions with physiological reductants such as ascorbate and NAD(P)H and found rate constants on the order of ~104-105 M−1s−1 for common reductants [Jackson et al., 2006]. Significantly, these rate constants are one or two orders of magnitude slower than the reaction with glutathione. Taken together it would appear that the oxidative chemistry of nitroxyl itself is rather specific for thiols, although cellular antioxidants and nitrogenous species such as amines, indoles and purines may also be targets as well.
At lower pH’s (4-6) HNO derived from Angeli’s salt has been shown to have an enhanced oxidation capacity [Ivanova et al., 2003]. This has been proposed to be due to the spontaneous liberation of hydroxyl radicals from hyponitrous acid, the dimerization product of nitroxyl (Reaction 10), but since Angeli’s salt generates increasing amounts of NO along with HNO at lower pH [Bonner and Ravid, 1975], interpretation of this system is potentially complex.
| (10) |
Although the formation of hydroxyl radicals from hyponitrous acid has been known for some time [Buchholz and Powell, 1965], the physiological relevance of this pathway is questionable as it is unlikely that concentrations of HNO could reach a level high enough to drive the dimerization reaction and generate sufficient hyponitrous acid [Miranda et al., 2003]. Based upon relative concentrations and reactivities, HNO would seem more likely to be drawn off into other reactions with thiols, metals, NO and, possibly, O2.
HNO can also mediate O2-independent oxidation chemistry via reaction with NO leading to the generation of hyponitrite radical anion (ONNO·−). Seddon and coworkers [1973] found that ONNO·− could decompose to generate HO· (Reaction 11), a reaction proposed originally by Buchholz and Powell [1965].
| (11) |
Poskrebyshev et al. [2003] reported that ONNO·− itself is a strong one-electron oxidant. In support of this, the presence of NO and HNO together has been observed to form more hydroxylated benzoate products [Ivanova et al., 2003] and more effectively inhibit the activity of a thiol dependent transcription factor [Cook et al., 2003] than HNO alone. Thus, it appears that HNO can serve as an oxidant either alone or in he presence of NO. However, the biological relevance of this chemistry remains to be determined.
Nitroxyl anion (NO−) reacts at diffusion-controlled rates with dioxygen to produce peroxynitrite, but, as discussed above, little nitroxyl anion is expected to be present in most pharmacological and experimental settings. The reaction of HNO and O2 has been studied [Miranda et al., 2001, 2002, 2005a] and its oxidative profile appears to be distinct from peroxynitrite. The product(s) of the reaction of nitroxyl and dioxygen have not been unambiguously identified however, so determination of the relevance of this pathway is awaiting further research.
3. HNO donors
3.1 Angeli’s salt
As mentioned previously, the dimerization of HNO with subsequent generation of N2O and water (Reaction 3) precludes the use of pure HNO in most chemical and, especially, biological studies. Fortunately, there are donor species that are amenable for both chemical and biological work. Herein, only a brief overview of several classes of HNO-donor is given. For a more thorough treatment of this topic, an excellent review by Miranda and coworkers [2005b] is available. The most convenient, studied and utilized HNO donor is sodium trioxodinitrate or Angeli’s salt (Na2N2O3) [Angeli, 1903; Bonner and Hughes, 1988; Bonner and Ravid, 1975]. Although fairly stable at high pH, Angeli’s salt will spontaneously decompose in neutral solution to generate one equivalent each of HNO and NO2− (Reaction 12).
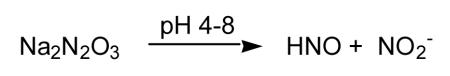 |
(12) |
Interestingly, at acidic pH, Angeli’s salt generates NO [for example, Dutton et al., 2004] and care must be taken when interpreting studies carried out under acidic or slightly acidic conditions. Moreover, the fact that one equivalent of NO2− is co-produced from Angeli’s salt decomposition may confound experimental interpretation, especially considering the recently published reports of NO2− physiology. However, control experiments using fully decomposed Angeli’s salt are typically run in most studies and will account for possible NO2− effects as well as possible impurities. Regardless, Angeli’s salt has been important in the discovery of most of the biology/pharmacology of HNO and numerous studies will be mentioned later which relied on it as the HNO source.
3.2 Piloty’s acid and derivatives
N-Hydroxysulfonamides (Piloty’s acid and its analogs and derivatives) are another class of HNO donor that has been examined in some detail (Reaction 13).
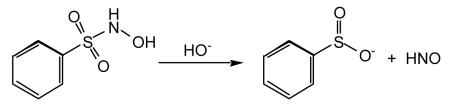 |
(13) |
One problem in the use of Piloty’s acid, as well as many of the derivative and/or related compounds, is the fact that significant HNO release typically occurs only at basic pH [Bonner and Ko, 1992]. Thus, under physiological conditions, many of these compounds do not release HNO readily and, in fact, can be oxidized and become NO donors [Zamora et al., 1995]. The pH restriction to HNO release has, therefore, precluded significant use of these compounds in biological studies.
3.3 HNO-Generating diazeniumdiolates
Although primarily known as NO donors, diazenium diolates (often referred to as “NONOates”) can be designed to generate HNO as well (Reaction 14).
| (14) |
In fact Angeli’s salt can be considered to be a type of diazenium diolate with an oxygen atom replacing the amine function (−ON(O)=NO−). NO-Releasing diazenium diolates are made using secondary amines [for example, Keefer et al., 1996]. However, diazenium diolates made with primary amines (for example, isopropyl amine) can release HNO at neutral pH and have been shown to possess biological activity akin to Angeli’s salt [Miranda et al., 2005c]. The byproduct of primary amine diazenium diolate decomposition is a nitrosamine (shown as a deprotonated tautomer in Reaction 14) [Miranda et al., 2005d; Dutton et al., 2006], a species of some toxicological concern [for example, Hecht, 1998].
3.4 Nitrosocarbonyls
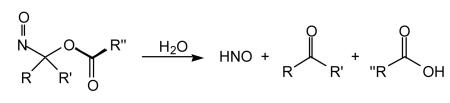
Compounds that generate nitrosocarbonyls represent another HNO donor class of compounds. The nitrosocarbonyl functionality is inherently unstable in aqueous solution as it spontaneously hydrolyzes to HNO and the corresponding carboxylic acid (Reaction 15).
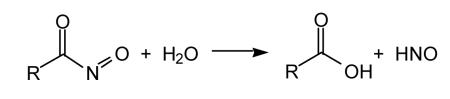 |
(15) |
In situ generation of nitrosocarbonyls can occur via retro-Diels Alder [for example, Ensley and Mahadevan, 1989; Zeng et al., 2004] or oxidation of hydroxamic acids [for example, Atkinson et al., 1996], among others [Miranda et al., 2005b and references therein]. To date, nitrosocarbonyl precursors that generate HNO in situ have not been used extensively for biological studies. However, there is no a priori reason that they could not be developed as efficient HNO-donor drugs.
3.5 Hydroxylamine
Two-electron reduction of HNO gives hydroxylamine (NH2OH). Thus, it would be expected that 2-electron oxidation of NH2OH could serve as a source of HNO. Indeed, NH2OH has been proposed as a source of HNO [Miranda et al., 2005b] and the possibility of oxidative conversion of NH2OH to HNO must always be considered when interpreting studies involving NH2OH. To date, the use of NH2OH as a precursor to HNO has not been exploited as a strategy for HNO generation in biological studies.
3.6 Cyanamide
Recent and provocative work indicating the possible utility of HNO as a treatment for heart failure and as a pre-conditioning agent to protect against ischemia-reperfusion toxicity (among other effects) has led to a rise in interest in HNO-donors as pharmacological agents (vide infra). However, it should be noted that the anti-alcoholism drug cyanamide (used in Europe, Canada and Japan as Dipsan®, Abstem®, Temposil® or Cyanamide Yoshitomi®) inhibits an enzyme in the metabolic pathway for ethanol via the bioactivation to HNO [for example, DeMaster et al., 1998]. In vivo activation of cyanamide can occur by oxidation by the enzyme catalase, leading to the generation of a fleeting intermediate, N-hydroxycyanamide, which spontaneously decomposes to release HNO and cyanide (Reaction 16).
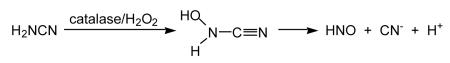 |
(16) |
3.7 Acyloxy nitroso compounds
It is important for a variety of HNO donors to be developed as replacements and/or compliments to Angeli’s salt as the primary HNO of choice for biological/pharmacological studies. This will serve to confirm that the biological activity is due to HNO, rather than other species associated with Angeli’s salt decomposition (or even the donor itself). Moreover, from a medicinal chemistry perspective, Angeli’s salt is not easily derivatized or modified. Of particular note in this regard are the acyloxy nitroso compounds recently developed by the King lab [Sha et al., 2006] (Reaction 17).
 |
(17) |
These compounds are easily synthesized from oxidation of oximes and, most importantly, appear to be capable of releasing HNO under physiological conditions. The release of HNO can be envisioned to occur either by spontaneous hydrolysis of the ester or via esterase activity. Moreover, the release rate of HNO can be varied via judicious choice of the appending R, R’ and R” groups. Clearly, the structural flexibility associated with this class of HNO-donors may prove to be important in the future development of drugs with the proper pharmacokinetics and distribution profile.
4. HNO Detection
Probably the most important (and asked) questions regarding HNO biology are: “Is HNO made endogenously and, if so, is it part of an endogenous and regulated signaling system?” Currently, there are no definitive answers to these questions. The single greatest obstacle to answering these, and other, questions regarding HNO biology is the lack of a sensitive and specific assay for HNO. Many proposed assays for HNO are either insensitive or equivocal. For example, since the dimerization/dehydration of HNO gives N2O (Reaction 3), detection of N2O has been used (primarily in chemical studies) as an indication of the presence of HNO [for example, Fukuto et al., 1992]. However, since this is a second order reaction that is highly unlikely under biological (or pharmacological) concentrations of HNO, this assay is not amenable to studies attempting to identify HNO as an endogenous species. Moreover, N2O generation can occur via mechanisms of nitrogen oxide decomposition that do not involve free HNO [For example, Yoo and Fukuto, 1995; Cho et al., 2003]. Other methods have been proposed or used for detection of HNO at high concentrations [for example, Bonner and Akhtar, 1981; Shoeman and Nagasawa, 1998; Bari et al., 2003; Bazylinski and Hollocher, 1985b] but all of these lack senstivity or specificity. More recently, several reports indicate that detection of the reaction product of HNO with thiols and/or thiol proteins can serve as a “fingerprint” for HNO [Shen and English, 2005; Donzelli et al., 2006]. Importantly, in these studies the generation of a sulfinamide product was reported (Reactions 4 and 6), a product that would be unique to HNO chemistry. Another recent report indicates that Mn(III) porphyrins may be capable of selectively binding to HNO and thus discriminate between HNO and NO [Marti et al., 2005]. It appears that progress is certainly being made in the development of tools to “hunt” for HNO in biological systems. However, to date there has yet to be any unequivocal evidence for the endogenous generation of HNO in mammalian systems.
5. HNO Pharmacology
5.1 Endogenous HNO generation?
As mentioned above, it has yet to be established that HNO is an endogenously generated signaling species, akin to other small molecule, endogenously synthesized signals like NO, H2O2 [for example, Forman et al., 2004], carbon monoxide (CO) [Kim et al., 2006] and hydrogen sulfide (H2S) [Wang, 2003]. As will be discussed below, the potency by which HNO elicits many of its pharmacological actions and the apparent selectivity it has towards some of its established biological targets would suggest that the actions of pharmacologically administered HNO are the result of existing signaling pathways that are already set up to respond to it. Moreover, it may make sense that Nature would exploit the unique and selective chemistry of HNO for some purpose considering that other nitrogen oxides are already known to be generated and used as signaling molecules. Of course, these types of arguments are spurious and contrived and only useful for providing the impetus for future studies intent on determining whether HNO is endogenously generated and/or part of normal signaling processes. Thus, although of vital importance and significant impact to the fields of small molecule signaling and nitrogen oxide biology, the issue of HNO biosynthesis will not be addressed further in this review. Instead, we focus on the fascinating and potentially important pharmacology of HNO.
5.2 HNO and Alcoholism
The first report of HNO serving as a drug was from work out of the Nagasawa lab demonstrating that the anti-alcoholic drug cyanamide was actually a prodrug for HNO. In a series of elegant and important studies, they showed that cyanamide was oxidatively bioactivated (for example, by catalase/H2O2) to generate HNO that, in turn, was responsible for the inhibition of the enzyme aldehyde dehydrogenase via modification of the active site cysteine thiolate [DeMaster et al., 1982; DeMaster et al., 1983; DeMaster et al., 1984; DeMaster et al., 1985; Nagasawa et al., 1990; Nagasawa et al., 1992; DeMaster et al., 1998]. This was one of the first indications that HNO was thiophilic and could disrupt/regulate the actions of a thiol protein. Interestingly, this drug releases cyanide ion as part of the bioactivation chemistry (Reaction 15). However, perusal of the literature indicates that cyanide-based toxicity associated with pharmacological use of cyanamide does not appear to be a problem, providing some evidence that HNO is an extremely potent inhibitor of aldehyde dehydrogenase, active at low enough concentrations to avoid cyanide poisoning. The thiophilicity of HNO has become an established biochemical property and, indeed, many of it actions may be due to interactions with thiol proteins. Considering the high concentration of GSH in all cells (1-11 mM), it may be expected that pharmacological application of an HNO donor would lead to a loss of GSH or that high levels of drug would be required to overcome scavenging by GSH. In the case of aldehyde dehydrogenase inhibition by cyanamide, this does not appear to be the case. One study found that doses of cyanamide that inhibited aldehyde dehydrogenase in cell culture did not deplete GSH [Hammond and Fry, 1999].
5.3 HNO and Thiol Proteins
As mentioned earlier, HNO has a great propensity to react with thiols and thiol proteins. In fact, based on the kinetics of the reaction of HNO with thiols, it was predicted that a major fate of HNO in biological systems would be reaction with GSH [Miranda et al., 2003] and thiol reactivity has been proposed as a means of discriminating between HNO and NO [Pino and Feelisch, 1994]. Indeed, high concentrations of Angeli’s salt (1-3 mM) caused a near complete loss of intracellular GSH in cells [Wink et al., 1998]. However, more recent work indicates that HNO can disrupt/regulate the activity of thiol proteins without significantly altering the GSH levels or the GSH/GSSG redox status of the cell. Lopez and coworkers [2005] reported that extremely low concentrations of Angeli’s salt were capable of inhibiting yeast glycolysis without lowering levels of GSH or altering the GSH/GSSG ratio. They further determined that the effect on glycolysis was likely the result of inhibition of the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a classic cysteine thiolate dehydrogenase. Thus, HNO was able to inhibit the thiol enzyme GAPDH without depleting GSH, indicating that HNO can be very selective and have specific protein targets even in the presence of huge relative concentrations of GSH. Other studies have also demonstrated the ability of HNO to react with and disrupt the actions of thiol proteins. For example, the cysteine protease papain is inhibited by HNO [Vaananen et al., 2005]. Another cysteine protease cathepsin P is also markedly inhibited at relatively low concentrations of Angeli’s salt [Vaananen et al., 2006]. Moreover, HNO appears to be a much more potent inhibitor of cathepsin B compared to other nitrogen oxides (NO and ONOO−) and capable of enzyme inhibition at levels that do not exhibit any cytotoxicity.
The disruption/regulation of thiol proteins by NO typically requires either O2, O2-derived species (e.g. O2−), or another oxidizing species (i.e. an oxidized metal) since thiols are known to be modified by more oxidized nitrogen oxide congeners like NO2, ONOO−, N2O3 or the equivalent of nitrosonium (NO+), in the case of a metal-mediated process [for example, Fukuto et al., 2000 and references therein]. (It should be noted, however, that a recent report indicates that thiols of a very specific structural motif may react directly with NO [Zhao and Houk, 2006]). On the other hand, HNO is capable of reacting directly with thiols (Reactions 4-6) as was demonstrated in an in vitro biological system when HNO was found to inhibit a thiol-containing yeast transcription factor in an oxygen-independent fashion [Cook et al., 2003]. This is an important distinction between HNO and NO as HNO does not require an “activation” event to react with thiols and the kinetics of these reactions are expected to be first order in HNO.
5.4 Vasorelaxant effects of HNO
Evidence has been accumulating over the last decade showing that HNO donors such as Angeli’s salt are powerful vasorelaxants. This well-established effect has been demonstrated in large conduit arteries [Ellis et al., 2000; Wanstall et al., 2001; Fukuto et al., 1992], in the intact pulmonary vascular bed [DeWitt et al., 2001] and in vivo in rabbits [Ma et al., 1999] and dogs [Paolocci et al, 2001; Paolocci et al., 2003]. Still, major questions concerning HNO vascular effects remain. First, HNO is thought to induce vasorelaxation both directly and through its possible conversion to NO. It is not known which mechanism is most prevalent and where one or the other takes place remains controversial. In some settings HNO-induced dilation appears to be cGMP-dependent, implying nitroxyl-to-NO conversion. Indeed, despite studies in large arteries showing HNO-mediated vasorelaxation accompanied by a rise in cGMP that was attenuated by inhibition of soluble guanylyl cyclase (sGC) [Fukuto et al. 2002], Dierks and Burstyn [1996] have suggested that NO is the only nitrogen oxide able to directly activate sGC. Moreover, it has been reported that the enzyme copper-zinc superoxide dismutase (CuZnSOD) increases the vasorelaxation by HNO via oxidative conversion of HNO to NO by the enzyme-bound cupric ion [Hobbs et al., 1994]. That endogenous copper can contribute to AS-induced vasorelaxation was further suggested by the ability of the copper chelator, diethyldithiocarmate, to produce significant inhibition of AS-relaxing effects in rat aorta [Pino and Feelisch, 1994]. More recently, Martin’s group has reported that oxidation of nitroxyl to NO by copper ions is possible. However, these investigators suggested that if endogenous copper contribute to the tissue-dependent oxidation of nitroxyl to NO, then the magnitude of this is contribution is small [Nelli et al., 2000]. Nevertheless, a number of candidates have proposed to act as intracellular oxidants to catalyze the HNO to NO conversion, including superoxide dismutase [Schimdt et al. 1996; Murphy and Sies, 1991], flavins [Fukuto et al., 1993], quinones [Buyukafsar et al., 2001] and cytochrome P450 [Nelli et al., 2001]. Thus, the HNO-to-NO conversion to produce a cGMP-dependent vasorelaxation appears biologically accessible and will be dependent on the presence and activity of these, and likely other, systems.
HNO does not elevate circulating cGMP levels when given intravenously to large mammals [Paolocci et al. 2001]. Although this seems to conflict with several of the previously mentioned studies indicating that the vasorelaxant effects of HNO can be due to elevations in cGMP, it is possible that this apparent discrepancy is due to artifacts of in vitro systems (i.e. the presence of oxidants), differences in in vivo versus in vitro levels of HNO and, perhaps, the sensitivity of the cGMP detection methods employed in the in vivo large vessel studies. On the other hand, HNO donors may use alternative mechanisms in small resistance vessels. In rat small mesenteric resistance-like arteries, Irvine and coworkers have recently shown that HNO induces relaxation that is mediated by the activation of sGC and voltage-gated K+ (Kv) channels whose opening should ultimately lead to vascular smooth muscle cell hyperpolarization [Irvine et al., 2003]. These authors also suggested that Kv activation by HNO is, at least in part, independent of cGMP accumulation.
A second aspect that needs further investigation is the apparent ability of HNO donors to substantially affect venous capacitance probably even more than the arterial compartment [Paolocci et al., 2001; Miranda et al., 2005c]. It has been proposed that HNO adminstration to large animals elicits a calcitonin gene-related peptide (CGRP) response [Paolooci et al., 2001]. Interestinlgy, CGRP does not significantly alter pre-load and lowers only peripheral vascular resistance [Katori et al, 2005]. So, it remains to be established how HNO affects the different circulatory compartments.
5.5 HNO and platelet function
In a recent, thorough investigation on human platelet function by Bermejo and coworkers, it emerged that HNO, derived from Angeli’s salt, effectively inhibits human platelet aggregation, in a rapid and concentration dependent-manner [Bermejo et al., 2005]. While the potential therapeutic relevance of these findings (i.e. in the context of atherothrombotic syndromes) is self-evident, it is interesting to note that the inhibition of platelet aggregation seen with HNO appears to be general (i.e. independent of the receptor involved in the platelet response, when compared with the NO-donor sodium nitroprusside (SNP)). Moreover, although both SNP and HNO significantly increased platelet cGMP accumulation (in the presence of the general phosphodiesterase inhibitor IBMX), L-cysteine inhibited HNO-induced cGMP accumulation but only partly blocked HNO-induced inhibition on platelet aggregation. In contrast, L-cysteine potentiated both SNP-induced cGMP accumulation and inhibitory effects on platelet aggregation. This scenario is compatible with the evidence showing that L-cysteine enhances the relaxation of aortic rings by SNP via increased NO production [Wanstall et al., 2001]. At the same time, this study confirms that even at the intravascular level, some of the effects of HNO cannot be ascribed to its conversion to NO, extending and clarifying the results previously obtained by Mondoro et al. [2001] showing that HNO inhibits aggregation in platelets from patients with sickle cell disease, and erroneously attributed to the generation of NO from HNO.
5.5 HNO and myocardial contractility
Over the last few years, the pharmacological perspective of the potential utility of HNO donors has changed. Originally, they were considered primarily useful (at least with regards to the cardiovascular system) as vasodilators. However, more recently HNO-donors have been found to be capable of profoundly affecting myocardial contractility [Wink et al., 2003]. In 1999 came the first demonstration that HNO may affect myocardial contractility in the setting of ischemia-reperfusion [Ma et al., 1999]. HNO, derived from Angeli’s salt, given to reperfused myocardium (after ischemia) is detrimental, as opposed to the beneficial effects exerted by hemodynamically equi-effective doses of the NO donor, nitrosoglutathione (GSNO) (vide infra). This paper raised the question: “How does HNO modulate myocardial properties, namely contractility, under normal and failing conditions?”. Using a pressure-volume relationship approach to dissect primary effects on the heart from changes in vascular loading conditions [Senzaki et al., 2001], it was demonstrated for the first time that Angeli’s salt-derived HNO is capable of increasing the inotropic status of the heart while hastening ventricular relaxation and unloading the heart [Paolocci et al., 2001]. One example of the effect of Angeli’s salt-derived HNO on normal canine myocardium is illustrated in Fig. 1.
Figure 1.
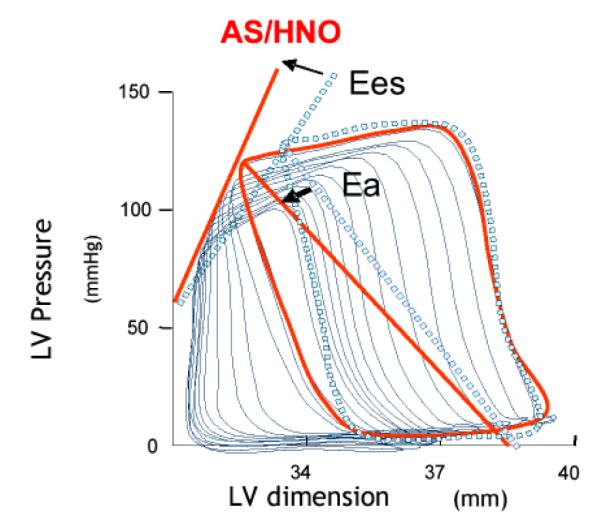
The slope relationship between end-systolic pressure and different end-diastolic volumes (Ees) gets steeper with HNO, indicating an increase in the intrinsic stiffness of the ventricle, i.e. a positive inotropic action that is independent from changes in cardiac volumes). In this example, the pre-load reduction typically seen with HNO (i.e. the shift of the P-V relation to the left) has been offset by volume repletion to demonstrate the persistence of HNO inotropy even at baseline filling volumes. As shown, peripheral vascular resistance also declined as indicated by the down-ward shift of arterial elastance (Ea in the figure). So, HNO contractile effects are load-independent (not baroreflex-mediated), and persistent during the infusion of Angeli’s salt, as indicated by the rise in Ees and in the other load-independent index of cardiac contractility (Eedd = slope of the relation between and dP/dtmax and end-diastolic volume, Fig. x B). HNO-mediated inotropy is not mimicked by an NO donor and was prevented by the HNO scavenger N-acetyl-L-cysteine (NAC). Moreover, the inotropy was not attenuated by a blocker of CGRP receptors. CGRP is a non-adrenergic/non-cholinergic (NANC) peptide endowed with several cardiovascular properties, including positive inotropy, at least in some species [Huang et al., 1999]. In a subsequent study, the effects of HNO were tested in dogs with tachypacing-induced congestive heart failure (CHF). HNO improved both contractility and relaxation, to an extent similar to that found for control dogs [Paolocci et al., 2003]. HNO inotropy was not blunted by sustained beta-adrenergic blockade, and further to this, was additive to beta-adrenergic agonists in elevating myocardial contractility. Positive inotropy, unloading cardiac action and peripheral vasodilation (to the same extent) are all desirable effects in CHF subjects. In current clinical practice, they are achieved via beta-agonists or phosphodiesterase inhibitors, to give inotropic support to the heart, and through nitrosovasodilators, to unload the heart. HNO combines both. From a mechanistic point of view, the fact that HNO infusion led to increased systemic levels of CGRP prompted the hypothesis that CGRP was the mediator of HNO positive inotropy. However, in a separate study, done in both normal and CHF dogs, Katori et al. [2005] demonstrated that CGRP inotropy is mediated by norepinephrine release from sympathetic efferent fibers, prevented by beta-blockers and blunted in CHF preparations. The latter is compatible with the down-regulation of the beta-adrenergic system in CHF [Wehrens and Marks, 2004]. Therefore, while CGRP release might, in part, explain HNO-induced vasodilation, it doesn’t account for HNO evoked inotropy.
Recently, in in vitro experiments, we [Tochetti et al., 2005] and others [Cheong et al., 2005] have shown that HNO elicits a prompt release of Ca2+ from cardiac ryanodine receptors (RyR2) and skeletal Ca2+ release channels (RyR1), respectively. In both cases, the effects were reversible upon the addition of a sulfhydryl reducing agent, dithiothreitol (DTT). HNO increased the single channel open probability of both purified RyR2 [Tocchetti et al., 2005] and RYR1 [Cheong et al., 2005]. Again, these effects were reversed upon the addition of DTT. Our preliminary data show that activation of RyR2 combined with the stimulation of sarcoplasmic reticulum (SR) Ca2+-ATPase by HNO, increasing Ca2+ re-uptake into the SR, are likely to induce and sustain positive inotropy and faster relaxation due to HNO in isolated murine myocytes [Tocchetti et al., 2005]. So, at the isolated myocyte level, Angeli’s salt-derived HNO enhances Ca2+ cycling at the SR level to increase contractility and does so in a cGMP- and cAMP-independent manner. Additional preliminary data shows that HNO is also capable of enhancing miofilament responsiveness to Ca2+, increasing myocardial contractility in rat cardiac trabecula [Dai et al., 2005]. As the increase in force development with HNO is greater than the rise in Ca2+ transients, Dai and coworkers proposed that HNO acts as a Ca2+ sensitizing agent. Altering intracellular redox conditions with DTT prevented HNO-induced augmentation in muscle force development, further confirming HNO affinity for strategically located thiols [Dai et al., 2005].
All in all, these in vitro data not only highlight some major differences in HNO signaling in the vasculature and in the cardiac muscle (i.e. cGMP-dependency) but also pave the way to further investigations aiming to identify the thiol “hot spots” that are likely to be the targets of HNO within the contractile machinery. Moreover, the fact that HNO inotropy appears to be independent of cAMP signaling makes HNO donors an ideal candidate for treating decompensated hearts, an alternative to current inotropic agents whose action is linked to a rise in intracellular cAMP levels. These agents (beta-agonists and phosphodiesterase inhibitors) not only are less effective in congestive heart failure, due to the well-known down-regulation of the beta-adrenergic system in this syndrome [Wehrens and Marks, 2004] but also when given chronically may alter intracellular Ca2+ homeostasis making the heart more prone to potentially fatal arrhythmias.
5.6 HNO and ischemic myocardium
The tissue injury (and mechanical dysfunction) occurring in the myocardium at the time in which coronary flow is restored after ischemiais known as myocardial reperfusion injury. Nitrogen oxides appears to play a major role in this setting [Jones and Bolli. 2006]. Still, some controversy persists as to whether NO is beneficial or detrimental to reperfused myocardium [Manukhina et al., 2006]. NO may protect myocardium by many ways i.e. increasing coronary flow, decreasing neutrophil accumulation, maintaining endothelial function, reducing myocardial oxygen consumption etc. [Manukhina et al., 2006]. In experimental models of infarction, drugs that are beneficial to the heart (such as statins, ACE inhibitors, and angiotensin-receptor blockers, etc.) appear to work through an enhanced NO biovailability [Jones and Bolli. 2006]. On the other hand, inhibition of NOS has been found to ameliorate myocardial reperfusion injury [Patel et al. 1993], mimicking ischemic preconditioning [Schulz and Wambolt, 1995] and suppressing nitrosative stress induced by reactive nitrogen species such as peroxynitrite [Yasmin et al., 1997]. It appears to be plausible that excess of NO can be harmful whereas lower concentrations can afford cardiac protection. Yet, altered tissue redox conditions (i.e. an imbalance between pro-oxidants and antioxidants) can also dictate when NO presence, particularly when in excess, can turn into something detrimental for the reperfused myocardium.
When compared to NO donors, the HNO releasing agent Angeli’s salt was found to exacerbate post-ischemic myocardial injury, in a dose-dependent manner [Ma et al., 1999]. No clear-cut mechanisms for the HNO-mediated exacerbation of injury were clearly identified in this study. However, one plausible explanation for HNO-induced injury can be searched in the evidence showing HNO ability to favor neutrophil accumulation [Vanuffelen et al., 1998]. As a matter of fact, in Ma’s study HNO tended to further enhance myeloperoxidase activity in the in vivo ischemic tissue. Another possible explanation is that HNO is also a powerful thiol oxidant and by this mechanism may further enhance increase in neutrophil adhesion prompted by oxygen free radical burst at reperfusion. Intriguingly, one-electron oxidation of HNO (by potassium ferricyanide), converting HNO to NO· in vitro, fully reverses the deleterious effects of HNO and leading to myocardial and endothelial protection similar to that observed with NO. Whether such conversion is likely to happen with endogenous oxidants in vivo remains unknown.
Although HNO can exacerbate ischemia-reperfusion injury, the HNO donor Angeli’s salt has also been shown to induce early preconditioning-like effects in isolated rat hearts [Pagliaro et al. 2003]. Ischemic preconditioning (IPC) is a powerful endogenous mechanism for myocardial protection against ischemia/reperfusion injury. The results of Pagliaro and coworkers showing that HNO affords myocardial protection to a degree similar to IPC but, perhaps, to a greater extent when compared to the NO donor DEA/NO hint to the possibility of some discrete signaling for HNO vs NO. In this study, HNO protection was completely prevented by NAC. Given the thiophilic nature of HNO, it is tempting to speculate that NAC prevented HNO targeting on strategically located thiols whose modification may the protective signaling cascade. What structures are targeted by HNO to afford protection is currently unknown but a recent, elegant study by Shiva and coworkers [2004] demonstrated that mitochondria are a possible target for HNO and the mechanisms of this interaction are quite distinct from those of NO or peroxynitrite, including the generation of NO, the modification of thiols and the inhibition of complexes I and II. Whether some of these mechanisms underlie HNO protection against ischemia is unclear at the moment. Though speculative, it may be possible that thiol modifications may, for instance, occur at the level of mitochondrial KATP channels [Cohen et al. 2000] or at the mitochondrial transition pore [Juhaszova et al. 2004] to trigger a protective signaling cascade. Another fascinating hypothesis is that CGRP mediates, at least in part, HNO’s ability to trigger protection. CGRP has been shown to afford protection per se [Li and Peng, 2002] and also mediate nitroglycerin-induced preconditioning effects [Hu et al. 1999].
The development of HNO donors as preconditioning agents is still in its early infancy. First, it remains to be firmly established whether other HNO donors, chemically un-related to Angeli’s, could also confer early-preconditioning-like effects. Preliminary data obtained with a newly characterized HNO donor IPA/NO appears to be consistent with what is seen with Angeli’s [Mancardi et al. 2004; Miranda et al., 2005c]. More importantly, these data highlight a potential greater ability of IPA/NO over AS to protect against irreversible myocardial injury and post-ischemic contractile dysfunction. Whether this feature is a function of an almost pure HNO donation from IPA/NO, as opposed to HNO-nitrite co-release by Angeli’s salt, is the subject of ongoing investigation. Finally, a totally unexplored area remains: the assessment of HNO releasing agents on the second window of protection, i.e. late preconditioning. Extensive literature is available showing the beneficial effects of NO, both endogenous and exogenous in this regard [Dawn and Bolli, 2002]. As for HNO, if its protective effects could be demonstrated in this setting too, then it will be a nice addition to the armamentarium of the potential beneficial cardiovascular effects already attributed to HNO.
5.7 HNO and the Nervous System
N-Methyl-D-aspartate (NMDA) receptors play a pivotal role in the regulation of neuronal communication and synaptic function in the central nervous system. HNO has been shown to interact with a thiol residue on the (NMDA) receptor leading to an attenuation of Ca2+ influx [Kim et al., 1999]. Since over-stimulation of the NMDA receptor has been implicated in the excitotoxicity associated with glutamate, the physiological outcome of this HNO action is to afford protection against glutamate-based excitotoxicity. However, Colton and coworkers reported that HNO was capable of blocking glycine-dependent desensitization of the NMDA receptor resulting in a net sensitization of the receptor [Colton et al., 2001]. Perhaps, the difference in O2 level can in part account for the divergent outcomes of these studies. However, both contributions hint to the ability of HNO to modify the activity of the NMDA receptor, either in a positive or a negative fashion.
5.8 HNO as an Antioxidant
As discussed previously, numerous studies have been published that indicate HNO and derived species can have prooxidant properties. However, the chemistry of HNO predicts that it should also function as an antioxidant. The H-NO bond dissociation energy is only about 50 kcal/mol, indicating that it should be a very good hydrogen atom donor with an ability to quench reactive radical species. Moreover, hydrogen atom abstraction from HNO results in the formation of NO, which is known to be a very good antioxidant due to its ability to also quench radical intermediates and coordinate redox metals [for example, Kaner et al, 1991; Wink et al., 1993; Hogg et al., 1993; Rubbo et al., 1994; Struck et al., 1995; Rubbo et al., 1995]. Recently, it was shown that HNO was capable of inhibiting lipid peroxidation both in yeast and in vitro model systems [Lopez et al., 2006]. It is hypothesized that HNO will inhibit free radical lipid oxidation (Reactions 18-20) by intercepting the initiating, lipid radical or lipid hydroperoxyl radical species (Reactions 21-23) with the incipient NO species acting similarly (Reactions 24-26).
| (18) |
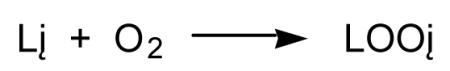 |
(19) |
| (20) |
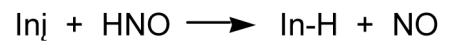 |
(21) |
| (22) |
| (23) |
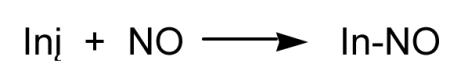 |
(24) |
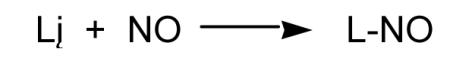 |
(25) |
| (26) |
One potentially important aspect of this study is the finding that HNO can exist and react in a lipid environment. Thus, it appears that HNO can act in both cytosolic and membrane compartments of cells. Although it is not known exactly how HNO will partition between lipid and aqueous environments, it’s structure may predict that it will partition favorably into nonpolar phases similar to other uncharged small molecules like NO and O2.
6. HNO Toxicology
As with all pharmacological agents (as well as physiological agents), there is an optimum therapeutic “window” where concentrations of the drug elicit a clinical benefit without exhibiting unbearable side effects and/or toxicity. As mentioned earlier, there is one currently used drug, cyanamide, whose mechanism of action is based on its ability to generate of HNO. Significantly, therapeutic use of cyanamide exhibits little inherent toxicity at pharmacological doses (in spite of the fact that it also generates cyanide ion!). Thus, there is no indication at this time that low levels of HNO will exhibit overt toxicity. However, considering the fact that there is only one established HNO producing drug and the development of others (vide supra) is still in its infancy, the possible toxicity of HNO from pharmacological applications is a topic of open discussion.
6.1 HNO-Mediated Thiol Depletion
One of the earliest studies of HNO toxicity was out of the Wink lab where the effects of the HNO donor Angeli’s salt on the viability of V79 fibroblasts was examined [Wink et al., 1998]. They found that HNO was capable of severely depleting cells of GSH and exhibited greater toxicity in an aerobic environment (as opposed to hypoxic conditions). Also, HNO was shown to cause double-stranded breaks in DNA. Importantly, these studies were carried out at 1-3 mM concentrations of Angeli’s salt, levels that are significantly higher than those likely attained in the previously discussed pharmacological applications. This study alludes to the possibility that HNO and O2 could react to generate an oxidant that was responsible for toxicity (vide supra).
6.2 HNO Neurotoxicity
The pro-oxidant properties of HNO have also been proposed to be responsible for the neurotoxicity of Angeli’s salt administered to rats via intranigral infusion [Vaananen et al., 2003]. In this study, Angeli’s salt was found to be neurotoxic to dopaminergic neurons with similar effects also seen in cell culture experiments. In another study using primary murine mixed cortical cells, Angeli’s salt at high concentrations (> 1 mM) was found to cause cell death [Hewett et al., 2005]. The toxicity of HNO was both acute and progressive (occurring after the dissipation of HNO) with the acute phase likely due to direct interactions of HNO with cellular components and the delayed/progressive phase due to excitotoxicity. The effect of HNO on motor and sensory neurons has also been examined. Intrathecal administration of Angeli’s salt to the lumbar spinal cord of rats resulted in motor neuron injury without effect on sensory neurons [Vaananen et al., 2004]. Significant effects on locomotor activity were seen only at the highest dose used (10 micromoles given once). Thus, there appears to be little doubt that HNO has the potential to be neurotoxic. However, at this time, it is difficult to determine exactly how HNO is toxic (i.e. what the primary cellular targets are) or whether the HNO administered at pharmacological doses (i.e. as a treatment for heart failure or as a preconditioning agent to protect from ischemia-reperfusion damage) will exhibit the type of toxicity discussed above.
6.3 HNO and Mitochondria
As species that is likely to be redox active under biological conditions [Dutton et al., 2005], it may be expected that HNO is capable of interacting with mitochondrial redox components. Although these interactions are not necessarily toxic in nature, we mention them here since the pharmacological utility of an HNO interaction with mitochondria has yet to be reported or proposed. NO has been reported to react with mitochondrial cytochrome C to produce HNO (krxn= 200 M−1 s−1) [Sharpe and Cooper, 1998]. Although the reaction is relatively slow, the HNO so formed might have functional consequences for mitochondrial activity. Bai and coworkers [2001] observed loss of mitochondrial membrane potential in the presence of Angeli’s salt. A recent study by Shiva et. al. [2004] examined the effect of Angeli’s salt-derived HNO on isolated rat liver mitochondria and demonstrated that it could be oxidized to NO by intact mitochondria. Previous studies have shown that mitochondrial components cytochrome C and ubiquinone could oxidize HNO forming NO [Buyukafsar et al., 2001]. Interestingly, under conditions where ubiquinone existed in the reduced state and could not accept electrons from HNO, conversion of HNO to NO was still observed [Shiva et al., 2004]. Consistent with the thiophilicitiy of HNO, multiple mitochondrial proteins exposed to HNO exhibited thiol modification (vide supra) when assayed via 2D gel electrophoreses, and this paralleled loss of mitochondrial activity. Another recent publication has invoked HNO in order to explain the observed inhibition of isolated rat brain mitochondra after co-treatment with a dopamine derivative and NO [Nunes et al., 2005]. The authors propose that the catechol moiety reduced NO to HNO that led to reversible inhibition of cytochrome oxidase. As noted previously, HNO has also been reported to effect the activity of another source of cellular energy, glycolysis [Lopez et al., 2005]. Interestingly, these authors didn’t observe any inhibition of mitochondrial oxygen uptake after HNO pretreatment and mitochondria deficient yeast were more susceptible to the toxic properties of HNO.
In addition to the typical respiratory components, other mitochondrial enzymes may potentially be susceptible to inhibition by HNO. The effect of HNO upon enzymes containing iron-sulfur clusters has been examined. Hughes’ group examined iron-sulfur cluster dependent hydrogen production in a bacterial cell model and showed that HNO was unique among the examined nitrogen oxides (NO2−, NO, NO+) in that it completely inhibited hydrogen production at concentrations 4 times lower than those which inhibited growth by 50% [Shaun et al., 1995]. SIN-1 was unreactive in this system, excluding the possibility of peroxynitrite mediating the observed effects. Additionally, Bastian et. al. [2002] used EPR to directly examine the reaction of HNO with iron-sulfer clusters. Aconitase was reversibly inhibited by 25% at 50 μM Angeli’s salt, and this was paralleled by a proportionate increase in EPR detectable modification of the cluster. Again, these effects were shown to be different from that of peroxynitrite.
6.4 HNO and DNA
The initial observation by Wink and coworkers [1998] of HNO-induced DNA double strand breaks in cultured fibroblasts was immediately followed by other reports of HNO-DNA interactions. Ohshima’s lab [Ohshima et al., 1999] demonstrated a time- and dose-dependent induction of HNO-mediated plasmid DNA single strand breaks which occurred more readily under acidic conditions (i.e. pH < 6) and without the need for oxygen. They also observed HNO-induced calf thymus DNA base oxidation and formation of oxidized products from oxidation of 2-deoxyribose. These effects decreased in the presence of ferricyanide, 4-hydroxy-TEMPO, hydroxyl radical scavengers, antioxidants, metal chelators, superoxide dismutase and catalase, pointing to the involvement of HNO not NO as species responsible for oxidation and alluding to the generation of hydroxyl radical (for example, Reactions 10, 11). Furthermore HNO-dependent DNA damage is enhanced in the presence of hydrogen peroxide (H2O2) and metals, suggesting that HNO also acts as a reductant generating potent oxidants derived from metal-mediated hydrogen peroxide activation [Chazotte-Aubert et al., 1999].
HNO also exhibits toxicity towards thymocytes and found to be partially decreased upon pretreatment with 4-hydroxy-TEMPO and ferricyanide (both which will convert HNO to NO) and two poly(ADP-ribose) polymerase (PARP) inhibitors, suggesting that the toxicity was due to HNO and involved PARP activation [Bai et al., 2001]. PARP, a DNA repair enzyme involved in both cell death and survival, is presumably activated via nitroxyl-induced nicks in DNA leading to PARP-dependent cell death. To be sure, the toxicity of HNO via PARP activation in thymocytes was only partially responsible for cell death as other mechanisms appear to exist as well. Interestingly, another study has shown that HNO can inhibit PARP activity in vitro, presumably via disruption of the zinc-finger domains [Sidorkina et al., 2003]. Thus, these contrasting reports indicate that the effect of HNO on PARP activation in cells may not be straightforward and dependent on experimental conditions.
7. Concluding remarks: The future of HNO as a pharmacological agent
Although an HNO-prodrug, cyanamide, is currently useful as a means for treating alcoholism, it is becoming increasingly clear that other potential therapeutic applications exist for HNO. From the shelf of the possible therapeutical uses, there is no doubt that the cardiovascular properties of HNO are among the most appealing, and, to date, the most investigated. HNO cardiotropic actions are attractive for, at least, three reasons. First, HNO appears to modulate contractility in a rather unique way that is enhancing intracellular Ca2+ cycling and miofilament sensitivity to Ca2+ without effecting Ca2+ homeostasis, at least in the short term. While future studies will test the efficacy and safety of HNO donors for treating diseased hearts, the experimental evidence collected so far holds great promise for HNO donors as an alternative to currently used cardiotropic agents. Second, exposure to small amounts of exogenous HNO may ultimately protect (precondition) the heart against ischemia-reperfusion. Finally, HNO donors not only induce vasodilation (reducing both pre- and after-load) but may also help to prevent atherosclerosis and vascular thrombosis.
Moreover, there appears to be a very distinct profile of action associated with HNO bioactivity, which is not apparent for NO. Although NO-donor drugs have now been used for over 150 years for the treatment of cardiovascular disorders such as angina pectoris and heart failure, there has been a spectacular failure in pharmacological manipulation of the NO synthetic pathway for therapeutic gain. This probably results from the ‘janus-faced’ biology of NO which holds true for a number of pathological conditions, exemplified in the cardiovascular system by ischaemic injury (e.g. myocardial infarction, stroke) and sepsis. In such cases, it appears as if there is a fine line between the cytoprotective (lower [NO]) and detrimental (higher [NO]) effects of NO, inferring that therapeutic intervention is likely to be hazardous. In marked contrast, and perhaps surprisingly, the biological targets of HNO seem to be considerably more restricted, thereby suggesting that in terms of novel therapy it is a more attractive commodity. This is also illustrated in the cardiovascular system where we and others have demonstrated the superior pharmacological activity of HNO (versus NO) in a number of animal models of disease. In addition, recent evidence suggests that HNO-donor drugs do not suffer from tachyphylaxis (at least in terms of their vasorelaxant activity), which may therefore offer an advance to organic nitrate-based therapy. It is also important to consider that many vascular and neurological disorders are associated with oxidative stress, well-characterized to result in impaired NO bioavailability/bioactivity. Under such circumstances, since the reaction of HNO with reactive oxygen-derived species (e.g. O2−) is negligible compared to NO, endogenous production or exogenous administration of HNO (or HNO donor drugs) is likely to maintain a cytoprotective influence that slows or reverses pathogenesis.
Footnotes
This review is dedicated to the career of Prof. Herbert T. Nagasawa, a pioneer in the field of HNO chemistry, biochemistry and pharmacology.
References
- Angeli A. Gazz. Chim. Ital. 1903;33(II):245. [Google Scholar]
- Atkinson RN, Storey BM, King SB. Reactions of acylnitroso compounds with amines: Production of nitroxyl (HNO) with the preparation of amides. Tet. Lett. 1996;37:9287–9290. [Google Scholar]
- Bai P, Bakondi E, Szabo E, Gergely P, Szabo C, Virag L. Partial protection by poly(ADP-ribose) polymerase inhibitors from nitroxyl-induced cytotoxity in thymocytes. Free Rad. Biol. Med. 2001;31:1616–23. doi: 10.1016/s0891-5849(01)00756-0. [DOI] [PubMed] [Google Scholar]
- Bari SE, Marti MA, Amorebieta VT, Estrin DA, Doctorovich F. Fast nitroxyl trapping by ferric porphyrins. J. Am. Chem. Soc. 2003;125:15272–15273. doi: 10.1021/ja036370f. [DOI] [PubMed] [Google Scholar]
- Bartberger MD, Fukuto JM, Houk KN. On the Acidity and Reactivity of HNO in Aqueous Solution and Biological Systems. Proc. Natl. Acad. Sci, USA. 2001;98:2194–2198. doi: 10.1073/pnas.041481598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proc. Natl. Acad. Sci., USA. 2002;99:10958–10963. doi: 10.1073/pnas.162095599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian NR, Foster MJP, Ballantyne M, Lu X. Nitrogen oxides, the good, the bad, and the ugly. Curr. Top. Biophys. 2002;26:115–127. [Google Scholar]
- Bazylinski DA, Hollocher TC. Evidence from the reaction between trioxodinitrate(II) and 15NO that trioxodinitrate(II) decomposes into nitrosyl hydride and nitrite in neutral aqueous solution. Inorg. Chem. 1985a;24:4285–4288. [Google Scholar]
- Bazylinski DA, Hollocher TC. Metmyoglobin and methemoglobin as efficient traps for nitrosyl hydride (nitroxyl) in neutral aqueous solution. J. Am. Chem. Soc. 1985b;107:7982–7986. [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide and peroxynitrite: The good the bad and the ugly. Am. J. Physiol. 1996;271(40):C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. Cell Physiol. [DOI] [PubMed] [Google Scholar]
- Bermejo E, Saenz DA, Alberto F, Rosenstein RE, Bari SE, Lazzari MA. Effect of nitroxyl on human platelets function. Thromb. Haemost. 2005;94:578–584. doi: 10.1160/TH05-01-0062. [DOI] [PubMed] [Google Scholar]
- Bonner FT, Ravid B. Thermal decomposition of oxyhyponitrite (sodium trioxodinitrate(II)) in aqueous solution. Inorg. Chem. 1975;14:558–563. [Google Scholar]
- Bonner FT, Akhtar MJ. Formation of nitrosotricyanonickelate (NiNO(CN)32−) in a direct displacement reaction. Inorg. Chem. 1981;20:3155–3160. [Google Scholar]
- Bonner FT, Hughes MN. The aqueous solution chemistry of nitrogen in low positive oxidation states. Comm. Inorg. Chem. 1988;7:215–234. [Google Scholar]
- Bonner FT, Ko Y. Kinetic, isotopic, and 15N NMR study of N-hydroxybenzenesulfonamide decomposition: An HNO source reaction. Inorg. Chem. 1992;31:2514–2519. [Google Scholar]
- Buchholz JR, Powell RE. The decomposition of hyponitrous acid. II The chain reactions. J. Am. Chem. Soc. 1965;87:2350–2353. [Google Scholar]
- Buyukafsar K, Nelli S, Martin W. Formation of nitric oxide from nitroxyl anion: role of quinones and ferricytochrome c. Br. J. Pharmacol. 2001;132:165–172. doi: 10.1038/sj.bjp.0703812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazotte-Aubert L, Oikawa S, Gilibert I, Bianchini F, Kawanishi S, Ohshima H. Cytotoxicity and site-specific DNA damage induced by nitroxyl anion (NO−) in the presence of hydrogen peroxide - Implications for various pathophysiological conditions. J. Biol. Chem. 1999;274:20909–20915. doi: 10.1074/jbc.274.30.20909. [DOI] [PubMed] [Google Scholar]
- Cheong E, Tumbev V, Abramson J, Salama G, Stoyanovsky DA. Nitroxyl triggers Ca2+ release from skeletal and cardiac sarcoplasmic reticulum by oxidizing ryanodine receptors. Cell Calcium. 2005;37:87–96. doi: 10.1016/j.ceca.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Cho JY, Dutton A, Miller T, Houk KN, Fukuto JM. Oxidation of N-hydroxyguanidines by copper(II): Model systems for elucidating the physiological chemistry of the nitric oxide biosynthetic intermediate N-hydroxy-L-arginine. Arch. Biochem. Biophys. 2003;417:65–76. doi: 10.1016/s0003-9861(03)00335-7. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: from adenosine receptor to KATP channel. Annu.Rev.Physiol. 2000;62:79–109. doi: 10.1146/annurev.physiol.62.1.79. [DOI] [PubMed] [Google Scholar]
- Colton CA, Gbadegesin M, Wink DA, Miranda KM, Espey MG, Vicini S. Nitroxyl anion regulation of the NMDA receptor. J. Neurochem. 2001;78:1126–1134. doi: 10.1046/j.1471-4159.2001.00509.x. [DOI] [PubMed] [Google Scholar]
- Cook NM, Shinyashiki M, Jackson MI, Leal FA, Fukuto JM. Nitroxyl-mediated disruption of thiol proteins: inhibition of the yeast transcription factor Ace1. Arch. Biochem. Biophys. 2003;410:89–95. doi: 10.1016/s0003-9861(02)00656-2. [DOI] [PubMed] [Google Scholar]
- Dai T, Tocchetti CG, Paolocci N, Gao WD. Increased Ca2+ responsiveness by HNO/NO− in cardiac muscle, American Heart Association 7th Scientific Sessions, November 12-16, 2005, Dallas. Circulation. 2005;112(17):U324–U324. 1359 Suppl. S. [Google Scholar]
- Dawn B, Bolli R. Role of nitric oxide in myocardial preconditioning. Ann.N.Y.Acad.Sci. 2002;962:18–41. doi: 10.1111/j.1749-6632.2002.tb04053.x. [DOI] [PubMed] [Google Scholar]
- DeMaster EG, Kaplan E, Shirota FN, Nagasawa HT. Metabolic activation of cyanamide by liver mitochondria, a requirement for the inhibition of aldehyde dehydrogenase enzymes. Biochem. Biophys. Res. Commun. 1982;107:1333–1339. doi: 10.1016/s0006-291x(82)80144-7. [DOI] [PubMed] [Google Scholar]
- DeMaster EG, Nagasawa HT, Shirota FN. metabolic activation of cyanamide to an inhibitor of aldehyde dehydrogenase in vitro. Pharmacol. Biochem. Behav. 1983;18:273–277. doi: 10.1016/0091-3057(83)90185-5. [DOI] [PubMed] [Google Scholar]
- DeMaster EG, Shirota FN, Nagasawa HT. The metabolic activation of cyanamide to an inhibitor of aldehyde dehydrogenase is catalyzed by catalase. Biochem. Biophys. Res. Commun. 1984;122:358–365. doi: 10.1016/0006-291x(84)90483-2. [DOI] [PubMed] [Google Scholar]
- DeMaster EG, Shirota FN, Nagasawa HT. Catalase mediated conversion of cyanamide to an inhibitor of aldehyde dehydrogenase. Alcohol. 1985;2:117–121. doi: 10.1016/0741-8329(85)90027-8. [DOI] [PubMed] [Google Scholar]
- DeMaster EG, redfern B, Nagasawa HT. Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem. Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- Dierks E, Burstyn J. Nitric Oxide, the Only Nitrogen Monoxide Redox Form Capable of Activating Soluble Guanylate Cyclase. Biochem. Pharmacol. 1996;51:1593–1600. doi: 10.1016/0006-2952(96)00078-0. [DOI] [PubMed] [Google Scholar]
- Donzelli S, Espey MG, Thomas DD, Mancardi D, Tocchetti CG, Ridnour LA, Paolocci N, King SB, Miranda KM, Lazzarino G, Fukuto JM, Wink DA. Discriminating formation of HNO from other reactive nitrogen oxide species. Free Rad. Biol. Med. 2006;40:1056–1066. doi: 10.1016/j.freeradbiomed.2005.10.058. [DOI] [PubMed] [Google Scholar]
- Doyle MP, Mahapatro SN, Broene RD, Guy JK. Oxidation and reduction of hemoproteins by trioxodinitrate(II). The role of nitrosyl hydride and nitrite. J. Am. Chem. Soc. 1988;110:593–599. [Google Scholar]
- Dutton AS, Fukuto JM, Houk KN. Mechanisms of HNO and NO Production from Angeli’s Salt: Density Functional and CBS-QB3 Theory Predictions. J. Am. Chem. Soc. 2004;126:3795–3800. doi: 10.1021/ja0391614. [DOI] [PubMed] [Google Scholar]
- Dutton AS, Suhrada CP, Miranda KM, Wink DA, Fukuto JM, Houk KN. Mechanism of pH-dependent decomposition of monoalkylamine diazeniumdiolates to form HNO and NO, deduced form th model compound mthylamine diazeniumdiolate, denstity functional theory and CBS-QB3 calculation. Inorg. Chem. 2006;45:2448–2456. doi: 10.1021/ic051505z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensley HE, Mahadevan S. Diels-Alder and ene reaction of nitrosyl and nitrosoformaldehyde. Tet. Lett. 1989;30:3255–3258. [Google Scholar]
- Farmer PJ, Sulc F. Coordination chemistry of the HNO ligand with hemes and synthetic coordination complexes. J. Inorg. Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Fedeli D, Damiani E, Greci L, Littarru GP, Falcioni G. Nitroxide radicals protect against DNA damage in rat epithelial cells induced by nitric oxide, nitroxyl anion and peroxynitrite. Mutat. Res. 2003;535:117–25. doi: 10.1016/s1383-5718(02)00296-6. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Fukuto J, Torres M. Redox Signaling - Chemistry Defines Which Reactive Oxygen and Nitrogen Species Can Act as Second Messengers. Am. J. Physiol. (Cell Physiology) 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Wallace GC, Hszieh R, Chaudhuri G. Chemical oxidation of N-hydroxyguanidine compounds. Biochem. Pharmacol. 1992;43:607–613. doi: 10.1016/0006-2952(92)90584-6. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Hobbs AJ, Ignarro LJ. Conversion of Nitroxyl (HNO) to Nitric Oxide in Biological Systems: The Role of Physiological Oxidants and Relevance to the Biological Activity of HNO. Biochem. Biophys. Res. Commun. 1993;196:707–713. doi: 10.1006/bbrc.1993.2307. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Cho JY, Switzer CH. The Chemical Properties of Nitric Oxide and Related Nitrogen Oxides, Chapt 2. In: Ignarro LJ, editor. Nitric Oxide Biology and Pathobiology. Academic Press; San Diego: 2000. pp. 23–40. [Google Scholar]
- Fukuto JM, Bartberger MD, Dutton AS, Paolocci N, Wink DA, Houk KN. The Physiological Chemistry and Biological Activity of Nitroxyl (HNO): The Neglected, Misunderstood and Enigmatic Nitrogen Oxide. Chem. Res. Toxicol. 2005a;18:790–801. doi: 10.1021/tx0496800. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Dutton AS, Houk KN. The Chemistry and Biology of Nitroxyl (HNO): A Chemically Unique Species with Novel and Important Biological Activity. ChemBioChem. 2005b;6:612–619. doi: 10.1002/cbic.200400271. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. Studies on relaxation of rabbit aorta by sodium nitrite: the basis for the proposal that the acid-activatable factor from bovine retractor penis is inorganic nitrite and the endothelium-derived relaxing factor is nitric oxide. In: Vanhoutte PM, editor. Vasodilatation: Vascular Smooth Muscle, Peptides, Autonomic Nerves, and Endothelium. Raven Press; New York, NY: 1988. pp. 401–414. [Google Scholar]
- Gladwin MT, Schechter AN, Kim_shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO, Kelm M, Wink DA, Espey MG, Oldfield EH, Pluta RM, Freeman BA, Lancaster JR, Jr., Feelisch M, Lundberg JO. The emerging biology of nitrite anion. Nature Chem. Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. [DOI] [PubMed] [Google Scholar]
- Gratzel M, Taniguchi S, Henglein A. A pulse radiolytic study of short-lived byproducts on nitric oxide reduction in aqueous solution. Ber. Bunsenges. Phys. Chem. 1970;74:1003–1010. [Google Scholar]
- Hammond AH, Fry JR. Effect of cyanamide on toxicity and glutathione depletion in rat hepatocyte cultures: differences between two dichloropropanol isomers. Chemico-Biol. Interact. 1999;122:107–115. doi: 10.1016/s0009-2797(99)00118-0. [DOI] [PubMed] [Google Scholar]
- Hausladen A, Gow A, Stamler JS. Flavohemoglobin denitrosylase catalyzes the reaction of a nitroxyl equivalent with molecular oxygen. Proc. Natl. Acad. Sci., USA. 2001;98:10108–10112. doi: 10.1073/pnas.181199698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- Hedberg JJ, griffiths WJ, Nilsson SJF, Hoog J-O. Reduction of S-nitrosoglutathione by human alcohol dehydrogenase 3 is an irreversible reaction as analyzed by electrospray mass spectrometry. Eur. J. Biochem. 2003;270:1249–1256. doi: 10.1046/j.1432-1033.2003.03486.x. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Espey MG, Uliasz TF, Wink DA. Neurotoxicity of nitroxyl: Insights into HNO and NO biochemical imbalance. Free Rad. Biol. Med. 2006;39:1478–1488. doi: 10.1016/j.freeradbiomed.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Hobbs AJ, Fukuto JM, Ignarro LJ. Formation of Free Nitric Oxide from L-Arginine by Nitric Oxide Synthase and Enhancement of Generation by Superoxide Dismutase. Proc. Natl. Acad. Sci., USA. 1994;91:10992–10996. doi: 10.1073/pnas.91.23.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg N, Kalyanaraman B, Joseph J, Struck A, Parthasarathy S. Inhibition of low-density lipoprotein oxidation by nitric oxide. FEBS Lett. 1993;334:170–174. doi: 10.1016/0014-5793(93)81706-6. [DOI] [PubMed] [Google Scholar]
- Hu CP, Li YJ, Deng HW. The cardioprotective effects of nitroglycerin-induced preconditioning are mediated by calcitonin gene-related peptide. Eur. J. Pharmacol. 1999;369:189–194. doi: 10.1016/s0014-2999(99)00050-3. [DOI] [PubMed] [Google Scholar]
- Huang MH, Knight PR, III, Izzo JL., Jr. Ca2+-induced Ca2+ release involved in positive inotropic effect mediated by CGRP in ventricular myocytes. Am. J. Physiol. 1999;276:R259–R264. doi: 10.1152/ajpregu.1999.276.1.R259. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, editor. Nitric Oxide Biology and Pathobiology. Academic Press; San Diego: 2000. [Google Scholar]
- Irvine JC, Favaloro JL, Kemp-Harper BK. NO− activates soluble guanylate cyclase and Kv channels to vasodilate resistance arteries. Hypertension. 2003;41:1301–1307. doi: 10.1161/01.HYP.0000072010.54901.DE. [DOI] [PubMed] [Google Scholar]
- Ivanova J, Salama G, Clancy RM, Schor NF, Nylander KD, Stoyanovsky DA. Formation of nitroxyl and hydroxyl radical in solutions of sodium trioxodinitrate: effects of pH and cytotoxicity. J. Biol. Chem. 2003;278:42761–8. doi: 10.1074/jbc.M305544200. [DOI] [PubMed] [Google Scholar]
- Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J. Mol. Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner J, Harel S, Granit R. Nitric oxide as an antioxidant. Arch. Biochem. Biophys. 1991;289:130–136. doi: 10.1016/0003-9861(91)90452-o. [DOI] [PubMed] [Google Scholar]
- Katori T, Hoover DB, Ardell JL, Helm RH, Belardi DF, Tocchetti CG, Forfia PR, Kass DA, Paolocci N. Calcitonin gene-related peptide in vivo positive inotropy is attributable to regional sympatho-stimulation and is blunted in congestive heart failure. Circ.Res. 2005;96:234–243. doi: 10.1161/01.RES.0000152969.42117.ca. [DOI] [PubMed] [Google Scholar]
- Keefer LK, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: Convenient nitric oxide dosage forms. Meth. Enzymol. 1996;268:281–292. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- Keshiive M, Singh S, Wishnok JS, Tannenbaum SR, Deen WM. Kinetics of S-nitrosation of thiols in nitric oxide solutions. Chem. Res. Toxicol. 1996;9:988–993. doi: 10.1021/tx960036y. [DOI] [PubMed] [Google Scholar]
- Kim WK, Choi YB, Rayudu PV, Das P, Asaad W, Arnelle DR, Stamler JS, Lipton SA. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO−. Neuron. 1999;24:461–469. doi: 10.1016/s0896-6273(00)80859-4. [DOI] [PubMed] [Google Scholar]
- Kim HP, Ryter SW, Choi AMK. CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006;46:411–49. doi: 10.1146/annurev.pharmtox.46.120604.141053. [DOI] [PubMed] [Google Scholar]
- Lemal DM, Rave TW. Diazenes from Angeli’s Salt. J. Am. Chem. Soc. 1965;87:393–394. [Google Scholar]
- Li YJ, Peng J. The cardioprotection of calcitonin gene-related peptide-mediated preconditioning. Eur.J.Pharmacol. 2002;442:173–177. doi: 10.1016/s0014-2999(02)01538-8. [DOI] [PubMed] [Google Scholar]
- Liochev SI, Fridovich I. The mode of decomposition of Angeli’s salt (Na2N2O3) and the effects thereon of oxygen, nitrite, superoxide dismutase and glutathione. Free Rad. Biol. Med. 2003;34:1399–1404. doi: 10.1016/s0891-5849(03)00111-4. [DOI] [PubMed] [Google Scholar]
- Lopez BE, Rodriguez CE, Pribadi M, Cook N, Shinyashiki M, Fukuto JM. Selective Inhibition of Yeast Glycolysis by Nitroxyl (HNO): Implications to HNO Pharmacology/Physiology. Arch. Biochem. Biophys. 2005;442:140–148. doi: 10.1016/j.abb.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Lopez BE, Shinyashiki M, Han TH, Fukuto JM. The antioxidant and/or oxidant actions of NO and HNO on yeast supplemented with polyunsaturated fatty acids. Nitric Oxide Biol. Chem. 2006:P027. (abstract), A26. [Google Scholar]
- Ma XL, Gao F, Liu GL, Lopez BL, Christopher TA, Fukuto JM, Wink DA, Feelisch M. Opposite effects of nitric oxide and nitroxyl on postischemic myocardial injury. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14617–14622. doi: 10.1073/pnas.96.25.14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manukhina EB, Downey HF, Mallet RT. Role of nitric oxide in cardiovascular adaptation to intermittent hypoxia. Exp. Biol. Med. (Maywood.) 2006;231:343–365. doi: 10.1177/153537020623100401. [DOI] [PubMed] [Google Scholar]
- Maraj SR, Khan S, Cui X-Y, Cammack R, Joannou CL, Hughes MN. Interactions of Nitric Oxide and Redox-related species with biological targets. Analyst. 1995;120:699–703. [Google Scholar]
- Marti MA, Bari SE, estrin DA, Doctorovich F. Discrimination of nitroxyl and nitric oxide by water-soluble Mn(III) porphyrins. J. Am. Chem. Soc. 2005;127:4680–4684. doi: 10.1021/ja044632n. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Espey MG, Yamada K, Krishna M, Ludwick N, Kim S, Jourd’heuil D, Grisham MB, Feelisch M, Fukuto JM, Wink DA. Unique oxidative mechanisms for the reactive nitrogen oxide species, nitroxyl anion. J. Biol. Chem. 2001;276:1720–1727. doi: 10.1074/jbc.M006174200. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Yamada K, Espey MG, Thomas DD, DeGraff W, Mitchell JB, Krishna MC, Colton CA, Wink DA. Further evidence for distinct reactive intermediates from nitroxyl and peroxynitrite: effects of buffer composition on the chemistry of Angeli’s salt and synthetic peroxynitrite. Arch. Biochem. Biophys. 2002;401:134–144. doi: 10.1016/S0003-9861(02)00031-0. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Fukuto JM, Wink DA. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc. Natl. Acad. Sci., USA. 2003;100:9197–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KM. The chemistry of nitroxyl (HNO) and implications in biology. Coord. Chem. Rev. 2005a;249:433–455. [Google Scholar]
- Miranda KM, Nagasawa HT, Toscano JP. Donors of HNO. Curr. Top. Med. Chem. 2005b;5:649–664. doi: 10.2174/1568026054679290. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Katori T, Torres de Holding CL, Thomas L, Ridnour LA, McLendon WJ, Cologna SM, Dutton AS, Champion HC, Mancardi D, Tocchetti CG, Saavedra JE, Keefer LK, Houk KN, Fukuto JM, Kass DA, Paolocci N, Wink DA. Comparison of the NO and HNO donating properties of diazeniumdiolates: primary amine adducts release HNO in vivo. J. Med. Chem. 2005c;48:8220–8228. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Dutton AS, Ridnour LA, Foreman CA, Ford E, Paolocci N, Katori T, Tocchetti CG, Mancardi D, Thomas DD, Espey MG, Houk KN, Fukuto JM, Wink DA. Mechanism of aerobic decomposition of Angeli’s salt (sodium trioxodinitrate) at physiological pH. J. Am. Chem. Soc. 2005d;127:722–731. doi: 10.1021/ja045480z. [DOI] [PubMed] [Google Scholar]
- Mondoro TH, Ryan BB, Hrinczenko BW, Schechter AN, Vostal JG, Alayash AI. Biological action of nitric oxide donor compounds on platelets from patients with sickle cell disease. Br. J. Haematol. 2001;112:1048–1054. doi: 10.1046/j.1365-2141.2001.02623.x. [DOI] [PubMed] [Google Scholar]
- Murphy ME, Sies H. Reversible conversion of nitroxyl anion to nitric oxide by superoxide dismutase. Proc. Natl. Acad. Sci., USA. 1991;88:10860–10864. doi: 10.1073/pnas.88.23.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa HT, DeMaster EG, Redfern B, Shirota FN, Goon DJW. Evidence for nitroxyl in the catalase-mediated bioactivation of the alcohol deterrent agent cyanamide. J. Med. Chem. 1990;33:3120–3122. doi: 10.1021/jm00174a001. [DOI] [PubMed] [Google Scholar]
- Nagasawa HT, Lee MJC, Kwon CH, Shirota FN, DeMaster EG. An N-hydroxylated derivative of cyanamide that inhibits yeast aldehyde dehydrogenase. Alcohol. 1992;9:349–353. doi: 10.1016/0741-8329(92)90031-5. [DOI] [PubMed] [Google Scholar]
- Nelli S, Hillen M, Buyukafsar K, Martin W. Oxidation of nitroxyl anion to nitric oxide by copper ions. Br. J. Pharmacol. 2000;131:356–362. doi: 10.1038/sj.bjp.0703550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelli S, McIntosh L, Martin W. Role of copper ions and cytochrome P450 in the vasodilator actions of the nitroxyl generator, Angeli’s salt, on rat aorta. Eur. J. Pharmacol. 2001;412:281–289. doi: 10.1016/s0014-2999(00)00845-1. [DOI] [PubMed] [Google Scholar]
- Nunes C, Almeida L, Laranjinha J. Synergistic inhibition of respiration in brain mitochondria by nitric oxide and dihydroxyphenylacetic acid (DOPAC): Implications for Parkinson’s disease. Neurochem. Int. 2005;47:173–182. doi: 10.1016/j.neuint.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Ohshima H, Gilibert I, Bianchini F. Induction of DNA strand breakage and base oxidation by nitroxyl anion through hydroxyl radical production. Free Rad. Biol. Med. 1999;26:1305–1313. doi: 10.1016/s0891-5849(98)00327-x. [DOI] [PubMed] [Google Scholar]
- Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolooci N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Rad. Biol. Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- Palmer RMJ, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature (London) 1987;327:524–526. doi: 10.1038/327524a0. 1987. [DOI] [PubMed] [Google Scholar]
- Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc.Natl.Acad.Sci.U.S.A. 2001;98:10463–10468. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolocci N, Katori T, Champion HC, St. John ME, Miranda KM, Fukuto JM, Wink DA, Kass DA. Positive inotropic and lusitropic effects of HNO/NO− in failing hearts: Independence from β-adrenergic signaling. Proc. Nat. Acad. Sci., USA. 2003;100:5537–5542. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VC, Yellon DM, Singh KJ, Neild GH, Woolfson RG. Inhibition of nitric oxide limits infarct size in the in situ rabbit heart. Biochem Biophys Res Commun. 1993;194:234–238. doi: 10.1006/bbrc.1993.1809. [DOI] [PubMed] [Google Scholar]
- Peyrot F, Fernandez BO, Bryan NS, Feelisch M, Ducrocq C. N-Nitroso Products from the Reaction of Indoles with Angeli’s Salt. Chem. Res. Toxicol. 2006;19:58–67. doi: 10.1021/tx050253b. [DOI] [PubMed] [Google Scholar]
- Pino RZ, Feelisch M. Bioassay discrimination between nitric oxide (NO) and nitroxyl (NO−) using cysteine. Biochem. Biophys. Res. Commun. 1994;201:54–62. doi: 10.1006/bbrc.1994.1668. [DOI] [PubMed] [Google Scholar]
- Poskrebyshev GA, Shafirovich V, Lymar SV. Hyponitrite radical, a stable adduct of nitric oxide and nitroxyl. J. Am. Chem. Soc. 2004;126:891–899. doi: 10.1021/ja038042l. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Squadrito GL. The chemistry of peroxynitrite: A product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 1995;268(12):L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. Lung Cell Mol. Physiol. [DOI] [PubMed] [Google Scholar]
- Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen species. Putting perspective on stressful biological situations. Biol. Chem. 2004;385:1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. J. Biol. Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- Rubbo H, Parthasarathy S, Barnes S, Kirk M, Kalyanaraman B, Freeman BA. Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation: Termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arch. Biochem. Biophys. 1995;324:15–25. doi: 10.1006/abbi.1995.9935. [DOI] [PubMed] [Google Scholar]
- Schmidt HHHW, Hofman H, Schindler U, Shutenko ZS, Cunningham DD, Feelisch M. No NO from NO synthase. Proc. Natl. Acad. Sci., USA. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R, Wambolt R. Inhibition of nitric oxide synthesis protects the isolated working rabbit heart from ischaemia-reperfusion injury. Cardiovasc. Res. 1995;30:432–439. [PubMed] [Google Scholar]
- Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001;15:1718–1726. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- Sha X, Isabell S, Patel RP, Day CS, King s. B. Hydrolysis of acyloxy nitroso compounds yields nitroxyl (HNO) J. Am. Chem. Soc. 2006 doi: 10.1021/ja062365a. published on web 07/12/2006. [DOI] [PubMed] [Google Scholar]
- Shafirovich V, Lymar SV. Nitroxyl and its anion in aqueous solutions: Spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc. Natl. Acad. Sci., USA. 2002;99:7340–7345. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafirovich V, Lymar SV. Spin-forbidden deprotonation of aqueous nitroxyl (HNO) J. Am. Chem. Soc. 2003;125:6547–6552. doi: 10.1021/ja034378j. [DOI] [PubMed] [Google Scholar]
- Sharpe MA, Cooper CE. Reactions of nitric oxide with mitochondrial cytochrome c: a novel mechanism for the formation of nitroxyl anion and peroxynitrite. Biochem J. 1998;332:9–19. doi: 10.1042/bj3320009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, English AM. Mass spectrometric analysis of nitroxyl-mediated protein modification: Comparison of products formed with free and protein-based cysteines. Biochemistry. 2005;44:14030–14044. doi: 10.1021/bi0507478. [DOI] [PubMed] [Google Scholar]
- Shiva SCJ, Ramachandran A, Ceaser EK, Hillson T, Brookes PS, Patel RP, Darley-Usmar VM. Mechanisms of the interaction of nitroxyl with mitochondria. Biochem J. 2004;379:359–366. doi: 10.1042/BJ20031758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoeman DW, Nagasawa HT. The reaction of nitroxyl (HNO) with nitrosobenzene gives cupferron (N-nitrosophenylhydroxylamine) Nitric Oxide Biol. Chem. 1998;2:66–72. doi: 10.1006/niox.1998.0166. [DOI] [PubMed] [Google Scholar]
- Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Inhibition of Poly(ADP-ribose) polymerase (PARP) by nitric oxide and reactive nitrogen oxide species. Free Rad. Biol. Med. 2003;35:1431–1438. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Singh SP, Wishnok JS, Keshive M, Deen WM, Tannenbaum SR. The chemistry of the S-nitrosoglutathione/glutathione system. Proc. Natl. Acad. Sci., USA. 1996;93:14428–14433. doi: 10.1073/pnas.93.25.14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer NY, Patel NK, Keszler A, Hogg N. Oxidation and nitrosylation of oxyhemoglobin by S-Nitrosoglutathione via nitroxyl anion. Free Rad. Biol. Med. 2003;35:1515–1526. doi: 10.1016/j.freeradbiomed.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Struck AT, Hogg N, Thomas JP, Kalyanaraman B. Nitric oxide donor compounds inhibit the toxicity of oxidized low-density lipoprotein to endothelial cells. FEBS Lett. 1995;361:291–294. doi: 10.1016/0014-5793(95)00178-c. [DOI] [PubMed] [Google Scholar]
- Sulc F, Immoos CE, Pervitsky D, Farmer PJ. Efficient trapping of HNO by deoxyhemoglobin. J. Am. Chem. Soc. 2004;126:1096–1101. doi: 10.1021/ja0376184. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Mower HF, Friesen MD, Gilibert I, Sawa T, Ohshima H. Nitration and nitrosation of N-acetyl--tryptophan and tryptophan residues in proteins by various reactive nitrogen species. Free Rad. Biol. Med. 2004;37:671–681. doi: 10.1016/j.freeradbiomed.2004.05.030. [DOI] [PubMed] [Google Scholar]
- Tocchetti CG, Wang W, Valdivia HH, Aon MA, Katori T, Zaccolo M, O’Rourke B, Froehlich JP, Cheng H, Paolocci N. Nitroxyl Anion: A novel thiol-sensitive positive inotrope that enhances SR Ca2+ release and uptake in murine cardiomyocytes, American Heart Association 7thScientific Sessions, November 12-16, 2005, Dallas. Circulation. 2005;112(17):U363–U363. 1553 Suppl. S. [Google Scholar]
- Vaananen AJ, Liebkind R, Kankuri E, Liesi P, Rauhala P. Angeli’s salt and spinal motor neuron injury. Free Rad. Res. 2004;38:271–282. doi: 10.1080/10715760410001659764. [DOI] [PubMed] [Google Scholar]
- Vaananen AJ, Kankuri E, Rauhala P. Nitric oxide-related species-induced protein oxidation: Reversible, irreversible and protective effects on enzyme function of papain. Free Rad. Biol. Med. 2005;38:1102–1111. doi: 10.1016/j.freeradbiomed.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Vaananen AJ, Salmenpera P, Hukkanen M, Rauhala P, Kankuri E. Cathepsin B is a differentiation-resistant target for nitroxyl (HNO) in THP-1 monocyte/macrophages. Free Rad. Biol. Med. 2006;41:120–131. doi: 10.1016/j.freeradbiomed.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Vanuffelen BE, Van Der ZJ, De Koster BM, Vansteveninck J, Elferink JG. Intracellular but not extracellular conversion of nitroxyl anion into nitric oxide leads to stimulation of human neutrophil migration. Biochem.J. 1998;330(Pt 2):719–722. doi: 10.1042/bj3300719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. The gasotransmitter role of hydrogen sulfide. Antiox. Redox Signal. 2003;5:493–501. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- Wanstall JC, Jeffery TK, Gambino A, Lovren F, Triggle CR. Vascular smooth muscle relaxation mediated by nitric oxide donors: a comparison with acetylcholine, nitric oxide and nitroxyl ion. Br. J. Pharmacol. 2001;134:463–472. doi: 10.1038/sj.bjp.0704269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Marks AR. Molecular determinants of altered contractility in heart failure. Ann.Med. 2004;36(Suppl 1):70–80. doi: 10.1080/17431380410032481. [DOI] [PubMed] [Google Scholar]
- Wink DA, Hanbauer I, Krishna MC, DeGraff W, Gamson J, Mitchell JB. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc. Nat. Acad. Sci., USA. 1993;90:9813–9817. doi: 10.1073/pnas.90.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wink DA, Mitchell JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Rad. Biol. Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Wink DA, Feelisch M, Fukuto J, Christodoulou D, Jourd’heuil D, Grisham M, Vodovotz V, Cook JA, Krishna M, DeGraff W, Kim S, Gamson J, Mitchell JB. The cytotoxicity of nitroxyl: Possible implications for the pathophysiological role of NO. Arch. Biochem. Biophys. 1998;351:66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- Wink DA, Miranda KM, Katori T, Mancardi D, Thomas DD, Ridnour L, Espey MG, Feelisch M, Colton CA, Fukuto JM, Pagliaro P, Kass DA, Paolocci N. Orthogonal properties of the redox siblings nitroxyl and nitric oxide in the cardiovascular system: a novel redox paradigm. Am. J. Physiol. (Heart Circ. Physiol.) 2003;285:H2264–H2276. doi: 10.1152/ajpheart.00531.2003. [DOI] [PubMed] [Google Scholar]
- Wong PSY, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, Nagasawa HT. Reaction between S-nitrosothiols and thiols: Generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- Woo HA, Chae HZ, Hwang SC, Yang KS, Kim K, Rhee SG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:592–594. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- Yasmin W, Strynadka KD, Schulz R. Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts. Cardiovasc Res. 1997;33:422–432. doi: 10.1016/s0008-6363(96)00254-4. [DOI] [PubMed] [Google Scholar]
- Yoo J, Fukuto JM. Oxidation of N-hydroxyguanidine by nitric oxide and possible generation of vasoactive species. Biochem. Pharmacol. 1995;50:1995–2000. doi: 10.1016/0006-2952(95)02098-5. [DOI] [PubMed] [Google Scholar]
- Zamora R, Grzesiok A, Weber H, Feelisch M. Oxidative release of nitric oxide accounts for guanylate cyclase stimulating. Vasodilator activity of Piloty’s acid: A comparison with Angeli’s salt. Biochem. J. 1995;312:333–339. doi: 10.1042/bj3120333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng B-B, Huang J, Wright MW, King SB. Nitroxyl (HNO) release from new functionalized N-hydroxyurea-derived acyl nitroso-9,10-dimethylanthracene cycloadducts. Bioorg. Med. Chem. Lett. 2004;14:5565–5568. doi: 10.1016/j.bmcl.2004.08.062. [DOI] [PubMed] [Google Scholar]
- Zhao Y-L, Houk KN. Thionitroxides, RSNHO·: The structure of a the SNO moiety in “S-Nitrosohemoglobin”, a possible NO reservoir and transporter. J. Am. Chem. Soc. 2006;128:1422–1423. doi: 10.1021/ja057097f. [DOI] [PMC free article] [PubMed] [Google Scholar]