

Abstract
Unlike alcohols, the reaction of C-nucleophiles with 2-O-benzyl-4,6-O-benzylidene-protected gluco- and mannopyranosyl thioglycosides is highly stereoselective providing the α-C-glycosides in the gluco-series and the β-C-glycosides in the manno-series. Conformational analysis of nucleophilic attack of putative intermediate glycosyl oxocarbenium ions suggests that the observed selectivities for C-glycoside formation can be explained by preferential attack on the opposite face of the oxocarbenium to the C2-H2 bond and that eclipsing interactions with this bond are the main stereodetermining factor. It is argued that the steric interactions in the attack of alcohols (sp3-hybridized O) and of typical carbon-based nucleophiles (sp2 C) on oxocarbenium ions are very different, with the former being less severe, and thus that there is no a priori reason to expect O- and C-glycosylation to exhibit parallel stereoselectivities for attack on a given oxocarbenium ion.
Introduction
C-Glycosides are important mimics of O-glycosides and accordingly numerous avenues have been developed for their formation among which, as with O-glycoside formation, conceptually the easiest and the most direct involve the Lewis acid-mediated attack of carbon-based nucleophiles on putative glycosyl cations or their equivalents.1 Several years ago prompted by computational results2 implicating donor-acceptor hydrogen bonding in the 4,6-O-benzylidene-directed β-mannosylation3 and α-glucsoylation3 reactions developed in our laboratories, we investigated the stereoselectivity of C-glycosylation reactions of 4,6-O-benzylidene-protected mannosyl and glucopyranosyl donors.4 We found that for 2,3-di-O-benzyl-4,6-O-benzylidene-protected gluco- and mannopyranosyl donors the same stereochemical trends were observed for the formation of O- and C-glycosides, namely the preferential formation of the α-isomers in the gluco- series and that of the β-isomers in the manno-series.4 Moreover, the same reversal of stereoselectivity was observed in the manno- series on replacing a 3-O-benzyl ether in the donor by a 3-O-carboxylate ester for both O-5 and C-6 glycoside formation. We concluded that there was likely a commonality of mechanism between the formation of the O- and C-glycosides, at least for the examples studied, and that donor-acceptor hydrogen bonding is likely not a major stereodirecting factor in the formation of the O-glycosides.4,6 Continuing the parallel between O-and C-glycosylation stereoselectivity, more recently we found that the N-acetyl-4-O,5-N-oxazolidinone-protected sialyl donors exhibit a strong equatorial preference in the formation of C-glycosides7 just as they were earlier shown to do for the O-glycosides.8 The presence of a 3-O-benzyl ether or related group is critical to the successful stereochemical outcome of 4,6-O-benzylidene-directed β-O- mannosylations and α-O-glucosylations as the corresponding 3-deoxy donors are far less selective (Fig. 1).9
Figure 1.

Influence of the 3-O-benzyl ether on glycoside formation in the gluco- and manno- series.
Accordingly, we anticipated a comparable loss of stereoselectivity when studying the formation of the 4,6-O-benzylidene-3-deoxy-C-glycosides in both the gluco- and manno- series and were therefore surprised to find high selectivity in both instances. These unexpected observations, and the further reflections on the analogies between C- and O-glycoside formation that they prompt, form the basis of this Article.
Results
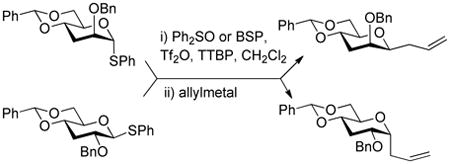
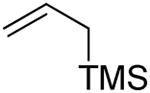
The 2,3-di-O-benzyl-4,6-O-benzylidene protected glycosyl donors 111 and 212 were prepared by known methods, as were the 3-deoxy analogs 3 and 4.9 Activation of 1 and 2 with the BSP and/or DPSO reagents3a,13 and triflic anhydride in the presence of the hindered non-nucleophilic base TTBP3a at −65 °C in dichloromethane was followed by the addition of allyltrimethylsilane, allyltributylstannane, or α-trimethylsiloxystyrene leading to the formation of the anticipated C-glycosides.
The results of these initial experiments are presented in Table 1 and, with the exception of minor differences in selectivity for entries 1 and 2,14 confirmed our previous observations on the selectivity of C-glycoside formation; namely that the β-C-glycosides are formed selectively in the mannose series and the α-C-glycosides in the glucose series. One noteworthy observation in this series of experiments was the formation of the double adduct 9 in the gluco-series (Table 1, entry 6) to which we return later.
Table 1.
Stereoselective C-glycoside formation in the gluco- and manno- series.
| ||||
|---|---|---|---|---|
| Entry | Donor | Nucleophile | Product | Yield |
| 1 | 1 |
|
|
57% |
| 2 | 1 |

|
|
60% |
| 3 | 1 |
|
|
76% |
| 4 | 2 |
|
|
56% |
| 5 | 2 |

|
|
58% |
| 6 | 2 |
|
|
40% |
| 40% | ||||
The same reaction conditions were then applied to the 3-deoxy donors 3 and 4, with the exception that in the activation of the 3-deoxy mannosyl donor 3 the BSP reagent was replaced by diphenyl sulfoxide as this combination was found to give cleaner activation in this series. The results of these addition reactions in the 3-deoxy series are presented in Table 2. Again, a double adduct was formed as a significant byproduct in the reaction of the gluco donor with the silyl enol ether (Table 2, entry 6).
Table 2.
Stereoselective C-glycoside formation in the 3-deoxy gluco- and manno- series.
| ||||
|---|---|---|---|---|
| Entry | Donor | Nucleophile | Product | Yield |
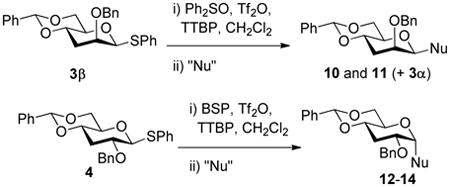
| 1 | 3 |
|
|
35% |
| 17% | ||||
| 2 | 3 |

|
|
58% |
| 3 | 3 |
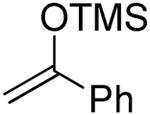
|
|
75% |
| 4 | 4 |
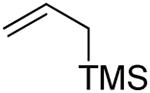
|
|
60% |
| 5 | 4 |

|
|
63% |
| 6 | 4 |
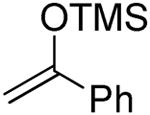
|
|
34% |
| 37% | ||||
The parallels between Tables 1 and 2 are obvious and indicate that, unlike the situation for O-glycosylation, the presence or absence of a benzyloxy group at the 3-position of the donor has little or no effect on the stereoselectivity of C-glycosylation with the three standard nucleophiles employed. With the manno-stereochemistry at C2, β-C-glycosides are formed whereas with the gluco-stereochemistry at the same position α-C-glycosides are formed. Formulated another way the nucleophile enters preferentially cis- to the C2-O2 bond in both stereochemical series.
Formation of Double Adducts
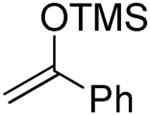
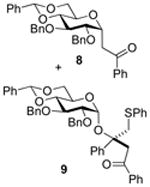
The formation of the double adducts 9 and 14 (Table 1 and 2, entries 6) as significant byproducts is interesting as it implies that the silyl enol ether is an ambident nucleophile and that attack via oxygen to form the vinyl glycosides15 15 and 16 competes with attack via carbon to form the C-glycosides 8 and 13, at least in the gluco- and deoxy gluco- series. Related vinyl glycosides derived from the ambident nucleophilicity of pinacolone trimethylsilyl enol ether were isolated in our initial study of C-glycoside formation, when they were also more significant in the gluco- than in the manno-series, and when they persisted in the reaction mixture presumably due to the steric bulk of the t-butyl group.4 Adducts 9 and 14 must be formed via the attack of a sulfenyl cation equivalent, a byproduct of the thioglycoside activation process, on 15 and 16, respectively, followed by quenching of the so-formed oxocarbenium ions 17 and 18, respectively, by a second equivalent of the silyl enol ether. Precedent for the reaction of other O-glycosyl oxocarbenium ions related to 17 and 18 with a variety of nucleophiles in preference to the obvious decomposition pathway to give glycosyl cations and a carbonyl compound, in the present case β-phenylthioacetophenone, is found on studies on the formation and coupling reactions of phenylthiomethyl glycosides.16 We tentatively assign adducts 9 and 14, both of which are formed with high selectivity (only one isomer was isolated in each case), the (R)-stereochemistry indicated on the grounds that of the two conformations of the vinyl glycosides possible, assuming the operation of the exo-anomeric effect, one is strongly favoured by the minimization of steric interactions between the aglycone and the anomeric hydrogen. Sulfenylation of this conformer will then afford the E-oxocarbenium ion, whereas sulfenylation of the minor isomer would provide the Z-isomer. Assuming the preference for an exo-anomeric effect-like conformation about the glycosidic bond, which minimizes steric interactions with the C2 benzyloxy group, attack on the exposed face of the E-isomer affords the assigned stereochemistry of 9 and 14 (Scheme 1). This model is consistent with that applied to rationalize inter alia diastereoselective Diels-Alder reactions of 1-glucopyanosyloxy dienes.17
Scheme 1.

Rationale for the stereoselective formation of 9 and 14.
We also note the formation of an inverted thioglycoside 3α from thioglycoside 3β in the 3-deoxy manno- series (Table 2, entry 1). As this phenomenon was only observed to a significant extent in the one example when the external nucleophile was the weakest among those employed in this study,18 we assume it arises from competing nucleophilic attack by thiol released in the course of the activation reaction on the glycosyl oxocarbenium intermediate. As the transition states for the formation of an S-glycoside are necessarily considerably different to those for the formation of a C-glycoside, being in principle more like but still looser than those for O-glycoside formation (vide infra); there is no reason, a priori, for C- and S-glycoside formation to exhibit the same face selectivity on a common oxocarbenium ion intermediate.
Mechanisms of O- and C-Glycoside Formation
The formation of O-glycosides is usually interpreted in terms of a continuum of mechanisms spanning the two extremes of pure SN1 and SN2 reactions, with most reactions considered to involve more or less tightly associated glycoyl oxocarbenium ion/counterion pairs depending on the protecting groups and reaction conditions employed.19 Kinetic enzymologists refer to exploded transition states to describe associative SN2-like transition states that nevertheless have considerable oxocarbenium ion character and longer partially formed or cleaved bonds than in simple SN2 transition states;20 such exploded transition states can be viewed as alternative representations of reactions occurring via nucleophilic attack on tight or intimate ion pairs. Direct spectroscopic evidence for the existence of glycosyl oxocarbenium ions as discrete intermediates in organic solution (or aqueous) media has yet to be obtained,19f,21 but indirect evidence exists in the form of glycosylation reactions exhibiting unimolecular kinetics in certain cases.19a,22 At the other end of the mechanistic continuum, a number of O-glycosylation reactions exhibiting bimolecular kinetics have recently been demonstrated.22c,23
With regard to the 4,6-O-benzylidene-directed O-glycosylations relevant to the work presented here, 13C primary kinetic isotope effect (KIE) measurements supported by DFT calculations indicate that the formation of the β-manno, β-gluco, and α-glucosides take place via associative mechanisms involving exploded transitions states in which the incoming alcohol displaces triflate from a glycosyl triflate of opposite configuration.24 On the other hand the 13C KIE measurements indicate that 4,6-O-benzylidene protected α-mannopyranosides are formed by a dissociative mechanism that approaches the intermediacy of a glycosyl oxocarbenium ion.24
With respect to the formation of the C-glycosides, the reaction of C-centered nucleophiles with putative glycosyl oxocarbenium ions was first identified as a viable entry into C-glycosides in 1973 by Hanessian and coworkers in the furanoside series.25 The method gained considerable momentum following the work of Kishi and co-workers who used allyltrimethylsilane as nucleophile in the pyranoside series and who interpreted their results in terms of pseudoaxial attack on a half-chair conformation of the intermediate glycosyl oxocarbenium ion.26 In more recent years, the Woerpel laboratory has studied extensively the reaction of a variety of glycosyl donors with allylsilanes, stannanes and silyl enol ethers,first in the deoxyfuranoside series,27 and subsequently in the pyranoside series,28 with the results interpreted in terms of nucleophilic attack on oxocarbenium ion intermediates. From these studies a series of conclusions emerged according to which i) glycosyl oxocarbenium ions adopt conformations which place electronegative substituents (C-O bonds) at all but the 2-position pseudoaxial when they afford the maximum through-space stabilization to the positively charged center – a conclusion that is corroborated by the extensive work of the Bols group on the differential stabilization of piperidinium cations by electronegative substituents in axial or equatorial bonds sites29 – and by important contributions from numerous other groups30 ii) that stereoelectronic control prevails in the attack of nucleophiles on glycosyl cations that adopt the half-chair conformation and leads preferentially to products in the chair conformation with the nucleophile in an axial position; iii) that in cases of steric hindrance to pseudoaxial axial attack on the lowest energy conformer of the glycosyl cation Curtin-Hammett kinetics come into play and pseudoaxial attack on less populated conformers may dominate; and iv) that as the reaction rate approaches the diffusion controlled limit in cases involving strong nucleophiles stereoelectronic control is eroded.
Regarding stereochemical parallels between O- and C-glycoside formation, in their initial work using a 2,3,5-tri-O-benzoyl protected ribofuranosyl donor and alkenes as nucleophiles Hanessian and co-workers noted the formation of the 1,2-trans-product (β-C-d-ribofuranoside) consistent with the stereochemical picture for O-glycoside formation.25 Subsequently Giannis and Sandhoff, adapting Kishi's allylsilane method to per-O-acetyl β-d-gluco and galactopyranose, found 1:1 α:β-mixtures of the C-glycosides on reaction in dichloromethane but very high α-selectivity in acetonitrile; a result that is clearly at odds with the stereochemical expectations for O-glycoside formation from the same donor.31 In contrast, in a later paper BeMiller and co-workers reported the reaction of per-O-acetyl-β-d-galactopyranose with allyltrimethylsilane and BF3OEt2 in nitromethane to give the β-C-galactoside.32 An unclear picture therefore emerges with respect to possible analogies between O- and C-glycoside formation, particularly with regard to what are typically considered participating protecting groups.
Conformational Analysis of Nucleophilic Attack on 4,6-O-Benzylidene-Protected Glycosyl Oxocarbenium Ions
The presence of the 4,6-O-benzylidene acetal group severely limits the range of conformations available to glycosyl oxocarbenium ions; in particular it excludes the possibility of “inverted” conformations such as the 3H4 half-chair.33 Computational work from the Whitfield laboratory2 corroborated by more recent computations in support of our primary 13C kinetic isotope effect studies24 suggests that any 4,6-O-benzylidene-protected mannopyranosyl oxocarbenium ion will adopt the B2,5 conformation. This conformation puts the C2-O2 bond in the minimally hindered bowsprit position and the C3-O3 bond in a pseudo-axial position from which it achieves maximum stabilization of the oxocarbenium ion. In the Stoddard projection33a of the pseudorotational itinerary for pyranoside rings the B2,5 conformation is flanked by the OS2 and 1S5 conformers to which any B2,5 oxocarbenium ion necessarily deforms following attack on its α- and β-faces, respectively. Therefore, adopting the oxocarbenium ion model for the formation of C-glycosides, the selective formation of the β-C-mannopyranosides is most reasonably explained by β-face attack on the B2,5 conformation of the oxocarbenium ion, in spite of the developing 1,3-diaxial interaction with the C3-O3 bond in the necessarily early transition state,34 leading initially to 1S5 conformer of the product in which the newly formed bond adopts a pseudoaxial position (Scheme 2). The alternative possibility of α-face attack, placing the isomeric product initially in the OS2 twist boat conformation with the newly formed bond pseudo-equatorial, suffers from a severe torsional interaction between the incoming nucleophile and the to the C2-H2 bond in the early transition state for this exothermic addition. In the 3-deoxy series, despite the absence of the through-space stabilization afforded to the cation by any C3-O3 bond, the B2,5 conformation with the sterically undemanding bowsprit 2-O-benzyl ether remains the more stable conformation of the oxocarbenium ion on which β-face attack is now all the more favoured because of the absent of a C3-substituent.
Scheme 2.

Nucleophilic attack on the B2,5 conformation of the 4,6-O-benzylidene-protected mannopyranosyl (X = OBn) and 3-deoxymannopyranosyl (X = H) oxocarbenium ions.
In the 4,6-O-benzylidene protected glucopyranosyl oxocarbenium ion, the B2,5 conformation is no longer favoured owing to the highly unfavourable positioning of the C2-O2 bond in a flagpole position. The preference is instead for the 4H3 half chair that retains the pseudoequatorial C2-O2 bond and upon which attack from the α-face in a pseudoaxial direction leads directly to the α-products observed in the preferred 4C1 chair conformation (Scheme 3). The alternative mode of attack on the β-face of the 4H3 cation leading to the β-product in a 1S3 twist boat conformation is disfavored by a torsional interaction between the incoming nucleophile and the pseudoaxial C2-H2 bond (Scheme 3). 1,3-Diaxial interactions between the incoming nucleophile and pseudoaxial C-H bonds exist for attack on either face of the 4H3 cation (H3 or H4) and as such are not considered to play a major role in face selectivity. As the C3 substituent is pseudoequatorial in the 4H3 half chair and does not hinder approach of the nucleophile, its removal is not expected to have a major influence on the stereoselectivity of reactions involving the 4,6-O-benzylidene protected glucopyranosyl cation.
Scheme 3.

Nucleophilic attack on the 4H3 conformation of the 4,6-O-benzylidene-protected glucopyranosyl (X = OBn) and 3-deoxyglucopyranosyl (X = H) oxocarbenium ions.
Overall, the stereoselectivity of C-glycoside formation in the 4,6-O-benzylidene series is adequately explained by nucleophilic attack on the preferred conformations of the intermediate glycosyl oxocarbenium ions, B2,5 for mannose and 4H3 for glucose, in such a way as to avoid torsional interactions between the incoming nucleophile and the C2-H2 bond in the necessarily early transition states. These torsional interactions, which are indicated in Figures 2 and 3, are directly comparable to those advanced by Boons and by Ito to rationalize the β-selectivity of arabinofuranosylation with donors carrying a 3,5-O-di-t-butylsilylene acetal or 3,5-O-(tetraisopropyl)disiloxane group.35
Figure 2.

Newman projections about C1-C2 showing alternative modes of attack on the 4,6-O-benzyidene-protected mannosyl and glucosyl oxocarbenium ions, indicating torsional interactions present in the disfavored modes.
Figure 3.

Representations of the transition states for antiperiplanar attack by π-type C-nucleophiles, and alcohols on the disfavored face of the glycosyl oxocarbenium ions (cf Figure 2, α-for manno- and β-for gluco) highlighting the influence of the C2-H2 bond. Of the two unhindered staggered conformations available for O-glycoside formation only the one taking account of any developing exo-anomeric effect is illustrated.
Discussion
If the oxocarbenium mechanism for C-glycoside formation is adopted along with the conformational analyses of Schemes 2 and 3, it is necessary to explain why the 2,3-di-O-benzyl-4,6-O-benzylidene protected mannopyranosyl oxocarbenium ion is β-selective toward the C-nucleophiles whereas it is moderately α-selective24 toward alcohols. Toward an understanding of this change in selectivity we note the difference in steric size between alkoxy and vinyl groups apparent in the steric A values, with the vinyl group being a simple model for the allyl-type C-nucleophiles employed in this study. The methoxy group has A value between 0.55 and 0.75 depending on the source and the t-butoxy group has the A value 0.75; the vinyl group on the other hand is unambiguously bigger with an A value of between 1.49 and 1.68.36 For related transition states with a comparable degree of advancement, those for the formation of C-glycosides will therefore necessarily be subject to greater steric hindrance than those for the formation of O-glycosides. Moreover the transition state requirements for C-glycoside formation with attack by π-electron-density and, we assume on the basis of standard models,37 antiperiplanar approach of the alkene to the oxocarbenium ion C=O bond are more rigid than those for O-glycoside formation which involve the use of electron density from an sp3 hybrid orbital and in which the alkyl moiety of the alkoxy group can orient itself away from the bulk of the oxocarbenium ion. Simply stated, in general alcohols will be less discerning than π-type C-nucleophiles in their attack on oxocarbenium ions and therefore predictably less selective. In particular, the different transition state requirements for O- and C-glycoside formation suggest that the eclipsing interactions with the C2-H2 suggested to be important in determining face selectivity in C-glycoside formation will be less important in O-glycoside formation leading to lower and perhaps even inverted selectivity (Fig 3).
We cannot completely exclude another possibility that invokes α-O-mannoside formation via the less populated 4H3 conformer of the oxocarbenium ion on which pseudoaxial attack on the α-face leads directly to the product in the 4C1 conformation (Scheme 4). The transition state for such a mode of attack can be considered to benefit from the developing anomeric stabilization of the nascent C-O bond. However, as the transition state for this exothermic reaction step is early and cation-like the stabilization gained from the anomeric effect can only be small, as persuasively argued recently by Cumpstey34 and previously by Sinnott.38 Accordingly, we do not consider it necessary to invoke such a pathway for α-O-mannoside formation.
Scheme 4. Alternative pathway for the formation of α-O-mannopyranosides.

Regarding the 3-deoxy series and the loss of selectivity in O-glycosylation but not in C-glycosylation, the models presented for C-glycosylation in Schemes 1 and 2 are adequate and no further discussion is required, but the same cannot be said for the O-glycosides. Computational work indicates that the selective formation of the β-O-mannopyranosides in the 4,6-O-benzylidene acetal protected series may be assisted by hydrogen bonding between the incoming acceptor and O3 of the donor,2a,2d,24 although it has most recently been suggested that such hydrogen bonds are an artifact of the computational method.39 Clearly such hydrogen bonds are not possible in the 3-deoxy series and any energetic contribution that they may have provided toward selectivity in the normal gluco- and manno- series will be lost leading to lower selectivity. A comparable argument cannot be advanced to explain the loss of α-selectivity in the 4,6-O-benzylidene protected glucopyranose series as the computations predict hydrogen bonding between the incoming acceptor and O2 of the donor in the formation of the α-glucoside24 and self-evidently this can persist in the 3-deoxy series. An alternative, perhaps more plausible explanation, invokes a change in mechanism away from associative and toward dissociative mechanisms for the complete set of O-glycoside formations owing to the removal of the electron-withdrawing C3-O3 bond and its destabilizing effect on the glycosyl oxocarbenium ion such as we have discussed previously.3 In such a hypothesis the differences in selectivity between O- and C-glycoside formation will arise from the less stringent transition state requirements for attack by alcohols than by C-nucleophiles as discussed above. The possibility that the 2-O-benzyl ether is less sterically bulky in the 3-deoxy series than in the fully substituted systems owing to a loss a buttressing interactions,40 is a valid consideration but one that is unlikely to be the origin of the diminished selectivity of O-glycoside formation in the 3-deoxy series as smaller protecting groups at the 2-position have been previously found to afford greater rather than lower β-selectivity in mannosylation.3 Finally, in an elegant study, Beaver and Woerpel have demonstrated that the simple alcohol ethanol attacks both faces of the 3,4,6-tri-O-benzyl glucopyranosyl oxocarbenium ion in an unselective manner and have provided evidence that is the consequence of a reaction taking place near the diffusion controlled limit when the selectivity simply reflects the relative probability of encounter of the two faces.28b Owing to the presence of usually two electron-withdrawing β-C-O bonds and their far greater steric bulk the nucleophilicity of typical carbohydrate alcohols is expected to be somewhat lower than that of ethanol and, thus, the probability of such a diffusion-controlled model operating for the 3-deoxy-O-glycosides small.
Conclusion
The study of C-glycoside formation in the 3-deoxy-4,6-O-benzylidene protected manno- and glucopyranoside series reveals no change in selectivity from the prototypical 3-O-benzyl series, an observation which is in contrast to O-glycoside formation where selectivity depends critically on the presence of a benzyloxy-type protecting group at the 3-position. These results are best understood in terms of subtle changes in mechanism between the formation of the O- and C-glycosides related to the different steric requirements for the attack of sp2 and sp3 hybridized nucleophiles on oxocarbenium ions.
Experimental Section
General
Glycosyl donors 1,11 2,12 3,9 and 4.9 were prepared by the literature methods. High resolution mass spectrometric measurements were made using an electrospray source coupled to a time of flight mass analyzer.
General Procedure for Glycosylation Using the BSP or DPSO/TTBP/Tf2O Systems
To a stirred solution of 0.05 M solution of donor, BSP or DPSO (1.2 equiv), TTBP (1.5 equiv) and 4 Å molecular sieves in dichloromethane was added at −65 °C Tf2O (1.2 equiv). The resulting mixture was stirred for 0.75 h before a solution of the acceptor (5 equiv) in dichloromethane (0.20 M) was slowly added. The resulting mixture was stirred at − 65 °C for 3 to 12 h. The reaction was quenched at − 65 °C by adding a saturated solution of NaHCO3. The temperature was allowed to warm to room temperature. The mixture was taken up in dichloromethane and filtered through Celite before phase separation. The organic phase was washed with a saturated solution of NaHCO3, dried over Na2SO4, filtered and concentrated. Purification by column chromatography eluting with hexanes/ethyl acetate 15:1 unless otherwise stated afforded the C-glycosides.
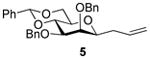
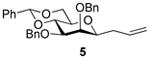
2,3-O-Di-benzyl-4,6-O-benzylidene-1-allyl-1-deoxy-β-d-mannopyranose (5)4
Compound (5) was prepared from 1 (100 mg) by the general glycosylation procedure and was obtained (50-52 mg) in 57 to 60% yield as a white solid: mp = 80.9 – 90.5 °C; [α]RTd – 26.0 (c = 1.0 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.52 (dd, J = 1.5, 7.5 Hz, 2H), 7.42 – 7.29 (m, 13H), 5.72 – 5.63 (m, 2H), 5.10 – 5.02 (m, 3H), 4.93 (d, J = 12.0 Hz, 1H), 4.77 (d, J = 12.5 Hz, 1H), 4.70 (d, J = 11.5 Hz, 1H), 4.30 – 4.24 (m, 2H), 3.86 (t, J = 10.0 Hz, 1) 3.81 (d, J = 2.0 Hz, 1H), 3.74 (dd, J = 2.5, 9.5 Hz, 1H), 3.46 (t, J = 7.0 Hz, 1H), 3.39 (dt, J = 5.0, 10.0 Hz, 1H), 2.47 (dt, J = 6.5, 14.0 Hz, 1H), 2.26 (dt, J = 7.0, 14.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 138.6, 138.4, 137.7, 134.3, 128.8, 128.5, 128.4, 128.3, 128.2, 127.7, 127.6, 127.5, 126.0, 117.5, 101.3, 80.7, 79.6, 79.5, 76.3, 75.0, 73.1, 72.0, 68.7, 35.5, 29.7; ESIHRMS calcd for C30H32O5Na [M + Na]+ 495.2147, found 495.2142.
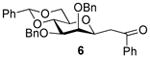
2,3-Di-O-benzyl-4,6-O-benzylidene-1-deoxy-1-(2-oxo-2-phenylethyl)-β- d-mannopyranose (6) 4
Compound (6) was prepared from 1 (100 mg) by the general glycosylation procedure and obtained in 76% yield (77 mg) as a colorless oil: [α]RTd– 10.4 (c = 0.75 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 7.0 Hz, 2H), 7.59 – 7.51 (m, 3H), 7.46 – 7.34 (m, 8H), 7.32 – 7.25 (m, 4H), 7.17 – 7.09 (m, 3H), 5.65 (s, 1H), 5.02 (d, J = 11.5 Hz, 1H), 4.96 (d, J = 12.5 Hz, 1H), 4.81 (d, J = 12.5 Hz, 1H), 4.61 (d, J = 12.0 Hz, 1H), 4.26 (ddt, J = 1.5, 6.5, 9.5, 2H), 4.15 (t, J = 6.0 Hz, 1H), 4.05 (d, J = 1.5 Hz, 1H), 3.88 (dd, J = 3.0, 9.5 Hz, 1H), 3.83 (t, J = 10.0 Hz, 1H), 3.48 (dt, J = 5.0, 10.0 Hz, 1H), 3.24 (dd, J = 5.5, 17.5 Hz, 1H), 3.13 (dd, J =7.5, 17.5 Hz, 1H); C NMR (125 MHz, CDCl3) δ 197.2, 138.5, 138.0, 137.7, 136.5, 133.3, 120.9, 128.8, 128.5, 128.4, 128.3, 128.2, 128.1, 127.8, 127.7, 127.6, 126.1, 101.4, 80.4, 79.4, 75.9, 75.6, 75.0, 73.3, 71.8, 68.6, 39.6; ESIHRMS calcd for C35H34O6Na [M + Na]+ 573.2252, found 573.2255.
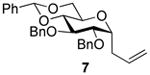
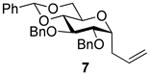
2,3-O-Di-benzyl-4,6-O-benzylidene-1-allyl-1-deoxy-α-d-glucopyranose (7)4
Compound (7) was prepared from 2 (100 mg) by the general glycosylation procedure and obtained in 56 to 58% yield (49-51 mg) as a white solid: mp = 90.8 – 91.2 °C; [α]RTd- 0.9 (c = 0.75 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.50 (dd, J = 1.5, 8.0 Hz, 2H), 7.41 – 7.27 (m, 13H), 5.81 – 5.73 (m, 1H), 5.13 (dd, J = 1.5, 17.0 Hz, 1H), 5.09 (d, J = 10.0 Hz, 1H), 4.92 (d, J = 11.0 Hz, 1H), 4.78 (d, J = 12.0 Hz, 1H), 4.64 (d, J = 11.5 Hz, 1H), 4.26 – 4.21 (m, 1H), 4.08 (dd, J = 5.5, 8.0 Hz, 1H), 3.90 - 3.86 (m, 1H), 3.76 (dd, J = 5.5, 8.5 Hz, 1H), 3.70 – 3.64 (m, 3H), 2.54 (t, J = 7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 138.6, 138.1, 137.4, 134.3, 128.4, 128.3, 128.2, 127.9, 127.8, 127.7, 127.6, 126.0, 117.2, 101.1, 82.7, 79.4,78.7, 74.9, 74.8, 73.6, 69.4, 63.4, 30.7; ESIHRMS calcd for C30H32O5Na [M + Na]+ 495.2147, found495.2138.
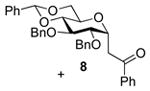
2,3-Di-O-benzyl-4,6-O-benzylidene-1-(2-oxo-2-phenylethyl)-α-d-glucopyranose (8) and [(1R)-2-Oxo-1,2-diphenyl-1-(phenylthiomethyl)ethyl] 2,3-di-O-benzyl-4,6-O-benzylidene-α-d- glucopyranoside (9)
Compounds (8)4 and (9) were obtained from 2 (100 mg) by the general glycosylation procedure. Purification by column chromatography eluting with hexanes/ethyl acetate 15:1 to 8:1 gave compounds 8 (40 mg) and 9 (40 mg) both in 40% yield.
Compound (8) was isolated as a colorless oil, [α]RTd + 17.6 (c = 0.75 M, CHCl3); 1H NMR (500 MHz, CDCl3)δ 7.90 (d, J = 7.5 Hz, 2H), 7.56 (dd, J = 7.0, 8.0 Hz, 1H), 7.51 – 7.43 (m, 4H), 7.40 – 7.35 (m, 5H), 7.34 – 7.26 (m, 8H), 5.57 (s, 1H), 4.96 – 4.93 (m, 2H), 4.82 (d, J = 12.0 Hz, 1H), 4.73 (d, J = 11.5 Hz, 1H), 4.63 (d, J = 17.6 Hz, 1H), 4.21 (dd, J = 3.0, 9.5 Hz, 1H), 3.91 – 3.83 (m, 2H), 3.72 – 3.64 (m, 3H), 3.50 – 3.46 (m, 1H), 3.29 (dd, J = 7.5, 16 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 197.2, 138.5, 137.7, 137.3, 137.0, 133.2, 128.9, 128.6, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 126.0, 101.2, 82.4, 78.8, 78.7, 74.8, 73.8, 72.0, 69.3, 64.7, 36.0; ESIHRMS calcd for C35H34O6Na [M + Na]+573.2253, found 573.2251.
Compound (9) was isolated as a colorless oil, [α]RTd + 28.0 (c = 0.25 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 7.0 Hz, 2H), 7.57 - 7.15 (m, 27H), 7.05 (t, J = 7.5 Hz, 1H), 5.46 (s, 1H), 5.08 (d, J = 3.5 Hz, 1H), 4.84 (d, J = 11.5, 1H), 4.74 (d, J = 11.0 Hz, 1H), 4.62 (d, J = 11.5 Hz, 1H), 4.50 (d, J = 12.0 Hz, 1H), 4.18 - 4.11 (m, 2H), 4.01 (d, J = 8.0 Hz, 1H), 3.98 (d, J = 12.0 Hz, 1H), 3.88 - 3.82 (m, 2H), 3.70 (d, J = 14.0 Hz, 1H), 3.50 - 3.43 (m, 2H), 3.67 (dd, J = 3.5, 9.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 195.8, 141.3, 138.9, 138.2, 137.6, 137.5, 137.1, 133.0, 129.5, 128.9, 128.6, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 127.4, 127.2, 126.1, 126.0, 101.2, 93.9, 82.7, 82.5, 79.6, 78.4, 75.2, 73.8, 68.9, 63.2, 45.9, 43.1, 29.7; ESIHRMS calcd for C49H46O7SNa [M + Na]+ 801.2862, found 801.2852.
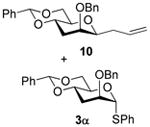
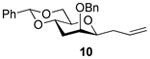
2-O-Benzyl-4,6-O-benzylidene-1-allyl-1,3-dideoxy- β-d-mannopyranose (10)
Compound (10) was prepared from 3 (80 mg) by the general glycosylation procedure and obtained in 35% to 59% yield (19-40 mg) as a colorless oil: [α]RTd + 1.7 (c = 0.75 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.49 (dd, J = 2.0, 8.0 Hz, 2H), 7.40 - 7.28 (m, 8H), 5.76 - 5.67 (m, 1H), 5.57 (s, 1H), 5.05 (ddd, J = 1.5, 10.0, 17.0 Hz, 2H), 4.74 (d, J = 12.0 Hz, 1H), 4.47 (d, J = 12.0 Hz, 1H), 4.27 (dd, J = 5.0, 10.5 Hz, 1H), 3.98 (m, 1H), 3.81 (t, J = 10.5 Hz, 1H), 3.65 (d, J = 1.0 Hz, 1H), 3.53 (dt, J = 1.5, 7.0 Hz, 1H), 3.44 (dt, J = 5.0, 10.0 Hz, 1H), 2.54 - 2.45 (m, 2H), 2.35 (dt, J = 7.5, 14.5 Hz, 1H), 1.67 (dt, J = 3.0, 13.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 137.9, 137.6, 134.4, 129.0, 128.4, 128.3, 128.1, 127.8, 126.1,117.3, 101.9, 80.1, 74.5, 74.1, 73.8, 71.2, 69.2, 35.5, 32.0, 29.7; ESIHRMS calcd for C23H26O4Na [M + Na]+ 389.1731, found 389.1733.
S-Phenyl 2-O-Benzyl-4,6-O-benzylidene-3-deoxy-α-d-thio-mannopyranoside (3α)
A colorless oil, was isolated from the standard glycosylation procedure (10 mg, 17%). [α]RTd - 34.1 (c = 1.0 M, CHCl3), 1H NMR (500 MHz, CDCl3) δ 7.41 (dd, J = 1.5, 8.0 Hz, 2H), 7.38 (dd, J = 1.5, 8.0 Hz, 2H), 7.32 – 7.20 (m, 11 H), 5.53 (s, 1H), 5.49 (s, 1H), 4.54 (dd, J = 12.0, 17.5 Hz, 2H), 4.26 (dt, J = 5.0, 10.0 Hz, 1H), 4.13 (dd, J = 5.0, 10.0 Hz, 1H), 4.07 – 4.02 (m, 1H), 3.93 (br s, 1H), 3.77 (t, J = 10.5 Hz, 1H), 2.82 – 2.38 (m, 1H), 1.96 (dt, J = 2.0, 13.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 137.6, 137.2, 134.1, 131.5, 129.1, 128.5, 128.3, 128.0, 127.7, 127.5, 126.2, 102.3, 86.2, 76.5, 74.3, 71.2, 69.0, 69.1, 30.7; ESIHRMS calcd for C26H26O4SNa [M + Na]+ 457.1450, found 457.1433.
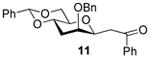
2-O-Benzyl-4,6-O-benzylidene-1,3-dideoxy-1-(2-oxo-2-phenylethyl)- β-d-mannopyranose (11)
Compound (11) was prepared from 3 (80 mg) by the general glycosylation procedure and obtained in 75% yield (62 mg) as a α/β > 1:20 mixture. The β anomer was isolated as a colorless oil: [α]RTd - 21.7 (c = 0.75 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.90 (dd, J 1.5, 8.5 Hz, 2H), 7.58 (t, J = 7.0 Hz, 1H), 7.50 (dd, J = 2.0, 8.0 Hz, 2H), 7.39 - 7.34 (m, 4H), 7.26 – 7.24 (m, 1H), 7.22 – 7.18 (m, 3H), 5.59 (s, 1H), 4.72 (d, J = 12.0 Hz, 1H), 4.35 (d, J = 12.0 Hz, 1H), 4.26 (dd, J = 3.0, 4.5 Hz, 2H), 3.98 (ddd, J = 4.5, 9.5, 13.5 Hz, 1H), 3.85 (t, J = 3.0 Hz, 1H), 3.78 (t, J = 10.5 Hz, 1H), 3.53 (ddd, J = 5.0, 9.0, 10.5 Hz, 1H), 3.30 (d, J = 7.0 Hz, 2H), 2.53 (dt, J = 3.5, 14.0 Hz, 1H), 1.84 – 1.78 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 197.6, 137.6, 136.8, 133.3, 129.0, 128.8, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.7, 126.1, 102,0, 76.2, 74.4, 74.0, 73.7, 71.2, 69.1, 39.6, 31.8, 29.7; ESIHRMS calcd for C28H28O5Na [M + Na]+ 467.1834, found 467.1830.
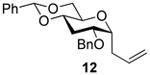
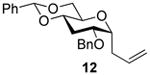
2-O-Benzyl-4,6-O-benzylidene-1-allyl-1,3-dideoxy-α-d-glucopyranose (12)
Compound (12) was prepared from 4 (80 mg) by the general glycosylation procedure and obtained in 60 to 63% yield (40-43 mg) as a white solid: mp = 101.2 - 101.8 °C; [α]RTd + 30.2 (c = 0.75 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.49 (dd, J = 2.0, 8.0 Hz, 2H), 7.40 - 7.30 (m, 8H), 5.89 -5.81 (m, 1H), 5.52 (s, 1H), 5.17 (dd, J = 9.0, 10.0 Hz, 2H), 4.58 (q, J = 12.0 Hz, 2H), 4.22 (dd, J = 4.3, 9.9 Hz, 1H), 4.12 (ddd, J = 3.8, 5.5, 7.3 Hz, 1H), 3.85 (ddd, J = 4.7, 5.5, 11.6 Hz, 1H), 3.63 (dd, J = 9.5, 10.5 Hz, 1H), 3.58 – 3.49 (m, 2H), 2.71 - 2.64 (m, 1H), 2.47 - 2.43 (m, 1H), 2.37 (dt, J = 4.3, 11.7 Hz, 1H), 1.85 (q, J = 1.6 Hz, 1H); C NMR (125 MHz, CDCl3) δ 138.0, 137.4, 134.5, 129.1, 128.5, 128.4, 127.8, 127.5, 126.1, 117.2, 101.7, 74.6, 74.0, 71.0, 69.7, 64.9, 30.8, 29.7, 28.9; ESIHRMS calcd for C23H26O4Na [M + Na]+ 389.1729, found 389.1725.
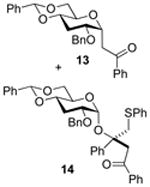
2-O -Benzyl-4,6-O-benzylidene-3-deoxy-1-(2-oxo-2-phenylethyl)-α-d-glucopyranose (13) and [(1R)-2-Oxo-1,2-diphenyl-1-(phenylthiomethyl)ethyl] 2-O-benzyl-4,6-O-benzylidene-3-deoxy-α-d- glucopyranoside (14)
Compounds (13) and (14) were obtained from 4 (80 mg) by the general glycosylation procedure. Purification by column chromatography eluting with hexanes/ethyl acetate 15:1 to 10:1 gave compounds 13 (28 mg) and 14 (30 mg) in 34 and 37% yield, respectively.
Compound (13) was isolated as a white solid, mp = 90.8 - 91.2 °C; [α]RTd + 32.4 (c = 0.5 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 7.5 Hz, 2H), 7.57 (t, J = 7.5 Hz, 1H), 7.48 - 7.45 (m, 4), 7.39 - 7.27 (m, 8H), 5.50 (s, 1H), 4.98 - 4.94 (m, 1H), 4.55 (s, 2H), 4.17 (, 1H), 3.89 (dt, J = 5.0, J = 12.5 Hz, 1H), 3.63 - 3.50 (m, 3H), 4.70 (d, J = 4.5 Hz, 1H), 3.37 (dd, J = 8.0, 16.5, 1H), 2.41 (dt, J = 4.0, 12.0, 1H), 1.85 (q, J = 12.0, 1H); 13C NMR (125 MHz, CDCl3) δ 197.6, 137.7, 137.3, 137.2, 133.1,129.1, 128.6, 128.5, 128.4, 128.2, 127.8, 127.6, 126.1, 101.7, 73.3, 71.6, 71.2, 69.7, 66.2, 34.5, 31.4,29.7; ESIHRMS calcd for C28H28O5Na [M + Na]+ 467.1834, found 467,1831.
Compound (14) was isolated as a colorless oil, [α]RTd + 57.6 (c = 0.5 M, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 7.5 Hz, 2H), 7.58 - 7.53 (m, 3H), 7.48 (dd, J = 2.0, 8.0 Hz, 2H), 7.42 (t, J = 8.0 Hz, 2H), 7.39 - 7.18 (m, 15H), 7.09 (t, J = 7.5 Hz, 1H), 5.39 (s, 1H), 5.05 (d, J = 3.5 Hz, 1H), 4.04 (s, 2H), 4.17 (d, J = 18.5 Hz, 1H), 4.07 - 4.00 (m, 3H), 3.76 (dd, J = 5.0, 15.0 Hz, 1H), 3.72 (d, J = 13.5 Hz, 1H), 3.43 - 3.34 (m, 3H), 2.19 - 2.16 (m, 1H), 1.97 (q, J = 12.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 195.8, 141.7, 138.0, 137.5, 137.4, 137.0, 133.0, 129.6, 129.1, 128.8, 128.6, 128.4, 128.3, 128.1, 127.9, 127.8, 127.7, 127.4, 127.2, 126.3, 125.9, 101.8, 92.5, 82.2, 76.9, 74.6, 71.0, 69.1, 64.8, 46.1, 43.1, 29.8; ESIHRMS calcd for C42H40O6SNa [M + Na]+ 695.2443, found 695.2449.
Supplementary Material
Acknowledgments
We thank the NIH (GM62160) for support of this work.
Footnotes
Supporting Information Available. Copies of the 1H and 13C NMR spectra of compounds 3α, and 9-14. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.a) Levy DE, Tang C. The Chemistry of C-Glycosides. Pergamon; Oxford: 1995. [Google Scholar]; b) Postema MHD, editor. C-Glycoside Synthesis. CRC; Boca Raton: 1995. [Google Scholar]; c Postema MHD, Calimente D. In: Glycochemistry: Principles, Synthesis, and Applications. Wang PG, Bertozzi CR, editors. Dekker; New York: 2001. pp. 77–131. [Google Scholar]
- 2.a) Nukada T, Bérces A, Wang L, Zgierski MZ, Whitfield DM. Carbohydr Res. 2005;340:841–852. doi: 10.1016/j.carres.2004.12.021. [DOI] [PubMed] [Google Scholar]; b) Ionescu AR, Whitfield DM, Zgierski MZ, Nukada T. Carbohydr Res. 2006;341:2912–2920. doi: 10.1016/j.carres.2006.09.027. [DOI] [PubMed] [Google Scholar]; c) Whitfield DM, Nukada T. Carbohydr Res. 2007;342:1291–1304. doi: 10.1016/j.carres.2007.03.030. [DOI] [PubMed] [Google Scholar]; d) Whitfield DM. Adv Carbohydr Chem Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]
- 3.a) Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]; b) Aubry S, Sasaki K, Sharma I, Crich D. Top Curr Chem. 2011;301:141–188. doi: 10.1007/128_2010_102. [DOI] [PubMed] [Google Scholar]
- 4.Crich D, Sharma I. Org Lett. 2008;10:4731–4734. doi: 10.1021/ol8017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crich D, Cai W, Dai Z. J Org Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 6.Crich D, Sharma I. J Org Chem. 2010;75:8383–8391. doi: 10.1021/jo101453y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noel A, Delpech B, Crich D. Org Lett. 2012;14:1342–1345. doi: 10.1021/ol300255q. [DOI] [PubMed] [Google Scholar]
- 8.a) Crich D, Li WJ. Org Chem. 2007;72:7794–7797. doi: 10.1021/jo7012912. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hsu CH, Chu KC, Lin YS, Han JL, Peng YS, Ren CT, Wu CY, Wong CH. Chem Eur J. 2010;16:1754–1760. doi: 10.1002/chem.200903035. [DOI] [PubMed] [Google Scholar]
- 9.Crich D, Vinogradova O. J Org Chem. 2006;71:8473–8480. doi: 10.1021/jo061417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The anomeric configuration of the thioglycosides is a matter of experimental convenience and ease of preparation; it does not affect anomeric selectivity.3
- 11.Lee YJ, Bark JY, Lee BL, Kang SS, Park HS, Jeon HB, Kim KS. Carbohydr Res. 2006;341:1708–1716. doi: 10.1016/j.carres.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Crich D, Cai W. J Org Chem. 1999;64:4926–4930. doi: 10.1021/jo990243d. [DOI] [PubMed] [Google Scholar]
- 13.Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Tetrahedron. 2004;60:1057–1064. [Google Scholar]
- 14.For the reaction of 1 with allyltrimethylsilane and allyltributylstannane to give 5 we previously reported yields of 58 and 61%, respectively, with α:β ratios of 1:5 and 1:8, respectively, at −60 °C. The formation of comparably minor amounts of the α-anomer of 5 in the present can not be excluded but these were not quantified.
- 15.a) Chenault HK, Castro A, Chafin LF, Yang J. J Org Chem. 1996;61:5024–5031. [Google Scholar]; b) Wang P, Haldar P, Wang Y, Hu H. J Org Chem. 2007;72:5870–5873. doi: 10.1021/jo070512x. [DOI] [PubMed] [Google Scholar]; c) Boons GJ, Heskamp B, Hout F. Angew Chem Int Ed. 1996;35:2845–2847. [Google Scholar]; d) Boons GJ, Isles S. J Org Chem. 1996;61:4262–4271. doi: 10.1021/jo960131b. [DOI] [PubMed] [Google Scholar]; e) Yuan J, Lindner K, Frauenrath H. J Org Chem. 2006;71:5457–5467. doi: 10.1021/jo0600510. [DOI] [PubMed] [Google Scholar]
- 16.Crich D, Yang F. Angew Chem Int Ed. 2009;48:8896–8899. doi: 10.1002/anie.200904168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Gupta RC, Slawin AMZ, Stoodley RJ, Williams DJ. J Chem Soc Chem Comm. 1986:1116–1118. [Google Scholar]; b) Larsen DS, Stoodley RJ. J Chem Soc Perkin Trans. 1989;1:1841–1852. [Google Scholar]; c) Stoodley RJ. Carbohydr Polymers. 1998;37:249–255. [Google Scholar]
- 18.Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66–77. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 19.a) Rhind-Tutt AJ, Vernon CA. J Chem Soc. 1960:4637–4644. [Google Scholar]; b) Kronzer FJ, Schuerch C. Carbohydr Res. 1973;27:379–390. [Google Scholar]; c) Lucas TJ, Schuerch C. Carbohydr Res. 39:1975. 39–45. doi: 10.1016/s0008-6215(00)82649-x. [DOI] [PubMed] [Google Scholar]; d) Marousek V, Lucas TJ, Wheat PE, Schuerch C. Carbohydr Res. 1978;60:85–96. [Google Scholar]; e) Lemieux RU, Hendriks KB, Stick RV, James K. J Am Chem Soc. 1975;97:4056–4062. [Google Scholar]; f) Bohé L, Crich D. CR Chimie. 2011;14:3–16. [Google Scholar]; g) Demchenko AV. In: Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Demchenko AV, editor. Wiley-VCH; Weinheim: 2008. pp. 1–27. [Google Scholar]
- 20.a) Sinnott ML. Chem Rev. 1990;90:1171–1202. [Google Scholar]; b) Zechel DL, Withers SG. Acc Chem Res. 2000;33:11–18. doi: 10.1021/ar970172+. [DOI] [PubMed] [Google Scholar]; c) Horenstein NA. Adv Phys Org Chem. 2006;41:275–314. [Google Scholar]
- 21.Saito K, Ueoka K, Matsumoto K, Suga S, Nokami T, Yoshida Ji. Angew Chem Int Ed. 2011;50:5153–5156. doi: 10.1002/anie.201100854. [DOI] [PubMed] [Google Scholar]
- 22.a) Wallace JE, Schroeder LR. J Chem Soc Perkin Trans. 1976;2:1632–1636. [Google Scholar]; b) Bülow A, Meyer T, Olszewski TK, Bols M. Eur J Org Chem. 2004:323–329. [Google Scholar]; c) Huang M, Retailleau P, Bohé L, Crich D. J Am Chem Soc. 2012;134 doi: 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Gouliaras C, Lee D, Chan L, Taylor MS. J Am Chem Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]; b) Wurst JM, Liu G, Tan DS. J Am Chem Soc. 2011;133:7916–7925. doi: 10.1021/ja201249c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang M, Garrett GE, Birlirakis N, Bohé L, Pratt DA, Crich D. Nat Chem. 2012;4:663–667. doi: 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogawa T, Pernet AG, Hanessian S. Tetrahedron Lett. 1973;14:3543–3546. [Google Scholar]
- 26.Lewis MD, Cha JK, Kishi Y. J Am Chem Soc. 1982;104:4976–4978. [Google Scholar]
- 27.a) Larsen CH, Ridgway BH, Shaw JT, Smith DM, Woerpel KA. J Am Chem Soc. 2005;127:10879–10884. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]; b) Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J Am Chem Soc. 1999;121:11208–12209. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- 28.a) Ayala L, Lucero CG, Romero JAC, Tabacco SA, Woerpel KA. J Am Chem Soc. 2003;125:15521–15528. doi: 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]; b) Beaver MG, Woerpel KA. J Org Chem. 2010;75:1107–1118. doi: 10.1021/jo902222a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lucero CG, Woerpel KA. J Org Chem. 2006;71:2641–2647. doi: 10.1021/jo0522963. [DOI] [PubMed] [Google Scholar]; d) Shenoy SR, Smith DM, Woerpel KA. J Am Chem Soc. 2006;128:8671–8677. doi: 10.1021/ja061110u. [DOI] [PubMed] [Google Scholar]; e) Smith DM, Woerpel KA. Org Biomol Chem. 2006;4:1195–1201. doi: 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]; f) Yang MT, Woerpel KA. J Org Chem. 2009;74:545–553. doi: 10.1021/jo8017846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Jensen HH, Bols M. Acc Chem Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]; b) Heuckendorff M, Pedersen CM, Bols M. Chem Eur J. 2010;16:13982–13994. doi: 10.1002/chem.201002313. [DOI] [PubMed] [Google Scholar]
- 30.a) Miljkovic M, Yeagley D, Deslongchamps P, Dory YL. J Org Chem. 1997;62:7597–7604. [Google Scholar]; b) Rhoad JS, Cagg BA, Carver PW. J Phys Chem A. 2010;114:5180–5186. doi: 10.1021/jp9100448. [DOI] [PubMed] [Google Scholar]; c) Bucher C, Gilmour R. Angew Chem Int Ed. 2010;49:8724–8728. doi: 10.1002/anie.201004467. [DOI] [PubMed] [Google Scholar]; d) Bucher C, Gilmour R. Synlett. 2011:1043–1046. [Google Scholar]
- 31.Giannis A, Sandhoff K. Tetrahedron Lett. 1985;26:1479–1482. [Google Scholar]
- 32.BeMiller JN, Yadav MP, Kalabokis VN, Myers RW. Carbohydr Res. 1990;200:111–126. [Google Scholar]
- 33.a) Stoddart JF. Stereochemistry of Carbohydrates. Wiley-Interscience; Toronto: 1971. [Google Scholar]; b) Bérces A, Whitfield DM, Nukada T. Tetrahedron. 2001;57:477–491. [Google Scholar]
- 34.Cumpstey I. Org Biomol Chem. 2012;10:2503–2508. doi: 10.1039/c2ob06696c. [DOI] [PubMed] [Google Scholar]
- 35.a) Zhu X, Kawatkar SP, Rao Y, Boons GJJ. Am Chem Soc. 2006;128:11948–11957. doi: 10.1021/ja0629817. [DOI] [PubMed] [Google Scholar]; b) Ishiwata A, Akao H, Ito Y. Org Lett. 2006;8:5525–5528. doi: 10.1021/ol062198j. [DOI] [PubMed] [Google Scholar]
- 36.Eliel EL, Wilen SH. Stereochemistry of Organic Compounds. Wiley; New York: 1994. [Google Scholar]
- 37.Gennari C. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon Press; Oxford: 1991. pp. 629–660. [Google Scholar]
- 38.Sinnott ML. Adv Phys Org Chem. 1988;24:113–204. [Google Scholar]
- 39.a) Whitfield DM. Carbohydr Res. 2012:191–195. doi: 10.1016/j.carres.2012.04.001. [DOI] [PubMed] [Google Scholar]; b) Whitfield DM. Carbohydr Res. 2012:180–190. doi: 10.1016/j.carres.2012.03.040. [DOI] [PubMed] [Google Scholar]
- 40.For interesting discussions on diastereoselectivity induced by the size, shape, and orientation of protecting groups at the 2-positions see: Kumar R, Whitfield DM. J Org Chem. 2012;77:3724–3739. doi: 10.1021/jo202563f.Ionescu AR, Whitfield DM, Zgierski MZ. Carbohydr Res. 2007;342:2793–2800. doi: 10.1016/j.carres.2007.09.007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.