

Abstract
The nuclear pore complex (NPC) consists of a conserved set of ~30 different proteins, termed nucleoporins, and serves as a gateway for the exchange of materials between the cytoplasm and nucleus. Tpr (translocated promoter region) is a component of NPC that presumably localizes at intranuclear filaments. Here, we show that Tpr knockdown caused a severe reduction in the number of nuclear pores. Furthermore, our electron microscopy studies indicated a significant reduction in the number of inner nuclear filaments. In addition, Tpr siRNA treatment impaired cell growth and proliferation compared to control siRNA-treated cells. In Tpr-depleted cells, the levels of p53 and p21 proteins were enhanced. Surprisingly, Tpr depletion increased p53 nuclear accumulation and facilitated autophagy. Our study demonstrates for the first time that Tpr plays a role in autophagy through controlling HSP70 and HSF1 mRNA export, p53 trafficking with karyopherin CRM1, and potentially through direct transcriptional regulation of autophagy factors.
Autophagy is a process for degrading intracellular constituents in lysosomes1,2,3. There are three types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy. Depending on the way of trafficking to the lysosome, macroautophagy, which for simplicity is denoted hereafter as autophagy, is the most comprehensively studied and is active in all cells1,2,4. After induction, membranous structures called phagophores or isolation membranes form from a variety of origins. As these membranes grow, they accumulate cytoplasmic components for digestion and form a double-membraned vesicle called an autophagosome, which entirely encapsulates the cargo1,2,4. Although active at basal levels in all cells as a homeostatic machinery for the breakdown of misfolded proteins and damaged organelles, the levels and cargoes of autophagy can adjust in response to a variety of stimuli2,4. For instance, the rate of autophagy can be elevated in response to starvation as a limited internal mechanism to fuel ATP synthesis until peripheral nutrients are available3,4.
Autophagy is also one of the major responses of cells to external or internal stimuli. As with any other major phenomenon in cell biology (such as division, differentiation and cell death), autophagy can be perturbed in cancer cells and is modulated by anticancer chemotherapies3,5. The progression to cancer is a multistage process6 that contains the perturbation of genes that stimulate cancer (oncogenes) and genes that naturally repress cancer (tumor suppressor genes)7. Accumulating evidence indicates that p53, the best-characterized human tumor suppressor protein, can control autophagy in a dual fashion, depending on its subcellular localization8,9. On the other hand, in response to various types of cellular stress, for example DNA damage or ribosomal stress, the levels of p53 rise above basal levels and accumulate in the nucleus, where p53 functions as a transcriptional activator of a series of genes involved in tumor suppression9. p53 has been shown to activate a number of genes that promote autophagy8,9. Amongst these genes is the damage-regulated autophagy modulator (DRAM), which belongs to a highly conserved family of proteins4,9. Despite the abundant information about p53 there are still question such as how is p53 trafficking into and out of the nucleus to activate downstream genes regulated? Could nuclear pore proteins regulate p53-induced autophagy? Or, could nucleoporins play a role in regulating autophagy through gene gating?10
The nuclear pore complex (NPC) is made of hundreds of copies of ~30 different proteins, called nucleoporins, and is merely the mediator of exchange between the nucleus and the cytoplasm in eukaryotic cells11,12. Several nucleoporins (Nups) are linked to cancer and metastasis6. Nucleoporin Tpr (translocated promoter region) was originally identified as the oncogenic activator of the met and trk proto-oncogenes13. Tpr is a component of NPC that presumably localizes at intranuclear filaments or nuclear baskets14,15.
The mammalian Tpr is a 267 kDa protein16. It consists of an ~1600-residue α-helical coiled-coil N-terminal domain and a highly acidic noncoiled 800-amino-acid carboxy terminus predicted to be unstructured17. Tpr has two homologs in yeast, Mlp1 and Mlp2, and one in Arabidopsis, AtTpr18. Mammalian Tpr directly binds to Nup15311,12. Tpr has been suggested to have a role in nucleocytoplasmic transport as a scaffolding element, in Erk2 translocation, mRNA export, unspliced RNA export, nuclear protein export, transcriptional telomeric chromatin organization, establishing perinuclear heterochromatin exclusion zones, SUMOylation (small ubiquitin-like modifier protein), and in controlling cellular senescence6,12,18. Recently, we have found that several nucleoporins are involved in mitotic spindle and kinetochores during mitosis19,20,21,22,23,24,25,26,27. In another study we identified that the molecular motor dynein complex transports nucleoporin Tpr during mitosis, and depleting Tpr causes a chromosome lagging phenotype28.
Here, we report the unexpected roles of Tpr in cell proliferation and in the autophagy machinery. Our new findings not only identified new autophagy regulators (nucleoporins and potentially nuclear trafficking factors), but also highlight additional complexity in autophagy control.
Results
Tpr depletion reduces nuclear pore complex protein marker (mAb414) expression
Recently, we found that Tpr interacts with the molecular motors dynein and dynactin, to regulate the spindle checkpoint proteins, Mad1 and Mad2, during cell division28. To further characterize the effect of Tpr knockdown on the cell cycle, cell fate and cell proliferation, we asked whether the primary reason for growth inhibition was due to Tpr depletion disrupting normal NPC components and eventually affecting the nuclear trafficking process. We found that in HeLa, HEK293T and A549 cell lines, Tpr RNAi-treated cells, which had a severely reduced Tpr immunofluorescence signal during interphase also showed reduced staining of the NPC marker, mAb414 (Fig. 1A). mAb414 recognizes mainly with Nup62, but also with other Phe-Gly (FG)-containing Nups, such as Nup358, Nup214/CAN, Nup153 and Nup98; all of which carry the O-linked N-acetylglucosamine modification29. Hence, we studied Tpr protein expression by immunoblotting. We found that the Tpr levels in total cell extract immunoblots were commonly less than 10%, compared with control proteins. The same blots were stripped and re-probed with mAb414 and α-tubulin antibodies, revealing that Nup62 expression was also dramatically reduced (α-tubulin was used as a loading control; Fig. 1B). Taken together, these results suggest that loss of Tpr may control NPC expression in mammalian cells to some extent.
Figure 1. Tpr knockdown leads to the reduction of nuclear pore proteins.
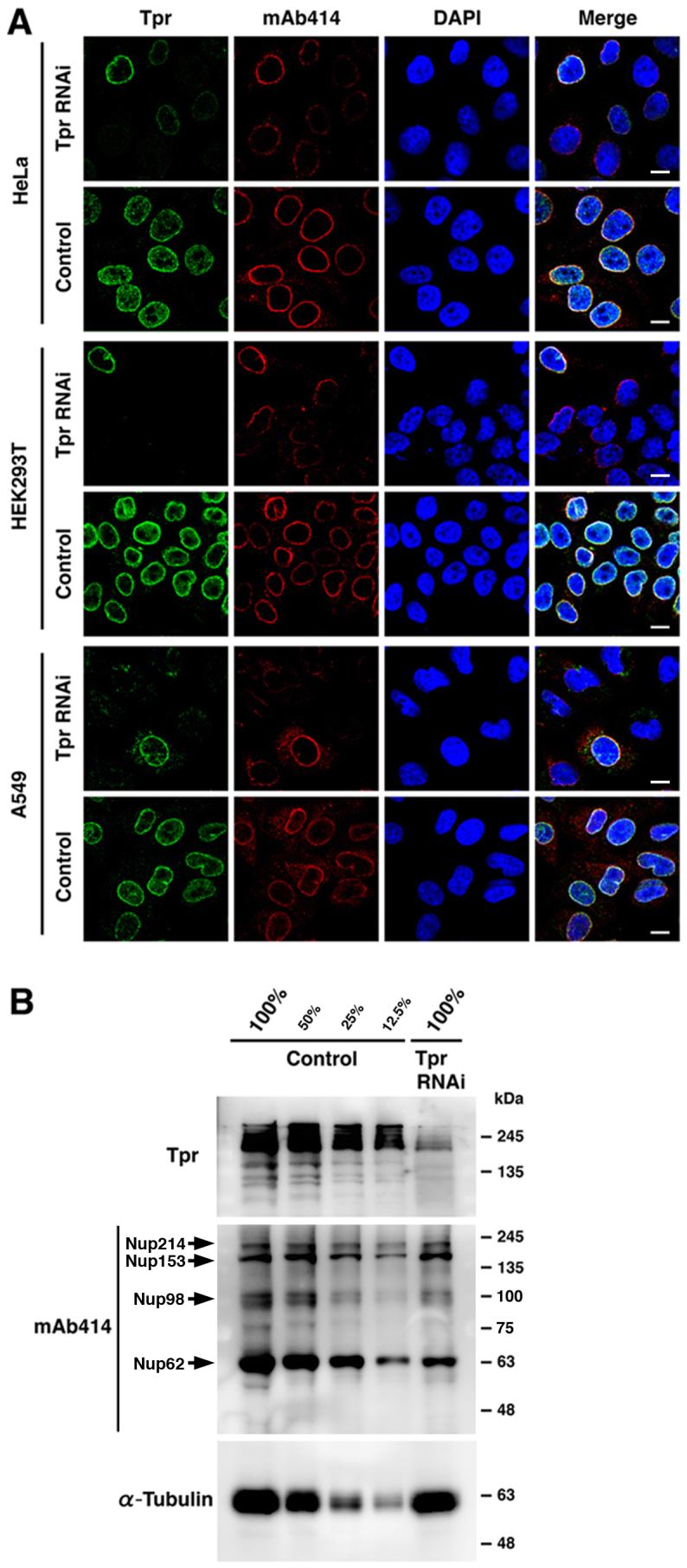
(A) HeLa, HEK293T, and A549 cells were transfected with either control siRNA duplex or Tpr siRNA (Tpr RNAi) for 72 h, and then Tpr and mAb414 (nuclear pore marker) were visualized by immunofluorescence following staining for nucleus with DAPI. Scale bars, 10 μm. (B) HeLa cells transfected with control or Tpr siRNA were analyzed by immunoblot analysis for Tpr and mAb414 expression (Arrows indicate corresponding Nups). Serial dilutions of whole cell extracts from control-transfected cells and cells treated with Tpr siRNA are shown. Representative results of three different experiments are shown.
Tpr depletion reduces NPC numbers and inhibits cell growth
The above data prompted us to further characterize the effect of Tpr knockdown on nano scale structural characteristics; we decided to investigate the effect of Tpr depletion on NPC numbers and morphology using high resolution electron microscopy (EM) during interphase. Next, we performed thin-section transmission electron microscopy (TEM) to gain more detailed physiological observations of the structural components of the HeLa cell NPC. In control siRNA cells (n = 27 cells), the nuclei retained typical nuclear envelopes and NPC structures between cytoplasmic and nuclear regions. Some nuclei had typical filamentous structures around the nuclear envelop (Fig. 2A left panel). In contrast, in Tpr-siRNA cells the nuclear envelope (n = 25 cells) had significantly reduced inner nuclear filaments (Fig. 2A right panel). From the set of micrographs shown in Fig. 2B and Fig. S1, we determined that there were 9.1 ± 1.01 nuclear pores/μm2 in control siRNA cells, which was close to published values30. Strikingly, however, we found that in Tpr-siRNA-treated cells, there were only 4.4 ± 1.20 nuclear pores/μm2 (triplicate experiments, P < 0.05; Fig. 2C). Furthermore, we found that Tpr siRNA-treatment impaired cell growth and proliferation compared to control siRNA-treated cells (Fig. 2D). These EM data on NPC numbers decreasing further supports our observations in mAb414 signal on the blots (Fig. 1B) and by cell staining decreases (Fig. 1A) following Tpr depletion. Taken together, we believe that the nucleoporin, Tpr, constitutes a central architectural element of nuclear pore formation and potentially regulates the cell growth process.
Figure 2. Nucleoporin Tpr is an important component of nuclear pores and cell growth.

(A) Electron microscopic images of nuclear pores (left panel, arrow indicates typical filamentous structure) in control HeLa cells. Scale bars, 120 nm. Electron microscopic images of nuclear pores (right panel, arrow indicates filamentous structure) in Tpr siRNA-transfected HeLa cells. Scale bars, 120 nm. Open circle indicates the nuclear pore. (B) Electron microscopy image of nuclear pores (arrows) in control (left panel) and Tpr siRNA-transfected (right panel) HeLa cells. Tangential section shows a frontal view of the pores. Scale bars, 0.25 µm. (C) Pore numbers of HeLa cells transfected with control or Tpr siRNAs were analyzed. The nuclear pores in control- or Tpr siRNA-transfected HeLa cells were counted in 9 square micrometers from the electron microscopic images. The data are presented as the means ± S.D. for triplicate determinations. bars, SD. *, P < 0.05 compared with control cells. (D) Cell growth after Tpr-depletion was determined by the trypan blue dye exclusion assay. HeLa cells were seeded at low density, transfected with control or Tpr siRNA, and grown for 96 h. The data are presented as the means ± S.D. for triplicate determinations.
Tpr depletion induces nuclear accumulation of p53
To further investigate the mechanistic consequences of growth inhibition induced by Tpr depletion, we examined the transcription factors p53 and p21. Consistent with a recent report31, we found that in nuclear extracts of Tpr-siRNA-treated cells p21 and p53 were upregulated compared to the control siRNA cells (α-tubulin and Histone H3) were used as a loading control; Fig. 3A, B). Interestingly, p53 and p21 appeared in an intense punctuated pattern in Tpr-siRNA-treated cells, but almost no signals were observed in the control siRNA cells. These results suggest that p53 and p21 nuclear trafficking may be blocked by Tpr depletion. We hypothesized that Tpr knockdown reduces nuclear pore formation and nucleo-cytoplasmic trafficking activities, thereby causing p53 and p21 nuclear accumulation and p53 activation, which induces specific downstream target genes of the p53 and p21 pathways. To prove this hypothesis, we carried out immunoprecipitation experiments. Immunoprecipitates were prepared from HeLa, HCT116 and SW480 cells, using an anti-Tpr, anti-p53, and normal rabbit IgG as a control. p21 (Waf1/Cip1/Sdi1) was initially identified as an inhibitor of cyclin-dependent kinases, a mediator of p53 in growth suppression, and a marker of cellular senescence32. p21 is also one of the p53 downstream targets32. Chromosome maintenance protein 1 (CRM1) was reported to facilitated p53 nuclear export33. To investigate possible mechanisms, we performed coimmunoprecipitation with HeLa lysates. We found that Tpr antibodies coimmunoprecipitated p53 and CRM1 (weak interaction), but not p21 (Fig. 3C and Fig. S2). Conversely, p53 antibodies coimmunoprecipitated Tpr and CRM1, but not p21 (Fig. 3C). These data suggest that Tpr associates with p53.
Figure 3. Tpr silencing facilitates nuclear accumulation of p53.

(A) HeLa cells were transfected with control or Tpr siRNA for 72 h, and then Tpr, p53 and p21 were visualized by immunofluorescence following nuclear DAPI staining. Scale bars, 10 µm. (B) HeLa cells transfected with control or Tpr siRNA were analyzed by immunoblot for Tpr, p53 and p21 expression. Representative results of three different experiments are shown. (C) HeLa cell lysates were immunoprecipitated with anti-Tpr, -p53, or nonspecific rabbit antibodies (IgG) followed by immunoblot analysis for Tpr, p53, p21 and CRM1 expression. Cell lysates were also immunoblotted as a control (Input). IP, immunoprecipitation.
Furthermore, to determine whether the observed Tpr-depletion-induced p53 nuclear localization phenotype is a different manifestation of the same defects, or whether the roles of Tpr can be uncoupled, we employed a rescue strategy by overexpressing GFP-Tpr (full length) in Tpr knockdown cells. 12 to 48 hours after transfection of GFP-Tpr into Tpr RNAi knockdown cells (Fig. 4A), the localization of p53 was partially restored; p53 cytoplasmic staining was found in GFP-Tpr-transfected Tpr RNAi cells (Fig. 4B, C). This result suggests that Tpr is one of the proteins that regulate p53 nuclear trafficking.
Figure 4. Tpr mediates p53 nuclear-cytoplasmic trafficking.

(A) Schedule of collecting cells after Tpr depletion following GFP-Tpr transfection. (B) Representative images of HeLa cells transfected with plasmids overexpressing GFP-Tpr (full-length) after Tpr depletion (see above schedule in A). Tpr and p53 were visualized by immunofluorescence following nuclear DAPI staining. Scale bars, 10 µm. White arrowheads indicate typical GFP-Tpr-transfected cells. (C) Quantitation of the subcellular localization of the p53 proteins. Localization was classified in two categories: nucleus and nucleus plus cytoplasm. Data are presented as the percentage of cells in each category, and the data are presented as the means ± S.D. for three different experiments, counting at least 100 transfected cells in each experiment.
Tpr depletion facilitates autophagy
The tumor suppressor p53 plays a critical role in safeguarding the integrity of the genome as well as being a vital mediator of cell death9. Tpr has been reported to induce p53 activation in mammalian cells, suggesting that p53 may be involved in Tpr-induced apoptosis and/or senescence31. To further characterize the Tpr-induced growth inhibition of HeLa cells (Fig. 2D) and the importance of the interaction with p53 (Fig. 3 and 4), we examined the apoptotic morphological changes in the cells. Surprisingly, when the cells were cultured with Tpr RNAi for 72 h, marked apoptotic morphological alterations, including cell shrinkage, membrane blebbing and nuclear fragmentation were not observed (data not shown). For quantitative analysis of Tpr RNAi-induced apoptosis, the HeLa cell population was assessed. Consistent with a recent report31, p21 was upregulated in Tpr-depleted cells (Fig. 3B), indicating that part of the p21 nuclear accumulation may be due to p53 activation. Moreover, depleting Tpr did not induce any significant staining in the TUNEL assay (Fig. S3A), or any dramatic changes in the DNA ladder assay of HeLa cells (Fig. S3B). Interestingly, analysis by electron microscopy revealed an accumulation of autophagic-like vesicles in Tpr-depleted cells (Fig. S3C). Because recent findings suggested that the p53 pathway is often involved in autophagy9,34, we further investigated the effect of Tpr depletion on autophagy. We examined the formation of autophagic vacuoles by staining cells with the specific autophagosome-detecting fluorescent dyes, monodansylcadaverine (MDC) and acridine orange (AO)35. As shown in Fig. 5A, the MDC and AO fluorescence intensity in Tpr-depleted cells was upregulated compared with the control cells. Furthermore, we found that the level of conversion from LC3-I to LC3-II, an important protein marker involved in autophagy, was increased in Tpr-depleted cells (Fig. 5B). The Tpr-depletion-induced upregulation of LC3 was confirmed by confocal microscopy with LC3 staining (Fig. 5C).
Figure 5. Tpr depletion induces autophagy.

(A) Top, MDC staining. HeLa cells were transfected with control or Tpr siRNA for 72 h, incubated with 50 µM MDC for 10 min, and then observed under a fluorescence microscope. Scale bars, 10 µm. Bottom, acridine orange staining. HeLa cells were transfected with control or Tpr siRNA for 72 h, stained with 1 µg/ml acridine orange, and examined under a fluorescence microscope. Scale bars, 10 µm. (B) HeLa cells transfected with control or Tpr siRNA were analyzed by immunoblot for LC3 expression. Representative results of three different experiments are shown. (C) HeLa cells were transfected with control or Tpr siRNA for 72 h, and then Tpr and LC3 were visualized by immunofluorescence following nuclear DAPI staining. Scale bars, 10 µm.
Because p53 associates with CRM1 for nuclear export36, we then sought to examine autophagy by depleting Tpr or CRM1, to distinguish the potential roles between Tpr and CRM1 in autophagy (Fig. 6). In control cells, we did not observe much autophagic cells (0.5 ±0.10 %). Interestingly, we found that depleting Tpr significantly enhanced autophagic cells (19.07 ±2.33 %) compared to the CRM1-depleted cells (11.4 ±1.60 %) (n>500 cells; p<0.05; Fig. 6A). Next, we transfected HeLa cells with siRNAs against TPR, CRM1 or control, for 72 h. Immunoblotting and immunofluorescence analysis revealed that LC3 upregulation was also found in CRM1 RNAi cells (Fig. 6B). Next, we performed the Tpr or CRM1 RNAi assay with confocal microscopy. We did not observe any major changes in localization between these two proteins immune-staining when either Tpr or CRM1 was knocked-down (Fig. 6C). On the other hand, in contrast to control RNAi cells, we observed slightly nuclear up-regulation of p53 in CRM1 RNAi cells (Fig. 6D). Recent studies have indicated that p53-induced autophagy requires induction of DRAM, a p53 target gene37. To further investigate whether the CRM1 regulation of p53 nuclear accumulation modulates autophagy through the p53 pathway, we tested the mRNA expression of several p53 target genes in CRM1-depleted HeLa cells by quantitative RT-PCR (qPCR) and immunoblotting. We found that an up-regulation of p53 and DRAM mRNA and protein expression (Fig. 6E-G). Moreover, we confirmed that p53 accumulation mainly in the nucleus after CRM1 knockdown via confocal microscopy observations (Fig. 6D).
Figure 6. CRM1 depletion also induces autophagy, but weaker than Tpr depletion.

(A) Quantification of acridine orange staining in Tpr or CRM1 siRNA transfected HeLa cells. HeLa cells were transfected with control, Tpr or CRM1 siRNA for 72 h, and were analyzed by acridine orange staining to determine the ratio of autophagy. At least 500 cells in each experiment were examined under a fluorescence microscope and the percentage of acridine orange staining cells was calculated. The data are presented as the means ± S.D. for three different experiments. *, P < 0.05. (B) HeLa cells transfected with control, Tpr, or CRM1 siRNA were analyzed by immunoblot for Tpr, CRM1, and LC3 expression. Representative results of three different experiments are shown. (C) HeLa cells were transfected with control, Tpr, or CRM1 siRNA for 72 h, and then Tpr and CRM1 were visualized by immunofluorescence following nuclear DAPI staining. Scale bars, 10 µm. (D) HeLa cells were transfected with control or CRM1 siRNA for 72 h, and then CRM1 and p53 were visualized by immunofluorescence following nuclear DAPI staining. Scale bars, 10 µm. (E) HeLa cells transfected with control or CRM1 siRNA were analyzed by immunoblot for CRM1, p53 and p21 expression. Representative results of three different experiments are shown. (F) HeLa cells were transfected with control or CRM1 siRNA for 72 h and mRNA levels were assayed by quantitative PCR. The data are presented as the means ± S.D. for triplicate determinations. bars, SD. (G) HeLa cells transfected with control or CRM1 siRNA were analyzed by immunoblot for CRM1 and DRAM expression. Representative results of three different experiments are shown.
To further investigate whether the Tpr also regulation of p53 nuclear accumulation modulates autophagy through the p53 pathway, we also tested the mRNA expression of several p53 target genes in Tpr-depleted HeLa cells by qPCR (Fig. 7A–C) and immuno-blotting (Fig. 7D). We found that p21, PUMA (p53 upregulated modulator of apoptosis) and DRAM mRNA were upregulated in Tpr-depleted cells (Fig. 7A), suggesting that depleting Tpr also enhanced p53 nuclear accumulation, which activated the p53-induced autophagy modulator, DRAM.
Figure 7. Tpr knockdown activates autophagic signals.

(A-C) HeLa cells were transfected with control or Tpr siRNA for 72 h and mRNA levels of p53 substrates (A), Tpr-related mRNA cargos (B) and autophagy factors (C) were assayed by quantitative PCR. The data are presented as the means ± S.D. for triplicate determinations. bars, SD. *, P < 0.05. (D) HeLa cells transfected with control or Tpr siRNA were analyzed by immunoblot for Tpr, DRAM, HSF1 and HSP70 expression. Representative results of three different experiments are shown.
Interestingly, human heat shock factors (HSF) also act as molecular chaperones in autophagy. Moreover, the HSF-Tpr interaction was reported to facilitate the export of stress-induced HSP70 mRNA38. To further examine the potential ability of Tpr to regulate HSP70 mRNA trafficking during autophagy, we performed qPCR assays. We found that both HSF1 and HSP70 mRNA were elevated in Tpr-depleted cells (Fig. 7B). The above data prompted us to further examine the direct nature of the ability of Tpr to activate autophagy, because Tpr directly associates with active transcription activities15,39. We examined the effects of Tpr depletion on several autophagy factors (LC3, Beclin1, Atg3, Atg5, Atg7 and Atg12) by qPCR. We found that Atg7 and Atg12 mRNA were significantly upregulated in Tpr-depleted cells (P < 0.05; Fig. 7C). To confirm our qPCR observations, we performed immunoblotting with antibodies against DRAM, HSF-1 and HSF70 in Tpr RNAi or control RNAi HeLa cells. Indeed, we found that these proteins were elevated in Tpr RNAi cells (Fig. 7D). More important, unlike the Tpr siRNA qPCR assay (Fig. 7A) and immuno-blotting (Fig. 7D), knockdown of CRM1 just slightly increased p21, PUMA mRNA expression (Fig. 6F) or p21 protein expression (Fig. 6E). Taken together, these results suggest that Tpr plays a role in autophagy through controlling HSP70 and HSF1 mRNA export, p53 trafficking with karyopherin CRM1, and potentially through direct transcriptional regulation of autophagy factors (Atg7 and Atg12).
Discussion
In this report, we discovered the unexpected roles of Tpr in cell proliferation and in the autophagy machinery (Fig. 8). Moreover, Tpr depletion resulted in reduction in nuclear pore formation and expression of NPC proteins.
Figure 8. A schematic diagram of Tpr-depleted nuclear pore complex.

Tpr-depletion causes the diminution of nuclear basket area, the pale pores and the pore number reduction. Tpr interacts with p53 to transport to the cytoplasm, thus Tpr silencing promotes p53 nuclear accumulation followed by inducing autophagy. Tpr also potentially regulates mRNA export and transcriptional activity. We propose Tpr as an essential component for nuclear pore formation that controls cellular programs such as autophagy.
Localization of Tpr using immuno-EM has given diverse results15,40. In an attempt to minimize variations due to differences in experimental procedures and to allow direct simple comparisons, we decided to perform conventional EM. Indeed, using conventional EM, NPCs were discovered in different eukaryotes between 1950 and the 1970s30,41,42. Interestingly, unlike earlier NPC EM images from the 1960-70s showing filament-like structures between nuclear envelopes30,41,42,43, recent Tpr-related EM studies demonstrated the nuclear envelope with “empty open holes”44. We wonder that these “empty open pores/holes” really reflect the 30-nucleoporin macro-complexes, and how normal cellular trafficking activities via these “empty holes” without any filamentous structures44.
Moreover, our data also challenges the principles of the current pore formation core components45,46. Are all 30 nucleoporins required to make a functional pore? Unlike the model concerning NPC core modules (Nup107-160 sub-complex and Nup205 sub-complex), here, for the first time, we provide evidence that Tpr, a non-core module, also has a vital role in nuclear pore formation, and governs cell growth and proliferation.
The next unexpected finding was the relationship between Tpr and autophagy (Fig. 8). Because the autophagy machinery is confined to the cytosol during interphase, it is difficult to envision how the nucleoporins are involved. Our qPCR and immunoblotting data clearly established the importance of Tpr in autophagy via three different pathways: governing transcription factors/protein export (p53), mRNA export (HSF1 and HSP70 mRNA), and as a novel transcription factor by itself (Fig. 5 and 7).
Our data indicates that Tpr is associated with p53 nuclear accumulation (Fig. 3 and 4). RNAi knockdown of Tpr, induced p53 nuclear accumulation, and activated p53 target genes (e.g., DRAM) that enhance autophagy36. In addition to Tpr, p53 also interacts with CRM137,47. We cannot completely rule out the possibility that autophagy induced by p53 may also be regulated by CRM1 or other nucleoporins. It is possible that CRM1-dependent nuclear export also participates in p53 nuclear accumulation-induced autophagy, although our data indicated that Tpr-depleted cells induced more autophagic cells compared to CRM1-depleted cells (Fig. 6A). Besides, unlike Tpr depletion, the mRNA up-regulation level of p21, PUMA in qPCR assay (Fig. 7A vs 6F) and the protein up-regulation level of p21 by immunoblotting (Fig. 3B vs 6E) after knockdown of CRM1 are not as high as Tpr depletion. One possible explanation is that Tpr not only responsible for p53 trafficking but also contributed in HSF1 and HSP70 mRNA trafficking, and transcriptional regulation of autophagy factors Atg7 and Atg12 (Fig. 8). Further investigation of this issue is vital in the near future. In addition, the detailed HSF1-Tpr mRNA shuttling mechanisms and involvement in autophagy remain to be determined, but the evidence suggest that HSF148 and HSP7049 participate in autophagy. Furthermore, we believe that a close link between NPC and gene gating, and gene regulation in higher eukaryotes. Knockdown of Mtor (the Drosophila homolog of mammalian Tpr) or Nup153 results in loss of the typical MSL (Male-specific lethal) X-chromosome staining and dosage compensation in Drosophila male cells, but not in female cells39. Our qPCR data on Atg7 and Atg12 mRNA upregulation also raise the possibility of an independent Tpr transcriptional role in the regulation of autophagy. Thus, additional studies in this area are required.
The data shown here provide evidence for the first time that nuceloporin Tpr and nuclear transport factor, CRM1, regulate autophagy. Identification of other Nups or karyopherins that are also involved in the autophagy machinery would be of great interest.
Methods
Mammalian cell culture
HeLa, HEK293T, and A549 cells were obtained from the American Type Culture Collection (ATCC). SW480 and HCT116 cells were a kind gift from Dr. Toshinari Minamoto (CRI, Kanazawa Univ.). All cell lines were cultured in DMEM (Invitrogen) with 10% Fetal Bovine Serum (FBS) and penicillin/streptomycin. All cell lines were maintained at 37°C in an air/5% CO2 incubator.
DNA constructs and RNA interference
The plasmid encoding full-length human Tpr, tagged with GFP, was a kind gift from Dr. Larry Gerace (The Scripps Research Institute). siRNA duplexes targeting Tpr (sc-45343), CRM1 (sc-35116) and control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology. siRNA transfections were performed using Lipofectamine 2000, following the manufacturer's protocol (Invitrogen). HeLa cells were imaged 72 h after transfection. If necessary, transfection efficiency was monitored with Block-iT (Invitrogen).
Growth curves
HeLa cells were seeded and transfected with Tpr siRNAs or control siRNAs. Cell growth was determined by the Trypan Blue dye exclusion assay.
Electron microscopy
HeLa cells adhered to culture dishes were washed with phosphate-buffered saline (PBS) and fixed in 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 20 min and then 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) overnight. They were then scraped off and sedimented at 200 g for 10 min. After rinsing in 0.1 M phosphate buffer (pH 7.4) buffer, the cells were post-fixed in 0.1 M phosphate buffer (pH 7.4) with 2% Osmium tetraoxide, dehydrated in ethanol (50, 70, 90, and 100% × 4; each for 1 h) at room temperature. The cells were consecutively incubated in a 2:1 and then a 1:2 (v/v) mixture of ethanol and epoxy resin at 20°C for 1 h each, followed by infiltration with pure epoxy resin at 20°C and polymerization at 70°C for 15 h. Sections of approximately 70 nm thickness were transferred onto 200-mesh copper grids without supporting film and stained with 2% uranyl acetate solution and then stained with lead stain solution before carbon vacuum deposition. Micrographs were recorded with a JEOL JEM-1200EX at 70–100 kV.
Antibodies, immunocytochemistry, and confocal microscopy
α-Tpr polyclonal and monoclonal antibodies were from Santa Cruz Biotechnology. In some experiments, an α-Tpr polyclonal antibody, which was a kind gift from Dr. Larry Gerace (Scripps Research Institute) was also used. α-mAb414 (MMS-120R) antibody was from COVANCE. α-LC3, α-p53, α-p21, α-HSF1 and α-HSP70 antibodies were from Cell Signaling Technology. α-CRM1 and α-Tubulin (DM1A) monoclonal antibody were from Sigma-Aldrich. α-DRAM was from Abcam. α-Histone H3 was from Millipore. Secondary antibodies were from Molecular Probes. For immunofluorescence, HeLa cells were washed in PBS and fixed for 10 min in 4% paraformaldehyde in PBS. Cells were then permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature. Samples were mounted onto coverslips with ProLong Gold Antifade reagent (Invitrogen) and were examined on a Zeiss LSM5 EXCITER confocal microscope, and all images were acquired using an aplan-Apochromat 63X with a 1.4-N.A. objective or at 100 × with a 1.4-N.A. objective.
Protein Extraction
For whole cell lysates, cells were washed twice with PBS and collected by scraping. Cell pellets were lysed in cold radioimmune precipitation assay buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% of Nonidet P40, 1 mM DTT and 10% glycerol) containing protein inhibitors. Samples were clarified by centrifugation (15,000 rpm in 4°C for 30 min). For nuclear protein fraction collection, cells were lysed in buffer A (10 mM Tris/HCl (pH 7.8), 10 mM NaCl and 1.5 mM MgCl2 supplemented with 0.2% Nonidet P40), then vigorously mixed. Samples were clarified by centrifugation (15,000 rpm in 4°C for 15 min) into cytoplasmic and nuclear fractions. The pellet were resuspended in buffer B (25 mM Tris/HCl (pH 7.8), 150 mM NaCl, 1mM EDTA and 0.5% Nonidet P40) and gently mixed at 4°C for 30 min. The aliquots of the supernatant were stored as nuclear protein extracts.
Western analysis
Equal amounts of protein were loaded in each lane. All protein samples were separated on either 8 or 12% SDS-PAGE gels and transferred to 0.2 µm polyvinylidene fluoride membrane (PVDF) at 25 mA overnight at 4°C. Then the membrane was blocked with 5% non-fat dried milk in PBS for 1 h at room temperature. The blocked membrane was incubated with diluted individual primary antibodies for 3 h at room temperature. After extensive washing to remove excess antibody from the membrane, either anti-rabbit or anti-mouse HRP-conjugated secondary antibody (1:5000) was added and incubated for 1 h at room temperature. Proteins were visualized using the enhanced chemiluminescence (ECL) system. Exposure and density of each band was determined on an ImageQuant™ LAS 4000 (GE Healthcare Life Sciences).
Autophagy detection
The cells were incubated with the autofluorescent agent monodansylcadaverine (MDC, 50 µM, Sigma) or the vital dye acridine orange (AO, 1 μg/mL, Sigma) at 37°C for 20 min, and then examined under a fluorescence microscope (Zeiss).
RT-PCR Analysis
RNA was prepared using TRIzol Reagent (Invitrogen), and cDNA was synthesized using the ThermoScript RT-PCR System (Invitrogen). Quantitative real-time PCR analysis was carried using the Thermal Cycler Dice Real Time System with the SYBR Premix Ex Taq (Takara). The sequence of primers used in this study was listed in Supplemental Table 1. Expression levels of genes analysed by qPCR were normalized relative to levels of GAPDH.
Apoptosis detection
DNA breaks were detected by the DeadEnd Fluorometric TUNEL System (Promega) according to the manufacturer’s protocol. Apoptotic cells were examined using a fluorescence microscope. DNA fragmentation was extracted using ApopLadder Ex kit (Takara) according to the manufacturer’s protocol. Fragmented DNA was separated by electrophoresis in a 2% agarose gel followed by ethidium bromide staining.
Statistical analysis
Data are expressed as means ± SD. Comparisons between groups were determined using the unpaired t test. P < 0.05 was considered statistically significant.
Author Contributions
T.F performed most of the experiments; E.T. performed the RT-PCR experiments. R.W. designed experiments and wrote the manuscript. All authors discussed the results and reviewed the manuscript.
Supplementary Material
Supplental FigS1-S3_figure legends_Table 1
Acknowledgments
T.F. was supported by Grants-in-Aid for Young Scientists (B) from MEXT Japan and by grants from the Astellas Foundation for Research on Metabolic Disorders, the Suzuken Memorial Foundation, and the Life Science Foundation of Japan. This work was also supported by Grants-in-Aid for Scientific Research on Innovative Areas, Grants-in-Aid for Challenging Exploratory Research and Grants-in-Aid for Scientific Research (B) from MEXT Japan, and by grants from the Asahi Glass Foundation, the Suzuken Memorial Foundation, the Sumitomo Foundation, the Kowa life science Foundation, the Mochida Memorial Foundation, the Sagawa Foundation, the Uehara Memorial Foundation, the Ichiro Kanehara foundation and the Takeda Science Foundation (to R. W.).
References
- Mizushima N., Levine B., Cuervo A. M. & Klionsky D. J. Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C. & Martens S. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol 24, 496–501 (2012). [DOI] [PubMed] [Google Scholar]
- Maiuri M. C. et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ 16, 87–93 (2009). [DOI] [PubMed] [Google Scholar]
- Mah L. Y., O'Prey J., Baudot A. D., Hoekstra A. & Ryan K. M. DRAM-1 encodes multiple isoforms that regulate autophagy. Autophagy 8, 18–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R., Karantza-Wadsworth V. & White E. Role of autophagy in cancer. Nat Rev Cancer 7, 961–967 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funasaka T. & Wong R. W. The role of nuclear pore complex in tumor microenvironment and metastasis. Cancer Metastasis Rev 30, 239–251 (2011). [DOI] [PubMed] [Google Scholar]
- Hanahan D. & Weinberg R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- Tasdemir E. et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10, 676–687 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri M. C. et al. Autophagy regulation by p53. Curr Opin Cell Biol 22, 181–185 (2010). [DOI] [PubMed] [Google Scholar]
- Blobel G. Gene gating: a hypothesis. Proc Natl Acad Sci U S A 82, 8527–8529 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martinez J. & Rout M. P. A jumbo problem: mapping the structure and functions of the nuclear pore complex. Curr Opin Cell Biol 24, 92–99 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E. J. & Wente S. R. Dynamic nuclear pore complexes: life on the edge. Cell 125, 1041–1053 (2006). [DOI] [PubMed] [Google Scholar]
- Peschard P. & Park M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene 26, 1276–1285 (2007). [DOI] [PubMed] [Google Scholar]
- Frosst P., Guan T., Subauste C., Hahn K. & Gerace L. Tpr is localized within the nuclear basket of the pore complex and has a role in nuclear protein export. J Cell Biol 156, 617–630 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krull S., Thyberg J., Bjorkroth B., Rackwitz H. R. & Cordes V. C. Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol Biol Cell 15, 4261–4277 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. J. & Cooper C. S. Nucleotide sequence analysis of human tpr cDNA clones. Oncogene 7, 383–388 (1992). [PubMed] [Google Scholar]
- Cordes V. C., Reidenbach S., Rackwitz H. R. & Franke W. W. Identification of protein p270/Tpr as a constitutive component of the nuclear pore complex-attached intranuclear filaments. J Cell Biol 136, 515–529 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman E., Medalia O. & Zwerger M. Functional architecture of the nuclear pore complex. Annu Rev Biophys 41, 557–584 (2012). [DOI] [PubMed] [Google Scholar]
- Nakano H. et al. Unexpected role of nucleoporins in coordination of cell cycle progression. Cell Cycle 10, 425–433 (2011). [DOI] [PubMed] [Google Scholar]
- Wong R. W. Interaction between Rae1 and cohesin subunit SMC1 is required for proper spindle formation. Cell Cycle 9, 198–200 (2010). [DOI] [PubMed] [Google Scholar]
- Wong R. W. An update on cohesin function as a ‘molecular glue’ on chromosomes and spindles. Cell Cycle 9, 1754–1758 (2010). [DOI] [PubMed] [Google Scholar]
- Wong R. W. & Blobel G. Cohesin subunit SMC1 associates with mitotic microtubules at the spindle pole. Proc Natl Acad Sci U S A 105, 15441–15445 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R. W., Blobel G. & Coutavas E. Rae1 interaction with NuMA is required for bipolar spindle formation. Proc Natl Acad Sci U S A 103, 19783–19787 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strambio-De-Castillia C., Niepel M. & Rout M. P. The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol 11, 490–501 (2010). [DOI] [PubMed] [Google Scholar]
- Funasaka T. et al. RNA export factor RAE1 contributes to NUP98-HOXA9-mediated leukemogenesis. Cell Cycle 10(2011). [DOI] [PubMed] [Google Scholar]
- Hashizume C., Nakano H., Yoshida K. & Wong R. W. Characterization of the role of the tumor marker Nup88 in mitosis. Mol Cancer 9, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume C. & Wong R. W. Structure and function of nuclear pore complex. Seikagaku 83, 957–965 (2011). [PubMed] [Google Scholar]
- Nakano H., Funasaka T., Hashizume C. & Wong R. W. Nucleoporin translocated promoter region (Tpr) associates with dynein complex, preventing chromosome lagging formation during mitosis. J Biol Chem 285, 10841–10849 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L. I. & Blobel G. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc Natl Acad Sci U S A 84, 7552–7556 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher H. W. & Cooper T. W. Electron microscope studies of the microvilli of HeLa cells. J Cell Biol 34, 569–576 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- David-Watine B. Silencing nuclear pore protein Tpr elicits a senescent-like phenotype in cancer cells. PLoS One 6, e22423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y. S., Qian Y. & Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 22, 1003–1012 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. G., Dawson J. & Sullivan D. M. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 83, 1021–1032 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden K. H. & Ryan K. M. p53 and metabolism. Nat Rev Cancer 9, 691–700 (2009). [DOI] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol 36, 2491–2502 (2004). [DOI] [PubMed] [Google Scholar]
- Kanai M. et al. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat Cell Biol 9, 1175–1183 (2007) [DOI] [PubMed] [Google Scholar]
- Crighton D. et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134 (2006). [DOI] [PubMed] [Google Scholar]
- Skaggs H. S. et al. HSF1-TPR interaction facilitates export of stress-induced HSP70 mRNA. J Biol Chem 282, 33902–33907 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendjan S. et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell 21, 811–823 (2006). [DOI] [PubMed] [Google Scholar]
- Byrd D. A. et al. Tpr, a large coiled coil protein whose amino terminus is involved in activation of oncogenic kinases, is localized to the cytoplasmic surface of the nuclear pore complex. J Cell Biol 127, 1515–1526 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall J. G. Observations on the nuclear membrane with the electron microscope. Exp Cell Res 7, 197–200 (1954). [DOI] [PubMed] [Google Scholar]
- Comes P. & Franke W. W. Composition, structure and function of HeLa cell nuclear envelope. I. Structural data. Z Zellforsch Mikrosk Anat 107, 240–248 (1970). [DOI] [PubMed] [Google Scholar]
- Watson M. L. Further observations on the nuclear envelope of the animal cell. J Biophys Biochem Cytol 6, 147–156 (1959). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krull S. et al. Protein Tpr is required for establishing nuclear pore-associated zones of heterochromatin exclusion. EMBO J 29, 1659–1673 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelz A., Debler E. W. & Blobel G. The structure of the nuclear pore complex. Annu Rev Biochem 80, 613–643 (2011). [DOI] [PubMed] [Google Scholar]
- Bilokapic S. & Schwartz T. U. 3D ultrastructure of the nuclear pore complex. Curr Opin Cell Biol 24, 86–91 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Watt P. J. & Leaner V. D. The nuclear exporter, Crm1, is regulated by NFY and Sp1 in cancer cells and repressed by p53 in response to DNA damage. Biochim Biophys Acta 1809, 316–326 (2011). [DOI] [PubMed] [Google Scholar]
- Alavez S., Vantipalli M. C., Zucker D. J., Klang I. M. & Lithgow G. J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472, 226–229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M. A. et al. PERK-dependent regulation of HSP70 expression and the regulation of autophagy. Autophagy 4, 364–367 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplental FigS1-S3_figure legends_Table 1