

Abstract
While PDZ domain-containing proteins represent cellular targets for several different viral oncoproteins, including human papillomavirus E6, human T-cell leukemia virus type 1 Tax, and human adenovirus E4-ORF1, the functional consequences for such interactions have not been elucidated. Here we report that, at the plasma membrane of cells, the adenovirus E4-ORF1 oncoprotein selectively and potently stimulates phosphatidylinositol 3-kinase (PI3K), triggering a downstream cascade of events that includes activation of both protein kinase B and p70S6-kinase. This activity of E4-ORF1 could be abrogated by overexpression of its PDZ-protein targets or by disruption of its PDZ domain-binding motif, which was shown to mediate complex formation between E4-ORF1 and PDZ proteins at the plasma membrane of cells. Furthermore, E4-ORF1 mutants unable to activate the PI3K pathway failed to transform cells in culture or to promote tumors in animals, and drugs that block either PI3K or p70S6-kinase inhibited E4-ORF1-induced transformation of cells. From these results, we propose that the transforming and tumorigenic potentials of the adenovirus E4-ORF1 oncoprotein depend on its capacity to activate PI3K through a novel PDZ protein-dependent mechanism of action.
Keywords: adenovirus, E4-ORF1, PDZ, PI3K, oncoprotein
Introduction
Human adenovirus type 9 (Ad9) is distinct among tumorigenic adenoviruses (Ads) in eliciting exclusively estrogen-dependent mammary tumors in rats (Javier et al., 1991) and in having as its primary oncogenic determinant the E4 region-encoded ORF1 (E4-ORF1) protein (Javier, 1994), rather than the E1 region-encoded E1A and E1B proteins (Thomas et al., 1999). In addition, a recombinant E1 region-deleted Ad5 vector engineered to ectopically express Ad9 E4-ORF1 induces solely mammary tumors, identical to those produced by Ad9, whereas the parental virus vector fails to generate tumors of any kind (Thomas et al., 2001). Thus, in animals, E4-ORF1 also largely determines the capacity of Ad9 to target tumorigenesis specifically within cells of the mammary gland.
Mutational analyses of the 125-amino-acid (aa) residue E4-ORF1 protein have identified three discrete regions (I, II, and III) essential for its cellular transforming functions (Weiss et al., 1997a). Although activities have not yet been ascribed to regions I and II, region III at the extreme carboxyl-terminus of E4-ORF1 defines a functional PDZ domain-binding motif that mediates interactions with a select group of cellular PDZ domain-containing proteins. PDZ domains are protein–protein interaction modules found in cellular factors that typically function in signal transduction (Sheng and Sala, 2001). To date, we have identified four E4-ORF1-associated PDZ proteins as the multi-PDZ protein MUPP1 (Lee et al., 2000) and the three membrane-associated guanylate kinase (MAGUK)-family proteins DLG, MAGI-1, and ZO-2 (Lee et al., 1997; Glaunsinger et al., 2000, 2001). MUPP1 consists of 13 PDZ domains and no other recognizable motifs, whereas the MAGUK proteins contain from three to five PDZ domains, either one SH3 domain or two WW domains, and a yeast guanylate kinase-homology domain. These domain structures comprising numerous protein-binding modules suggest that, like the Drosophila multi-PDZ protein InaD (Tsunoda and Zuker, 1999), E4-ORF1-associated PDZ proteins function as scaffolding proteins both to assemble receptors and cytosolic factors into supramolecular complexes and to localize these signaling complexes to the plasma membrane at specialized regions of cell–cell contact (Sheng and Sala, 2001).
High-risk human papillomavirus E6 and human T-cell leukemia virus type 1 Tax oncoproteins also possess PDZ domain-binding motifs at their carboxyl-termini (Lee et al., 1997). These motifs mediate binding to some E4-ORF1-associated PDZ proteins (Lee et al., 1997, 2000; Glaunsinger et al., 2000) and, moreover, are required for E6-induced transformation of rat 3Y1 fibroblasts (Kiyono et al., 1997) and for Tax-induced reversal of a block to cell-cycle progression caused by overexpression of DLG in mouse 3T3 cells (Suzuki et al., 1999). The latter observation should be interpreted cautiously, however, as DLG expressed at normal physiological levels has not yet been demonstrated to suppress cell-cycle progression in mammalian cells. Thus, while PDZ proteins represent common cellular targets for several different human virus oncoproteins, the functional consequences of these interactions are not yet understood.
As PDZ proteins typically function at the plasma membrane, it is reasonable to hypothesize that viral oncoproteins target these cellular factors to disregulate cell growth-regulatory signaling pathways originating from this site. Phosphatidylinositol 3-kinase (PI3K) is a key component of one such signaling cascade that normally becomes triggered by activation of tyrosine kinase and heterotrimeric G-protein-coupled membrane receptors (Blume-Jensen and Hunter, 2001). These receptors stimulate the activity of PI3K and its recruitment to the plasma membrane, where this lipid kinase phosphorylates phosphatidylinositides at position D3 of the inositol moiety. The resulting 3,4,5-phosphatidylinositide lipid products then act as second messengers to induce membrane translocation of the serine/threonine protein kinase Akt/protein kinase B (PKB) and stimulation of its kinase activity through phosphorylation of threonine-residue 308 (Thr308) by PDK1 and serine-residue 473 (Ser473) by an undetermined kinase. Activated PKB has been shown to promote both survival and proliferation of cells through its ability to control the activities of multiple downstream effectors, including proapoptotic Forkhead transcription factors, translation and cell-cycle progression regulator p70S6-kinase (S6K), and cyclin-dependent kinase inhibitor p27Kip1 (Yu and Sato, 1999; Medema et al., 2000). A critical antagonist of the PI3K signaling pathway is the tumor-suppressor protein PTEN, a lipid phosphatase that specifically removes the D3 phosphate from 3,4,5-phosphatidylinositide and thereby reverses the action of PI3K (Yamada and Araki, 2001). The presence of PTEN mutations in approximately 20% of human cancers and amplification of PI3K and PKB in some human tumors have established a central role for disregulated PI3K signaling in the development of human malignancies (Mills et al., 2001).
Our previous results showed that E4-ORF1 sequesters a large fraction of its PDZ-protein targets in the cytoplasm of cells (Glaunsinger et al., 2000, 2001; Lee et al., 2000). We now report that E4-ORF1 likewise complexes with its PDZ-protein targets at the plasma membrane to selectively activate PI3K and that this activity is required for the oncogenic potential of E4-ORF1. In addition to exposing a novel PDZ protein-dependent mechanism for PI3K stimulation, these findings are also significant in revealing for the first time a physiologically relevant functional consequence for interactions of a viral oncoprotein with its PDZ-protein targets.
Results
Selective stimulation of PI3K by wild-type (wt) but not transformation-deficient mutant E4-ORF1
The fact that cellular PDZ proteins generally function in signal transduction prompted experiments to assess whether E4-ORF1 activates any known signaling factors in cells. We initially focused our attention on a group of such proteins (PI3K, JNK1, β-catenin/TCF, NFκB, STAT3, and Notch) that have been implicated in transformation (Kimble and Simpson, 1997; Bowman et al., 2000; Tsatsanis and Spandidos, 2000; Uthoff et al., 2001; Vogt, 2001). By comparing their activities in E4-ORF1-expressing versus nonexpressing cells using a variety of approaches, we discovered that E4-ORF1 selectively stimulates PI3K (Figure 1) (unpublished data). For example, the PI3K activity in a CREF rat embryo fibroblast line stably expressing E4-ORF1 (CREF-E4-ORF1) was shown to be 2.5-fold higher than that in normal CREF cells (Figure 1a). This level of activation was comparable to the threefold increase in PI3K activity produced by stimulating CREF cells with the growth factor PDGF and was likewise abolished by the PI3K inhibitor wortmannin (Figure 1a).
Figure 1.

E4-ORF1 activates PI3K. (a) Elevated PI3K activity in E4-ORF1-expressing cells. An equivalent amount of extract from serum-starved CREF or CREF-E4-ORF1 cells was subjected to in vitro PI3K assays with PI substrate (bottom) as described in Materials and methods or to immunoblot analyses with anti-p85 antibodies (top). Indicated samples were untreated (−) or treated with 10 ng/ml PDGF and/or 100 nM wortmannin (wort). Results were compiled from three independent experiments. (b) PI3K lipid products accumulate at the plasma membrane of E4-ORF1-expressing cells. NIH 3T3 cells on coverslips in 6-cm dishes were lipofected with pGFP-AH or -AHR25C (0.5 μg each) and pGW1 encoding wt or the indicated mutant E4-ORF1 (50 ng each). Confluent serum-starved cells were visualized by fluorescence microscopy. Indicated cells were pretreated with 100 nM wortmannin (wort). For cells expressing E4-ORF1 mutants unable to activate PI3K, GFP-AH accumulated at variable levels in NIH 3T3 cell nuclei. Therefore, the brighter nuclear GFP-AH signal observed in the cell expressing T123D was not a general finding in these experiments. (c) Focus formation by wt and mutant E4-ORF1 proteins. CREF cells in 10-cm dishes were transfected with empty pJ4Ω (−) or pJ4Ω encoding wt or the indicated mutant E4-ORF1 (20 μg each) in triplicate. Foci were quantified 3 weeks post-transfection
The GFP-AH fusion protein (Watton and Downward, 1999), consisting of GFP linked to the pleckstrin-homology (PH) domain of PKB, was exploited to demonstrate similar PI3K stimulation by transiently expressed E4-ORF1 and to reveal the location of this event in cells. Owing to specific binding of this PH domain to PI3K lipid products, GFP-AH translocates from the nucleus/cytoplasm to the membrane upon activation of PI3K in cells. In agreement with results presented in Figure 1a, we demonstrated that, whereas GFP-AH transiently expressed alone exhibits an expected diffuse nuclear and cytoplasmic distribution in 3T3 cells, coexpression with E4-ORF1 causes this fusion protein to accumulate at the plasma membrane in greater than 75% of transfected cells (Figure 1b). This effect was inhibited by wortmannin and also was specific because E4-ORF1 failed to promote membrane translocation of GFP-AHR25C Figure 1b), which contains a mutant PH domain unable to bind PI3K lipid products (Watton and Downward, 1999).
We extended these results by also examining a collection of E4-ORF1 mutants that either lack focus-forming activity (IIIA, T123D, V125A, IA, IIA, IIB) or exhibit wt focus-forming activity (T108S) in CREF cells (Figure 1c) (Weiss et al., 1997a). Transformation-deficient mutants IIIA, T123D, and V125A have region III mutations that inactivate the PDZ domain-binding motif, whereas transformation-deficient mutants IA, IIA, and IIB have region I or II mutations that do not affect the function of this motif (Weiss and Javier, 1997). The T108S mutation is located outside of the three crucial regions of E4-ORF1. Modeling the E4-ORF1 polypeptide to the crystal structure of the related dUTPase enzyme (Weiss et al., 1997b) predicts that regions I and II lie in close proximity to each other within the native polypeptide (unpublished data), hinting that these sequences contribute to one additional E4-ORF1 functional domain having an undefined activity. Despite exhibiting wt protein expression (unpublished data), all transformation-deficient mutants failed to promote membrane translocation of GFP-AH in cells, whereas the control transformation-proficient mutant T108S behaved similar to wt E4-ORF1 in the assay (Figure 1b). These findings reveal an intimate link between E4-ORF1-mediated cellular transformation and PI3K activation at the plasma membrane and also indicate that both activities are dependent on the PDZ domain-binding motif and an undefined region I/II function of E4-ORF1.
PDZ protein-dependent recruitment of E4-ORF1 to the plasma membrane
The finding that E4-ORF1 caused accumulation of PI3K lipid products at the plasma membrane prompted analyses to investigate whether E4-ORF1 associates with PDZ proteins at this site in cells. In human TE85 cells stably expressing E4-ORF1 (TE85-E4-ORF1), this viral protein displayed almost continuous plasma membrane staining along regions of cell–cell contact where PDZ proteins typically accumulate as well as the previously described punctate cytoplasmic staining (Figure 2a) (Weiss et al., 1996). E4-ORF1 was also present discontinuously at regions of cell–cell contact along the plasma membrane and in the cytoplasm of both CREF-E4-ORF1 cells and 20-8 cells, which are derived from an Ad9-induced rat mammary tumor (Figure 2a). In addition, while exhibiting less punctate cytoplasmic staining than untagged E4-ORF1, fusion proteins consisting of GFP linked to the amino-terminus of E4-ORF1 revealed that wt E4-ORF1 and region I/II mutants (IA, IIA, IIB) can localize to the plasma membrane of CREF cells, whereas the GFP moiety alone and region III mutants (IIIA, V125A, T123D) cannot (Figure 2b) (unpublished data). These observations suggest that the PDZ domain-binding motif of E4-ORF1 promotes its recruitment into PDZ-protein complexes at the plasma membrane. Supporting this conclusion, we showed that endogenous ZO-2 and MUPP1 also localize to the plasma membrane at regions of cell–cell contact and, at these sites, display either extensive or partial co-localization, respectively, with GFP-tagged wt E4-ORF1 in TE85 (Figure 2c) and CREF cells (unpublished data). The utilization of an optimized paraformaldehyde fixation procedure in these experiments, as opposed to methanol fixation employed previously (Weiss et al., 1997a), substantially improved our ability to detect E4-ORF1, MUPP1, and ZO-2 at the plasma membrane. These findings are consistent with the notion that E4-ORF1 constitutively stimulates PI3K by binding to and subverting the activity of specific plasma membrane-associated PDZ-protein signaling complexes.
Figure 2.

E4-ORF1 and PDZ proteins localize to the plasma membrane. (a) E4-ORF1 localizes to the plasma membrane. TE85-E4-ORF1 cells, CREF-9ORF1-low cells (CREF-E4-ORF1 cells), or 20-8 mammary tumor cells were subjected to indirect IF assays with E4-ORF1 antibodies. In the middle and right panels, cell nuclei are circumscribed by dashed lines to aid visualization of discontinuous E4-ORF1 staining present along the membrane at regions of cell–cell contact. (b) The PDZ domain-binding motif of E4-ORF1 promotes its recruitment to the plasma membrane. CREF cells stably expressing GFP-tagged wt or mutant E4-ORF1 proteins were visualized by fluorescence microscopy. Results with GFP-IA or GFP-IIIA are representative of findings with region I/II mutants IIA and IIB or region III mutants T123D and V125A, respectively. (c) E4-ORF1 colocalizes with PDZ proteins at the plasma membrane. TE85 cells stably expressing GFP-tagged wt E4-ORF1 were subjected to indirect IF assays with either ZO-2 (top three panels) or MUPP1 (bottom three panels) antibodies and visualized by deconvolution fluorescence microscopy
PI3K-dependent activation of PKB by E4-ORF1 in cells
We next investigated whether in cells E4-ORF1 also elevates the activity of the PI3K downstream effector PKB. Our results indicated that transient expression of E4-ORF1 in COS7 cells causes a 10-fold increase in PKB activity (Figure 3a). This effect was stronger than the seven-fold induction in PKB activity produced by the RasV12 oncoprotein (Figure 3a). Additionally, E4-ORF1-mediated PKB stimulation was blocked by wortmannin in COS7 cells (Figure 3a) and, as determined by immunoblot analyses with antibodies that recognize activated PKB, was similarly inhibited in 3T3 cells by treatment with the PI3K inhibitor LY294002 or by overexpression of the p85 PI3K regulatory subunit (Figure 3b), which can act as a dominant negative inhibitor of class I PI3K (Rameh et al., 1995). Therefore, PI3K activation mediated by E4-ORF1 results in potent stimulation of PKB in cells. The observation that RasV12, but not E4-ORF1, could enhance the activity of ERK2 in COS7 cells (Figure 3c) or CREF cells (unpublished data) underscores the selective capacity of E4-ORF1 to activate the PI3K pathway.
Figure 3.

E4-ORF1 selectively activates PKB in a PI3K-dependent manner. (a) Stimulation of PKB activity by E4-ORF1. COS7 cells on 10-cm dishes were lipofected with pCDNA3-HA-PKB (1.6 μg), pGW1 encoding wt E4-ORF1 (6.4 μg), and pSG5-RasV12 (6.4 μg). Extracts (200 μg protein) from serum-starved cells were immunoprecipitated with anti-HA antibodies, and immunocomplexes were subjected to in vitro kinase assays with H2B substrate or to immunoblot analyses with anti-HA antibodies (inset panel). Indicated cells were treated with 100 nM wortmannin (wort). One representative experiment is shown. (b) Overexpression of p85 inhibits E4-ORF1-mediated PKB activation. NIH 3T3 cells on 6-cm dishes were lipofected with pGW1-HA-PKB (0.5 μg), pGW1 encoding wt E4-ORF1 (20 ng), and pCG-HA-p85 (2 μg). Serum-starved cells were lysed in RIPA buffer, and extracts (75 μg protein) were immunoblotted with anti-(P)Thr308 PKB or anti–HA antibodies. Indicated cells were treated with 50 μM LY294002 (LY). (c) E4-ORF1 fails to activate ERK2 in COS7 cells. COS7 cells on 10-cm dishes were lipofected with pCEP4-HA-ERK2 (1.6 μg), pGW1 encoding wt E4-ORF1 (6.4 μg), and pSG5–RasV12 (6.4 μg). Extracts (200 μg protein) from serum–starved cells were immunoprecipitated with anti-HA antibodies, and immunocomplexes were subjected to in vitro kinase assays with MBP substrate (bottom panel) or to immunoblot analyses with anti–HA antibodies (top panel). One representative experiment is shown. (d) Transformation by E4-ORF1 is intimately linked to its ability to activate PKB. CREF cells on 6-cm dishes were lipofected with pGW1 (−) and pGW1 encoding wt or the indicated mutant E4-ORF1 (right anf left panels) (2 μg each) or pCMVBam3–Neo (−) and pCMVBam3–Neo encoding wt or mutant T108S E4-ORF1 (center panel) (2 μg each). Serum-starved cells were lysed in sample buffer, and extracts (100 μg protein) were immunoblotted with anti-(P)Ser473 PKB, anti-PKB, or anti-E4-ORF1 antibodies. Indicated cells were treated with 100 nM wortmannin (wort) or 50 μM LY294002 (LY). (e) Stimulation of PKB by E4-ORF1 proteins derived from representative subgroup A–D human Ads. COS7 cells on 6–cm dishes were lipofected with pCMVBam3-Neo (−) and pCMVBam3-Neo plasmids encoding the indicated HA epitope-tagged E4-ORF1 (1.5 μg each). Serum-starved cells were lysed in RIPA buffer, and extracts (100 μg protein) were immunoblotted with anti-(P)Thr308 PKB, anti–PKB, or anti-HA antibodies. (f) E4-ORF1 activates PKB in Ad9-infected permissive cells. Serum-starved A549 cells were mock infected or infected at a multiplicity of 10 with wt Ad9 or mutant Ad9–IIIA. At the indicated times postinfection, extracts (75 μg protein) in sample buffer were immunoblotted with anti-(P)Ser473 PKB, anti-PKB, or anti–E4-ORF1 antibodies. (g) Constitutive activation of PKB in an Ad9-induced tumor cell line. Confluent serum-starved 20–8 cells or control CREF cells were lysed in RIPA buffer, and extracts (100 μg protein) were immunoblotted with anti-(P)Ser473 PKB or anti-PKB antibodies (top panels). CREF and 20-8 extracts were also immunoprecipitated with anti-E4-ORF1 antibodies, and recovered proteins were immunoblotted with the same antibodies (bottom panel). Indicated cells were untreated (−) or treated with 100 nM wortmannin (wort)
The relation between PKB activation and transformation by E4-ORF1 was assessed in CREF cells transiently expressing either wt or mutant E4-ORF1 proteins. Utilizing antibodies that recognize activated PKB, we demonstrated that wt E4-ORF1 and the control transformation-proficient mutant T108S, but none of the transformation-deficient mutants, are capable of stimulating PKB (Figure 3d). These results are fully concordant with the PI3K-activation phenotypes of these E4-ORF1 proteins (see Figure 1b). Also notable is that E4-ORF1 mutants IIIC and IIID, which have weak transforming activity owing to region III mutations that decrease but do not eliminate the function of the PDZ domain-binding motif (Weiss and Javier, 1997), showed a reduced capacity to activate PKB (Figure 3d). Other results showed that E4-ORF1 proteins encoded by Ad12, Ad3, Ad5, or Ad9, representative human Ads from subgroup A, B, C, or D, respectively, share the ability to stimulate PKB in COS7 cells (Figure 3e) in a PI3K-dependent manner (unpublished data), thereby revealing a common activity for these viral polypeptides.
We also tested whether E4-ORF1 stimulates PKB during a productive wt Ad9 infection of human A549 cells. Virus Ad9-IIIA encoding the mutant E4-ORF1 IIIA gene, which fails to activate PI3K or PKB in cells (see Figures 1b and 3d), was included as a negative control in the assays. We found that activated PKB was evident at 8 h postinfection with wt Ad9 and persisted at 24 h postinfection, but failed to accumulate at any time post-infection with Ad9-IIIA, despite the fact that both viruses exhibited comparable E4-ORF1 protein expression and replication in A549 cells (Figure 3f) (unpublished data). Furthermore, unlike control CREF cells, 20-8 mammary tumor cells displayed constitutively high levels of activated PKB that was ablated by wortmannin (Figure 3g), suggesting that disregulated PI3K signaling also contributes to Ad9-induced mammary tumorigenesis.
Abrogation of E4-ORF1-mediated PKB activation by overexpression of PDZ proteins
As our findings with E4-ORF1 mutants indicated that activation of PI3K and PKB by E4-ORF1 depends on its interactions with cellular PDZ proteins, we expected that overexpression of MUPP1, MAGI-1, ZO-2, or DLG would affect the ability of E4-ORF1 to stimulate this signaling cascade. The results showed that over-expression of each wt PDZ protein individually suppresses E4-ORF1-mediated PKB stimulation in 3T3 cells (Figures 4a–d). This effect precisely correlated with the capacities of the PDZ proteins to complex with E4-ORF1 because PDZ-protein deletion mutants able to bind E4-ORF1 (MUPP1−PDZ10−13, MAGI-1-ΔPDZ1, −ΔPDZ2, −ΔPDZ3, −ΔPDZ4, −ΔPDZ5, −ΔPDZ3+4, −ΔPDZ3+5, −ΔPDZ4+5, ZO-2-NT) (Glaunsinger et al., 2000, 2001; Lee et al., 2000) blocked E4-ORF1-mediated PKB activation, whereas deletion mutants unable to bind E4-ORF1 (MUPP1−ΔPDZ7+10, −PDZ11−13, MAGI-1-ΔPDZ1+3, ZO-2-ΔPDZ1, DLG-ΔPDZ1+2) (Glaunsinger et al., 2000, 2001; Lee et al., 1997, 2000) did not (Figures 4a–d). These results provide additional evidence demonstrating that interactions with one or more PDZ-protein targets are crucial for E4-ORF1 to activate PI3K signaling in cells.
Figure 4.

Overexpression of PDZ proteins inhibits E4-ORF1-mediated PKB activation. NIH 3T3 cells on 6-cm dishes were lipofected with pGW1–HA-PKB or pGW1-myc-PKB (0.5 μg), pGW1 encoding wt E4-ORF1 (20 ng), and pGW1 encoding HA epitope-tagged or untagged wt or indicated mutant (a) MUPP1 (2.5 μg), (b) MAGI-1 (0.5 μg), (c) ZO-2 (0.5 μg), or (d) DLG (1 μg). Serum-starved cells were lysed in RIPA buffer, and extracts (60 μg protein) were immunoblotted with anti-HA, anti-myc, and/or DLG antibodies and also with anti-(P)Thr308 PKB or anti-(P)Ser473 PKB antibodies
PI3K-dependent stimulation of S6K by E4-ORF1
One of the downstream effectors of PKB is the regulator of translation and cell-cycle progression S6K (Cantrell, 2001). In COS7 cells, E4-ORF1 and RasV12 comparably increased the activity of S6K by three-fold (Figure 5a). E4-ORF1-mediated S6K stimulation showed an expected sensitivity to wortmannin, as well as to rapamycin (Figure 5a), a specific inhibitor of the protein kinase mTOR required for activation of S6K (Dufner and Thomas, 1999). Using antibodies that recognize activated S6K (Dufner and Thomas, 1999), we also showed that, compared to control CREF cells, both CREF-E4-ORF1 cells and 20-8 mammary tumor cells have constitutively high levels of activated S6K that could be eliminated by wortmannin or rapamycin (Figure 5b,c). Our inability to detect constitutively activated S6K in CREF lines stably expressing transformation-deficient mutants IA, IIA, IIB, and IIIA (Figure 5b) further implicated this activity in E4-ORF1-mediated transformation.
Figure 5.

E4-ORF1 activates S6K. (a) Activation of S6K enzymatic activity by E4-ORF1. COS7 cells on 6-cm dishes were lipofected with PMT2-HA-p70 S6K (0.15 μg), pGW1 encoding wt E4-ORF1 (50 ng), and RasV12 (0.1 μg). Extracts (200 μg protein) from serum-starved cells were immunoprecipitated with anti-HA antibodies. Recovered proteins were subjected to in vitro kinase assays with an RRRLSSLRA peptide substrate or immunoblotted with anti-HA antibodies (inset panel). Indicated cells were treated with 100 nM wortmannin (wort) or 20 ng/ml rapamycin (rapa). One representative experiment is shown. (b) The transforming potential of E4-ORF1 is linked to its ability to activate S6K. Serum-starved CREF cells stably expressing wt E4-ORF1, the indicated mutant E4-ORF1, or no E4-ORF1 (−) were lysed in RIPA buffer, and extracts (85 μg protein) were immunoblotted with anti-(P)Thr389 S6K, anti-(P)Thr421/Ser424 S6K, anti-S6K, or anti-E4-ORF1 antibodies. Indicated cells were treated with 20 ng/ml PDGF, 50 μM LY294002 (LY), or 20 ng/ml rapamycin (rapa). (c) Constitutive activation of S6K in an Ad9-induced mammary tumor cell line. Extracts (100 μg protein) from 20-8 mammary tumor cells or control CREF cells in RIPA buffer were immunoblotted with anti-(P)Thr389 S6K or anti-S6K antibodies. Indicated cells were untreated (−) or treated with either 100 nM wortmannin (wort) or 20 ng/ml rapamycin (rapa)
Inactivation of FKHRL1 and downregulation of p27kip1 in E4-ORF1-expressing cells
Activated PKB phosphorylates and inactivates FKHRL1, a Forkhead-family transcription factor that promotes programmed cell death and blocks cell-cycle progression by stimulating expression of proapoptotic factors (Brunet et al., 1999) and the cyclin-dependent kinase inhibitor p27kip1 (Medema et al., 2000), respectively. In 3T3 cells transfected with a luciferase reporter plasmid containing a Forkhead-responsive promoter, we found that expression of wt FKHRL1 induces an approximately twofold increase in luciferase activity (Figure 6a). Coexpression with E4-ORF1 blocked the activity of wt FKHRL1 but not constitutively active mutant FKHRL1 TM (Figure 6a), which cannot be phosphorylated by PKB (Brunet et al., 1999). The results of immunoblot analyses with antibodies that recognize phosphorylated, inactive FKHRL1 (Brunet et al., 1999) showed that, compared to control CREF cells, CREF-E4-ORF1 cells have elevated levels of phosphorylated FKHRL1 that were substantially diminished by LY294002 (Figure 6b). Expression of p27kip1 was also downregulated in CREF-E4-ORF1 cells in a PI3K-dependent manner (Figure 6c).
Figure 6.
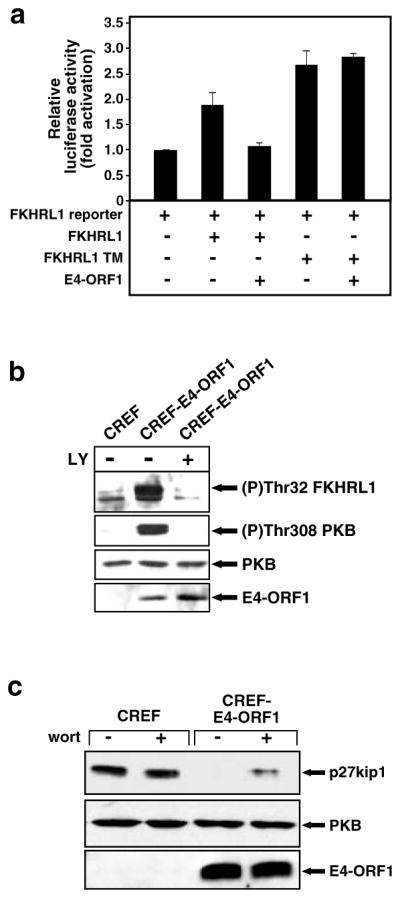
E4-ORF1 inactivates FKHRL1 and downregulates p27kip1. (a) Inactivation of FKHRL1 by E4-ORF1. NIH 3T3 cells on 6-cm dishes were lipofected with pGL3-3xFHRE (1 μg), pEF-lacZ (0.25 μg), pECE–HA–FKHRL1 wt or TM (1 μg each), and pGW1 encoding wt E4-ORF1 (0.75 μg). Extracts of serum–starved cells were prepared and assayed for luciferase activity. Results are compiled from three independent experiments. (b) E4-ORF1 promotes phosphorylation of FKHRL1. Confluent serum-starved CREF or CREF-E4-ORF1 cells were lysed in RIPA buffer, and extracts (60 μg protein) were immunoblotted with anti-(P)Thr32 FKHRL1, anti-(P)Thr308 PKB, anti-PKB, or anti–E4-ORF1 antibodies. Indicated cells were untreated (−) or treated with 50 μM LY294002 (c) Decreased p27kip1 levels in E4-ORF1-expressing cells. Confluent serum-starved CREF lines described above in (b) were lysed in RIPA buffer, and 20 μg or 100 μg of protein from cell extracts was immunoblotted with anti-p27kip1 or anti–PKB antibodies, respectively. Indicated cells were untreated (−) or treated with 100 nM wortmannin (wort)
Inhibition of E4-ORF1-induced transformation by LY294002 or rapamycin
Our findings with E4-ORF1 mutants suggested that transformation by wt E4-ORF1 depends on its ability to activate PI3K in cells. In an attempt to provide direct support for this idea, we tested whether the LY294002 can block transformation by E4-ORF1 in CREF cells. Our results showed that LY294002 greatly diminishes the ability of CREF-E4-ORF1 cells to form colonies in soft agar (Figure 7a), and also causes a 25-fold decrease in E4-ORF1-mediated focus formation on CREF cells (Figure 7b). Rapamycin has been reported to block transformation by constitutively activated forms of PI3K and PKB but not by 11 other oncoproteins (Aoki et al., 2001), so we also examined this drug in our transformation assays. Rapamycin likewise abrogated E4-ORF1-mediated soft-agar growth and focus formation in CREF cells (Figure 7a,b). It is important to note that, whereas the low doses of drugs employed in these assays robustly inhibited both transformed growth properties and activation of PI3K and/or S6K induced by E4-ORF1 in CREF cells, neither the viability nor growth of normal CREF cells was appreciably affected under these conditions (unpublished data). These findings demonstrate that the transforming potential of E4-ORF1 depends on its ability to activate the PI3K pathway in cells.
Figure 7.

LY294002 or rapamycin blocks transformation by E4-ORF1. (a) Soft-agar growth inhibition of E4-ORF1-expressing CREF cells by LY294002 and rapamycin. An equal number of CREF-E4-ORF1 cells or control CREF cells was suspended in soft agar without (−) or with the indicated amount of LY294002 (LY) or rapamycin (rapa) for 2 weeks and then photographed. (b) Inhibition of E4-ORF1-mediated focus formation by LY294002 and rapamycin. CREF cells on 10-cm dishes were lipofected with pJ4Ω or pJ4Ω plasmid encoding wt E4-ORF1 (8 μg each). At 1-week posttransfection, indicated cells were cultured in a medium containing either 10 μM LY294002 (LY) or 5 ng/ml rapamycin (rapa). Foci were quantified 3-weeks post–transfection. Error bars indicate range of focus numbers obtained on duplicate dishes. (c) Soft-agar growth inhibition of Ad9-induced mammary tumor cells by LY294002 and rapamycin. An equal number of 20-8 mammary tumor cells or control CREF cells was analysed as described above in (a)
Association of PI3K activation by E4-ORF1 with Ad9-induced mammary tumorigenesis
In an effort to similarly, directly link PI3K activation by E4-ORF1 to Ad9-induced mammary tumorigenesis, we investigated whether LY294002 and rapamycin can reverse the transformed growth properties of 20-8 mammary tumor cells. Like CREF-E4-ORF1 cells, 20-8 cells grew efficiently in soft agar yet, in the presence of either drug, this growth was substantially reduced (Figure 7c), albeit more efficiently, with LY294002 than with rapamycin. To provide additional evidence relating PI3K activation by E4-ORF1 to Ad9-induced mammary tumorigenesis, we determined the tumorigenic potentials of a collection of E4-ORF1 mutant Ad9 viruses. The results revealed that Ad9 viruses coding for E4-ORF1 mutants unable or having a limited capacity to stimulate the PI3K pathway fail to elicit tumors of any kind in rats (Table 1), despite the fact that such viruses display wt E4-ORF1 protein expression and replication in A549 cells (unpublished data). In contrast, the control Ad9 virus coding for E4-ORF1 mutant T108S, which stimulates the PI3K pathway similar to wt E4-ORF1 (see Figure 3d), behaved like wt Ad9 in generating mammary tumors in 100% of infected female rats (Table 1). Collectively, these findings strongly argue that E4-ORF1-mediated stimulation of PI3K is required for Ad9-induced mammary tumorigenesis in animals.
Table 1.
Mammary tumorigenic phenotypes of E4-ORF1 mutant Ad9 viruses
| Ad9 E4-ORF1 gene | Tumorigenic potential in context of Ad9a (No. of rats that developed tumors/rats infected with virus)
|
|
|---|---|---|
| Females | Males | |
| wt | 3/3 | 0/2 |
| T108S | 4/4 | 0/3 |
| Region I/II mutants | ||
| IA | 0/5 | 0/3 |
| IIA | 0/4 | 0/3 |
| IIB | 0/5 | 0/4 |
| Region III mutants | ||
| IIIA | 0/7 | 0/4 |
| T123D | 0/5 | 0/2 |
| V125A | 0/5 | 0/2 |
| IIIC | 0/5 | 0/3 |
| IIID | 0/5 | 0/4 |
Newborn Wistar–Furth rats were injected subcutaneously with 7 × 107 PFU of virus and monitored by palpation for mammary tumor development for eight months
Discussion
In this paper, we demonstrated that interactions between the Ad9 E4-ORF1 oncoprotein and cellular PDZ proteins result in constitutive stimulation of cellular PI3K, as well as its downstream effectors PKB and S6K (Figures 1, 3, and 5). These findings are significant in uncovering a new PDZ-protein-dependent mechanism for the activation of the PI3K signaling pathway. Consistent with the established link between disregulation of this signaling pathway and cancer, we also showed that the ability of E4-ORF1 to activate PI3K is required both for E4-ORF1-mediated transformation of cells and for Ad9-induced mammary tumorigenesis in animals (Figure 7 and Table 1). The fact that stimulation of the PI3K cascade engenders a potent survival signal in cells may indicate that E4-ORF1 likewise functions during the Ad life cycle to block programmed cell death.
One question arising from our results is whether PI3K activation by E4-ORF1 depends on its ability to bind one or more of its PDZ-protein targets. Certain observations suggest but do not prove that full PI3K activation by E4-ORF1 may result from the sum of its interactions with all these PDZ proteins. First, E4-ORF1 colocalizes with both MUPP1 and ZO-2 at the plasma membrane (Figure 2c) where E4-ORF1-mediated PI3K activation occurs (Figure 1c). Additionally, MAGI-1 and DLG have been shown to localize at the plasma membrane of cells (Muller et al., 1995; Nishimura et al., 2000), and our preliminary findings suggest that E4-ORF1 similarly colocalizes with these two PDZ proteins at this site. While our results indicate that binding of E4-ORF1 to PDZ proteins is necessary for PI3K stimulation, these interactions are clearly not sufficient because this activity is abolished by disruptive mutations within either the PDZ-domain-binding motif or region I/II of E4-ORF1 (Figures 1b and d). The function of region I/II is not presently understood, but E4-ORF1 region I/II mutants, unlike region III mutants, retain the ability to bind PDZ proteins and to become recruited to the plasma membrane (Figure 2b). Therefore, we speculate that region I/II may bind an unidentified cellular factor(s) or confer E4-ORF1 with an undetermined enzymatic activity, either function of which could be envisioned to subvert the activity of targeted PDZ-protein complexes for selective stimulation of PI3K.
While E4-ORF1 clearly must activate PI3K signaling to transform cells, this viral protein likely has at least one additional activity crucial for this process. In this regard, although constitutively activated forms of PI3K or PKB have been shown to transform some cells (Chang et al., 1997; Klippel et al., 1998; Mirza et al., 2000), such proteins lack the potent transforming activity of E4-ORF1 in CREF cells (unpublished data). Moreover, subgroup A–C Ad E4-ORF1 proteins stimulate PI3K (Figure 3e) and have limited transforming potential (Weiss et al., 1997b), but only subgroup D Ad9 E4-ORF1 can both transform CREF cells and promote tumors in animals (Javier, 1994; Weiss et al., 1997b). Regarding the latter observation, we recently linked the unique oncogenic properties of Ad9 E4-ORF1 to a select interaction with the candidate tumor-suppressor protein ZO-2 (Glaunsinger et al., 2001). E4-ORF1 was found to sequester a large fraction of ZO-2, as well as MUPP1 and MAGI-1, in the cytoplasm of CREF cells (Glaunsinger et al., 2000, 2001; Lee et al., 2000). Further considering that DLG is the mammalian homolog of the Drosophila discs-large tumor-suppressor protein (Woods and Bryant, 1991), we speculate that the PDZ-protein targets of E4-ORF1 exist both at the membrane and in the nucleus/cytoplasm and that these two separate protein pools have distinct functions, either to organize signaling complexes or to suppress inappropriate cell proliferation, respectively. Studies reporting the existence of membrane and nuclear forms of several different MAGUK proteins, including ZO-2, are consistent with this idea (Gottardi et al., 1996; Hsueh et al., 2000; Islas et al., 2002). Thus, the oncogenic potential of E4-ORF1 may result from its combined capacities to activate PI3K by modifying PDZ-protein complexes at the plasma membrane and to inhibit antiproliferative signals by sequestering nuclear/cytoplasmic forms of the PDZ proteins.
We do not understand how E4-ORF1 selectively stimulates the PI3K signaling pathway. As PDZ proteins typically link membrane receptors to cytosolic signaling factors, one possibility is that the PDZ-protein targets of E4-ORF1 organize membrane protein complexes that function specifically to modulate signal transduction both to and from PI3K. Selective PI3K stimulation by E4-ORF1 might therefore occur through its ability to disregulate the activity of such specialized complexes. Regarding the possible nature of these protein complexes, PDZ proteins have been associated with monomeric/heterotrimeric G proteins and receptor tyrosine kinases (Lou et al., 2001), which can transduce activation signals to PI3K (Blume-Jensen and Hunter, 2001), and also with the tumor-suppressor protein PTEN, which can antagonize activation signals emanating from PI3K. Interestingly, two recent studies demonstrated that the MAGI-1-related proteins MAGI-2 and MAGI-3 are able to bind PTEN and to increase its ability to repress PI3K signaling (Wu et al., 2000a, b). We have found that MUPP1, MAGI-1, and DLG can likewise form complexes with PTEN (unpublished data). Thus, an intriguing scenario is that potent PDZ-protein-dependent stimulation of the PI3K signaling pathway by E4-ORF1 involves not only activation of PI3K but also inhibition of PTEN.
Materials and methods
Plasmids
E4-ORF1 genes were introduced into pJ4Ω (Weiss et al., 1997a), pBABE-puro (Morgenstern and Land, 1990), pGW1 (British Biotechnologies), and pEGFP-C3 (Clontech) expression plasmids. pCMVBam3-Neo plasmids expressing E4-ORF1 (Weiss et al., 1997a, b) and pGW1 plasmids expressing amino-terminal HA epitope-tagged MAGI-1, MAGI-1ΔPDZ1, MAGI-1ΔPDZ3, MAGI-1ΔPDZ1+3, MAGI-1Δ5PDZ, ZO-2, ZO-2ΔPDZ1, MUPP1, or MUPP1ΔPDZ7+10 (Glaunsinger et al., 2000, 2001; Lee et al., 2000) were described previously. Plasmids were also generously supplied by Julian Downward (pSG5-RasV12, pGFP–AH, pGFP–AHR25C), Melanie Cobb (pCEP4–HA-ERK2), James Woodgett (pCDNA3-HA-PKB), Arnold Levine (pSP72–RasV12), Lewis Williams (pCG–HA–p85), Joseph Avruch (pMT2–HA–p70S6K), Michael Greenberg (pECE–HA–FKHRL1 wt, pECE–HA-FKHRL1 TM, pGL3–(3x)FHRE, pEF–lacZ), Kyung-Ok Cho (pGW1–HA–SAP97), and Guy James (pCDNA3-MAGI-1b–ΔPDZ2 (aa 625–702 deleted), −ΔPDZ4 (aa 953–1025 deleted), −ΔPDZ5 (aa 1043–1116 deleted), −ΔPDZ3+4, −ΔPDZ3+5, −ΔPDZ4+5). After attachment of an HA–epitope tag at their amino–termini, MAGI–1b deletion–mutant cDNAs were introduced into pGW1, as were cDNAs coding for RasV12, MUPP1−PDZ10−13 (aa 1531–2054), MUPP1−PDZ11−3 (aa 1701–2054), SAP97ΔPDZ1+2 (aa 226–368 deleted), ZO-2-NT (aa 1–572), and amino-terminal myc epitope–tagged PKB.
Cells
Cells were maintained in culture medium (DME supplemented with gentamicin and FBS). CREF and TE85 lines stably expressing wt or mutant Ad9 E4-ORF1 were isolated by selection with a puromycin-resistance marker. With the exception of CREF-9ORF1-low cells, which express low levels of the E4-ORF1 protein (Weiss et al., 1997a), pBABE-puro expressing wt Ad9 E4-ORF1 was used to establish CREF-E4-ORF1 lines. The 20-8 cell line was derived from an Ad9-induced rat mammary tumor.
Cell transfections and treatments
Transfections were performed according to manufacturers’ recommendations using Fugene6 (Roche), Lipofectamine or Lipofectamine plus (Invitrogen Life), or by the calcium phosphate precipitation method (Kingston, 1989). Experimental analyses were routinely conducted 48 h post-transfection. In some experiments, cells were serum starved by incubation in culture medium lacking serum for 16–24 h, at which time, cells were treated for 10–20 min with indicated doses of PDGF-BB (Gibco), wortmannin (Sigma), LY294002 (Cell Signaling Technologies), rapamycin (Sigma), or DMSO vehicle (untreated) as a control.
Antisera and antibodies
Rabbit polyclonal antibodies to Ad9 E4-ORF1, MUPP1, ZO-2, and DLG were described (Javier, 1994; Lee et al., 1997, 2000; Glaunsinger et al., 2001). Antibodies purchased from Cell Signaling Technologies (anti-Akt, anti-phospho-Akt (Ser473), anti-phospho-Akt(Thr308), anti-S6 Kinase, anti-phospho-S6 Kinase(Thr389), anti-phospho-S6 Kinase(Thr421/Ser424), anti-phospho-FKHRL1(Thr32)), Transduction Labs (anti-p27kip1), Upstate (anti-PI3K p85), Covance (16B12 anti-HA), Santa Cruz (9E10 anti-myc), Southern Biotechnology Associates Inc. (horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse IgG), and Molecular Probes (goat anti-rabbit IgG conjugated to either Alexa Fluor 594 or FITC) were used according to manufacturers’ recommendations.
Extract preparation, immunoprecipitations, and immunoblot assays
Methods for preparation of cell extracts in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) or NETN buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40) supplemented with protease inhibitors and phosphatase inhibitors have been described (Lee et al., 1997). Protein concentrations in cell extracts were determined by the Bradford method (Bradford, 1976). Immunoprecipitation and immunoblot analyses were carried out as described previously (Weiss et al., 1996).
In vitro lipid kinase assays
In vitro lipid kinase assays were performed on total cell extracts as described previously (Susa et al., 1992). A Storm Molecular Dynamics Phosphorimager with ImageQuant software was used to quantify the amount of [32P]-labeled D3–phosphorylated phosphatidylinositol produced in each reaction.
In vitro protein kinase assays
Immunocomplexes of PKB, ERK2, or S6K were subjected to in vitro kinase assays with the appropriate protein or peptide substrate by standard methods (Pelech and Krebs, 1987; Coso et al., 1995; Franke et al., 1995). 32P-labeled protein substrates were separated by SDS-PAGE and quantified with a Storm Molecular Dynamics phosphorimager. 32P-labeled phosphorylated RRRLSSLRA peptide substrate (Upstate Biotech) bound to P81 ion-exchange cellulose (Whatman) was quantified using a Beckman LS 3801 scintillation counter.
Fluorescence microscopy analyses
Immunofluorescence assays were performed as described previously (Weiss et al., 1997a), except that cells grown on coverslips were fixed for 10 min at RT in 4% paraformaldehyde (Polysciences, Inc.) and then permeablized for 5 min at RT in 0.5% Triton X–100. Coverslips were mounted onto slides with Slowfade Light (Molecular Probes), and cells were visualized with a Zeiss Axioplan 2 epifluorescence microscope and photographed with a CoolSnap HQ CCD camera (Photometrics). Deconvolution microscopy was carried out on a Zeiss Axiovert S100 TV microscope and a Deltavision Restoration Microscopy System. Z-series digital images were deconvolved with the Deltavision constrained iterative algorithm.
Luciferase assays
Cell extracts were subjected to luciferase assays using the Luciferase Assay System (Promega) as recommended by the manufacturer. Luciferase activities were normalized to β-galactosidase activity generated by pEF-lacZ, which was included as an internal control plasmid in each transfection. β-Galactosidase assays were performed by standard methods (Kingston, 1989).
Focus and soft-agar growth assays
Focus and soft-agar growth assays were carried out as described previously (Javier, 1994; Thomas et al., 1999).
Mutant virus construction, virus infections, and virus tumor assays
Ad9 E4-ORF1 mutant genes were reintroduced into the natural genomic location of an Ad9 infectious plasmid, from which virus was recovered, amplified, and titered as previously described (Thomas et al., 2001). For tumor studies, newborn Wistar–Furth rats (Harlan Sprague–Dawley, Indianapolis, IN, USA) were subcutaneously inoculated with virus on both flanks (Thomas et al., 2001). Caring and handling of animals were in accordance with the institutional guidelines.
Acknowledgments
We thank Lyuba Varticovski for suggestions on performing PI3K assays. Predoctoral fellowships from the Viral Oncology Training Grant (T32 CA09197) to KKF and DLT, the US Army Breast Cancer Training Grant (DAMD17-94-J4204) to SSL, RSW, and IJL, and the Molecular Virology Training Grant (T32 AI07471) to BG helped to support this study. This research was funded by grants to RTJ from the National Cancer Institute (RO1 CA58541) and from the US Army (DAMD17-97-1-7082).
References
- Aoki M, Blazek E, Vogt PK. Proc Natl Acad Sci USA. 2001;98:136–141. doi: 10.1073/pnas.011528498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Bowman T, Garcia R, Turkson J, Jove R. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Cantrell DA. J Cell Sci. 2001;114:1439–1445. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, Cantley LC, Roberts TM, Vogt PK. Science. 1997;276:1848–1850. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Dufner A, Thomas G. Exp Cell Res. 1999;253:100–109. doi: 10.1006/excr.1999.4683. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. Oncogene. 2000;19:5270–5280. doi: 10.1038/sj.onc.1203906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaunsinger BA, Weiss RS, Lee SS, Javier R. EMBO J. 2001;20:5578–5586. doi: 10.1093/emboj/20.20.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottardi CJ, Arpin M, Fanning AS, Louvard D. Proc Natl Acad Sci USA. 1996;93:10779–10784. doi: 10.1073/pnas.93.20.10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh YP, Wang TF, Yang FC, Sheng M. Nature. 2000;404:298–302. doi: 10.1038/35005118. [DOI] [PubMed] [Google Scholar]
- Islas S, Vega J, Ponce L, Gonzalez-Mariscal L. Exp Cell Res. 2002;274:138–148. doi: 10.1006/excr.2001.5457. [DOI] [PubMed] [Google Scholar]
- Javier R, Raska K, Jr, Macdonald GJ, Shenk T. J Virol. 1991;65:3192–3202. doi: 10.1128/jvi.65.6.3192-3202.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javier RT. J Virol. 1994;68:3917–3924. doi: 10.1128/jvi.68.6.3917-3924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimble J, Simpson P. Annu Rev Cell Dev Biol. 1997;13:333–361. doi: 10.1146/annurev.cellbio.13.1.333. [DOI] [PubMed] [Google Scholar]
- Kingston RE. In: Current Protocols in Molecular Biology. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Greene Publishing Associates and Wiley-Interscience; New York, NY: 1989. [Google Scholar]
- Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. Proc Natl Acad Sci USA. 1997;94:11612–11616. doi: 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Escobedo MA, Wachowicz MS, Apell G, Brown TW, Giedlin MA, Kavanaugh WM, Williams LT. Mol Cell Biol. 1998;18:5699–5711. doi: 10.1128/mcb.18.10.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Glaunsinger B, Mantovani F, Banks L, Javier RT. J Virol. 2000;74:9680–9693. doi: 10.1128/jvi.74.20.9680-9693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Weiss RS, Javier RT. Proc Natl Acad Sci USA. 1997;94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Yano H, Lee F, Chao MV, Farquhar MG. Mol Biol Cell. 2001;12:615–627. doi: 10.1091/mbc.12.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Mills GB, Lu Y, Fang X, Wang H, Eder A, Mao M, Swaby R, Cheng KW, Stokoe D, Siminovitch K, Jaffe R, Gray J. Semin Oncol. 2001;28:125–141. doi: 10.1016/s0093-7754(01)90290-8. [DOI] [PubMed] [Google Scholar]
- Mirza AM, Kohn AD, Roth RA, McMahon M. Cell Growth Differ. 2000;11:279–292. [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller BM, Kistner U, Veh RW, Cases-Langhoff C, Becker B, Gundelfinger ED, Garner CC. J Neurosci. 1995;15:2354–2366. doi: 10.1523/JNEUROSCI.15-03-02354.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W, Iizuka T, Hirabayashi S, Tanaka N, Hata Y. J Cell Physiol. 2000;185:358–365. doi: 10.1002/1097-4652(200012)185:3<358::AID-JCP6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Pelech SL, Krebs EG. J Biol Chem. 1987;262:11598–11606. [PubMed] [Google Scholar]
- Rameh LE, Chen CS, Cantley LC. Cell. 1995;83:821–830. doi: 10.1016/0092-8674(95)90195-7. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sala C. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- Susa M, Keeler M, Varticovski L. J Biol Chem. 1992;267:22951–22956. [PubMed] [Google Scholar]
- Suzuki T, Ohsugi Y, Uchida-Toita M, Akiyama T, Yoshida M. Oncogene. 1999;18:5967–5972. doi: 10.1038/sj.onc.1203008. [DOI] [PubMed] [Google Scholar]
- Thomas DL, Schaack J, Vogel H, Javier R. J Virol. 2001;75:557–568. doi: 10.1128/JVI.75.2.557-568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DL, Shin S, Jiang BH, Vogel H, Ross MA, Kaplitt M, Shenk TE, Javier RT. J Virol. 1999;73:3071–3079. doi: 10.1128/jvi.73.4.3071-3079.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsanis C, Spandidos DA. Int J Mol Med. 2000;5:583–590. doi: 10.3892/ijmm.5.6.583. [DOI] [PubMed] [Google Scholar]
- Tsunoda S, Zuker CS. Cell Calcium. 1999;26:165–171. doi: 10.1054/ceca.1999.0070. [DOI] [PubMed] [Google Scholar]
- Uthoff SM, Eichenberger MR, McAuliffe TL, Hamilton CJ, Galandiuk S. Mol Carcinogen. 2001;31:56–62. doi: 10.1002/mc.1039. [DOI] [PubMed] [Google Scholar]
- Vogt PK. Oncogene. 2001;20:2365–2377. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- Watton SJ, Downward J. Curr Biol. 1999;9:433–436. doi: 10.1016/s0960-9822(99)80192-4. [DOI] [PubMed] [Google Scholar]
- Weiss RS, Javier RT. J Virol. 1997;71:7873–7880. doi: 10.1128/jvi.71.10.7873-7880.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RS, Gold MO, Vogel H, Javier RT. J Virol. 1997a;71:4385–4394. doi: 10.1128/jvi.71.6.4385-4394.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RS, Lee SS, Prasad BV, Javier RT. J Virol. 1997b;71:1857–1870. doi: 10.1128/jvi.71.3.1857-1870.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RS, McArthur MJ, Javier RT. J Virol. 1996;70:862–872. doi: 10.1128/jvi.70.2.862-872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods DF, Bryant PJ. Cell. 1991;66:451–464. doi: 10.1016/0092-8674(81)90009-x. [DOI] [PubMed] [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, Wood J, Ross C, Sawyers CL, Whang YE. Proc Natl Acad Sci USA. 2000a;97:4233–4238. doi: 10.1073/pnas.97.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky LA. J Biol Chem. 2000b;275:21477–21485. doi: 10.1074/jbc.M909741199. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Araki M. J Cell Sci. 2001;114:2375–2382. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- Yu Y, Sato JD. J Cell Physiol. 1999;178:235–246. doi: 10.1002/(SICI)1097-4652(199902)178:2<235::AID-JCP13>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]