

Abstract
Previous studies have shown ubiquitin activating enzyme E1 to be sensitive to adduction through both Michael addition and SN2 chemistry in vitro. E1 presents a biologically important putative protein target for adduction due to its role in initiating ubiquitin based protein processing and the involvement of impaired ubiquitin protein processing in two types of familial Parkinson's disease. We tested whether E1 is susceptible to xenobiotic-mediated electrophilic adduction in vivo and explored the potential contribution of E1 adduction to neurodegenerative events in an animal model. N,N-Diethyldithiocarbamate (DEDC) was administered to rats using a protocol that produces covalent cysteine modifications in vivo and brain E1 protein adducts were characterized and mapped using shotgun LC-MS/MS. E1 activity, global and specific protein expression, and protein carbonyls were used to characterize cellular responses and injury in whole brain and dorsal striatal samples. The data demonstrate that DEDC treatment produced S-(ethylaminocarbonyl) adducts on Cys234 and Cys179 residues of E1 and decreased the levels of activated E1 and total ubiquitinated proteins. Proteomic analysis of whole brain samples identified expression changes for proteins involved in myelin structure, antioxidant response and catechol metabolism, systems often disrupted in neurodegenerative disease. Our studies also delineated localized injury within the striatum as indicated by decreased levels of tyrosine hydroxylase, elevated protein carbonyl content, increased antioxidant enzyme and α-synuclein expression, as well as enhanced phosphorylation of tau and tyrosine hydroxylase. These data are consistent with E1 having similar susceptibility to adduction in vivo as previously reported in vitro and support further investigation into environmental agent adduction of E1 as a potential contributing factor to neurodegenerative disease. Additionally this study supports the predictive value of in vitro screens for identifying sensitive protein targets that can be used to guide subsequent in vivo experiments.
Introduction
Electrophilic chemical species generated from the metabolism of xenobiotics and endogenous oxidative processes can react with nucleophilic sites of proteins to generate covalent adducts that impair function through altering conformation or blocking active sites.1–3 Loss of function mutations in single proteins underlie a variety of inherited diseases, suggesting that selective adduction of critical proteins, if accompanied by loss of function, may also contribute to the risk for disease. However, relative to the current state of knowledge regarding the role that DNA adducts play in carcinogenesis comparatively few biologically important target proteins have been identified for environmental agents, and the mechanisms through which protein adduction can mediate adverse biological consequences are still evolving. Thus, identifying susceptible proteins for adduction by environmental agents and delineating how specific adduction can contribute to human disease are topical questions.
Multiple experimental approaches have been used to address these questions but characterizing the specificity and distribution of adducts on a proteome wide basis within biological systems remains an analytical challenge. Earlier investigations using radiolabeling and two-dimensional gel-based proteomics supported the notion of non-random modification of proteins by electrophiles within biological systems but were limited by the number of proteins that could be identified and the capacity to characterize the structure and location of these adducts.4–6 However, more recent advancements in affinity chromatography enrichment and multi dimensional LC/MS/MS coupled with biotin labeled electrophiles have made it possible to identify modified proteins and map adducts to specific amino acids within proteins.7–10 Interestingly, studies of cellular fractions of HEK293 cells demonstrated that a relatively small number of 90 cytoplasmic proteins were reproducibly modified at specific nucleophilic sites by electrophiles through both Michael addition and bimolecular nucleophilic substitution (1SN2).11 It was proposed that those proteins might represent a subset of redox sensitive proteins containing hyper reactive cysteines. If so they present a logical pool of putative targets for reactive species derived from environmental agents or generated during oxidative stress.
Among the 90 proteins identified to be susceptible to adduction was ubiquitin activating enzyme E1 (E1).11 This protein presents an important potential protein target for adduction in many cell types due to its role in initiating ubiquitin based protein-processing and thus having the potential to compromise many and varied cellular functions including protein trafficking, transcription factor activity, protein degradation, and multiple signaling pathways.12, 13 More specifically, two familial forms of Parkinson's disease are associated with loss of function mutations in a ubiquitin ligase, Parkin, and a dual function deubiquintinating and ligase enzyme, ubiquitin carboxy-terminal hydrolase L1.14 Dopaminergic cell lines also appear to exhibit an increased susceptibility to E1 inhibition suggesting that the nigrostriatal system may be a sensitive region for compromised E1 function.15 In this study we tested whether E1 has similar susceptibility to xenobiotic-mediated electrophilic adduction in vivo as that observed in vitro and explored the potential contribution of E1 adduction to neurodegenerative processes. N,N-diethyldithiocarbamate (DEDC) was selected as a test agent based upon its recognized metabolism to electrophilic species and previous studies reporting that DEDC and its dimethyl analog inhibit E1 activity and proteasome function in vitro.15 DEDC was administered to rats using an established protocol known to generate covalent cysteine modifications in vivo and the protein adducts present in brain E1 digests characterized and mapped using shotgun LC-MS/MS.16 An initial assessment of the biological significance of in vivo E1 adduction was performed through evaluating E1 activity and using proteomic and biochemical endpoints to assess oxidative stress and injury within whole brain and specifically in the dorsal striatum. The results demonstrate that E1 is modified through electrophilic adduction in vivo at the same residue identified on human E1 in vitro and that this adduction is associated with inhibition of E1 and alterations of biological processes known to be disrupted in neurodegenerative disease states.
Materials and Methods
Reagents and supplies
2ML4 Alzet® osmotic pumps were obtained from Braintree Scientific (Braintree, MA). Sodium N,N-diethyldithiocarbamate (DEDC) was obtained from Alfa Aesar (Ward Hill, MA). Bovine serum albumin (BSA), EDTA, sodium chloride, dithiothreitol and tris-base were obtained from Sigma-Aldrich (St. Louis, MO). Protease and phosphatase inhibitors were purchased from Amersham Biosciences (Piscataway, NJ) and Sigma-Aldrich (Saint Louis, MO). Dulbecco's PBS (pH 7.4) was purchased from MP Biomedicals (Irvine, CA). All HPLC grade solvents were purchased from Fisher Scientific (Pittsburgh, PA). Dynabeads®Protein G was purchased from Invitrogen Corporation (Carlsbad, CA).
Animals and exposures
All treatments and procedures using animals were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Vanderbilt University. Twenty adult male Sprague-Dawley rats were obtained from Harlan Bioproducts (Indianapolis, IN) and caged at Vanderbilt University animal facilities in a temperature controlled room (21–22 °C) with a 12 h light-dark cycle, supplied with Purina Lab Diet 5001, a segment of 4 in diameter PVC tubing for environmental enrichment and water ad libitum. After a 10–14 day acclimatization period, ten animals were assigned to either a control or DEDC-exposed group. The exposure group was administrated DEDC at 0.3 mmol/kg/day using intra-abdominal 2mL 4-week Alzet® osmotic pumps (Braintree Scientific, Braintree, MA) surgically implanted under anesthesia (100 mg/Kg ketamine with 8 mg/Kg xylazine ip). Exposure duration was 4 weeks (n=6) except for the animals used for the differential gel electrophoresis proteomic analyses (n=4) in which the osmotic pumps were replaced after 4 weeks to extend the exposure period to 8 weeks. The selected exposure periods were based upon protocols previously established to produce detectable levels of DEDC-derived cysteine adducts and to allow for the development of subchronic neurodegenerative effects.16 Controls were implanted with pumps delivering phosphate buffered saline at pH 7.4. The average starting body weight of all animals was 313 ± 9 g (SEM).
Collection and preparation of whole brain and striatal proteins
At the end of 4 weeks exposure, control and exposed animals were deeply anesthetized with pentobarbital (100 mg/Kg body weight, ip) and perfused through the left ventricle of the heart with PBS (pH 7.4) and tissues collected. The brains were removed, the hemispheres separated with a sagittal cut and immediately frozen in liquid nitrogen and stored at −80 °C until further analysis.
Whole brain protein samples were prepared from approximately 150 mg of frozen brain (~ 2mm thick midline sagittal section of the left hemisphere) that was powdered in a mortar and pestle containing liquid nitrogen. Total proteins were extracted using 0.7 mL cold TNE buffer (2 mM EDTA, 150 mM NaCl, 50 mM Tris-base, 2 mM DTT, and NP-40 (1%, v/v)), containing protease inhibitors (P-2714 Sigma-Aldrich, St. Louis, MO, and 80-6501-23 protease inhibitor mix, Amersham Biosciences, Piscataway, NJ) and phosphatase inhibitors (P-2850 and P-5726 Sigma-Aldrich). The homogenate was sonicated in an ice/water bath for 3 min and centrifuged at 40,485×g for 40 min at 4 °C. The supernatant was collected and stored at −80 °C. Protein concentration in the supernatant of the tissue samples was measured by a modified Bradford method17 using bovine serum albumin (BSA) as the standard.
Dorsal striatum protein samples were dissected from frozen coronal slices of the left-brain hemisphere and proteins were extracted with cold TNE lysis buffer using a sonicator (Sonic Dismembrator Model 60, Fisher Scientific). After centrifugation at 28,928×g for 30 min at 4 °C, the supernatant was collected and stored at −80 °C. Protein concentration in the supernatant was measured by a modified Bradford method using BSA as the standard.17
Isolation of E1 protein and characterization of covalent modifications by LC/MS/MS
The E1 protein was isolated from brain protein extracts of DEDC exposed and control rats. Briefly, 1.5 mg of Dynabeads®Protein G were incubated with 3 μL of ubiquitin activating enzyme (N-terminus) E1 antibody (PW8385, Enzo Life Sciences, Plymouth Meeting, PA) for 10 min at room temperature with rotation. Bound bead/Protein G/antibody complex was washed twice. Then 450 μL of pooled whole brain tissue extracts (n=5 per group) containing 5 mg protein was added, mixed by pipetting and incubated overnight at 4 °C with rotation. The captured bead/Protein G/antibody/E1 complex was washed several times, and antibody/E1 complex was eluted by boiling at 70 °C for 10 min in 20 μl of reducing sample loading buffer (5 μL of NUPAGE-LDS sample buffer, 2 μL of 0.5 M DTT, 13 μL deionized water). The antibody/E1 complex was separated on a 10% denaturing SDS-PAGE gel and proteins visualized by colloidal Coomassie blue. The protein band corresponding to E1 with apparent molecular weight of approximately 110 kDa was excised. In gel digestion with trypsin, elastase, and subtilysin was performed and the resulting peptides separated by capillary high performance liquid chromatography coupled directly to the inlet of a ThermoFisher LTQ-orbitrap. Full scan MS data were collected using high-resolution scans within the orbitrap while MS/MS spectra were collected in the LTQ portion of the instrument. MS/MS spectra were extracted from the instrument data file using the ScanSifter program. Spectra were compared to the rat database (UniprotKB v155) using SEQUEST considering dynamically the differential mass shifts of 71.0368 and 99.13 on cysteine residues.18 Based on number of identifications to a reversed protein version of the rat database, IDPicker was used to filter identifications to a false discovery rate of 5%.
Analysis of protein-conjugated ubiquitin
To assess the potential of DEDC to inhibit protein ubiquitination, the ubiquitinated protein levels in whole brain protein samples were analyzed by slot blot. Equal amounts of brain protein (as determined by modified Bradford assay) were bound to Immobilon-P membranes (Millipore, MA) using the Bio-Dot® Blot apparatus (BioRad, CA) in triplicate for each control and exposed animal. A standard curve also was generated on the membrane using bovine serum albumin (BSA). The amount of protein in each slot was then quantified by densitometry using the MemCode™Reversible Protein Stain Kit for PVDF membranes (Pierce, IL) and the BSA standard curve. The membranes were blocked in blocking buffer (5% nonfat powdered skim milk in tris buffered saline, pH 7.4 (TBS)) at room temperature for 1 h and then probed with a mouse monoclonal antibody conjugated to horseradish peroxidase specific for protein-conjugated ubiquitin, (PW0150 Enzo Life Sciences, PA). To determine the amount of ubiquitinated protein, as well as in immunoblots described below unless otherwise noted, the membranes were incubated with Western Lightning Chemiluminescence Reagent Plus (Perkin-Elmer LAS, Inc, Boston, MA) substrate according to the manufacturer's directions, and exposed to Kodak X-Omat Blue XB-1 film. The films were scanned with a GS-700 densitometer and analyzed using Quantity One 1-D Analysis Program version 4.1 (Bio-Rad, CA). The optical density values were then normalized to mg of protein on the membrane quantified using the BSA standard curve and statistical comparisons performed using the two-tailed unpaired t test.
Analysis of activated E1 protein
E1 activity was assessed by measuring ubiquitinated E1 using a previously published approach.19 Aliquots containing equal amounts of whole brain protein (10 μg) obtained from DEDC exposed (n=6) and control (n=5) rats were separated by SDS PAGE and transferred to PVDF membranes. Nonspecific binding sites were blocked in blocking buffer at room temperature for 1 h. After blocking, the membranes were probed with a primary polyclonal rabbit antibody directed toward the E1 ubiquitin activating enzyme (PW8385 Enzo Life Sciences, PA). After washing, the membranes were incubated with an anti-rabbit peroxidase conjugated secondary antibody (A-8275, Sigma, St. Louis, MO). The E1 protein was visualized using chemiluminecence as two bands migrating with molecular weights of approximately 110 and 117 kDa. The ratio of the activated higher molecular weight ubiquitinated E1 species to the nonactivated lower molecular weight species was calculated using densitometry. Comparison of the mean ratios for the two groups was performed using the unpaired t-test.
E1 Inhibition in vitro by ethylisocyanate
To assess the potential of E1 adduction to inhibit ubiquitin activation recombinant human His6-ubiquitin activating enzyme (10 nM) (Boston Biochem, Boston, MA) was incubated with and without the putative ultimate alkylating species of DEDC, ethylisocyanate (5 nM), for 30 min in reaction buffer consisting of 50 mM Tris-HCl and 50 mM NaCl, pH 7.4, with 0.1 mM DTT and 0.1 mg/ml BSA (n = 4 for each condition). After incubation the activity of E1 was assayed by addition of ubiquitin (10 nM) and MgCl2/ATP (5mM) and incubation at 37 °C for 30 minutes. The amount of the ubiquitin-E1 conjugated species (~117 kDa) was determined by western blot using anti conjugated ubiquitin antibody and chemiluminescence detection as described above in Analysis of protein-conjugated ubiquitin. The ethylisocyanate incubated sample E1 band intensities were expressed as a percentage of the band intensities obtained for E1 without ethylisocyante.
Two-dimensional difference gel electrophoresis
Approximately 2 mm midsagittal sections of right hemisphere frozen brain were powdered with a mortar and pestle in liquid nitrogen and proteins extracted in chilled TNE buffer (2mM EDTA, 150 mM NaCl, 50mM Tris-base, 2 mM DTT, NP-40 (1%, v/v), containing phosphatase inhibitor cocktail I (100 μl/10 ml of TNE buffer), phosphatase inhibitor cocktail II (100 μl/10 ml of TNE buffer), protease inhibitor cocktail (20 μl/10 ml of TNE buffer), and protease inhibitor mix (100 μl/10 ml of TNE buffer), and subjected to sonication at 0 °C for 1 min. Protein expression levels were determined by difference gel electrophoresis (DIGE) using NHS-reactive cydyes (cy2, cy3 and cy5) and a mixed-sample internal standard methodology essentially as described previously20; all reagents and equipment for DIGE were from GE Healthcare (Piscataway, NJ). A pooled reference standard was comprised of equal aliquots from each of the eight samples, and labeled en masse with cy2. Pairs of cy3/5-labeled samples were then mixed with an equal portion of the cy2-labeled standard mixture, and the resulting tripartite mixtures were co-resolved using four 2D-gel separations. Two different isoelectric focusing ranges of pH 4–7 and pH 7–11 (24 cm IPG strips) were analyzed using a manifold-equipped IPGphor II IEF cell according to the manufacture's recommended focusing protocols. Following focusing and second dimensional separation by large-format (24×20cm 12% SDS-PAGE) the individual components of the samples were imaged using mutually exclusive excitation and emission wavelengths for the Cy2, Cy3, and Cy5 dyes.20 Control:standard and exposed:standard ratios were calculated for each resolved protein and then normalized across the 4 DIGE gels in each set of experiments, i.e. pH 4–7 and pH 7–11. Protein features with abundance changes ≥ 1.3 fold increase or decrease and p values less than 0.05 (Student's unpaired t test) were identified and analyzed by MALDI-TOF peptide mapping and TOF/TOF tandem mass spectrometry following trypsin digestion.
Western analysis for heme oxygenase 1 (HO-1), Cu-Zn superoxide dismutase 1 (SOD-1), dopamine transporter (DAT), tyrosine hydroxylase (TH), phospho-ser40-tyrosine hydroxylase (phospho-TH), phospho-tau ser-396/404 (phospho-tau) and α-synuclein
Equal amounts (10–30 μg) striatal proteins were loaded and separated by 10% SDS-PAGE along with molecular weight markers (Novex®Sharp Pre-Stained Protein Standards (MW 3.5–260 kDa), MagicMark™XP Western Standard, Invitrogen Carlsbad, CA). Following electrophoresis, separated proteins were transferred onto an Immobilon-P membrane or Immobilon-PSQ for α-synuclein using an XCell II™ Blot Module (Invitrogen, Carlsbad, CA). Nonspecific binding sites were blocked with blocking buffer and the membranes were then incubated with the following primary antibodies; SOD-1 (FL-154: sc-11407, Santa Cruz Biotechnology, Santa Cruz, CA, dilution 1:5,000), HO-1 (OSA-111, Stressgen, Ann Arbor, MI, dilution 1:2,000), TH (TH: AB151, Millipore Inc., Billerica, MA, dilution 1:3,000), phospho-TH (TH phospho-ser40: KAP-TK125, Stressgen, Ann Arbor, MI, dilution 1:2,500), anti-dopamine transporter (N-terminal) (DAT: D6944, Sigma, St. Louis, MO, dilution 1:5,000), and α-synuclein (α-syn: BD Biosciences, Franklin Lakes, NJ, dilution 1:500), overnight at 4 °C. Striatal levels of phospho-tau were determined similarly by western blot using PHF-1 antibody that was generously provided to us by Dr. Peter Davies of Albert Einstein College of Medicine. Concurrently, with the above primary antibodies, membranes were also probed with primary anti-actin antibody (A-2066, rabbit anti-actin, Sigma, St. Louis, MO, dilution 1:5,000). After washing the membranes were incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (A-8275: anti-rabbit HRP, Sigma, St. Louis, MO, dilution 1:10,000 or SC-2314: donkey anti-mouse HRP, Santa Cruz Biotechnology, Santa Cruz, CA, dilution 1:10,000). Proteins were visualized by chemiluminescence. The presence of SOD-1 (MW ≈ 19 kDa) and HO-1 (MW ≈ 32 kDa) were confirmed by comparing the migration of positive control SOD-1 (bovine liver, Alexis Biochemicals, San Diego, CA) or HO-1 (rat liver microsome extract, Stressgen, Ann Arbor, MI). The presence of beta actin (MW = 42 kDa) was confirmed by comparison to the molecular weight standard. Protein levels were determined by densitometry and the optical density of each target protein was normalized to the optical density of beta actin within the same sample.
Protein carbonyl determination
The protein carbonyl content of samples and standards was determined by the fluoresceinamine-cyanoborohydride method using immunochemical detection as previously described.21 Briefly, 50 μg of whole brain or 25 μg of striatum protein were treated with fluoresceinamine (12 μL of 0.25 M) and sodium cyanoborohydride (10 μL of 0.4 M) for 1 hour at 37°C. Protein was precipitated at room temperature with ethanol:water:chloroform (4:3:1, v/v) washed 5 times with acidified ethanol:ethyl acetate (1:1) for 5 min at 37°C followed by centrifugation (13,000 rpm, 10 min) and then solubilized in 200 μL sodium hydroxide (0.1 N) for 15 min at 37°C. Treated proteins (controls or DEDC exposed) were bound to Immobilon-P membranes (Millipore, MA) using the Bio-Sot® Blot apparatus (BioRad, CA). Four replicates per sample containing approximately 0.25 μg of protein per well were loaded. Protein carbonyls were detected by chemiluminescence using the CDP-Star Universal Alkaline Phophatase kit (Sigma-Aldrich Co., MO) and quantified by densitometry. The protein carbonyl content of samples was determined from a standard curve generated using oxidized BSA for which carbonyl content was determined spectrophotometrically (ε=86,000 M−1cm−1 at 490 nm) using a Shimadzu UV-2401 PC. Oxidized BSA standards were prepared by incubating 10 mg of BSA dissolved in 1 mL of 20 mM TrisHCl (pH 7.4) with 100 mM Fe2+ and 100 mM hydrogen peroxide, at room temperature for 1 hour. Reduced BSA was prepared by mixing oxidized-BSA (10 mg/mL) with 5 mg of sodium borohydride for 30 min at 37°C. The quantity of protein bound to the PVDF membrane was determined using the MemCode™Reversible Protein Stain Kit for PVDF membranes (Pierce, IL) using BSA as standards.
Striatal dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) analysis
Biogenic amines were quantified by HPLC in samples obtained from the right dorsal striatum of control rats and rats exposed to DEDC through services provided by the Center for Molecular Neuroscience Neurochemistry Core Laboratory at Vanderbilt University. Punch biopsies from striatal coronal sections were homogenized using a tissue dismembrator in 150 μL of 0.1 M TCA, which contained 10 mM sodium acetate, 0.1 mM EDTA, 5ng/mL isoproterenol (as internal standard), and 10.5% methanol (pH 3.8). Following centrifugation at 10,000×g for 20 min, supernatant was removed and stored at 80 °C. The pellet was used to determine protein content using BCA Protein Assay Kit purchase from Pierce Chemical Company (Rockford, IL). Before injection into the HPLC the supernatant was thawed and centrifuged for 20 minutes. Samples of the supernatant were analyzed for biogenic monoamines. Biogenic amines were determined by a specific HPLC assay utilizing an Antec Decade II electrochemical detector set at a potential of 500mV and operated at 33 °C. Twenty μl samples of the supernatant were injected using a Water 717+ autosampler onto a Phenomenex Nucleosil (5u, 100A) C18 HPLC column (150 × 4.60 mm). Biogenic amines were eluted with a mobile phase consisting of 89.5% 0.1M TCA, 10−2 M sodium acetate, 10−4 M EDTA and 10.5 % methanol (pH 3.8). Solvent was delivered at 0.6 ml/min using a Waters 515 HPLC pump. HPLC control and data acquisition were managed by Millennium 32 software. The biogenic amines dopamine (DA) and 3,4-dihydroxyphenylacetic acid (DOPAC) were quantified using internal standards with electrochemical detection after separation by HPLC and reported as ng/mg protein.
Statistical Analyses
Unpaired one-tailed and two-tailed student's t-tests were performed using Prism 4 (Graphpad Software, Inc.) except for proteomics expression comparisons that used Decyder software version 6.5 (GE Healthcare Bio-Sciences). Statistical significance was taken to be p< 0.05 unless otherwise noted. Treatment groups consisted of n=4 for the 2D DIGE proteomic analyses and n=6 for all other analyses. However analyses using SDS PAGE were limited to 11 lanes so that an n=6 and n=5 were used for the exposed and control groups, respectively.
Results
Covalent Modification of E1 Activating Enzyme
Characterization of E1 Protein Covalent Modifications Produced by DEDC In Vivo
To determine if any of the expected potential adduction modifications were present on UBA1 after treatment with DEDC at 0.3mmol/Kg for 4 weeks, data dependent LC-MS/MS analysis of peptides derived from the UBA1 region of the gel-resolved immunopurified UBA1 were analyzed. These demonstrated excellent coverage of rat ubiquitin-activating enzyme E1 (UBA1_RAT) (70% by amino acid - see supplementary data for regions covered). Mass spectral data were also searched for modified peptides exhibiting masses consistent with the generation of the S-(aminocarbonyl) cysteine adducts (+99 Da for diethyl and +71 Da for monoethyl). This allowed for identification of the S-ethylaminocarbonyl adduct (+71.0368 Da) on Cys234 (DNPGVVTCLDEAR peptide - m/z 730.3502) and Cys179 (VGEFCHSR peptide - m/z 503.2346). In addition to the differential parent mass, localization is evidenced on the Cys234 peptide by a +71 Da shift of the y6–y12 and b8–b12 fragments and on the Cys179 peptide a +71 Da on the y7–y4 and b5–b7 fragments (Figure 1 and supplementary data). Ions at m/z of the parent peptide +71Da on either DNPGVVTCLDEAR or VGEFCHSR peptides derived from E1 were not observed from control animals. (see supplementary material).
Figure 1.

Covalent modification on Cys234 and Cys179 of E1 activating enzyme. MS/MS spectra obtained using a ThermoFisher LTQ-orbitrap of the S-(ethylaminocarbonyl) adduct on (A) Cys234 of E1 protein isolated from DEDC treated rats (+71 Da on the y6-y12 and b8-b12 fragments of DNPGVVTCLDEAR) and (B) Cys179 of E1 protein isolated from DEDC treated rats (+71 Da on the y7-y4 and b5-b7 fragments of VGEFCHSR).
Inhibition of E1 Activating Enzyme
Total Protein Ubiquitination
To explore potential impacts of in vivo E1 adduction, total ubiquitinated protein levels were measured using slot blot. There was a significant decrease in whole brain levels obtained from DEDC exposed rats relative to controls (two-tailed unpaired t-test p < 0.05, n = 6). The mean optical density values for conjugated ubiquitin normalized to μg of protein on the membrane are shown in Figure 2A.
Figure 2.

Inhibition of E1 activating enzyme by ethylisocyanate, the putative alkylating species of DEDC. (A) Mean values (+SE) of protein conjugated ubiquitin were quantified by slot blot and normalized to the total amount of protein bound to the membrane for each sample (see methods for details). (* p < 0.05 two-tailed unpaired t-test with n= 6) (B) Activated (E1-Ub) and non-activated (E1) species of E1 ubiquitin activating enzyme were separated by SDS-PAGE and visualized by western blot. The mean ratio E1-Ub/E1 values (+SE) for each treatment group are shown. (**p < 0.01 two-tailed unpaired t-test with n= 6 for exposed and n= 5 for control) (C) The mean value (+SE) of E1 activity incubated with ethylisocyanate measured as E1-Ub protein expressed as a percent of control. (**p < 0.01 two-tailed unpaired t-test with n= 4).
Levels of Activated E1
Because E1 and ubiquitinated E1 (Ub-E1) protein resolve as two bands migrating with molecular weights of approximately 110 and 117 kDa (Figure 2B) with the higher molecular weight band representing the activated form Ub-E1, it was possible to infer the level of E1 activation.15, 19 The mean ratios of the activated higher molecular weight species Ub-E1 to the non-activated lower molecular weight species were calculated by densitometry and are shown in Figure 2B. Comparison of the mean ratios for the two groups showed a significant decrease in the level of activated E1 in DEDC exposed animals relative to controls (two-tailed unpaired t-test p < 0.01, n = 5 controls and n = 6 exposed).
E1 Inhibition In Vitro by Ethylisocyanate
The mean value of ubiquitin conjugated E1 protein pre-incubated with the putative ultimate alkylating species of DEDC, ethylisocyanate, expressed as a percent of control is shown in Figure 2C. Incubation of recombinant human ubiquitin activating enzyme E1 with the ethylisocyanate produced a significant decrease of approximately 50% in activated E1 (two-tailed unpaired t-test p < 0.01, n = 4).
Cellular Responses and Oxidative Stress
Brain Protein Expression Assessed by Proteomics
We used a two-tiered approach to explore the biological impact of E1 adduction and inhibition in the brain. Global assessments were initially performed through evaluation of changes in protein levels of whole brain samples followed by subsequent evaluation of select biochemical endpoints of localized perturbations within the dorsal striatum. Changes in protein expression levels in DEDC-treated vs. control brains using 2D DIGE indicated 26 protein features with an increase or decrease expression change larger than 1.3 fold (p<0.05) that were subsequently identified by mass spectrometry. Nineteen features corresponding to 10 non-redundant proteins were identified in this manner, 9 of which were up regulated and 1 down regulated (see Table 1 for separations using isoelectric focusing between pH 4–7 and Table 2 for separations using isoelectric focusing between pH 7–11 representative annotated gels showing the approximate isoelectric points and molecular weights are included in the supplementary material). Among the proteins demonstrating increased levels were myelin structural proteins (myelin basic protein and 2', 3'-cyclic-nucleotide 3'-phosphodiesterase), antioxidant proteins (glutathione transferase pi, peroxiredoxin-6 and peroxiredoxin-2) and an enzyme involved in catechol synthesis (dihydropteridine reductase).
Table 1.
Proteins exhibiting significantly altered expression in DEDC exposure group vs. controls identified using pH 4–7 isoelectric focusing.
| Average ratio DEDC vs. controlsa | t-testb (p <) | Accession No.c | Protein identificationd | Protein |
|---|---|---|---|---|
|
| ||||
| 1.39 | .05 | PYGB_RAT P53534 | 129,24,0,24% | Glycogen phosphorylase, brain form-Rattus norvegicus |
|
| ||||
| 1.47 | .01 | DPYL2_RAT P47942 | 95,16(1),3,31% | Dihydropyrimi dinase-related protein 2-Rattus norvegicus |
|
| ||||
| 1.62 | .01 | ENOA_RAT P04764 | Alpha enolase-Rattus norvegicus | |
| 1.69 | .01 | |||
| 1.37 | .05 | 54,9(1),1,23% | ||
|
| ||||
| 1.37 | .01 | PRDX6_RAT O35244 | 84,5,1,20% | Peroxiredoxin-6 |
|
| ||||
| 1.28 | .01 | PRDX2_RAT P35704 | 42,7,1,27% | Peroxiredoxin-2 Rattus norvegicus |
average volume ratios for each protein spot identified
level of significance determined by students t-test using DeCyder software version 6.5 using the mixed sample internal standard methodology
Swiss-Prot database
Imported from MASCOT and GPS Explorer. Data include predicted MW and pI to correlate with gel position (see supplementary material for annotated gel images), and the additional four scores: the four numbers associated with the protein identifications: e.g., 951 ,16(1)2 ,33 ,31%4 represent:
1MOWSE score. MOlecular Weight SEarch score, a statistic calculated by the MASCOT database search algorithm. The 95th percentile cutoff is calculated for each search and indicated at the bottom of each analysis summary. About 5% of random matches could yield higher MOWSE scores. See matrixscience.com for more details.
2peptide #. number of peptide ion masses matched (number of un-matched ions).
3ms2 #. number of matching peptide ions with MS/MS data.
4(%coverage). Percent of the amino acids accounted for by the matching peptides
Table 2.
Proteins significantly altered expression in DEDC exposure group vs. controls identified using pH 7–11 isoelectric focusing.
| Average ratio DEDC Level vs. controlsa | t-testb(p <) | Accession No.c | Protein identificationd | Protein |
|---|---|---|---|---|
|
| ||||
| −1.94 | .01 | ECHA_RAT Q64428 | Hydroxyacyl-Coenzyme Adehydrogenase | |
| −1.91 | .01 | 77,15(l),0,23% | ||
| −1.79 | .01 | 56,12(1),2,12% | ||
| −2.01 | .01 | |||
|
| ||||
| 1.22 | .05 | CN37_RAT P13233 | 243,11,9,23% | 2',3'-cyclic-nucleotide 3'-phosphodiesterase-Rattus norvegicus |
| 1.3 | .05 | 288,16,7,30% | ||
| 1.33 | .05 | 249,17,6,33% | ||
| 1.25 | .05 | 120,12,4,23% | ||
|
| ||||
| 1.32 | .05 | DHPR_RAT P11348 | 86,6,3,18% | Dihydropteridine reductase- Rattus norvegicus |
|
| ||||
| 1.33 | .05 | GSTP1_RAT P04906 | 71,7,1,30% | Glutathione S-transferase P- Rattus norvegicus |
|
| ||||
| 1.38 | .05 | MBP_RAT P02688 | 273,16,7,59% | Myelin basic protein S- Rattus norvegicus |
| 1.4 | .05 | 145,12,3,48% | ||
average volume ratios for each protein spot identified
level of significance determined by students t-test using DeCyder software version 6.5 using the mixed sample internal standard methodology
Swiss-Prot database
Imported from MASCOT and GPS Explorer. Data include predicted MW and pi to correlate with gel position (see supplementary material for annotated gel images), and the additional four scores: the four numbers associated with the protein identifications: e.g., 951 ,16(1)2 ,33 ,31%4 represent:
1MOWSE score. MOlecular Weight SEarch score, a statistic calculated by the MASCOT database search algorithm. The 95th percentile cutoff is calculated for each search and indicated at the bottom of each analysis summary. About 5% of random matches could yield higher MOWSE scores. See matrixscience.com for more details.
2peptide #. number of peptide ion masses matched (number of un-matched ions).
3ms2 #. number of matching peptide ions with MS/MS data.
4(%coverage). Percent of the amino acids accounted for by the matching peptides
Oxidative Stress and Injury
To begin to assess global versus regional levels of oxidative stress both whole brain and striatal samples were analyzed for protein carbonyl content as an index of oxidative protein damage (Figure 3A). Samples from the striatum showed a significant increase (p < 0.05 two-tailed unpaired t-test n = 5 control and n = 6 exposed) in protein carbonyl content for exposed rats whereas there was no difference in the levels of protein carbonyls in the whole brain samples. To further characterize oxidative stress within the striatum, levels of HO-1 and SOD1 were determined using western blot immunoassay. The mean optical density values expressed as a ratio to actin within the same sample are shown in Figure 3B. The expression levels for both of these antioxidant enzymes were significantly increased (p < 0.05 one-tailed unpaired t-test n = 5 control and n = 6 exposed) at the protein level in DEDC exposed animals.
Figure 3.

Oxidative stress and injury. (A) Protein carbonyls were reacted with fluoresceinamine and quantified by immuno slot blot as nmol carbonyl/mg protein. Mean values (+SE) are shown for protein samples isolated from whole brain and striatal samples. (*p < 0.05 two-way unpaired t-test n= 6). (B) Protein levels of heme oxygenase 1 (HO-1) and (C) copper zinc super oxide dismutase (SOD1) in the striatum were determined by western blot and expressed as the ratio of their optical density to actin within the same sample. (*p < 0.05 one-way unpaired t-test n= 5 control and n= 6 exposed).
Striatal Dopamine Metabolism
Striatal TH Protein Expression
The striatum was selected for more detailed evaluation based upon our proteomic data suggestive of altered catechol metabolism, the localized increase of oxidative injury within this region and previous report that identified decreased TH fiber staining in DEDC treated mice.15 To assess loss of dopaminergic terminals the TH levels in the striatum were determined by western blot and the normalized optical density mean values are shown in Figure 4A. TH levels were significantly decreased in DEDC exposed animals (p < 0.05 two-tailed unpaired t-test).
Figure 4.

Striatal catechol metabolism. (A) Striatal tyrosine hydroxylase (TH) levels were determined by western blot and the mean optical density values (+SE) expressed as a ratio to actin used as an intra sample standard are shown. (*p < 0.05 two-way unpaired t-test n= 5 for control and n = 6 for exposed). (B) Levels of dopamine (DA) and 3,4-dihydroxyphenylacetic acid (DOPAC) were determined by HPLC with electrochemical detection as ng/mg of total protein in samples obtained from the striatum of control and exposed animals. The mean values (+SE) expressed as percent of control values (SE) of DA 158.8 (8.8) and DOPAC 10.9 (0.9) are shown. (*p < 0.05 one-way unpaired t-test n= 6). (C) Tyrosine hydroxylase activity was assessed by quantifying the level of tyrosine hydroxylase phosphorylated at serine 40 using western blot and the mean values for treatment groups expressed as the ratio to actin used as an intra sample standard are shown. (**p < 0.01 two-way unpaired t-test n= 5 for control and n = 6 for exposed)
Striatal Dopamine and DOPAC Levels
To further assess dopaminergic toxicity, biogenic amines were quantified in punch biopsies prepared from coronal sections of striatum obtained from control rats and rats exposed to DEDC. The biogenic amines DA and DOPAC were quantified and the mean values and the ratio of DOPAC to DA expressed as a percentage of controls are shown in Figure 4B. Decreases in DOPAC were the only significant changes observed in the DEDC treated animals.
Activated Tyrosine Hydroxylase
Phosphorylation of TH at ser-40 is an important post translation modification contributing to the activation of TH.22 To assess the relative activity of striatal TH in the two treatment groups the levels of TH phosphorylated at ser-40 were determined by western blot. The mean optical densities of phospho-TH normalized to internal actin are shown in Figure 4C. Although levels of total TH were lower in DEDC exposed animals, there were significantly greater amounts of phospho-TH in the exposed animals (p < 0.01 two-tailed unpaired t-test).
Alterations in Striatal Protein Processing
Dopamine Transporter (DAT), α-Synuclein and Phosphorylation of Tau
To assess biochemical events conducive to neurodegenerative injury within the striatum alterations in processing of the specific dopaminergic neuron marker DAT and molecular events facilitating protein aggregation including expression of α-synuclein and phosphorylation of tau were evaluated. Protein levels were determined by western blot and the normalized optical density mean values obtained for DAT, α-synuclein and phospho-tau are shown in Figure 5A, 5B and 5C respectively. The levels of all three proteins were significantly increased relative to controls ((α-synuclein p < 0.05) (DAT and phospho-tau p < 0.01) two-tailed unpaired t-test).
Figure 5.
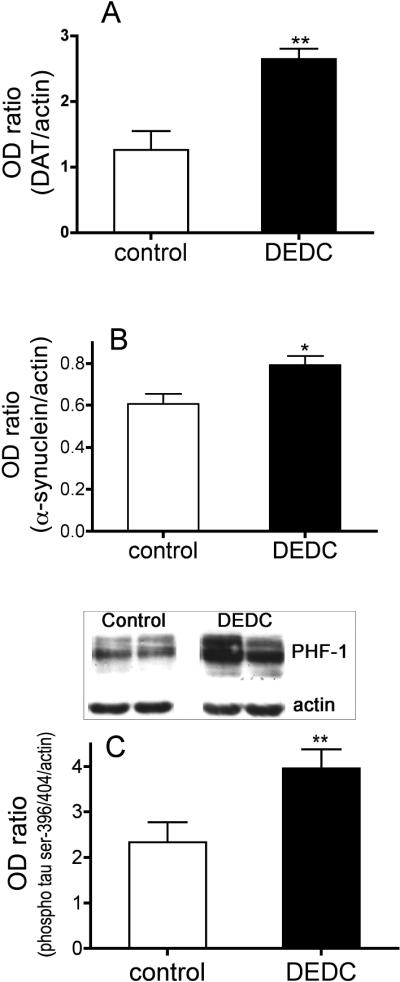
Altered protein processing and expression in the striatum. (A) The expression level of dopamine transporter protein (DAT) was determined by western blot and shown as the mean (+SE) ratio to actin within the same sample. (B) The expression level of α-synuclein was determined by western blot and shown as the mean (+SE) ratio to actin within the same sample. (C) The degree of tau phosphorylation was assessed through western blot assay probing for tau phosphorylated at serine 396 and 404 using PHF-1 antibody. The mean optical density values (+SE) expressed as a ratio to actin are shown. For all panels (*p < 0.05 and **p < 0.01 two-way unpaired t-test n = 5 for control and n = 6 for exposed groups).
Discussion
The goal of this study was to assess E1 as a target for electrophilic adduction in vivo, map susceptible sites for adduction and characterize the effects of adduction on E1 function. We identified two residues sensitive to electrophilic adduction by DEDC, Cys234 and Cys179. The Cys234 location is identical to that reported for human E1 in vitro whereas the modified Cys179 on rat E1 represents an additional modification.11 Several possible factors may have contributed to the generation of this additional adduct on rat E1 in vivo. These include small differences in amino acid sequence between human and rat E1 which share 96% sequence homology, the longer duration of exposure used for the in vivo experiment and potential differences in the reactivity of the electrophiles used in the two experiments. Further investigation will be required to determine whether Cys179 is also a susceptible site in human E1.
Both residues of rat E1 were modified with the same S-(ethylaminocarbonyl) moiety exclusively even though DEDC has the potential to generate both S-(N,N-dialkyl) or S-(N-monoalkyl) adducts (Scheme 1).23, 24 The most likely proximate alkylating species for the observed S-(ethylaminocarbonyl) adduct is ethylisocyanate that presumably is generated through facile decomposition of the monoethyl thiocarbamate sulfoxide metabolite. Exclusive formation of the S-(ethylaminocarbonyl) adduct suggests that the ethylisocyanate metabolite is selective for E1, possibly due to its reactivity and/or its generation in close proximity to E1 within the cytoplasm. Generation of this ethylisocyanate metabolite in vivo is also supported by a previous study that detected an S-(ethylaminocarbonyl) adduct on the active site cysteine of low Km aldehyde dehydrogenase (ALDH) in rats treated with disulfiram, the bis disulfide of DEDC.25
Scheme 1.

Major metabolic pathways leading to protein adduct formation. N,N-diethyldithiocarbamate (DEDC) is S-methylated and undergoes several oxidation steps to generate the sulfoxide and sulfone metabolites capable of reacting with protein cysteine residues to form S-(diethylaminocarbonyl) cysteine adducts. Alternatively DEDC metabolites can be dealkylated and oxidatively metabolized to the N-monalkyl sulfoxide that can either react directly with a protein cysteine or undergo facile decomposition to ethylisocyante prior to reacting with a cysteine to generate the S-(ethylaminocarbonyl) cysteine adduct observed on Cys234 and Cys179 of E1.
Interestingly, E1 was not modified at its active site cysteine by DEDC but decreased levels of activated E1 and total ubiquitinated protein were observed consistent with the Cys234 and Cys179 adducts preventing activation of E1 and diminishing overall protein ubiquitination. This interpretation was also supported by the ability of ethylisocyanate to inhibit recombinant E1 activation in vitro. The canonical catalytic mechanism of E1 involves activation of E1 through binding and adenylation of ubiquitin followed by generation of a thioester linkage between ubiquitin and the active site E1 cysteine. Activation of E1 is followed by binding and adenylation of a second molecule of ubiquitin and then transfer of ubiquitin from the E1 active site to an E2 conjugase. Both Cys234 and Cys179 are located within the first catalytic cysteine domain (FCCH) of E1, a subdomain conserved in human E1 that spans from residues 175–265 and forms one wall of a broad deep groove unique to eukaryotic E1 (Figure 6). This groove functions to distinguish ubiquitin from other ubiquitin like proteins through binding the ubiquitin globular domain.13 Additionally, correct binding in the FCCH region is essential for positioning of ubiquitin for adenlyation and thioester bond linking of ubiquitin to the active site cysteine located within the second catalytic cysteine domain of E1. The observed decrease in activated E1 suggests that DEDC interfered with one of the initial steps of E1 function involving either ubiquitin binding, adenylation or thioester formation.
Figure 6.

Relative locations of adducted residues. Neither human nor rat E1 structures were available at the time of writing but computer modeling has supported a high degree of similarity between the quarternary structures of human E1 (P22314) and Saccharomyces cerevisiae E1 (22515).47 This yeast E1 has ~50% overall sequence homology and 88% sequence homology within the ubiquitin binding region with human E1 and rat E1. (A) The structure of yeast E1 activating enzyme was obtained from the Protein Database (PDB ID: 3CMM), and is presented as a model for canonical E1 structure. The common domains of E1s are colored as follows: the inactive adenylation domain (IAD) in blue, the active adenylation domain (AAD) in red, ubiquitin fold domain (UFD) in orange, the four helix bundle domain (4HB) in aqua, the first catalytic cysteine half domain (FCCH) in purple, and the second catalytic cysteine half domain (SCCH) in green. The S-(ethylaminothiocarbonyl) adducts on amino acid residues 179 and 234 were located within the FCCH domain of rat E1 (purple). The active site cysteine Cys600 (white) is located in the SCCH domain. (B) The decreased levels of activated E1 obtained from DEDC exposed animals suggest that covalent modification of residues within the FCCH region by DEDC may inhibit either the binding or adenylation of ubiquitin (Ub), shown in yellow, or formation of the thioester linkage of Ub to Cys600.
Our 2D-DIGE analyses of whole brain protein levels identified several significantly altered proteins among which were several consistent with neurodegenerative processes that pointed toward striatal involvement. The elevated scavenging proteins, glutathione S-tranferase pi and the two peroxiredoxins suggested increased oxidative stress and glutathione S-transferase pi has been proposed as an important protein in modulating the progression of Parkinson's disease.26 Due to its role in catechol metabolism the increased dihydropteridine reductase also implicated striatal involvement.27 Although TH catalyzes the rate-limiting step of catecholamine synthesis, TH activity is governed by several factors including levels of end product, TH phosphorylation state and availability of its cofactor, tetrahydrobiopterin.22 Dihydropteridine reductase regenerates tetrahydrobiopterin and increased levels of this protein have been measured in the substantia nigra pars compacta of Parkinson's disease patients consonant with a compensatory response attempting to maintain catecholamine levels.28, 29
The protein carbonyl levels provided evidence for regional differences in oxidative injury with the striatum being a region more severely affected. Proteasome inhibition has been linked to increased oxidative cellular damage including the accumulation of oxidized proteins suggesting that inhibition of E1 can contribute to the accumulation of protein carbonyls; and because the nigrostriatal system exhibits a high basal level of oxidative stress it may be a region more susceptible to this consequence of compromised E1 function.30,31 To better assess the striatal injury we performed more focused evaluations of biochemical markers within the striatum that demonstrated significant changes in three hallmark biological processes disrupted in Parkinson's disease. These included; oxidative stress, dopaminergic toxicity and biochemical processes facilitating protein aggregation. In agreement with the enhanced protein carbonyl content observed in this region, our data also showed elevated levels of two antioxidant enzymes linked to the antioxidant response element, HO-1 and SOD1. Dopaminergic toxicity was evidenced by perturbations in two dopaminergic terminal markers, TH and DAT, which exhibited a significant decrease and increase, respectively. Although TH protein levels were decreased, consistent with the loss of dopaminergic nerve terminals, our analysis of biogenic amines did not demonstrate a concomitant decrease in dopamine content. Similar apparently conflicting relationships have been reported for TH and dopamine in animal models that were interpreted to represent an early stage of injury at which time dopamine levels could still be maintained in the presence of lower total TH by increasing the pool of activated TH.32 To assess this possibility in our study, we quantified levels of activated TH (phospho-TH). Despite the decreased total TH protein in DEDC exposed animals, there was increased phospho-TH, suggesting that together with other compensatory responses, e.g., increased dihydropteridine reductase expression, TH activity was sufficiently compensated to maintain normal DA levels in the DEDC treated animals.22 Comparable early stages of injury have also been reported for mice administered DEDC in which motor deficits and decreased TH striatal staining were observed in the absence of nigral cell loss.15
Although indicative of perturbations in normal biology within dopaminergic terminals the increased levels of DAT in our study appear contradictory to a loss of dopaminergic terminals. However, increased levels of DAT have also been observed in PC12 cells exposed to rotenone suggesting that, at least in early stages of injury, DAT may undergo increased expression or decreased clearance.33 One possible interpretation is that compromised E1 function may have contributed to an accumulation of DAT since ubiquitination of DAT is thought to play a role in trafficking and targeting of misfolded DAT for degradation.34–37 Assessment of this possibility will require analysis of the cellular location and functionality of DAT in future studies. However, if the elevated DAT is functional it could contribute to the reported increased uptake of MPP+ through this transporter and enhanced toxicity of MPTP when it is co-administered with DEDC.38
The third biological process that we evaluated that is common to several neurodegenerative diseases was protein aggregation. The two proteins we examined, α-synuclein and tau, have been associated with the etiology of Parkinson's disease. Investigations have established a role for oligomeric α-synuclein in the development of Parkinson's disease and recent genome wide association studies have associated single nucleotide polymorphisms in tau with an increased risk for sporadic Parkinson's disease.39, 40 Critical factors promoting oligomerization of α-synuclein include increased protein concentration and phosphorylation at ser-129.41 Similarly, increased expression and phosphorylation of tau at ser-396 and 404 contribute to its aggregation into nonfunctional oligomers and neurofibrillary tangles.42, 43 Thus the increased expression of α-synuclein and phosphorylation of tau observed in the DEDC treated animals favor α-synuclein and tau oligomerization within this brain region and could potentially contribute to neurodegenerative change.
In conclusion the data are consistent with E1 presenting a similar target for electrophilic adduction in vivo to that identified in vitro and that DEDC exposure can promote degenerative change within the nigrostriatal system. E1 adduction compromised E1's enzymatic function and the reactivity of E1's Cys234 and Cys179 suggests that other endogenous and xenobiotic derived electrophiles can covalently modify these sites, and previous reports have shown that thiocarbamate herbicides, other dialkyldithiocarbamates and the benzimadazole fungicides can all give rise to alkylating species having the potential to adduct E1 analogous to DEDC.44–46 A potential hazard for compromised ubiquitin based protein processing supported by previous studies and familial forms of Parkinson's disease is degenerative change within the nigrostriatal system. This suggests that DEDC-mediated E1 inhibition contributed to the nigrostriatal injury observed in the present study and raises the possibility that multiple environmental agents may compromise E1 function and contribute to a cumulative risk for development of Parkinson's disease over the life of an individual. To assess this possibility further investigations are needed to determine the contribution of E1 inhibition to striatal injury, to delineate the downstream events of E1 inhibition contributing to striatal injury and to evaluate the susceptibility of E1 to adduction by other xenobiotic and endogenous electrophiles. More generally the present study also serves as an example of how protein targets identified using in vitro screens can be filtered using knowledge of their biological function and role of associated pathways in heritable diseases to guide in vivo studies to further our understanding of how specific adduction may contribute to disease.
Supplementary Material
Acknowledgements
We are grateful to Dr. Peter Davies of Albert Einstein College of Medicine for providing the PHF-1 antibody and to Diana Neely for her guidance in collecting the striatal punch biopsies.
Funding Sources This work was supported by the National Institutes of Environmental Health Science grants R01 ES019969 (WMV), P30 ES00267 The Vanderbilt Center in Molecular Toxicology, and T32 ES007028 Training in Environmental Toxicology (SWC).
Abbreviations
- SN2
bimolecular nucleophilic substitution
- DEDC
N,N-diethyldithiocarbamate
- E1
ubiquitin activating enzyme E1
- PVC
polyvinyl chloride
- BSA
bovine serum albumin
- PVDF
polyvinyliidene fluoride
- TBS
tris buffered saline
- TBST
tris buffered saline with tween
- DIGE
difference gel electrophoresis
- cy2
cyanine
- cy3
indocarbocyanine
- cy5
indodicarbocyanine
- MALDI-TOF
matrix assisted laser desorption time of flight
- HO-1
hemoxygenase 1
- SOD-1
Cu-Zn superoxide dismutase 1
- DAT
dopamine transporter
- TH
tyrosine hydroxylase
- phosphor-TH, phospho-ser40-tyrosine hydroxylase, phospho-tau
phosphor-tau ser 396/404
- DOPAC
3,4-dihydroxyphenylacetic acid
- DA
dopamine
- FCCH
first catalytic cysteine domain
- SCCH
second catalytic cysteine domain
- IAD
inactive adenylation domain
- AAD
active adenylation domain
- UFD
ubiquitin fold domain
- 4HB
four helix bundle domain
- Ub
ubiquitin
Footnotes
Supporting Information Available: The LC/MS/MS peptide coverage obtained for the tri enzyme digests of E1 and the masses for the fragmentation spectra obtained for the S-(ethylaminocarbonyl) adducted and non adducted DNPGVVTCLDEAR and VGEFCHSR peptides containing Cys234 and Cys179, respectively, as well as annotated 2D-DIGE gels showing the approximate isoelectric points and molecular weights of the proteins having altered expression are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR, Hinson JA. Selective protein covalent binding and target organ toxicity. Toxicol Appl Pharmacol. 1997;143:1–12. doi: 10.1006/taap.1996.8074. [DOI] [PubMed] [Google Scholar]
- (2).Liebler DC, Guengerich FP. Elucidating mechanisms of drug-induced toxicity. Nat Rev Drug Discov. 2005;4:410–420. doi: 10.1038/nrd1720. [DOI] [PubMed] [Google Scholar]
- (3).Pumford NR, Halmes NC. Protein targets of xenobiotic reactive intermediates. Annu Rev Pharmacol Toxicol. 1997;37:91–117. doi: 10.1146/annurev.pharmtox.37.1.91. [DOI] [PubMed] [Google Scholar]
- (4).Lame MW, Jones AD, Wilson DW, Dunston SK, Segall HJ. Protein targets of monocrotaline pyrrole in pulmonary artery endothelial cells. J Biol Chem. 2000;275:29091–29099. doi: 10.1074/jbc.M001372200. [DOI] [PubMed] [Google Scholar]
- (5).Lin CY, Isbell MA, Morin D, Boland BC, Salemi MR, Jewell WT, Weir AJ, Fanucchi MV, Baker GL, Plopper CG, Buckpitt AR. Characterization of a structurally intact in situ lung model and comparison of naphthalene protein adducts generated in this model vs lung microsomes. Chem Res Toxicol. 2005;18:802–813. doi: 10.1021/tx049746r. [DOI] [PubMed] [Google Scholar]
- (6).Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- (7).Spiess PC, Deng B, Hondal RJ, Matthews DE, van der Vliet A. Proteomic profiling of acrolein adducts in human lung epithelial cells. J Proteomics. 2011;74:2380–2394. doi: 10.1016/j.jprot.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chavez JD, Wu J, Bisson W, Maier CS. Site-specific proteomic analysis of lipoxidation adducts in cardiac mitochondria reveals chemical diversity of 2-alkenal adduction. J Proteomics. 2011;74:2417–2429. doi: 10.1016/j.jprot.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lin D, Saleh S, Liebler DC. Reversibility of covalent electrophile-protein adducts and chemical toxicity. Chem Res Toxicol. 2008;21:2361–2369. doi: 10.1021/tx800248x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liebler DC. Protein damage by reactive electrophiles: targets and consequences. Chem Res Toxicol. 2008;21:117–128. doi: 10.1021/tx700235t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dennehy MK, Richards KA, Wernke GR, Shyr Y, Liebler DC. Cytosolic and nuclear protein targets of thiol-reactive electrophiles. Chem Res Toxicol. 2006;19:20–29. doi: 10.1021/tx050312l. [DOI] [PubMed] [Google Scholar]
- (12).Kirkin V, Dikic I. Role of ubiquitin- and Ubl-binding proteins in cell signaling. Curr Opin Cell Biol. 2007;19:199–205. doi: 10.1016/j.ceb.2007.02.002. [DOI] [PubMed] [Google Scholar]
- (13).Lee I, Schindelin H. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell. 2008;134:268–278. doi: 10.1016/j.cell.2008.05.046. [DOI] [PubMed] [Google Scholar]
- (14).Greenamyre JT, Hastings TG. Biomedicine. Parkinson's--divergent causes, convergent mechanisms. Science. 2004;304:1120–1122. doi: 10.1126/science.1098966. [DOI] [PubMed] [Google Scholar]
- (15).Chou AP, Maidment N, Klintenberg R, Casida JE, Li S, Fitzmaurice AG, Fernagut PO, Mortazavi F, Chesselet MF, Bronstein JM. Ziram causes dopaminergic cell damage by inhibiting E1 ligase of the proteasome. J Biol Chem. 2008;283:34696–34703. doi: 10.1074/jbc.M802210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tonkin EG, Valentine HL, Zimmerman LJ, Valentine WM. Parenteral N,N-diethyldithiocarbamate produces segmental demyelination in the rat that is not dependent on cysteine carbamylation. Toxicol Appl Pharmacol. 2003;189:139–150. doi: 10.1016/s0041-008x(03)00093-0. [DOI] [PubMed] [Google Scholar]
- (17).Ramagli LS. Quantifying protein in 2-D PAGE solubilization buffers. Methods Mol Biol. 1999;112:99–103. doi: 10.1385/1-59259-584-7:99. [DOI] [PubMed] [Google Scholar]
- (18).Eng JK, Fischer B, Grossmann J, Maccoss MJ. A fast SEQUEST cross correlation algorithm. J Proteome Res. 2008;7:4598–4602. doi: 10.1021/pr800420s. [DOI] [PubMed] [Google Scholar]
- (19).Jahngen-Hodge J. Regulation of Ubiquitin-conjugating Enzymes by Glutathione Following Oxidative Stress. Journal of Biological Chemistry. 1997;272:28218–28226. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- (20).Viquez OM, Valentine HL, Friedman DB, Olson SJ, Valentine WM. Peripheral nerve protein expression and carbonyl content in N,N-diethlydithiocarbamate myelinopathy. Chem Res Toxicol. 2007;20:370–379. doi: 10.1021/tx6003453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- (22).Fujisawa H, Okuno S. Regulatory mechanism of tyrosine hydroxylase activity. Biochem Biophys Res Commun. 2005;338:271–276. doi: 10.1016/j.bbrc.2005.07.183. [DOI] [PubMed] [Google Scholar]
- (23).Hu P, Jin L, Baillie TA. Studies on the metabolic activation of disulfiram in rat. Evidence for electrophilic S-oxygenated metabolites as inhibitors of aldehyde dehydrogenase and precursors of urinary N-acetylcysteine conjugates. J Pharmacol Exp Ther. 1997;281:611–617. [PubMed] [Google Scholar]
- (24).Jin L, Davis MR, Hu P, Baillie TA. Identification of novel glutathione conjugates of disulfiram and diethyldithiocarbamate in rat bile by liquid chromatography-tandem mass spectrometry. Evidence for metabolic activation of disulfiram in vivo. Chem Res Toxicol. 1994;7:526–533. doi: 10.1021/tx00040a008. [DOI] [PubMed] [Google Scholar]
- (25).Shen ML, Johnson KL, Mays DC, Lipsky JJ, Naylor S. Determination of in vivo adducts of disulfiram with mitochondrial aldehyde dehydrogenase. Biochem Pharmacol. 2001;61:537–545. doi: 10.1016/s0006-2952(00)00586-4. [DOI] [PubMed] [Google Scholar]
- (26).Shi M, Bradner J, Bammler TK, Eaton DL, Zhang J, Ye Z, Wilson AM, Montine TJ, Pan C, Zhang J. Identification of glutathione S-transferase pi as a protein involved in Parkinson disease progression. Am J Pathol. 2009;175:54–65. doi: 10.2353/ajpath.2009.081019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Musacchio JM, D'Angelo GL, McQueen CA. Dihydropteridine reductase: implication on the regulation of catecholamine biosynthesis. Proc Natl Acad Sci U S A. 1971;68:2087–2091. doi: 10.1073/pnas.68.9.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Basso M, Giraudo S, Corpillo D, Bergamasco B, Lopiano L, Fasano M. Proteome analysis of human substantia nigra in Parkinson's disease. Proteomics. 2004;4:3943–3952. doi: 10.1002/pmic.200400848. [DOI] [PubMed] [Google Scholar]
- (29).Basso M, Giraudo S, Lopiano L, Bergamasco B, Bosticco E, Cinquepalmi A, Fasano M. Proteome analysis of mesencephalic tissues: evidence for Parkinson's disease. Neurol Sci. 2003;24:155–156. doi: 10.1007/s10072-003-0106-2. [DOI] [PubMed] [Google Scholar]
- (30).Lee MH, Hyun DH, Jenner P, Halliwell B. Effect of proteasome inhibition on cellular oxidative damage, antioxidant defences and nitric oxide production. Journal of Neurochemistry. 2001;78:32–41. doi: 10.1046/j.1471-4159.2001.00416.x. [DOI] [PubMed] [Google Scholar]
- (31).Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003;53(Suppl 3):S26–36. doi: 10.1002/ana.10483. discussion S36–28. [DOI] [PubMed] [Google Scholar]
- (32).McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- (33).Sai Y, Wu Q, Le W, Ye F, Li Y, Dong Z. Rotenone-induced PC12 cell toxicity is caused by oxidative stress resulting from altered dopamine metabolism. Toxicol In Vitro. 2008;22:1461–1468. doi: 10.1016/j.tiv.2008.04.019. [DOI] [PubMed] [Google Scholar]
- (34).Jiang H, Jiang Q, Feng J. Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J Biol Chem. 2004;279:54380–54386. doi: 10.1074/jbc.M409282200. [DOI] [PubMed] [Google Scholar]
- (35).Miranda M, Wu CC, Sorkina T, Korstjens DR, Sorkin A. Enhanced ubiquitylation and accelerated degradation of the dopamine transporter mediated by protein kinase C. J Biol Chem. 2005;280:35617–35624. doi: 10.1074/jbc.M506618200. [DOI] [PubMed] [Google Scholar]
- (36).Miranda M, Dionne KR, Sorkina T, Sorkin A. Three ubiquitin conjugation sites in the amino terminus of the dopamine transporter mediate protein kinase C-dependent endocytosis of the transporter. Mol Biol Cell. 2007;18:313–323. doi: 10.1091/mbc.E06-08-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. RNA interference screen reveals an essential role of Nedd4-2 in dopamine transporter ubiquitination and endocytosis. J Neurosci. 2006;26:8195–8205. doi: 10.1523/JNEUROSCI.1301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Irwin I, Wu EY, DeLanney LE, Trevor A, Langston JW. The effect of diethyldithiocarbamate on the biodisposition of MPTP: an explanation for enhanced neurotoxicity. Eur J Pharmacol. 1987;141:209–217. doi: 10.1016/0014-2999(87)90265-2. [DOI] [PubMed] [Google Scholar]
- (39).Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- (40).Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Uversky VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- (42).Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson's disease models. Faseb J. 2009;23:2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Frasier M, Walzer M, McCarthy L, Magnuson D, Lee JM, Haas C, Kahle P, Wolozin B. Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol. 2005;192:274–287. doi: 10.1016/j.expneurol.2004.07.016. [DOI] [PubMed] [Google Scholar]
- (44).Staub RE, Quistad GB, Casida JE. Mechanism for benomyl action as a mitochondrial aldehyde dehydrogenase inhibitor in mice. Chem Res Toxicol. 1998;11:535–543. doi: 10.1021/tx980002l. [DOI] [PubMed] [Google Scholar]
- (45).Staub RE, Quistad GB, Casida JE. S-methyl N-butylthiocarbamate sulfoxide: selective carbamoylating agent for mouse mitochondrial aldehyde dehydrogenase. Biochem Pharmacol. 1999;58:1467–1473. doi: 10.1016/s0006-2952(99)00239-7. [DOI] [PubMed] [Google Scholar]
- (46).Zimmerman LJ, Valentine HL, Valentine WM. Characterization of S-(N,N-Dialkylaminocarbonyl)cysteine Adducts and Enzyme Inhibition Produced by Thiocarbamate Herbicides in the Rat. Chem Res Toxicol. 2004;17:258–267. doi: 10.1021/tx034209c. [DOI] [PubMed] [Google Scholar]
- (47).Brahemi G, Burger AM, Westwell AD, Brancale A. Homology Modelling of Human E1 Ubiquitin Activating Enzyme. Lett Drug Des Discov. 2010;7:57–62. doi: 10.2174/157018010789869316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.