

Abstract
RNA helicase A (RHA) promotes multiple steps of HIV-1 RNA metabolism during viral replication, including transcription, translation, and the annealing of primer tRNA3Lys to the viral RNA. RHA is a member of the DExH subclass of RNA helicases that uniquely contains two double-stranded RNA binding domains (dsRBDs) at its N terminus. Here, we performed a genome-wide analysis of the interaction of RHA with HIV-1 RNA both in vitro, using fluorescence polarization, and during viral replication, using an RNA-protein coprecipitation assay. In vitro, RHA binds to all the isolated regions of the HIV-1 RNA genome tested, with Kd (equilibrium dissociation constant) values ranging from 44 to 178 nM. In contrast, during viral replication, RNA-protein coprecipitation assays detected only a major interaction of RHA with the 5′-untranslated region (5′-UTR) and a minor interaction with the Rev response element (RRE) of HIV-1 RNA. Since RHA does not associate well with all the highly structured regions of HIV-1 RNA tested in vivo, the results suggest that other viral or cellular factors not present in vitro may modulate the direct interaction of RHA with HIV-1 RNA during virus replication. Nevertheless, a role for duplex RNA as a target for RHA binding in vivo is suggested by the fact that the deletion of either one or both dsRBDs eliminates the in vivo interaction of RHA with HIV-1 RNA. Furthermore, these mutant RHAs do not promote the in vivo annealing of tRNA3Lys to viral RNA, nor are they packaged into virions, demonstrating that the dsRBDs are essential for the role of RHA in HIV-1 replication.
INTRODUCTION
RNA helicases are ubiquitous in nature and have been shown to be involved in all aspects of RNA metabolism. They are classified into five different superfamilies, of which superfamily 2 (SF2) is the largest (7). RNA helicase A (RHA), also known as DHX9, is an SF2 DExH box protein that can unwind both double-stranded RNA (dsRNA) and double-stranded DNA (dsDNA) by utilizing any of the four ribo- or deoxyribonucleotide triphosphates (31, 57). RHA contains 7 domains (depicted in Fig. 3A). The core helicase domain is located at the center and is composed of two subdomains, the DExH subdomain (named after a signature amino acid sequence in RNA helicases, which is DEIH in RHA) and the HELICc subdomain (35). The core helicase domain can bind and hydrolyze nucleotide triphosphates (NTPs) and is composed of 8 motifs that are conserved in all members of the DExD/H box helicases (14). The residues N terminal to the core helicase domain contain two dsRNA binding domains (dsRBDs). The C terminus of RHA is characterized by a stretch of repeated arginine and glycine-glycine (RGG) residues that have a higher binding affinity for single-stranded RNA (ssRNA) than for dsRNA (10, 45, 57). Between the core helicase and the RGG domains are the helicase-associated domain (HA2) and the oligonucleotide/oligosaccharide binding (OB-fold) that was defined by its homology with the Saccharomyces cerevisiae protein Prp43p (51).
Fig 3.

Abilities of mutant RHAs containing deletions of either one or both dsRBDs to bind to viral RNA in vivo. An RNA-protein coprecipitation assay was performed, as described in the Fig. 2 legend, with recombinant plasmids expressing mutant RHAs. (A) Schematic representation of the domain structure of wild-type or mutant RHA. The proteins were tagged with 6×His at the N terminus. The dotted line represents the deleted region. (B) Western blotting with an anti-6×His tag antibody detected the expression of His-tagged wild-type or mutant RHA in cotransfected cells. The 6×His tag peptide only was not resolved in Western blots. (C) Western blotting with an anti-6×His tag antibody detected His-tagged wild-type or mutant RHA in precipitates. (D) PCR amplification products from the indicated RNA preparations with or without incubation with reverse transcriptase (+RT or −RT, respectively), using primer set P1-F/R or P13-F/R, which is specific for the 5′-UTR or RRE of HIV-1 RNA, respectively.
RHA was reported to participate in a diverse set of cellular processes involving RNA, including the activation of transcription (1, 2, 18 32, 37, 50) and stimulating translation (22), and was found to be associated with the Rev-Rev response element (RRE) complex (36), the nuclear pore complex (19), and the RNA-induced silencing complex (RISC) involved in the RNA interference (RNAi) pathway (47). RHA can also act as a sensor for pathogenic dsRNA in myeloid dendritic cells and, through its interaction with IPS-1, activates the pathway leading to the production of alpha/beta interferon (IFN-α/β) and proinflammatory cytokines (58). The stimulatory effect of RHA on translation might involve direct binding to the highly structured 5′-untranslated region (5′-UTR) in retrovirus mRNA, with a possible unwinding of this region to facilitate ribosomal scanning (22). However, for other target cellular mRNAs, RHA's stimulatory effect on translation appears to result from RHA's recruitment to the translational machinery via its interaction with other mRNA binding proteins such as Lin28 (26) or an La ribonucleoprotein domain family member, LARP6 (34). These examples make it clear that the roles of RHA in regulating multiple stages of RNA metabolism are likely to be regulated by its interaction with specific proteins and/or RNA structures.
RHA has been found to participate widely in the replication of viruses such as hepatitis C virus (HCV) (23, 24), foot-and-mouth disease virus (30), influenza A virus (33), spleen necrosis virus (SNV), reticuloendotheliosis virus A (REV-A), and human T-cell leukemia virus type 1 (HTLV-1) etc. (9). In particular, RHA is involved in HIV-1 replication at multiple steps, including the transcription (17), nuclear export (32), and translation (8) of viral mRNA, and is packaged into HIV-1 virions (48). RHA was also shown to play a role in promoting the annealing of tRNA3Lys, the primer for reverse transcription in HIV-1, to viral RNA (56). In this report, we used fluorescence polarization (FP) and an RNA-protein coprecipitation assay (5) to carry out a genome-wide analysis of RHA binding to HIV-1 RNA in vitro and in vivo, respectively. This study demonstrates that while RHA binds to many regions of the HIV-1 RNA genome in vitro, RHA interacts primarily with the 5′-UTR in vivo, with a more weak interaction detected with the RRE region. The results also demonstrate the importance of the dsRBDs for the binding of RHA to viral RNA in vivo and, in particular, for promoting both RHA packaging into virions and tRNA3Lys annealing to the viral RNA.
MATERIALS AND METHODS
Cell culture.
293T, a human embryonic kidney cell line, was cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with l-glutamine (Gibco), penicillin-streptomycin (Gibco), and 10% fetal bovine serum. 293E cells are a stably transfected 293 cell line that produces Epstein-Barr virus nuclear antigen 1 (EBNA1) (12). The cells were adapted to grow in suspension in defined medium, without serum, and EBNA1 promotes the amplification of plasmids containing the replication origin region (OriP) of Epstein-Barr virus, which results in high expression levels of proteins coded for by these plasmids.
Plasmids.
Recombinant plasmids were generated by PCR. The gel-purified PCR products were digested with NotI and BstEII and then cloned into plasmid pTT5-SH5-RHA (56). Plasmid pTT5-SH5-RHA contains the full-length human RHA coding sequence under the control of a cytomegalovirus (CMV) promoter and was also used as a parent template for PCRs. All recombinant plasmids were verified by performing restriction mapping and DNA sequencing. Recombinant plasmid pRHAΔaa1-100 or pRHAΔdsRBD1-2 was generated by PCR using primer pair RHA-F8(301)/RHA(BstEII)1026-R or RHA-F2(901)/RHA(BstEII)1026-R, respectively. pRHAΔaa134-250 was constructed by fusing the PCR product of primer pair RHA-F1 (4)/RHA-d2-R(399) to the PCR product of primer pair RHA-d2-F(751)/RHA(BstEII)1026-R. The primer pairs used for PCR are listed in Table 1.
TABLE 1.
Primers used for PCR to construct recombinant plasmids
| Primer | Sequence (5′–3′)a |
|---|---|
| RHA-F8(301) | CGGGCGGCCGCCGCTGAAGGAGATTTACCAA |
| RHA(BstEII)1026-R | CCAAGGATTCCAGTTGGATTG |
| RHA-F1(4) | CGGGCGGCCGCCGGTGACGTTAAAAATTTTC |
| RHA-d2-R(399) | ACCATAGCCAGAGGCCCCT |
| RHA-d2-F(751) | AGGGGCCTCTGGCTATGGTTACCATCTTGGAGTGGTT |
| RHA-F2(901) | CGGGCGGCCGCCCCTGAAGATCCTTCTGTG |
NotI restriction sites are in boldface type.
Expression and purification of proteins.
N-terminally 6×His-tagged full-length RHA (referred to as the wild type in this study) was expressed and purified from 293E cells, as described previously (56). Glutathione S-transferase (GST) was expressed in 293E cells and purified by using high-performance glutathione-Sepharose (GE Healthcare). The protein was eluted with 33 mM reduced glutathione (Fisher Scientific) in elution buffer containing 50 mM Tris-HCl (pH 8.0) and 10% glycerol. Purified RHA and GST were dialyzed against a solution containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 20 mM KCl, 2 mM MgCl2, 2 mM dithiothreitol, and 10% glycerol and then stored at −80°C.
RNA substrates.
The HIV-1 genomic RNA fragments were synthesized by in vitro transcription using T7 RNA polymerase (T7 MEGAscript kit; Ambion), as described previously in detail (56). The DNA templates for the in vitro transcription of HIV-1 RNAs were amplified by PCR from a vector containing the sequences for HIV-1 BH10 using primer sets listed in Table 2. RNA was refolded by first heating the RNA to 85°C and then allowing the RNA to slowly cool to room temperature.
TABLE 2.
Primers used for PCR to amplify DNA templates for in vitro transcription
| Primer | Sequence (5′–3′)a | RNAb |
|---|---|---|
| HIV1-Forward | CGCTAATACGACTCACTATAGGGAGAGGTCTCTCTGGTTAGACC | HIV-1 RNA(1–104) |
| HIV1-Reverse | CACTACTTGAAGCACTCAAGGCAA | |
| HIV2-Forward | CGCTAATACGACTCACTATAGGGAGATGTGCCCGTCTGTTGTGT | HIV-1 RNA(105–277) |
| HIV2-Reverse | GCTCCTCTGGTTTCCCTTTCGCTTTCA | |
| HIV3-Forward | CGCTAATACGACTCACTATAGGGAGATCTCTCGACGCAGGACTC | HIV-1 RNA(228–386) |
| HIV3-Reverse | TTCCCATCGATCTAATTC | |
| HIV4-Forward | CGCTAATACGACTCACTATAGGGAGAAAAGGGCTGTTGGAAATG | HIV-1 RNA(1565–1708) |
| HIV4-Reverse | GGGGCTGTTGGCTCTGGTCTGCTCTGAA | |
| HIV5-Forward | CGCTAATACGACTCACTATAGGGAGAAGACTTCTGGGAAGTTCA | HIV-1 RNA(2386–2543) |
| HIV5-Reverse | CATTGTTTATACTAGGTATGGTA | |
| HIV6-Forward | CGCTAATACGACTCACTATAGGGAGAGGGCAAGTCAGATTTACC | HIV-1 RNA(2928–3212) |
| HIV6-Reverse | GGGCACCCCTCATTCTTGCATA | |
| HIV7-Forward | CGCTAATACGACTCACTATAGGGAGAGGCAACATATCTATGAAA | HIV-1 RNA(5268–5395) |
| HIV7-Reverse | CGAGTAACGCCTATTCTGCTATGTC | |
| HIV8-Forward | CGCTAATACGACTCACTATAGGGAGACACAAGAAGTAGTATTGG | HIV-1 RNA(6045–6270) |
| HIV8-Reverse | TTTTTTATCTCTCCTTTCTCCATT | |
| HIV9-Forward | CGCTAATACGACTCACTATAGGGAGAAGGAGCTTTGTTCCTTGG | HIV-1 RNA(7348–7581) |
| HIV9-Reverse | AGGAGCTGTTGATCCTTTAGG |
The T7 promoter region is underlined.
The numbers in brackets indicate the nucleotide position of RNA sequences relative to the transcription start site of HIV-1 BH10 genomic RNA as 1.
RNA labeling by FTSC.
The 3′ terminus of the RNA was labeled with fluorescein-5-thiosemicarbazide (FTSC), as described previously (11), with minor modifications. The RNA was incubated in a volume of 200 μl containing 2.5 mM and 100 mM sodium acetate (NaOAc) (pH 5.0) for 1 h on ice and then precipitated with 1 volume of 2-propanol, washed with 70% ethanol, dissolved in 200 μl of 100 mM NaOAc (pH 5.0) and 0.5 mM FTSC, and incubated on ice overnight. The FTSC-labeled RNA was precipitated in ethanol, redissolved in 25 mM Tris-HCl (pH 7.5), further purified by using Microspin G-25 columns (GE Healthcare), and then stored at −20°C.
RNA labeling with 32pCp and native polyacrylamide gel electrophoresis.
Radioactive RNAs were prepared by labeling with [5′-32P]Cytidine 3′,5′-bis(phosphate) (32pCp) using T4 RNA ligase (Fermentas), further purified by using Microspin G-25 columns (GE Healthcare), and then separated in a native 6% polyacrylamide gel. Radioactive RNA bands were visualized by using a PhosphorImager instrument.
FP assay.
Twenty-microliter reaction mixtures containing 10 nM FTSC-labeled RNA and the indicated amounts of RHA or GST protein in binding buffer (10 mM Tris-HCl [pH 8.0], 50 mM KCl, 2 mM MgCl2, 2 mM dithiothreitol, 2.5 mM NaH2PO4, 15 mM NaCl, 4% glycerol) were transferred into 96-well black PS HE microplates (Molecular Devices). Fluorescence polarization (FP) assays were performed in quadruplicate by using a Synergy 4 multimode microplate reader (BioTek) to measure the polarization change. The equilibrium dissociation constant (Kd) was obtained by fitting the binding curves using KaleidaGraph, as previously described (29).
Western blot analysis.
Total viral or cell lysates were prepared and then separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, blotted onto a nitrocellulose membrane, and probed with appropriate antibodies. After extensive washing, horseradish peroxidase-conjugated secondary antibodies (Sigma) were applied, and the membrane was washed again, developed with a chemiluminescence reagent (Perkin-Elmer), and visualized on Kodak X-ray film. The primary antibodies used for Western blotting included rabbit anti-HIV reverse transcriptase antibody, mouse anti-CAp24 antibody (NIH AIDS Research and Reference Reagent Program), β-actin monoclonal antibody (MAb) (Sigma), RHA MAb (M01; Abnova Inc.), and polyhistidine MAb (Sigma).
RNA-protein coprecipitation assays.
The RNA-protein coprecipitation assay was developed to pull down RNA-protein complexes and was performed as previously described (5), except that the RNA-protein complex containing 6×His-tagged RHA was pulled down by using Ni-nitrilotriacetic acid (NTA) agarose instead of antibody. Briefly, 293T cells were cotransfected with a BH10 vector and a plasmid expressing 6×His-tagged wild-type or mutant RHA. Twenty-four hours later, cells were fixed in 1% (wt/vol) formaldehyde (Bioshop) for 10 min at 37°C, followed by two washes with ice-cold phosphate-buffered saline. Cells were lysed in 2 ml of lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM KCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 50 mM NaF, 1,000 U/ml SUPERase-in [Ambion], and protease inhibitor cocktail tablets [Roche]) for 10 min on ice before extensive sonicaion. After extract clearing by centrifugation, supernatants were recovered in a clean microcentrifuge tube for use in RNA-protein coprecipitation experiments. Input RNA (2%) was stored at −80°C until the samples were eluted. Salmon sperm DNA- and mammalian RNA-saturated Ni-NTA agarose was added to each sample, and the samples were incubated for 2 h at 4°C with rotation to capture RHA. Samples were then centrifuged, and Ni-NTA agarose was washed with cold wash buffer (50 mM Tris-HCl [pH 7.5], 150 mM KCl, 1% NP-40, 0.25% sodium deoxycholate). Recombinant proteins were eluted in 250 mM imidazole solution (pH 7.4) diluted with 2× dilution buffer (100 mM Tris-HCl, pH.5, 10 mM EDTA, 20 mM dithiothreitol, 2% SDS, 2,000 U/ml SUPERase-in), incubated at 70 °C for 5 h, and then extracted with TRIzol reagent to isolate RNA. After DNase treatment, the RNA sample was subjected to reverse transcription (RT)-PCR analysis. Purified inputs were used as a control.
RT-PCR.
RT was carried out by using the Superscript II kit (Invitrogen), according to the manufacturer's protocol. RNA was prepared by using TRIzol (Invitrogen), treated with DNase I (Ambion), and then reverse transcribed by using primers specific for HIV-1 transcripts. PCR was then performed by using Taq DNA polymerase (Bio Basic Canada Inc.). The amplification was started with an initial denaturation step at 94°C for 5 min followed by 25 cycles at 94°C for 30 s, 52°C for 30 s, and 72°C for 20 s. The final extension step was done at 72°C for 5 min. The PCR products were analyzed in a 1.5% agarose gel containing ethidium bromide. The primers used for RT and PCR are listed in Table 3.
TABLE 3.
Primers used for RNA-protein coprecipitation assays
| Primer | Sequence (5′–3′) |
|---|---|
| P1-F | GGTCTCTCTGGTTAGACCAG |
| P1-R | GTCTGAGGGATCTCTAGTTA |
| P2-F | GACTCGGCTTGCTGAAGCGC |
| P2-R | TTTTTTCCCATCGATCTAAT |
| P3-F | ACACAGTGGGGGGACATCAA |
| P3-R | TTCCTGCTATGTCACTTCCC |
| P4-F | GGAAAAAGGGCTGTTGGAAA |
| P4-R | GTGGGGCTGTTGGCTCTGGT |
| P5-F | TCAATTAGGAATACCACATC |
| P5-R | GGTGTCTCATTGTTTATACT |
| P6-F | TTACCCAGGGATTAAAGTAA |
| P6-R | TCATAATACACTCCATGTAC |
| P7-F | GAAAGAACCCATAGTAGGAG |
| P7-R | TGTAACTCAGTTTTCTGATT |
| P8-F | TGTAGTAGCAAAAGAAATAG |
| P8-R | GCTACATGAACTGCTACCAG |
| P9-F | GATTGGGGGGTACAGTGCAG |
| P9-R | TTTGCTGGTCCTTTCCAAAG |
| P10-F | TTGGCTCCATGGCTTAGGGC |
| P10-R | TCTGTCGAGTAACGCCTATT |
| P11-F | GGTAAATGTGACAGAAAATT |
| P11-R | TTCTTCAAATCAGTGCACTT |
| P12-F | ACAGCTGAACCAATCTGTAG |
| P12-R | GCTCTACTAATGTTACAATG |
| P13-F | AGGAAGCACTATGGGCGCAG |
| P13-R | AGCTGCTTGATGCCCCAGAC |
| P14-F | GCTAAAGAATAGTGCTGTTA |
| P14-R | AGCAAAATCCTTTCCAAGCC |
| P15-F | TAGCATTTCATCACATGGCC |
| P15-R | TGCAGGATCTGAGGGCTCGC |
Primer extension.
The tRNA3Lys-viral RNA template was extracted from purified HIV-1 particles. The extension ability of tRNA3Lys annealed in vivo to viral RNA was measured as described previously (20). Reactions were carried out with a volume of 20 μl containing 50 mM Tris-HCl (pH 7.8), 100 mM KCl, 10 mM MgCl2, 10 mM dithiothreitol, 0.16 μM [α-32P]dCTP, 50 ng of HIV-1 reverse transcriptase, and an RNase inhibitor (Ambion). After incubation for 15 min at 37°C, the samples were precipitated with 2-propanol and were separated on a denaturing 6% polyacrylamide gel. The relative amounts of extended tRNA3Lys were determined by using a PhosphorImager instrument. The relative amounts of viral RNA in the reaction mixtures were also determined by measuring the ability of a DNA oligonucleotide (5′-TCTAATTCTCCCCCGCTTAATACTGACGCT-3′) annealed at room temperature to the viral RNA to prime a 1-base dCTP extension.
Knockdown of endogenous RHA with siRNA.
Endogenous RHA in 293T cells was knocked down by using small interfering RNA (siRNA) oligonucleotides, as described previously (56). Levels of endogenous RHA were measured by Western blotting using anti-RHA antibodies. The same membranes were probed with β-actin MAb (Sigma), to measure levels of β-actin as an internal control.
Statistical analysis.
The Student t test was employed in statistical analyses. The lowest level of significance was set at a P value of <0.05.
RESULTS
Binding of RHA to different regions within the HIV-1 genomic RNA in vitro.
We first examined in vitro RHA binding to RNA fragments representing different regions within the BH10 HIV-1 genome. These regions included the trans-activation response element (TAR)-poly(A), the primer binding site (PBS) stem-loop, the leader, the GagPol frameshift site, and the Rev response element (RRE) as well as regions coding for reverse transcriptase, Vpr, and gp120. These RNAs were synthesized by in vitro transcription, and as shown in Fig. 1A, one-dimensional (1D) PAGE of 32pCp-labeled RNAs separated in native 6% polyacrylamide gels indicated the presence of one species. In order to obtain quantitative binding data, fluorescence polarization (FP) was used to measure the interaction between viral RNA end labeled with FTSC and increasing amounts of full-length RHA protein that was purified from 293E cells (56). The efficiency of RNA labeling by FTSC was examined by measuring the fluorescence emission of 10 nM FTSC-labeled RNA at a wavelength of 528 nm. As shown in Fig. 1B, all the RNAs tested were labeled by FTSC at similar levels. The equilibrium dissociation constant (Kd) for the RHA-viral RNA interaction was then determined from the FP data, as previously described (29). Glutathione S-transferase (GST), also isolated from 293E cells, was used as a non-RNA binding negative control. The conditions for equilibrium binding were determined by varying the incubation temperatures and times, and since equilibrium was reached for all RNA fragments tested at room temperature within 5 min, all reactions were run at room temperature for 15 min. In contrast with the lack of GST binding to RNA, RHA bound to all HIV-1 RNA regions tested (Fig. 1C), with Kd values ranging from 44 nM to 178 nM.
Fig 1.

Ability of RHA to bind to different regions of HIV-1 genomic RNA. (A) Native gel electrophoresis of synthetic HIV-1 RNA fragments. The HIV-1 RNAs were synthesized by in vitro transcription, labeled with 32pCp, separated in native 6% polyacrylamide gels, and visualized by using a PhosphorImager instrument. (B) Labeling of HIV-1 RNA by FTSC. The fluorescence emission at a wavelength of 528 nm was measured for 10 nM FTSC-labeled HIV-1 RNAs. (C) FP was used to determine the binding affinities of RHA for the different regions of the HIV-1 genomic RNA. The GST protein was used in place of RHA as a negative control. FTSC (10 nM)-labeled viral RNA was incubated with various amounts of RHA or GST in the absence of ATP, and the FP was then measured. The FP values obtained are plotted relative to the corresponding values that were obtained at a zero protein concentration, which are presented as a value of 1.0.
Binding of RHA to HIV-1 RNA during viral replication.
In order to identify sites in the HIV-1 RNA bound by RHA during viral replication, an RNA-protein coprecipitation assay was used (5). 293T cells were cotransfected with a BH10 vector containing full-length BH10 HIV-1 proviral DNA and a plasmid expressing His-tagged RHA. As a negative control, 293T cells were also cotransfected with a BH10 vector and a plasmid expressing only the 6×His tag. At 24 h posttransfection, cells were exposed to formaldehyde cross-linking, lysed, and sonicated. His-tagged RHA was precipitated from the cell lysate by using Ni-NTA agarose. Western blot analysis verified that His-tagged RHA was expressed in cells (Fig. 2A) and precipitated (Fig. 2B). Total RNA (as the input) and RNA isolated from the precipitates were harvested and analyzed by RT-PCR with primers specific for different regions of the HIV-1 RNA (Fig. 2C). All RNA-protein coprecipitation results are presented relative to input values. The HIV-1 transcripts were detected by RT-PCR from total RNA preparations. RT-PCR detected the HIV-1 transcripts containing sequence of the 5′-UTR (primer pairs P1-F/R and P2-F/R) or RRE (primer pair P13-F/R) in RHA-coprecipitated RNA but not in His-tag-coprecipitated RNA. No amplification products were detected in reaction mixtures that lacked reverse transcriptase (−RT). The results demonstrate that RHA binds to specific regions of the HIV-1 RNA during viral replication, with the 5′-UTR of HIV-1 RNA being the major target for RHA.
Fig 2.
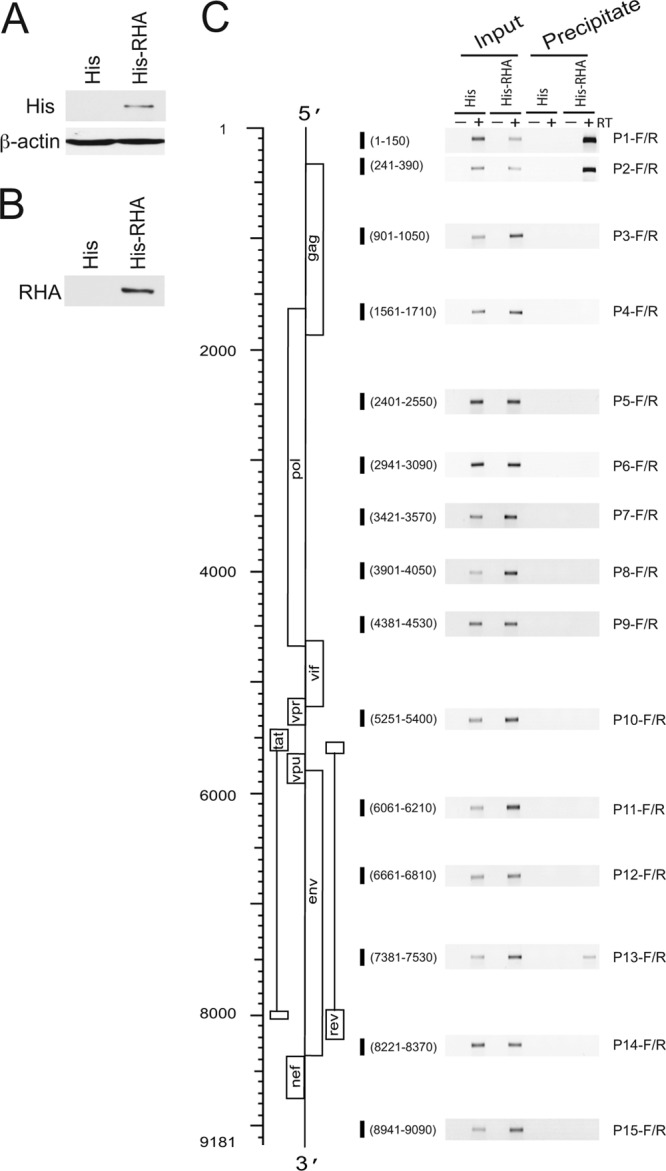
RHA binding to viral RNA in vivo using an RNA-protein coprecipitation assay. 293T cells were cotransfected with a BH10 vector containing full-length HIV-1 BH10 proviral DNA and a plasmid expressing His-tagged RHA or only the 6×His tag. Twenty-four hours later, cells were cross-linked, lysed, and sonicated. The cell lysates were subjected to coprecipitation with Ni-NTA agarose. RNA was isolated from cell lysates (input) or precipitates and was subjected to RT-PCR analysis. (A) Western blotting with an anti-His tag antibody detected the expression of His-tagged RHA in cotransfected cells. The 6×His tag peptide only was not resolved in Western blots. (B) Western blotting with an anti-RHA antibody detected His-tagged RHA in precipitates. (C) RT-PCR amplification products from the indicated RNA preparations. The schematic shown on the left is HIV-1 genomic RNA. Numbers indicate the nucleotide positions relative to the transcription start site (position +1). The open reading frames in HIV-1 RNA are indicated. Shown on the right are the amounts of input (0.1%) or coprecipitated viral RNA, as measured by RT-PCR using primers specific for the different regions of the HIV-1 RNA. Shorter vertical bold lines represent the nucleotide sequences in the HIV-1 genome that were amplified by RT-PCR. RT-PCR was performed in the presence (+) or absence (−) of reverse transcriptase.
Role of the dsRBDs in RHA binding to HIV-1 RNA during viral replication.
RHA is characterized by two dsRBDs at the N terminus. We evaluated the role of the dsRBDs in the binding of RHA to HIV-1 RNA during viral replication by performing RNA-protein coprecipitation experiments, in which 293T cells were cotransfected with a BH10 vector containing full-length BH10 HIV-1 proviral DNA and a plasmid, either pRHAΔaa1-100, pRHAΔaa134-250, or pRHAΔdsRBD1-2, which codes for an N-terminally His-tagged mutant RHA protein lacking dsRBD1, dsRBD2, or both dsRBD1 and dsRBD2, respectively (Fig. 3A). Western blotting verified that these mutant RHAs were expressed in cells (Fig. 3B) and were precipitated (Fig. 3C). RNA was then isolated and analyzed by RT-PCR. As shown in Fig. 3D, RT-PCR detected HIV-1 transcripts in total RNA (input) and in wild-type RHA-coprecipitated RNA, using primer sets P1-F/R and P13-F/R, which are specific for the HIV-1 5′-UTR and RRE regions, respectively. However, no HIV-1 transcripts were detected in the precipitates of mutant RHAs. These results demonstrate that both dsRBDs are essential for the binding of RHA to HIV-1 RNA during viral replication.
The dsRBDs are required for RHA to promote tRNA3Lys annealing to viral RNA.
RHA is packaged into HIV-1 particles (48) and also promotes the annealing of tRNA3Lys to viral RNA (56). Because dsRBDs are necessary for the binding of RHA to HIV-1 RNA in vivo, we investigated whether the deletion of either or both dsRBDs could affect these two processes. 293T cells were first transfected with siRNARHA to knock down endogenous RHA and were then cotransfected 16 h later with a BH10 vector coding for BH10 of HIV-1 and a plasmid coding for His-tagged wild-type RHA or His-tagged mutant RHA lacking either one or both dsRBDs. siRNARHA does not reduce the expression level of exogenous wild-type or mutant RHA, since the mRNAs lack the siRNARHA target sequences (56). Forty-eight hours later, the viral particles were purified, and cell lysates were prepared. Western blot analysis of cell lysates probed with an anti-6×His tag antibody verified the expression of exogenous wild-type or mutant RHA in the presence of siRNARHA (Fig. 4A), but only wild-type exogenous RHA was packaged into virus particles (Fig. 4B). Thus, mutant RHAs lacking either one or both dsRBDs were not detectable in the virus, indicating that both dsRBDs are necessary for the packaging of RHA into viral particles.
Fig 4.
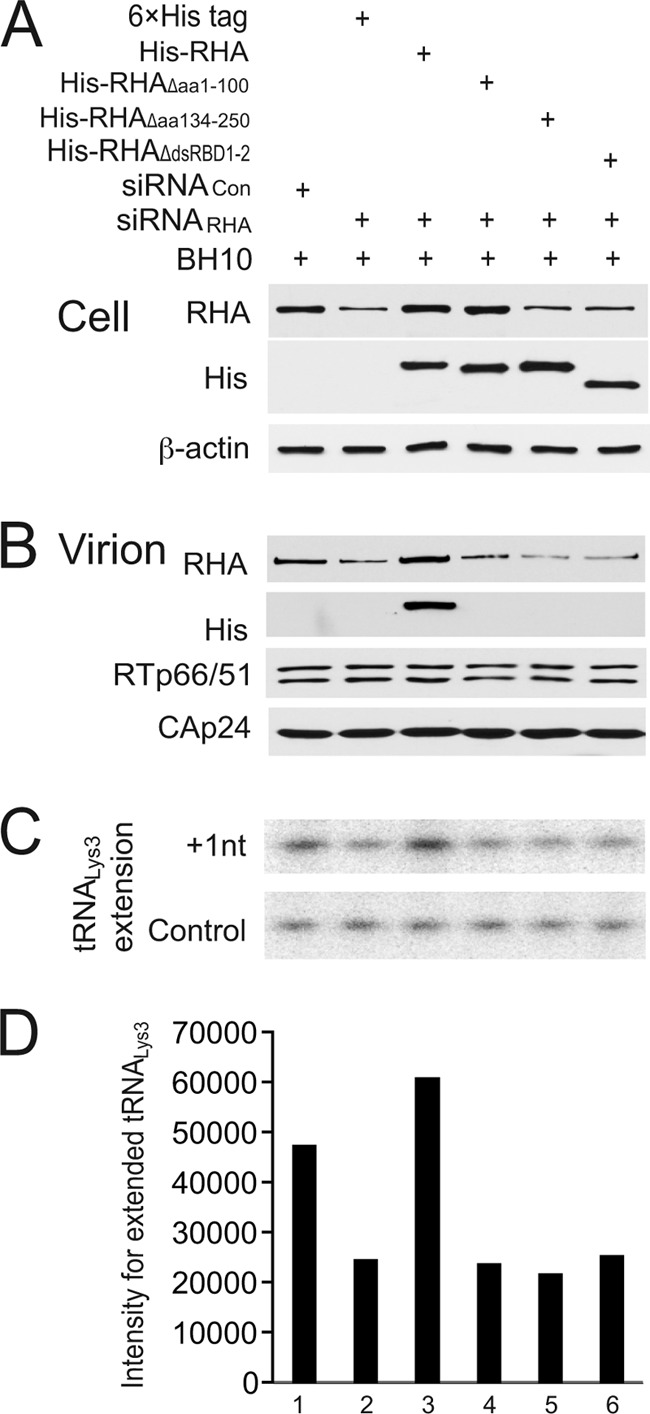
Role of dsRBDs in the in vivo promotion of tRNA3Lys annealing to HIV-1 RNA by RHA. 293T cells were first transfected with siRNACon or siRNARHA and were then cotransfected 16 h later with a BH10 vector coding for HIV-1 BH10 and a plasmid expressing either the 6×His tag or His-tagged wild-type or mutant RHA lacking either one or both dsRBDs. (A) Western blots of cell lysates probed with antibodies to RHA, the 6×His tag, or β-actin. While the anti-RHA MAb efficiently recognized both wild-type RHA and mutant RHA missing dsRBD1, it was not able to efficiently detect RHA missing dsRBD2 or both dsRBD1 and dsRBD2. (B) Western blots of viral lysates, containing equal amounts of CAp24, probed with antibodies to RHA, the 6×His tag, CAp24, or RTp66/p51. (C) One-nucleotide extension assay (+1-nt extension) to measure tRNA3Lys-primed initiation of reverse transcription. Total viral RNA was isolated, and tRNA3Lys annealed to viral RNA was extended by 1 nt by using reverse transcriptase ([32P]dCTP). The extended tRNA3Lys products were resolved by 1D PAGE and detected by using a PhosphorImager instrument. The control gel represents the +1-nt extension of a DNA primer annealed to viral RNA downstream of PBS and was used to show that equal amounts of viral RNA were used in each extension reaction mixture. (D) The values of the +1-nt-extended tRNA3Lys products, quantitated by using a PhosphorImager instrument, are presented graphically. Shown are values representative of 3 independent experiments.
The amount of tRNA3Lys annealed to viral RNA can be examined by a 1-nucleotide (nt) extension assay of tRNA3Lys by reverse transcriptase. Deproteinized total viral RNA was used as the source of primer tRNA3Lys annealed in vivo to viral genomic RNA, and the incorporation of the first deoxynucleoside triphosphate (dNTP), [32P]dCTP, was measured (56). Using this assay, it was shown that the knockdown of endogenous RHA by siRNARHA reduces the annealing of tRNA3Lys to viral RNA by approximately 50% and that this defect in tRNA3Lys annealing was rescued by the expression of exogenous RHA whose mRNA product lacks siRNARHA target sequences (56). Since the mRNAs encoding the mutant RHA lacking either one or both dsRBDs in this report also lack siRNARHA target sequences, we performed the 1-nt extension assay to examine the ability of these mutant RHAs to rescue reduced tRNA3Lys annealing by siRNARHA. Equal amounts of viral genomic RNA used in these reaction mixtures were first quantitated by dot blot hybridization, and this quantitation was further checked by annealing a DNA primer complementary to viral RNA sequences downstream of the primer binding site (PBS) and extending it by 1 nt (Fig. 4C, control). The 1D PAGE data shown in Fig. 4C were quantitated by using a PhosphorImager instrument, and the results are shown graphically in Fig. 4D. tRNA3Lys priming was not decreased by siRNARHA in the presence of exogenous wild-type RHA (Fig. 4C, lane 3), but mutant RHAs lacking either one or both dsRBDs were not able to replace reduced endogenous RHA and promote annealing. Thus, these results indicate that dsRBDs are necessary for RHA to promote the annealing of tRNA3Lys to viral RNA.
DISCUSSION
RHA has been found to be involved in multiple steps of HIV-1 replication, and in this report, we have examined how RHA interacts with HIV-1 RNA. Initially, we examined the ability of RHA to bind to isolated RNA fragments rather than these same sequences within the context of the entire HIV-1 RNA genome. The lengths of these fragments were somewhat heterogeneous, since we tried to include the stem-loop structures for each region previously predicted by SHAPE analysis (53). We found that RHA can bind all the isolated HIV-1 RNA fragments, with Kd values ranging from 40 to 178 nM (Fig. 1). However, RNA-protein coprecipitation analyses indicated that during viral replication in the cell, RHA could be detected only in association with two viral RNA regions, i.e., primarily with the 5′-UTR and, to a much lesser extent, the RRE (Fig. 2). While associations with other regions may have been below our level of detection, these results indicate that RHA binds to very specific regions in vivo.
Various roles for RHA in regulating the functions facilitated by sequences within the 5′-UTR of HIV-1 have been reported. For example, RHA was found to be associated with the TAR stem-loop in HIV-1 RNA, and the overexpression of RHA results in an increase in HIV mRNA accumulation (17). RHA was also reported previously to play a role in promoting the annealing of tRNA3Lys to PBS (56). A better translational efficiency of retroviral mRNA is achieved by RHA binding to R/U5 sequences within the 5′-UTR, perhaps resulting in the unwinding of this structurally complex region to facilitate ribosomal scanning (22).
Similarly, the interaction of RHA with the RRE region in HIV-1 RNA was also implicated in playing a role in the HIV-1 life cycle. Rev binds to the RRE, and Rev function involves the export of unspliced and partially spliced mRNA out of the nucleus (43). It was reported previously that the overexpression of RHA resulted in an increased ratio of unspliced and partially spliced to fully spliced HIV-1 RNAs (32). Furthermore, RHA was identified in the spliceosome (46), suggesting that RHA may facilitate the release of unspliced and partially spliced RNAs from the spliceosome.
Comparisons of RHA binding to HIV-1 RNA in vitro and in vivo suggest the presence of factors in the cell that can modulate RHA binding that are not present in the in vitro binding experiments. Nevertheless, duplex regions in the RNA still appear to be a major binding target for RHA. Thus, when the dsRBDs are deleted, RHA binding to viral RNA in vivo is inhibited (Fig. 3), favoring the hypothesis that the dsRBDs are the main domains that mediate the binding of RHA to substrate RNA and that the main target for RHA binding would be a duplex RNA region. The importance of dsRBDs for the interaction of RHA with RNA is suggested by the fact that dsRBDs are critical for RHA to promote the translational efficiency of HIV-1 mRNA (45), and, in contrast to wild-type RHA, mutant RHAs lacking either one or both dsRBDs are unable to be packaged into virus particles or to promote the annealing of tRNA3Lys to viral RNA (Fig. 4).
The mode of interaction of the dsRBD with dsRNA has been elucidated through analyses of the crystal structure of a complex that comprises a short synthetic 10-bp RNA duplex and a dsRBD of X1rbpa (49) or the entire RNase III (6) or through nuclear magnetic resonance (NMR) analyses of a complex between the RNA stem-loop and the dsRBD of Staufen (44) or Rnt1 (52, 55). In all these cases, the dsRBD adopts an α-β-β-β-α topology and binds to dsRNA through interactions with the phosphodiester backbone and 2′-OH groups (49), suggesting that the binding of dsRBDs to RNA in vitro lacks apparent sequence specificity (16). This further supports the idea that other viral or cellular factors may modulate the specific binding of RHA to the substrate duplex RNA in vivo.
It is not clear why the 5′-UTR binds much more strongly to RHA in vivo than does the RRE, since both the 5′-UTR (4, 13, 15, 21, 27, 38–40, 54) and the RRE (3, 25, 28, 41, 42, 53) have been shown to have a high content of double-stranded RNA regions by using several different methods to determine their structures. We have also shown that these two regions have similar binding affinities for RHA in vitro (Fig. 1). Whether this difference in binding reflects an altered duplex content of cytoplasmic viral RNA in vivo or a change in the RHA structure that alters its ability to bind to these RNA regions, the results imply the presence of other viral or cellular factors that promote such changes in the RNA or RHA structure. An example of RHA binding being directed through its interaction with other proteins in mammalian cells is the interaction between RHA and the stem cell protein Lin28. Lin28 recognizes specific sequences in a subset of mRNAs, and its direct interaction with RHA is believed to result in the recruitment of RHA to the translational machinery, resulting in the stimulation of translation (26). A similar phenomenon was observed for LARP6, a member of the La ribonucleoprotein domain family, where LARP6 was shown to bind to both specific sequences in the 5′-UTR of collagen mRNA and to RHA, thereby directing RHA to this site (34). Directed RHA binding may also occur when RHA binds to the transcription factors CREB binding protein (CBP) and RNA polymerase II (2).
ACKNOWLEDGMENT
This work was supported by a grant from the Canadian Institutes of Health.
Footnotes
Published ahead of print 26 September 2012
REFERENCES
- 1. Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. 1998. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 19:254–256 [DOI] [PubMed] [Google Scholar]
- 2. Aratani S, et al. 2001. Dual roles of RNA helicase A in CREB-dependent transcription. Mol. Cell. Biol. 21:4460–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Battiste JL, et al. 1996. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science 273:1547–1551 [DOI] [PubMed] [Google Scholar]
- 4. Baudin F, et al. 1993. Functional sites in the 5′ region of human immunodeficiency virus type 1 RNA form defined structural domains. J. Mol. Biol. 229:382–397 [DOI] [PubMed] [Google Scholar]
- 5. Bittencourt D, Auboeuf D. 2012. Analysis of co-transcriptional RNA processing by RNA-ChIP assay. Methods Mol. Biol. 809:563–577 [DOI] [PubMed] [Google Scholar]
- 6. Blaszczyk J, et al. 2004. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure 12:457–466 [DOI] [PubMed] [Google Scholar]
- 7. Bleichert F, Baserga SJ. 2007. The long unwinding road of RNA helicases. Mol. Cell 27:339–352 [DOI] [PubMed] [Google Scholar]
- 8. Bolinger C, Sharma A, Singh D, Yu L, Boris-Lawrie K. 2010. RNA helicase A modulates translation of HIV-1 and infectivity of progeny virions. Nucleic Acids Res. 38:1686–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolinger C, et al. 2007. RNA helicase A interacts with divergent lymphotropic retroviruses and promotes translation of human T-cell leukemia virus type 1. Nucleic Acids Res. 35:2629–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burd CG, Dreyfuss G. 1994. Conserved structures and diversity of functions of RNA-binding proteins. Science 265:615–621 [DOI] [PubMed] [Google Scholar]
- 11. Busch S, Kirsebom LA, Notbohm H, Hartmann RK. 2000. Differential role of the intermolecular base-pairs G292-C(75) and G293-C(74) in the reaction catalyzed by Escherichia coli RNase P RNA. J. Mol. Biol. 299:941–951 [DOI] [PubMed] [Google Scholar]
- 12. Cachianes G, et al. 1993. Epstein-Barr virus-derived vectors for transient and stable expression of recombinant proteins. Biotechniques 15:255–259 [PubMed] [Google Scholar]
- 13. Clever JL, Miranda D, Jr, Parslow TG. 2002. RNA structure and packaging signals in the 5′ leader region of the human immunodeficiency virus type 1 genome. J. Virol. 76:12381–12387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cordin O, Banroques J, Tanner NK, Linder P. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37 [DOI] [PubMed] [Google Scholar]
- 15. Damgaard CK, Andersen ES, Knudsen B, Gorodkin J, Kjems J. 2004. RNA interactions in the 5′ region of the HIV-1 genome. J. Mol. Biol. 336:369–379 [DOI] [PubMed] [Google Scholar]
- 16. Fierro-Monti I, Mathews MB. 2000. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem. Sci. 25:241–246 [DOI] [PubMed] [Google Scholar]
- 17. Fujii R, et al. 2001. A role of RNA helicase A in cis-acting transactivation response element-mediated transcriptional regulation of human immunodeficiency virus type 1. J. Biol. Chem. 276:5445–5451 [DOI] [PubMed] [Google Scholar]
- 18. Fuller-Pace FV. 2006. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 34:4206–4215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gatfield D, et al. 2001. The DExH/D box protein HEL/UAP56 is essential for mRNA nuclear export in Drosophila. Curr. Biol. 11:1716–1721 [DOI] [PubMed] [Google Scholar]
- 20. Guo F, Saadatmand J, Niu M, Kleiman L. 2009. Roles of Gag and NCp7 in facilitating tRNA(Lys)(3) annealing to viral RNA in human immunodeficiency virus type 1. J. Virol. 83:8099–8107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harrison GP, Miele G, Hunter E, Lever AM. 1998. Functional analysis of the core human immunodeficiency virus type 1 packaging signal in a permissive cell line. J. Virol. 72:5886–5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hartman TR, et al. 2006. RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 13:509–516 [DOI] [PubMed] [Google Scholar]
- 23. He QS, et al. 2008. Comparisons of RNAi approaches for validation of human RNA helicase A as an essential factor in hepatitis C virus replication. J. Virol. Methods 154:216–219 [DOI] [PubMed] [Google Scholar]
- 24. Isken O, et al. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13:1675–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iwai S, Pritchard C, Mann DA, Karn J, Gait MJ. 1992. Recognition of the high affinity binding site in rev-response element RNA by the human immunodeficiency virus type-1 rev protein. Nucleic Acids Res. 20:6465–6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin J, et al. 2011. Evidence that Lin28 stimulates translation by recruiting RNA helicase A to polysomes. Nucleic Acids Res. 39:3724–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kasprzak W, Bindewald E, Shapiro BA. 2005. Structural polymorphism of the HIV-1 leader region explored by computational methods. Nucleic Acids Res. 33:7151–7163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kjems J, Brown M, Chang DD, Sharp PA. 1991. Structural analysis of the interaction between the human immunodeficiency virus Rev protein and the Rev response element. Proc. Natl. Acad. Sci. U. S. A. 88:683–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kovaleski BJ, et al. 2006. In vitro characterization of the interaction between HIV-1 Gag and human lysyl-tRNA synthetase. J. Biol. Chem. 281:19449–19456 [DOI] [PubMed] [Google Scholar]
- 30. Lawrence P, Rieder E. 2009. Identification of RNA helicase A as a new host factor in the replication cycle of foot-and-mouth disease virus. J. Virol. 83:11356–11366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee CG, Hurwitz J. 1992. A new RNA helicase isolated from HeLa cells that catalytically translocates in the 3′ to 5′ direction. J. Biol. Chem. 267:4398–4407 [PubMed] [Google Scholar]
- 32. Li J, et al. 1999. A role for RNA helicase A in post-transcriptional regulation of HIV type 1. Proc. Natl. Acad. Sci. U. S. A. 96:709–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin L, et al. 2012. Identification of RNA helicase A as a cellular factor that interacts with influenza A virus NS1 protein and its role in the virus life cycle. J. Virol. 86:1942–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manojlovic Z, Stefanovic B. 2012. A novel role of RNA helicase A in regulation of translation of type I collagen mRNAs. RNA 18:321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marchler-Bauer A, et al. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229 doi:10.1093/nar/gkq1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Naji S, et al. 2012. Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Mol. Cell. Proteomics 11:M111.015313 doi:10.1074/mcp.M111.015313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakajima T, et al. 1997. RNA helicase A mediates association of CBP with RNA polymerase II. Cell 90:1107–1112 [DOI] [PubMed] [Google Scholar]
- 38. Ooms M, Verhoef K, Southern E, Huthoff H, Berkhout B. 2004. Probing alternative foldings of the HIV-1 leader RNA by antisense oligonucleotide scanning arrays. Nucleic Acids Res. 32:819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paillart JC, et al. 2004. First snapshots of the HIV-1 RNA structure in infected cells and in virions. J. Biol. Chem. 279:48397–48403 [DOI] [PubMed] [Google Scholar]
- 40. Paillart JC, Skripkin E, Ehresmann B, Ehresmann C, Marquet R. 2002. In vitro evidence for a long range pseudoknot in the 5′-untranslated and matrix coding regions of HIV-1 genomic RNA. J. Biol. Chem. 277:5995–6004 [DOI] [PubMed] [Google Scholar]
- 41. Pallesen J, Dong M, Besenbacher F, Kjems J. 2009. Structure of the HIV-1 Rev response element alone and in complex with regulator of virion (Rev) studied by atomic force microscopy. FEBS J. 276:4223–4232 [DOI] [PubMed] [Google Scholar]
- 42. Phuphuakrat A, Auewarakul P. 2003. Heterogeneity of HIV-1 Rev response element. AIDS Res. Hum. Retroviruses 19:569–574 [DOI] [PubMed] [Google Scholar]
- 43. Pollard VW, Malim MH. 1998. The HIV-1 Rev protein. Annu. Rev. Microbiol. 52:491–532 [DOI] [PubMed] [Google Scholar]
- 44. Ramos A, et al. 2000. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 19:997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ranji A, Shkriabai N, Kvaratskhelia M, Musier-Forsyth K, Boris-Lawrie K. 2011. Features of double-stranded RNA-binding domains of RNA helicase A are necessary for selective recognition and translation of complex mRNAs. J. Biol. Chem. 286:5328–5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rappsilber J, Ryder U, Lamond AI, Mann M. 2002. Large-scale proteomic analysis of the human spliceosome. Genome Res. 12:1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robb GB, Rana TM. 2007. RNA helicase A interacts with RISC in human cells and functions in RISC loading. Mol. Cell 26:523–537 [DOI] [PubMed] [Google Scholar]
- 48. Roy BB, et al. 2006. Association of RNA helicase A with human immunodeficiency virus type 1 particles. J. Biol. Chem. 281:12625–12635 [DOI] [PubMed] [Google Scholar]
- 49. Ryter JM, Schultz SC. 1998. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 17:7505–7513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toretsky JA, et al. 2006. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 66:5574–5581 [DOI] [PubMed] [Google Scholar]
- 51. Walbott H, et al. 2010. Prp43p contains a processive helicase structural architecture with a specific regulatory domain. EMBO J. 29:2194–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Z, Hartman E, Roy K, Chanfreau G, Feigon J. 2011. Structure of a yeast RNase III dsRBD complex with a noncanonical RNA substrate provides new insights into binding specificity of dsRBDs. Structure 19:999–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Watts JM, et al. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460:711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilkinson KA, et al. 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 6:e96 doi:10.1371/journal.pbio.0060096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu H, Henras A, Chanfreau G, Feigon J. 2004. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc. Natl. Acad. Sci. U. S. A. 101:8307–8312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xing L, Liang C, Kleiman L. 2011. Coordinate roles of Gag and RNA helicase A in promoting the annealing of formula to HIV-1 RNA. J. Virol. 85:1847–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang S, Grosse F. 1997. Domain structure of human nuclear DNA helicase II (RNA helicase A). J. Biol. Chem. 272:11487–11494 [DOI] [PubMed] [Google Scholar]
- 58. Zhang Z, Yuan B, Lu N, Facchinetti V, Liu YJ. 2011. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J. Immunol. 187:4501–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]