

Abstract
DNA is increasingly used as an important tool in programming the self-assembly of micrometer- and nanometer-scale particles. This is largely due to the highly specific thermoreversible interaction of cDNA strands, which, when placed on different particles, have been used to bind precise pairs in aggregates and crystals. However, DNA functionalized particles will only reach their true potential for particle assembly when each particle can address and bind to many different kinds of particles. Indeed, specifying all bonds can force a particular designed structure. In this paper, we present the design rules for multiflavored particles and show that a single particle, DNA functionalized with many different “flavors,” can recognize and bind specifically to many different partners. We investigate the cost of increasing the number of flavors in terms of the reduction in binding energy and melting temperature. We find that a single 2-μm colloidal particle can bind to 40 different types of particles in an easily accessible time and temperature regime. The practical limit of ∼100 is set by entropic costs for particles to align complementary pairs and, surprisingly, by the limited number of distinct “useful” DNA sequences that prohibit subunits with nonspecific binding. For our 11 base “sticky ends,” the limit is 73 distinct sequences with no unwanted overlaps of 5 bp or more. As an example of phenomena enabled by polygamous particles, we demonstrate a three-particle system that forms a fluid of isolated clusters when cooled slowly and an elastic gel network when quenched.
Keywords: multifunctional, thermodynamics
A defining feature of DNA nanotechnology is the ability of DNA single strands to bind selectively only with complementary strands (1–8). Identical particles coated with identical DNA strands can be joined together by adding to the suspension a linker strand that attaches to the two coatings (9, 10). Such structures have been used for immunoassays (11), particle aggregation, and formation of crystalline structures, typically Face Centered Cubic (FCC) (12). Use has also been made of different particles, A and B, functionalized with cDNA strands (13). This configuration, where A-A and B-B bonds do not occur but A-B bonds do (14–16), has been exploited to form more complex crystals, such as BCC or CsCl structures (12, 17). Over the past several years, there has been a great deal of progress in modeling the DNA-mediated interparticle interaction and making quantitative comparisons with experiments (16, 18–23). Although nanoscale particles are typically coated with tens to hundreds of DNA molecules, and micrometer-scale colloids can be coated with 104–105 DNA strands, there has been little work on coating particles with more than one type of DNA sequence on the same particle. Allowing these particles to be “polygamous,” to specifically bind to a particular set of other particles, enables not only the fabrication of more complex crystals but the design of more general programmed structures. For rigid structures, specifying each interparticle bond specifically is sufficient to define the object (24, 25). The construction can therefore be set by coating each particle with the DNA strands that only link to other specific particles.
In this article, we outline the design rules by which polygamous particles can be made and demonstrate, in the case of four different coatings, that one particle can bind to four different types of particles without mutual interference. We then address the limitations of polygamous particles: How many different flavors we can have on each particle while maintaining its ability to attract and mate with other particles? One might suppose that if a particle can accommodate 105 DNA strands, it can be coated with 105 different sequences to bind to 105 different particles, not all at the same time, of course, but a total of 105 potential mates. However, even though a DNA strand can bind to its complementary strand when suspended in solution, two DNA strands attached to the surfaces of two different spheres can only bind when the spheres are in particular configurations. The result is a substantial entropy cost that has to be taken into account in the binding energy of the DNA. By contrast, when many identical DNA strands coat the particles uniformly, bonds can form in any orientation. Diluting the surface coverage of each sequence restricts the configurations and increases the entropy cost. We also require that subsequences do not pair with subsequences on wrong chains or to form hairpins. Avoiding mutual interference of subsequences greatly limits the number of available “flavors.” For example, the longer the length of sticky end DNA, the fewer sequences avoid five-base interferences that would hybridize above 0 °C. As a result, there is a practical limit of ∼100 different strand flavors for our 2-μm particles. We show both experimentally and theoretically how the melting temperature changes as sequences are diluted, and we calculate the number of distinct sequences of N bases avoiding M overlaps. Finally, we demonstrate how we can use such polygamous particles to synthesize an elementary system with properties that cannot be achieved by traditional monogamous particles, a system that gels when temperature is quenched and forms isolated clusters when cooled slowly.
Polygamous Particles
Particles and DNA Structures.
Our basic construct for this study is shown schematically in Fig. 1A. A DNA double strand is functionalized with a biotin molecule. The 5′ end of a DNA single strand, 61 nt long, is connected to the biotin group by a flexible polymer PEG spacer. On the other end, the single strand is terminated by an 11-base “sticky end,” 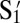 , S1, and T in Fig. 1A. A 49-base complementary strand makes a rigid double helix between the PEG spacer and the active sticky end. Spheres are coated with streptavidin, which can bind irreversibly at our operating temperatures to biotinylated DNA (19, 20, 26, 27). The number of DNA binding sites ranges from 6,000 for 1-μm spheres to 70,000 for 2-μm spheres depending on the vendor and batch. The sites may contain a single type of DNA sequence or a number of different DNA sequences randomly distributed on the surface. The ratio of different sequences is set by their solution ratio before functionalizing the surface. Details of colloid preparation and stabilization as well as relative sequence concentration studies are found in studies by Dreyfus et al. (19, 20). The sticky ends reside about L = 15 nm from their binding site on the surface. Their surface density is ∼1 strand/(13 nm)2. The maximum number of bonds that can geometrically form between complementary sticky ends on different particles is ∼200.
, S1, and T in Fig. 1A. A 49-base complementary strand makes a rigid double helix between the PEG spacer and the active sticky end. Spheres are coated with streptavidin, which can bind irreversibly at our operating temperatures to biotinylated DNA (19, 20, 26, 27). The number of DNA binding sites ranges from 6,000 for 1-μm spheres to 70,000 for 2-μm spheres depending on the vendor and batch. The sites may contain a single type of DNA sequence or a number of different DNA sequences randomly distributed on the surface. The ratio of different sequences is set by their solution ratio before functionalizing the surface. Details of colloid preparation and stabilization as well as relative sequence concentration studies are found in studies by Dreyfus et al. (19, 20). The sticky ends reside about L = 15 nm from their binding site on the surface. Their surface density is ∼1 strand/(13 nm)2. The maximum number of bonds that can geometrically form between complementary sticky ends on different particles is ∼200.
Fig. 1.

Several kinds of DNA-mediated colloidal self-assembly systems. (A) Watson–Crick system: Colloids can bind specifically to complementary particles but not to the same species. (B) Particles coated with two different kinds of DNA. (C) Particles coated with four different kinds of DNA that can address four different kinds of particles. (D) Two-shell system. An X is first surrounded by Y’s to form a complete shell (green), and the green shell is then surrounded by Z’s (blue shell) to form, finally, a two-shell cluster.
Synthesis and Design Rules.
To manufacture polygamous particles, special attention needs to be paid to the design of the complementary pairs of DNA. The hybridization of DNA strands involves competition between an enthalpic contribution, which includes hydrogen bonding and hydrophobic interactions, and an entropic contribution, which includes the loss of configurational entropy when two flexible single strands make one more rigid double strand. Hence, the binding free energy depends on temperature and the dissociation, or melting temperature, depends on the base sequence matches and mismatches. Thus, two rules emerge for designing the sequences of DNA on polygamous particles (6, 7):
i) Over the temperature range of the experiments, only complementary pairs of DNA should hybridize. The melting temperatures of noncomplementary pairs of DNA should be set below the working range, namely, below 0 °C. This step is to ensure that each DNA flavor of a polygamous particle is specific and that no nonspecific DNA links are allowed between particles.
ii) Folding structures for all the DNA sticky ends must be minimized. Secondary structures, such as loops and hairpins, reduce the number of active ends, and hence the binding free energy and melting temperature (26, 28–31).
These rules might look trivial. However, enumerating the distinct sequences that obey these rules requires calculation or computation. A method to find all the complementary pairs of DNA sequences following the design rules is as follows:
i) List all the possible DNA sequences with the specific length N. (In our case, the length of sticky end DNA is 11.) This will allow 4N different DNA sequences.
ii) Eliminate the sequence pairs that are “partially” (see Number of “Useful” DNA Sequences of Length N) complementary to any of the rest of DNA sequences. This step is to enforce rule i.
iii) Eliminate the sequence pairs that are partially complementary to themselves. This step is to enforce rule ii.
Number of “Useful” DNA Sequences of Length N.
We would like to design DNA sequences that bind only to their complementary sequences using the complete N bases. We want to avoid any hairpins or improper binding of subsequences above a minimum temperature considerably below our characteristic melting curves. Because the melting temperature for 5-bp dsDNA is about 0 °C, and about 10 °C for 6-bp dsDNA, we would like to avoid any inadvertent 5-bp sequence overlaps in our 11-bp sticky ends. First, we treat the general problem of avoiding M-bp sequences in N-bp strings. The number of M-bp sequences is 4M. We want to avoid any palindromes here because they can lead to strands sticking to themselves or forming hairpins. The number of palindromes is 4M/2 if M is even. There are no DNA palindromes if M is odd. [A DNA palindrome read left to right is complementary to the sequence read right to left (e.g., ACGT is complementary to TGCA but ACXGT cannot be complementary to TGXCA because no base is its own complement).] The number of M-bp words in an N-bp sequence is (N − M + 1). We require all these M-bp words to be different. We also require that the complementary strand be read in the same 3′ → 5′ direction as the original strand to have different M-letter words. Thus, each “useful” sequence depletes 2 × (N − M + 1) sequences or words of M bp from the total number of 4M − 4M/2 for M even or 4M for M odd. The number, Pmax, of useful sequences is then
 |
 |
where ⌊…⌋ is the integer part function. The numbers above are upper limits; it is not evident that Pmax distinct sequences can be found. For N = 11 and M = 5, Pmax = 73; thus, for our experimental conditions and requirements, we would be limited to 73 different flavors. Eliminating 5-bp overlaps ensures that there are no 6-bp, 7-bp, or higher overlaps. This allows us to work at any temperature above ∼0 °C. If we relax our conditions and allow 5-bp overlaps but no 6-bp overlaps, Tm ≈ 10 °C (where Tm is the melting temperature), we have for N = 11 and M = 6, Pmax = 336. Then, comparison must be done with the actual melting temperature of the sequences, and we would have to be careful not to allow the system to cool below ∼10 °C. It seems that a practical limit of P ∼100 is reasonable.
Example.
To manufacture polygamous particles of two and four different flavors, four sequences and their complements need to be designed with regard to the two rules expressed earlier. The sequences we generate and use are as follows:
S1: 5′-GTAGAAGTAGG-3′
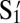 : 5′-CCTACTTCTAC-3′
: 5′-CCTACTTCTAC-3′S2: 5′-GATGGATTAGG-3′
 : 5′-CCTAATCCATC-3′
: 5′-CCTAATCCATC-3′S3: 5′-GTATTCGAGTT-3′
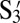 : 5′-AACTCGAATAC-3′
: 5′-AACTCGAATAC-3′S4: 5′-ATAGATTCCGA-3′
 : 5′-TCGGAATCTAT-3′
: 5′-TCGGAATCTAT-3′
An Internet-based application, the UNAFold Web Server, is used to predict the melting temperatures of DNA to give a check of the sequences (32–34). Because the melting temperatures of DNA are sensitive to the concentrations of sodium and DNA sequences, we choose [Na+] = 73.4 mM and DNA concentration = 0.012 μM, which are the conditions for the following experiments. The melting temperatures for all possible pairs of DNA sequences are shown in Table 1. There are no unwanted associations above 0 °C. The melting temperatures of the secondary structures of all DNA sequences are listed at the bottom of Table 1. We find that hairpins are also suppressed above 0 °C. Table 1 shows that we can easily find DNA sequences obeying the design rules for P ≪ Pmax.
Table 1.
Melting temperatures of DNA pair hybridization (°C) determined from the UNAFold Web Server with 73.4 mM sodium and 0.012 micromolar DNA
| S1 |  |
S2 | 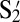 |
S3 | 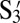 |
S4 |  |
|
| S1 | −214 | 18 | −151 | −68 | −76 | −147 | −66 | −66 |
 |
— | −214 | −68 | −177 | −99 | −69 | −80 | −78 |
| S2 | — | — | −146 | 19 | −138 | −101 | −77 | −39 |
 |
— | — | — | N/A | −96 | −102 | −45 | −50 |
| S3 | — | — | — | — | −54 | 20 | −56 | −55 |
 |
— | — | — | — | — | −54 | −56 | −55 |
| S4 | — | — | — | — | — | — | −52 | 19 |
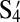 |
— | — | — | — | — | — | — | −51 |
| Folding | −41 | −54 | −175 | N/A | −48 | N/A | N/A | −81 |
Test of Mutual Interference.
Our first experiment is to determine whether the presence of several different DNA flavors on the same surface interferes with binding of complementary sequences on different particles. To study this, particles are coated with two different kinds of active DNA at moderate concentrations. The remaining surface sites are filled with neutral DNA (T). The neutral DNA strands are poly-dT oligomers (19, 20). Three species of colloidal particles, A, B, and C, are manufactured in such a way that each species can address the other two species as shown in Fig. 1B. Species A is covered with 23% S1, 18% S2, and 59% T. Species B is covered with 20% S3, 18%  , and 62% T. Species C is covered with 23%
, and 62% T. Species C is covered with 23%  , 20%
, 20% 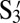 , and 57% T. The relative concentrations have been adjusted to match the melting temperatures of pairs of particles. To quantify the melting temperature, the “singlet fraction,” the fraction of unbound particles, is measured as a function of temperature. As predicted by our design, the melting curves of Watson–Crick-like colloidal pairs A + B, A + C, and B + C are essentially identical as shown in Fig. 2. The presence of additional active strands does not change/modify/affect the association of particle pairs or aggregates. Finally, we mix A, B, and C particles in equal amounts and measure the fraction of unbound particles of any species. Because the attraction strength between each pair (A + B, B + C, and A +C) is similar, one might expect that the aggregation behavior and the melting curve of all particles mixed together would be similar to the aggregation behavior of each pair. Surprisingly, the results show that the melting curve for all particles mixed together is different from the Watson–Crick pairs. The melting temperature, Tm, defined as f(Tm) ≡ 0.5, is ∼0.8 °C higher than the melting temperature of each pair as shown in Fig. 2.
, and 57% T. The relative concentrations have been adjusted to match the melting temperatures of pairs of particles. To quantify the melting temperature, the “singlet fraction,” the fraction of unbound particles, is measured as a function of temperature. As predicted by our design, the melting curves of Watson–Crick-like colloidal pairs A + B, A + C, and B + C are essentially identical as shown in Fig. 2. The presence of additional active strands does not change/modify/affect the association of particle pairs or aggregates. Finally, we mix A, B, and C particles in equal amounts and measure the fraction of unbound particles of any species. Because the attraction strength between each pair (A + B, B + C, and A +C) is similar, one might expect that the aggregation behavior and the melting curve of all particles mixed together would be similar to the aggregation behavior of each pair. Surprisingly, the results show that the melting curve for all particles mixed together is different from the Watson–Crick pairs. The melting temperature, Tm, defined as f(Tm) ≡ 0.5, is ∼0.8 °C higher than the melting temperature of each pair as shown in Fig. 2.
Fig. 2.

Melting behaviors of particles that can address two different particles: AB (pink), BC (yellow-green), AC (cyan), and ABC (black). The dots are the experimental data. The solid curves are the model plots.
There are two reasons for this shift. (i) A system with three components and three interactions (A + B + C) has more binding configurations than any of the paired systems (A + B, A + C, or B + C), each of which has two components and one interaction. In the A + B system, each particle can bind to 1/2 of the other particles in the system. In the A + B + C system, each particle can bind to 2/3 of the other particles in the system. These extra binding configurations cause the A + B + C system to have a higher Tm than the A + B, B + C, or A + C system (SI Text). (ii) Importantly, there is an additional binding energy when A, B, and C form a triangle with three bonds, (AB, BC, and AC) rather than an open structure with two bonds (e.g., AB and BC or AC and CB or BA and AC). The melting curves of A + B, A + C, B + C, and A + B + C are plotted along with the model predictions in Fig. 2 (SI Text). According to our calculations, the observed shift of ∼0.8 °C is ∼0.2 °C from i and ∼0.6 °C from ii.
Polygamous Experiments.
Although the three mutually attractive particles show aggregation in separate pairs as well as collectively, because the particles are optically identical, we cannot show directly that one particle has paired specifically with a number of different particles. For this demonstration, we need labeled particles. Polygamous particles D are coated with four different flavors of DNA corresponding to S1, S2, S3, and S4 as shown in Fig. 1C. We then made four particles, E, F, G, and H, complementary only to the sequences on D and not to each other. E, F, G, and H are coated with 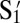 ,
,  ,
, 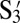 , and
, and 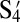 , respectively, and can be distinguished by fluorescence and size as shown in Fig. 1C. The buffer is also dyed with fluorescein; thus, our nonfluorescent, polygamous particle D can be identified as the black object in the fluorescent environment.
, respectively, and can be distinguished by fluorescence and size as shown in Fig. 1C. The buffer is also dyed with fluorescein; thus, our nonfluorescent, polygamous particle D can be identified as the black object in the fluorescent environment.
Results.
We check that each particle pairing works properly. Fig. 3A shows that D binds separately to each of E, F, G, and H at room temperature. In Fig. 3B, we compare the aggregation of a mixture of all five particles and the four monogamous spouses without the polygamous one. Clusters form in the presence of the polygamous D, but there are only unbound particles when E, F, G, and H are suspended in solution without D. Aggregates form when there are a sufficient number of polygamous particles to bridge clusters (or to share partners.) If we reduce the number of polygamous D particles relative to the number of other species, only single clusters form with D surrounded by its partners. An example is shown in Fig. 3C, where a cluster of E, G, H, and 2 F’s is bound to a polygamous D.
Fig. 3.

Polygamous particles that can address four particles. (A) First control experiment: D can bind to each of E, F, G, and H separately. (B) Second control experiment: E, F, G, and H cannot form any aggregation without D. (C) D attached to four different particles E, F, G, and H simultaneously.
Rotational Entropy.
Our two- and four-partner studies indicate that particles can be coated with several flavors of DNA and that the different sequences bind independently, without interference, to their complements on other particles. The question then arises as to how many different flavors can be put on a particle and what the cost is. Naively, one might expect that the limit is simply set by the number of DNA strands that can be attached to a particle. Given the right concentration and buffer, two cDNA strands can hybridize in solution; thus, given the same concentration and buffer conditions, should two complementary particles with a single DNA strand on each not be able to bind? The problem lies in additional entropy costs. Each sphere with particle radius Rp has rotational entropy corresponding to 4π of solid angle. For DNA strands of length L with sticky end length l, overlap and binding can occur only when two strands are less than 2L + l apart. For particles with a surface separation of h as shown in Fig. 1A, the active patch around each DNA on each particle has radius [(L + l/2)2 − (h/2)2]1/2. The active fraction of the surface area covered by a DNA is ϕ = π[(L + l/2)2 − (h/2)2]/[4π(Rp + h/2)2] (a detailed explanation of the rotational entropy is provided in SI Text). With one strand on each of two spheres, binding only occurs for a limited number of relative orientations corresponding to overlapping patches. Suppose that the active patches on a sphere cover ϕ = 1/10 of the area of the sphere. The number of angular orientations allowed for binding two spheres is ϕ2 = 1/100, one-hundredth of the number of orientations allowed for unbound spheres. The entropy loss is ΔSr = kBln(ϕ2), where kB is Boltzmann’s constant. Depending on ϕ, this type of reduction can bring the hybridization temperature from above 40 °C to below 0 °C.
How Many Flavors at What Cost?
Experiments.
There are other factors that influence the binding or aggregation of DNA-coated colloids. Here, we chose to study theoretically and experimentally the effect of reducing the DNA coverage. Using a single flavor and its complement suffices. We coat our particles with cDNA S1 and 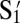 strands to form a Watson–Crick paired system as shown in Fig. 1A. We mix Watson, coated with S1, and Crick, coated with
strands to form a Watson–Crick paired system as shown in Fig. 1A. We mix Watson, coated with S1, and Crick, coated with 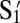 , homogeneously in equal amounts and measure the melting curves of the colloidal aggregation. The experiment is performed for χ = 0.025, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.75, and 1. Here, χ is the fraction of active strands,
, homogeneously in equal amounts and measure the melting curves of the colloidal aggregation. The experiment is performed for χ = 0.025, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.75, and 1. Here, χ is the fraction of active strands, 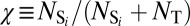 , where
, where  is the number of strands of sequence Si (e.g., S1,
is the number of strands of sequence Si (e.g., S1, 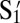 , …) and NT is the number of inert poly-dT strands. The melting curves and the melting temperatures for each χ are shown in Fig. 4 A and B. The results show that the melting temperature decreases from 50.3 °C for χ = 1–22 °C for χ = 0.025. The transition width of the melting curves increases from 0.8 °C for χ = 1–5 °C for χ = 0.025. The more sticky end DNA strands there are on particle surfaces, the higher the melting temperature and the sharper the melting transition of the colloidal aggregation will be.
, …) and NT is the number of inert poly-dT strands. The melting curves and the melting temperatures for each χ are shown in Fig. 4 A and B. The results show that the melting temperature decreases from 50.3 °C for χ = 1–22 °C for χ = 0.025. The transition width of the melting curves increases from 0.8 °C for χ = 1–5 °C for χ = 0.025. The more sticky end DNA strands there are on particle surfaces, the higher the melting temperature and the sharper the melting transition of the colloidal aggregation will be.
Fig. 4.

Melting behaviors of polygamous particles with different coverage of a flavor. (A) Melting curve of an A-B system for different coverage χ. From left (red) to right (blue), χ = 0.025, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.75, and 1, respectively. The solid lines are the melting curves determined from Eq. 1, whereas the dots are the experimental data. (B) Melting temperature as a function of the DNA sticky end coverage χ. The blue dots are the experimental data. The solid line is the melting temperature, Tm, determined from Eq. 1. The dashed lines indicate 6.9% measurement error of the total DNA surface coverage on the particles. (Inset) Melting temperatures as a function of the sticky end DNA coverage χ in a log (χ)-linear (Tm) plot.
Model.
To model the system quantitatively, we consider the chemical equation, C1 + Ci ⇋ Ci+1, where Ci indicates the number density of clusters with i particles. A detailed explanation of the model and its parameters is provided in SI Text. Because the system is a two-component system, in thermal equilibrium, we can solve the chemical equation and find that
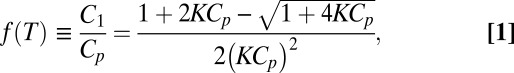 |
where Cp = 0.01 μm−2 is the total particle concentration,  is the equilibrium constant, Aw = [2π(2Rp)](2b) is the “wiggling area” of bound particles, Rp = 980 nm is our particle radius, b ≈ 0.34 nm is the spacing of bases along a dsDNA (35, 36), and ΔFp is the binding energy of a pair of particles (19, 20, 37, 38):
is the equilibrium constant, Aw = [2π(2Rp)](2b) is the “wiggling area” of bound particles, Rp = 980 nm is our particle radius, b ≈ 0.34 nm is the spacing of bases along a dsDNA (35, 36), and ΔFp is the binding energy of a pair of particles (19, 20, 37, 38):
 |
where ΔF0 = ΔH0 − T(ΔS0 + ΔSp), ΔH0 = −328,000 J/mol, ΔS0 = −966 J/mol of K, and ΔSp ≈ −11 kB. ΔSp is the configurational entropy loss of a DNA double strand with one end bound on a particle surface, on binding to a DNA double strand on a complementary particle. Before binding, the free “sticky” end of each DNA strand can explore a hemisphere of radius the strand length. After the sticky ends bind, they are confined to a ring. Nb is the number of sticky ends that can bind between two particles, and gb is the number of sticky ends on one particle that an opposing sticky end can reach. Nb and gb depend on the surface-to-surface particle separation, h (taken as L + l/2 ≈ 16.8 nm, which is about the half of the length of our dsDNA link), and the DNA surface coverage χ. At high coverage, Nb and gb are both proportional to χ, whereas at low coverage, Nb and gb are both proportional to χ2. We perform a computation to determine Nb and gb as a function of χ. ΔSr is the particle rotational entropic cost between the unbound and bound states for a pair of complementary particles (SI Text). The ΔSr term was introduced in the work of Biancaniello et al. (18). For high coverage, there is little loss in rotational entropy for the particles. Whatever rotational configuration they have when apart will allow binding when they are together; thus, ΔSr ∼ 0. At low coverage, where only a fraction of the surface is covered, the binding rotational configurations are reduced from the configurations of free particles and ΔSr becomes significant. For a DNA strand of length L = 15 nm with sticky ends of the length l ≈ 3.6 nm, surface separation h, spherical particle of radius Rp, and Nt total DNA binding sites on the particle, each DNA again has a fractional active area of ϕ = π[(L + l/2)2 − (h/2)2]/[4π(Rp + h/2)2]. The fractional area coverage of n = Ntχ DNAs randomly placed on the surface is 1 − (1 − ϕ)n, and the entropic cost of binding two such particles is ΔSr = 2kB ln[1 − (1 − ϕ)n].
Comparison of Model and Experiments.
From gb, Nb, and Eqs. 1 and 2, we can determine the singlet fraction as a function of temperature T, and we plot it in Fig. 4A for each DNA coverage χ. Fig. 4B shows the comparison of the model with the experimental data for melting temperature, Tm, vs. coverage, χ. Within experimental error, the model and the data are consistent. For high coverage, the variation of Tm with χ is dominated by the number of DNA bonds, Nb, and the degeneracy of interparticle binding for each DNA sticky end (gb). At low coverage, Nb ∼ 1 and gb ∼ 1. Here, the variation of Tm with coverage comes from the loss of rotational entropy of particles, ΔSr.
Dual-Phase Materials
To demonstrate the utility of polygamous particles, we design a two-shell system as shown in Fig. 1D. There are three species in the system: X, Y, and Z. To form a two-shell–like cluster as shown in Fig. 1D, the melting temperature of X-Y, TXY, must be higher than the melting temperature of Y- Z, TYZ. Hence, based on our model, the coverages of S3 on X and 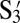 on Y have to be adjusted to be higher than the coverages of S1 on Y and
on Y have to be adjusted to be higher than the coverages of S1 on Y and 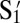 on Z. Hence, X is coated with 100% S3. Y, which is the polygamous particle in this system, is coated with 75%
on Z. Hence, X is coated with 100% S3. Y, which is the polygamous particle in this system, is coated with 75% 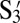 , 10% S1, and 15% T. Z is coated with 50%
, 10% S1, and 15% T. Z is coated with 50% 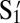 and 50% T. Particle concentrations of X, Y, and Z are nX = 0.006 μm−2, nY = 0.03 μm−2, and nZ = 0.06 μm−2, respectively. The melting curves of the system, fXYZ, can be predicted by our model by superposing the melting curves of X-Y, fXY and Y-Z, fYZ, with the correct weight:
and 50% T. Particle concentrations of X, Y, and Z are nX = 0.006 μm−2, nY = 0.03 μm−2, and nZ = 0.06 μm−2, respectively. The melting curves of the system, fXYZ, can be predicted by our model by superposing the melting curves of X-Y, fXY and Y-Z, fYZ, with the correct weight:  . The plot of the model is shown in Fig. 5A. Unlike the melting curves of usual binary systems, like Fig. 4A, the melting curve is a two-step function. The first step is due to the Y-Z melting, and the second step is due to the X-Y melting. A comparison of the model prediction and the measured melting curve is shown in Fig. 5A (SI Text, SI Thermodynamic Model of Dual-Phase Materials). The melting temperatures of X-Y and Y-Z are TXY ≈ 47 °C and TYZ ≈ 41 °C, respectively. Our model can be used to predict not only the binary system but the multistep melting curves for the polygamous system.
. The plot of the model is shown in Fig. 5A. Unlike the melting curves of usual binary systems, like Fig. 4A, the melting curve is a two-step function. The first step is due to the Y-Z melting, and the second step is due to the X-Y melting. A comparison of the model prediction and the measured melting curve is shown in Fig. 5A (SI Text, SI Thermodynamic Model of Dual-Phase Materials). The melting temperatures of X-Y and Y-Z are TXY ≈ 47 °C and TYZ ≈ 41 °C, respectively. Our model can be used to predict not only the binary system but the multistep melting curves for the polygamous system.
Fig. 5.

Dual-phase (fluid/gel) system. (A) Melting curve of an X-Y-Z system. The blue dots are data. The red curve is the model plot. (B) Gel synthesized by one-step quenching. The yellow dashed line is the melting temperature of XY. The cyan dashed line is the melting temperature of YZ. (C) Fluid synthesized by two-step cooling.
The dual-phase nature of the system is shown in Fig. 5 B and C. First, we heat the sample to 52 °C, where all clusters are melted, for 5 min. Then, we quench the sample to 23 °C, at which point X binds to Y and Y binds to Z, for 160 min. The system is similar to a usual binary system. Particles aggregate and form a branched percolating network as shown in Fig. 5B. All clusters in the system are immobile and not diffusive. The system is a gel. In contrast, if we cool the system to 43 °C, which is between TXY and TYZ, for 120 min, X will absorb all the Y’s in the solution and form a cluster with X as a core and Y’s as the shell. Then, we cool the system to 23 °C, which is below both TXY and TYZ, for 160 min. At this stage, Z’s will stick to the one-shell cluster, saturate the periphery of the cluster, and form the second shell as shown in Fig. 1D. The one-shell clusters diffuse too slowly to aggregate before being coated by the Z’s. After that, the system will only have several two-shell clusters and some excess individual Z particles. Although some cluster-cluster bridging is unavoidable, this aggregation is too little to percolate. Fig. 5C shows the system with several inert clusters and individual Z particles. Because each object (cluster or individual particle) is inert to the other, the system is mobile and diffusive. The system behaves like the fluid.
Using polygamous particles, we have made a dual-phase system whose connectivity, and hence rheology, is history/protocol-dependent. The system is designed so that its structural and physical properties depend on the cooling process. Basically, a few X particles sequester enough Y particles to inhibit percolation of the Y-Z system. Slow or two-step cooling yields a fluid phase with disconnected clusters. A quench yields a percolating rigid gel.
Conclusions
Our experimental study shows that a coverage of 0.025 allows aggregation at room temperature. Thus, a polygamous particle with 1/0.025 = 40 flavors of DNA could operate conveniently. For our particles, the limit for DNA to operate normally would be 0 °C, where water freezes; here, the coverage could be as low as ∼0.001 or ∼1,000 flavors. In fact, changing the salt concentration of the buffer would allow any coverage down to a single strand per flavor. However, the number of distinct DNA sequences that avoid unwanted nonspecific subsequence binding is surprisingly strongly limited. If we want to eliminate any unwanted 5-bp overlaps in our 11-bp sticky ends, we are limited to 73 flavors. Hence, our study suggests that a practical limit for polygamous particles is about 100 flavors per system due to the intrinsic properties of DNA sequence combination and hybridization. Of course, for particles of the same size in direct contact, the maximum number of partners a particle can have is 12, but a set of particles with particular properties (e.g., color, dielectric constant, conductivity) could be programmed to associate with 40 different particles or 40 different sites in a structure. For particles of different sizes, there is no limit to the number of, for example, small partners a large particle can have. For immunology or other bulk assays, such polygamous particles could quickly separate a host of other particles from suspension. For colloidal architecture, many repeating motifs could be bound to different places on the structure.
Supplementary Material
Acknowledgments
We thank Colm Kelleher for technical assistance with the experiments. We acknowledge partial support from National Aeronautics and Space Administration under Grant NNX08AK04G and the Materials Research Science and Engineering Center under Grant DMR-0820341 for particle development and characterization. N.C.S. acknowledges support from National Institutes of Health Grant GM-29554 for DNA synthesis and characterization, and K.-T.W., L.F., and P.M.C. acknowledge support for thermal and optical data and analysis from Department of Energy–Basic Energy Sciences Grant DE3C0007991.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1207356109/-/DCSupplemental.
References
- 1.Seeman NC. DNA in a material world. Nature. 2003;421(6921):427–431. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- 2.Aldaye FA, Palmer AL, Sleiman HF. Assembling materials with DNA as the guide. Science. 2008;321(5897):1795–1799. doi: 10.1126/science.1154533. [DOI] [PubMed] [Google Scholar]
- 3.Yan H, Zhang X, Shen Z, Seeman NC. A robust DNA mechanical device controlled by hybridization topology. Nature. 2002;415(6867):62–65. doi: 10.1038/415062a. [DOI] [PubMed] [Google Scholar]
- 4.Winfree E, Liu F, Wenzler LA, Seeman NC. Design and self-assembly of two-dimensional DNA crystals. Nature. 1998;394(6693):539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 5.Yurke B, Turberfield AJ, Mills APJ, Jr, Simmel FC, Neumann JL. A DNA-fuelled molecular machine made of DNA. Nature. 2000;406(6796):605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 6.Seeman NC. Nucleic acid junctions and lattices. J Theor Biol. 1982;99(2):237–247. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 7.Nilsen TW, Grayzel J, Prensky W. Dendritic nucleic acid structures. J Theor Biol. 1997;187(2):273–284. doi: 10.1006/jtbi.1997.0446. [DOI] [PubMed] [Google Scholar]
- 8.Wang T, et al. Self-replication of information-bearing nanoscale patterns. Nature. 2011;478(7368):225–228. doi: 10.1038/nature10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tkachenko AV. Morphological diversity of DNA-colloidal self-assembly. Phys Rev Lett. 2002;89(14):148303. doi: 10.1103/PhysRevLett.89.148303. [DOI] [PubMed] [Google Scholar]
- 10.Jones MR, et al. DNA-nanoparticle superlattices formed from anisotropic building blocks. Nat Mater. 2010;9(11):913–917. doi: 10.1038/nmat2870. [DOI] [PubMed] [Google Scholar]
- 11.Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature. 1996;382(6592):607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 12.Park SY, et al. DNA-programmable nanoparticle crystallization. Nature. 2008;451(7178):553–556. doi: 10.1038/nature06508. [DOI] [PubMed] [Google Scholar]
- 13.Maye MM, Nykypanchuk D, van der Lelie D, Gang O. DNA-regulated micro- and nanoparticle assembly. Small. 2007;3(10):1678–1682. doi: 10.1002/smll.200700357. [DOI] [PubMed] [Google Scholar]
- 14.Crocker JC. Nanomaterials: Golden handshake. Nature. 2008;451(7178):528–529. doi: 10.1038/451528a. [DOI] [PubMed] [Google Scholar]
- 15.Milam VT, Hiddessen AL, Crocker JC, Graves DJ, Hammer DA. DNA-driven assembly of bidisperse, micron-sized colloids. Langmuir. 2003;19:10317–10323. [Google Scholar]
- 16.Valignat MP, Theodoly O, Crocker JC, Russel WB, Chaikin PM. Reversible self-assembly and directed assembly of DNA-linked micrometer-sized colloids. Proc Natl Acad Sci USA. 2005;102(12):4225–4229. doi: 10.1073/pnas.0500507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nykypanchuk D, Maye MM, van der Lelie D, Gang O. DNA-guided crystallization of colloidal nanoparticles. Nature. 2008;451(7178):549–552. doi: 10.1038/nature06560. [DOI] [PubMed] [Google Scholar]
- 18.Biancaniello PL, Kim AJ, Crocker JC. Colloidal interactions and self-assembly using DNA hybridization. Phys Rev Lett. 2005;94(5):058302. doi: 10.1103/PhysRevLett.94.058302. [DOI] [PubMed] [Google Scholar]
- 19.Dreyfus R, et al. Simple quantitative model for the reversible association of DNA coated colloids. Phys Rev Lett. 2009;102(4):048301. doi: 10.1103/PhysRevLett.102.048301. [DOI] [PubMed] [Google Scholar]
- 20.Dreyfus R, et al. Aggregation-disaggregation transition of DNA-coated colloids: Experiments and theory. Phys Rev E Stat Nonlin Soft Matter Phys. 2010;81(4 Pt 1):041404. doi: 10.1103/PhysRevE.81.041404. [DOI] [PubMed] [Google Scholar]
- 21.Rogers WB, Crocker JC. Direct measurements of DNA-mediated colloidal interactions and their quantitative modeling. Proc Natl Acad Sci USA. 2011;108(38):15687–15692. doi: 10.1073/pnas.1109853108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mognetti BM, Leunissen ME, Frenkel D. Controlling the temperature sensitivity of DNA-mediated colloidal interactions through competing linkages. Soft Matter. 2012;8(7):2213–2221. [Google Scholar]
- 23.Mognetti BM, et al. Predicting DNA-mediated colloidal pair interactions. Proc Natl Acad Sci USA. 2012;109(7):E378–E379, author reply E380. doi: 10.1073/pnas.1119991109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arkus N, Manoharan VN, Brenner MP. Minimal energy clusters of hard spheres with short range attractions. Phys Rev Lett. 2009;103(11):118303. doi: 10.1103/PhysRevLett.103.118303. [DOI] [PubMed] [Google Scholar]
- 25.Arkus N, Manoharan VN, Brenner MP. Deriving finite sphere packings. SIAM J Discrete Math. 2011;25(4):1860–1901. [Google Scholar]
- 26.Leunissen ME, et al. Switchable self-protected attractions in DNA-functionalized colloids. Nat Mater. 2009;8(7):590–595. doi: 10.1038/nmat2471. [DOI] [PubMed] [Google Scholar]
- 27.Leunissen ME, et al. Towards self-replicating materials of DNA-functionalized colloids. Soft Matter. 2009;5(12):2422–2430. [Google Scholar]
- 28.Bonnet G, Krichevsky O, Libchaber A. Kinetics of conformational fluctuations in DNA hairpin-loops. Proc Natl Acad Sci USA. 1998;95(15):8602–8606. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace MI, Ying L, Balasubramanian S, Klenerman D. FRET fluctuation spectroscopy: Exploring the conformational dynamics of a DNA hairpin loop. J Phys Chem B. 2000;104(48):11551–11555. [Google Scholar]
- 30.Antao VP, Tinoco I., Jr Thermodynamic parameters for loop formation in RNA and DNA hairpin tetraloops. Nucleic Acids Res. 1992;20(4):819–824. doi: 10.1093/nar/20.4.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ying L, Wallace MI, Klenerman D. Two-state model of conformational fluctuation in a DNA hairpin-loop. Chem Phys Lett. 2001;334(1–3):145–150. [Google Scholar]
- 32.Dimitrov RA, Zuker M. Prediction of hybridization and melting for double-stranded nucleic acids. Biophys J. 2004;87(1):215–226. doi: 10.1529/biophysj.103.020743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Markham NR, Zucker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:577–581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Markham NR, Zuker M. Bioinformatics: Structure, Function and Applications. Totowa, NJ: Humana Press; 2008. Vol 2, Chap 1, pp 3–31. [Google Scholar]
- 35.Bates AD, Maxwell A. DNA Topology. New York: Oxford Univ Press; 2005. pp. 1–23. [Google Scholar]
- 36.Watson JD, Crick FHC. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 37.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA. 1998;95(4):1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charbonneau P, Frenkel D. Gas-solid coexistence of adhesive spheres. J Chem Phys. 2007;126(19):196101. doi: 10.1063/1.2737051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.