

Abstract
Alzheimer’s disease (AD) is the most common form of dementia among the elderly. In AD patients, memory loss is accompanied by the formation of beta-amyloid plaques and the appearance of tau in a pathological form. Given the lack of effective treatments for AD, the development of new management strategies for these patients is critical. The continued failure to find effective therapies using molecules aimed at addressing the anti-beta amyloid pathology has led researchers to focus on other non-amyloid-based approaches to restore memory function. Promising non-amyloid related candidate targets include phosphosdiesterases (PDEs), and indeed, Rolipram, a specific PDE4 inhibitor, was the first compound found to effectively restore cognitive deficits in animal models of AD. More recently, PDE5 inhibitors have also been shown to effectively restore memory function. Accordingly, inhibitors of other members of the PDE family may also improve memory performance in AD and non-AD animal models. Hence, in this review, we will summarize the data supporting the use of PDE inhibitors as cognitive enhancers and we will discuss the possible mechanisms of action underlying these effects. We shall also adopt a medicinal chemistry perspective that leads us to propose the most promising PDE candidates on the basis of inhibitor selectivity, brain distribution, and mechanism of action.
Keywords: Clinical trial, cGMP, mild cognitive impairment, pCREB, GSK3, PKG, p-tau, sildenafil, tadalafil
A significant number of people develop cognitive impairments with increasing age. Mild cognitive impairment is characterized by problems that affect memory, language or other essential cognitive functions, but that do not interfere with an individual’s daily life. Dementia is characterized by the progressive global deterioration of cognitive abilities, including memory and other intellectual functions, to an extent that does interfere with daily life. Alzheimer’s disease (AD) is the most common form of dementia among older people. Clinically, AD is characterized by the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles in the brain, structures that provoke neuronal dysfunction and cell death.
The increasing number of AD patients associated with the aging of the population makes the development of new disease management/treatment strategies critical. The search for effective AD management has been largely based on the amyloid (Aβ) hypothesis, mainly focusing on reducing the number of senile plaques, although with little success to date. There is a growing belief that current approaches in AD treatment begin far too late to significantly slow down the progress of the disease or to delay the onset of the most severe symptoms. Indeed, the continued failure of these therapies suggests that new approaches must consider alternative, nonamyloid strategies, to restore memory function.
Interestingly, specific phosphodiesterase (PDE) inhibitors have been shown to improve memory performance in different animal models of AD. PDE inhibitors regulate signaling pathways by elevating levels of cAMP and/or cGMP, which may ultimately promote gene transcription by activating the cAMP response element-binding (CREB).1,2 CREB-dependent gene expression underlies long-term memory formation and persistent long-term potentiation (LTP), which are indicators of synaptic plasticity and strength.3,4 In the hippocampus, this probably occurs through the formation of new synaptic connections.5,6 In AD, memory impairment appears to significantly precede the death of nerve cells, implying that neuronal dysfunction may be responsible for the pathophysiology of early stage AD. Thus, by activating the CREB signaling pathway, PDE inhibitors may ameliorate AD symptoms by restoring synaptic function. Moreover, other CREB-independent mechanisms seem to act in synergy to restore cognitive impairment in AD.
1. PDEs in the Rodent and Human Brain
The cyclic nucleotide phosphodiesterases (PDEs) comprise a group of enzymes that control the rate of hydrolysis of cAMP and cGMP. Hence, these enzymes are important regulators of the signals mediated by these second messenger molecules. Eleven different PDE families (PDE1–PDE11) have been described, each typically comprising multiple isoforms and splice variants, leading to a total of more than 100 proteins.7 One key question that arises is whether the different PDE variants play specific roles in distinct physiological and pathological processes. To determine the specific function of these isoforms in the central nervous system (CNS), several studies have quantified the mRNA or protein of different PDE isoforms in the brain.8,9 The regional distribution of these isoforms suggests that these enzymes play an important role in neurons, regulating intracellular signaling cascades triggered by neurotransmitters that bind to specific cell surface receptors. Their mechanism of action is straightforward, as they control the intracellular concentration of the two aforementioned second messengers, cAMP and/or cGMP. In fact, classification of PDEs is largely based on substrate specificity (cAMP, cGMP, or dual: see Figure 1). Although most PDE isoforms are expressed in the brain (PDE1, PDE2, PDE3, PDE4, PDE5A, PDE7A, PDE7B, PDE8B, PDE9A, PD10A, and PDE11A), their level of expression varies from one region to another, and in some cases even between neurons of the same region. For example, PDE5 and PDE1 are localized in the cerebellum but only in Purkinje neurons, while PDE1B is localized in specific subsets of Purkinje cells.10
Figure 1.
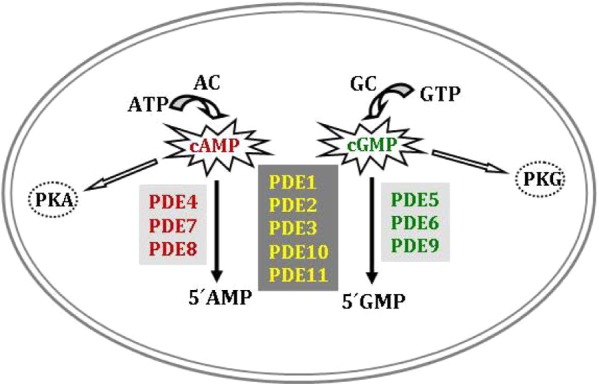
Substrate specificity of the different PDEs. In green: cAMP-specific PDEs, in red: cGMP-specific PDEs, and in yellow: cAMP/cGMP-specific PDEs. AC: adenyl cyclase, GC: guanylyl cyclase, PKA: protein kinase A, PKG: protein kinase G.
PDE6 has not been detected in the CNS, as its expression is restricted to the retina and pineal gland.7 In contrast to other PDEs, neither PDE6 nor PDE3 appears to play a significant role in neural function and each is expressed at low levels in the CNS.8,11 Although PDE3 was thought to be present at low levels in the CNS, inhibitors of this PDE activate the CREB signaling pathway in the brain (see below) and recent evidence suggests that PDE3B is expressed in proopiomelanocortin and neuropeptide Y neurons in the mouse hypothalamus.12
Three subtypes of PDE1 have been described that exhibit comparable distribution patterns in the CNS: 1A, 1B, and 1C. The areas in which these subtypes have been analyzed included the hippocampus, cerebral cortex, thalamus, and striatum, with particularly high levels of PDE1B in the caudate nucleus and the nucleus accumbens.8,13 The expression of PDE1 in the hippocampus and cerebral cortex suggests that this enzyme may play a role in memory formation and storage. Indeed, the PDE1 inhibitor vinpocetine has beneficial effects on cognitive function in rats with streptozotocin-induced learning and memory impairments,14 and in animal models of mental retardation. (15)
Several studies of murine and/or human brain tissue have demonstrated that PDE2A is widely expressed in the brain, with the strongest expression in the cortex, striatum, and hippocampus.8,11,16−18 The expression of PDE2A in the hippocampus suggests a role in memory formation, which has been demonstrated in vivo through the effects of specific PDE2 inhibitors in different memory tasks.19,20
PDE4 is widely expressed in the CNS21−24 and there are four genes (PDE4A, 4B, 4C, and 4D) that encode over 20 different variants of this enzyme. (25) The exact role of PDE4 in higher neural functions remains unknown but the development of selective inhibitors led PDE4 to be considered as a therapeutic target for diseases that cause cognitive impairment (reviewed in ref (26)).
Several discrepancies have arisen regarding the presence of PDE5 in the rodent and primate CNS. While some authors have suggested that PDE5 is not detectable in the human brain,17 others detected PDE5 mRNA in the human cortex, hippocampus, and striatum through techniques that included Northern blotting and real-time polymerase chain reaction.8,27 While high levels of PDE5 have been found in mouse Purkinje neurons,10 the levels of PDE5 mRNA in the hippocampus and cortex of the human brain appear to be very low relative to those of other PDEs, such as PDE2.8 The distribution of PDE5 in the CNS has been recently reassessed given the improvements reported in memory performance following treatment with PDE5 inhibitors both in wild type animals28,29 and in animal models of AD.30−32 In situ hybridization revealed the expression of PDE5 mRNA in several areas of the mouse and primate brain, including the hippocampus and cerebral cortex. In the hippocampus, PDE5 transcripts are expressed in the pyramidal cell layers of CA1, CA3 and in the granule cells of the dentate gyrus. These data suggest an important role of this PDE in neural activity related to cognition, supporting the hypothesis that PDE5 inhibitors affect memory-related processes. (32)
Two genes encode PDE7, PDE7A, and PDE7B, each giving rise to several splice variants of which PDE7A1 and PDE7B2 are found in the brain.7 While low levels of PDE7A mRNA are detected in several brain areas, including the hippocampus, cortex, and striatum,8,33 PDE7B expression in the CNS is stronger than that found in other tissues. Regions expressing high levels of PDE7B include the caudate nucleus, the nucleus accumbens, the cortex, and the hippocampus.8 Moreover, pharmacological inhibition of PDE7 has been proposed to protect against inflammation caused by neuronal death following spinal cord injury.34
The PDE8 family is also coded by two genes, PDE8A and PDE8B, whose protein products are specific for cAMP. PDE8A is ubiquitous, while the expression of PDE8B is restricted to the brain and thyroid.7 The highest levels of PDE8 in the brain are found in caudate, cortex, and hippocampus. To date, no data regarding the effects of PDE8 inhibitors have been reported.
Members of the PDE9 family appear to show the highest affinity for cGMP. Only one gene for this PDE has been identified (PDE9A), although multiple mRNA variants are expressed.7 Although widely expressed in a variety of tissues, high levels of this isoform are detected in the brain,8,35 particularly in the Purkinje cells of the cerebellum and the caudate nucleus.8 Based on the relatively high levels in the CNS, it has been have suggested that this enzyme may be an important regulator of cGMP signaling in the brain.35 Moreover, in several animals models, beneficial effects of PDE9 inhibitors have been reported on cognitive function, which appear to be mediated by increased levels of this second messenger.36
Only one gene (PDE10A) has been identified for PDE10, although four variants may be generated.7 PDE10 transcripts are present in a number of brain areas (cerebellum, thalamus, hippocampus, striatum, and spinal cord8,37−39), and since the strongest expression is in the caudate nucleus, they may fulfill a specific role in striatal function.40 In fact, specific inhibitors of PDE10 are currently being developed to treat Parkinson's disease and schizophrenia.41 In addition, decreased PDE10 mRNA and protein levels have been described in the striatum of patients of Huntington's disease.42 Based on its localization in the hippocampus and the increases in PDE10A after LTP induction,43 a role for this protein in memory and cognition has also been proposed. This PDE may represent an important target to treat cognitive deficits associated to schizophrenia.44
The PDE11 family contains one identified gene (PDE11A), which encodes one of the less abundant PDEs in the tissues analyzed.8 Although low levels of PDE11A have been described in the cortex, caudate nucleus, nucleus accumbens, cerebellum and hippocampus,8 a recent study demonstrated that PDE11A is enriched in the ventral hippocampus, and that its deletion induces psychiatric disease-related phenotypes. (45)
In conclusion, many members of the PDE family are present in the CNS, particularly in areas implicated in learning and memory processes. As these enzymes control the concentration of two second messengers that are crucial for neural function, PDE inhibitors represent promising candidate drugs for the treatment of altered cognition states.
2. Cognitive Enhancement by PDE Inhibitors in Animal Models
Several selective PDE inhibitors have been shown to improve learning in rodent models of cognition. Both cAMP and cGMP are important second messengers in the mature brain that are directly involved in time-dependent events of memory consolidation.46 Activation of the cAMP-PKA pathway cascade triggers the activation of transcription factors such as CREB, inducing the gene transcription required to consolidate learning and memory.47,48 Moreover, recent findings have also linked the cGMP pathway to cognition. Basal levels of cGMP are higher in the newborn brain than in the adult brain, and they decrease with age, a decline thought to be the consequence of increased expression of cGMP-dependent PDEs.49 Inhibition of PDEs that specifically mediate the hydrolysis of cGMP in the brain (mainly isoforms 2, 5, and 9) results in increases in the levels of this cyclic nucleotide, which in turn may prevent the onset of senile dementia.50 In addition to inhibitors of PDE2, PDE5, and PDE9, which are specific for cGMP, inhibitors of the cAMP-specific PDEs (PDE1, PDE4, and PDE10) have been shown to boost memory in certain animal models.9
Vinpocetine is a specific PDE1 inhibitor that has been tested as a memory enhancer, and it has been found to facilitate LTP,51 improve memory retrieval in passive avoidance in rats,52 and enhance cognitive performance in humans.53 Recent studies demonstrated that vinpocetine also improves synaptic plasticity in a model of fetal alcohol spectrum disorders, where there is impaired cortical development and sensory function.15,54
The effect of PDE2 inhibitors on memory has been tested using the selective inhibitor, Bay 60-7550, which improves memory in young and aged rats when administered at specific time points (1 h before and 3 h after the first trial in the object recognition test46). Similarly, Bay 60-7550 reverses NMDA antagonist-induced memory deficits in mice and attenuates memory deficits caused by acute tryptophan depletion in rats.19,20,46,55,56 These results suggest that inhibition of PDE2 augments neuronal cGMP levels under conditions of guanylate cyclase stimulation.19
The differential distribution of the four PDE4 subtypes (PDE4A-D) in the brain57 reflects their distinct roles in the CNS, which may be attributed to the regulation of cAMP/CREB signaling. Based on its predominant expression in hippocampal CA1,57 PDE4D appears to be the main subtype involved in mediating memory consolidation and LTP.58,59 Indeed, mice lacking PDE4D display enhanced long-term hippocampal-dependent memory. Furthermore, the inhibition of PDE4 by rolipram in PDE4D-deficient mice did not alter memory, indicating a key role of PDE4D in memory processes. However, it should be noted that mice lacking PDE4B do not exhibit altered cognition in acquisition and consolidation processes,60,61 although they do display impaired reversal learning.62 The cognitive effects of PDE4 inhibitors, primarily the prototypic PDE4 inhibitor rolipram, have been widely studied. Rolipram has beneficial effects on hippocampal-dependent memory tasks, such as the object recognition task, as well as on passive avoidance learning and in fear conditioning memory paradigms.63−65 In addition, positive effects on prefrontal cortex-dependent memory in young rats and monkeys have also been reported.66 Interestingly, opposite effects of rolipram have been described in rat and monkey models of aging, which may reflect the differential PKA regulation in the CNS of different species.66 Other beneficial effects of rolipram on memory have been demonstrated in animals with memory deficits induced by a number of manipulations.59,65,67−71 Yet despite these findings, the use of rolipram to treat cognitive-related chronic disease in humans is limited by its side effects, which include nausea and headaches. Another PDE4 inhibitor, MK 0952, enhances cognition in preclinical studies,72 which prompted a clinical trial in patients with mild-to-moderate Alzheimer’s disease.73 To the best of our knowledge, a phase II study has been completed, although no results have yet been released. Associated side effects represent a major drawback to the use of PDE4 inhibitors as memory enhancers, and they include emesis,74 which appears to be PDE4D-dependent.75 However, 3-cyclopentyloxy-4-methoxybenzaldehyde (GEBR-7b), a recently developed novel and selective PDE4D inhibitor, is 3–10 times more potent as a memory enhancer than rolipram, and it can exert these effects at doses that do not produce emesis-like symptoms in rodents.76 Inhibition of brain PDE4 without emetic side effects may also be achieved by allosteric modulation (see section 8). It should be however noted that it is not entirely clear whether PDE4 inhibition alone is the sole basis of emesis. For instance, the emetic in the ferret cannot be explained by PDE4D selectivity or differences in PDE4D inhibitor potency (see Spina for details).77
Selective PDE5 inhibitors, such as sildenafil, tadalafil, and vardenafil, are currently used for erectile dysfunction and for chronic human pulmonary hypertension. In the past decade, evidence has accumulated that suggests beneficial effects of PDE5 inhibitors in cognition, both in physiological and pathological conditions (see below). Memory enhancement following sildenafil treatment has been demonstrated in a variety of behavioral paradigms, both in rodents and primates.58,78−83 In a one-trial learning paradigm (object recognition and passive avoidance), sildenafil improved early consolidation processes (less than 3 h), but no effect was observed when the drug was administered before or 3 h after training, indicating that sildenafil has no effect on acquisition and late consolidation processes. A recent study demonstrated that sildenafil improves spatial memory in the Morris Water Maze (MWM) test when administered after the last training trial on each day of the acquisition phase of the test.84 In addition to its effects on hippocampal-dependent memory, sildenafil improves performance of a prefrontal-cortex-dependent cognition test in monkeys,58 and attenuates memory impairment induced by nitric oxide synthase inhibition,55,80 hyperammonemia,81 blockade of muscarinic cholinergic receptors,85 diabetes conditions, and electroconvulsive shock. (86)
In addition to PDE5, PDE6 and PDE9 are specific for cGMP, with the latter exhibiting the greatest affinity for cGMP (Km = 170 nM),87 highlighting its importance in the regulation of cGMP downstream signaling pathways. Two selective PDE9 inhibitors have been developed and tested in higher-function tasks, BAY73-6691 and PF-04447943. Interestingly, these compounds behave as enhancers of memory and synaptic plasticity, reversing memory deficits and attenuating forgetting.36,88,89 BAY73-6691 appears to be more effective in facilitating LTP in hippocampal slices from aged versus young rats, while PF-04447943 attenuates the loss of hippocampal spine density in the Tg2576 AD model, and it is currently under clinical development for the treatment of AD.90 Indeed, a phase 2 clinical trial has been completed, although no results have yet been disclosed (Pfizer, 2009; ClinicalTrials.gov Identifier: NCT00930059).
Recently, PDE10 inhibitors have attracted interest for the treatment of CNS disorders, particularly schizophrenia44 and Huntington's disease.91 The expression of PDE10 in the striatum suggests that this protein may be more closely involved in striatal functions than those controlled by the hippocampus or prefrontal cortex.42,92 Papaverine, a selective PDE10 inhibitor, impairs memory in rats when administered by the intracerebroventricular (i.c.v.) route both before and after learning.93 By contrast, in rats treated with the NMDA receptor antagonist phencyclidine (PCP), PDE10 inhibitors improve information processing and attention.41 In general, behavioral deficits in papaverine-treated mice resemble those of PDE10A deficient mice,94 probably reflecting striatal cell activation.91,95
In summary, cGMP and/or cAMP signaling pathways appear to play an important role in the processes underlying learning and memory. Indeed, a recent study demonstrated that the beneficial effects of rolipram and sildenafil on spatial memory are reversed to control levels by hippocampal inhibition of PKA and PKG, respectively.84 Moreover, increases in either cAMP or cGMP may promote CREB phosphorylation (see section 5), a key factor in regulating the expression of plasticity-associated genes (i.e., genes that trigger long-lasting changes in synaptic plasticity).48,96,97 Since cGMP/cAMP have vasodilator effects, increased blood flow and glucose metabolism may also contribute to the effects of PDE inhibitors on memory.98,99
3. PDE Inhibitors in the Treatment of Alzheimer’s Disease
To evaluate the therapeutic value of PDEs in AD, it is important to determine the expression of the different enzymes in the human brain, both in normal and pathological conditions. In situ hybridization has demonstrated marked region-specific alterations in the expression of PDE7 and PDE8 in AD.100 While PDE7B mRNA expression remains unchanged, PDE7A transcripts are reduced and PDE8B mRNA levels increase in hippocampal regions in advanced stages of AD.100 A more recent study failed to report a change in the expression of two cGMP PDEs (PDE2 and PDE9) in AD brain samples.17 Moreover, expression of all but one of the PDE4 isoforms is reduced or unchanged in AD brains.101 Interestingly, expression of a short isoform, PDE4D1, augments in AD, suggesting that this isoform may represent a specific target to enhance memory consolidation in AD patients.101 However, further studies are required to determine whether these changes in PDE expression are a cause or consequence of AD.
Over a decade ago, one of the first studies investigating the effects of PDE inhibitors in AD in humans reported a negative outcome, demonstrating that the PDE1 inhibitor vinpocetine failed to improve cognition or slow the rate of decline in AD patients.102 However, more recent studies in the search for drugs that may alleviate or reverse AD symptoms have confirmed PDE inhibitors to be promising therapeutic tools. The effectiveness of repeated administration of PDE3 (cilostazol), PDE4 (rolipram), or PDE5 (sildenafil or tadalafil) inhibitors has been demonstrated in several mouse models of AD.30−32,103−105 As each of these models harbors distinct genetic alterations, the potential of PDE inhibitors in the treatment of AD is high indeed, although the underlying mechanism remains to be fully elucidated (see section 5).
4. PDEs and PDE Inhibitors in Mouse Models of Alzheimer’s Disease
Cilostazol is a PDE3 inhibitor that is used as an antiplatelet agent in the treatment of subcortical vascular disease. This drug increases cAMP levels, probably by promoting the phosphorylation of CREB,60 which is known to play a crucial role in memory enhancement and synaptic plasticity.3,5 Cilostazol also increases cerebral blood flow and exerts antioxidative effects. Based on these properties, this compound has been tested in mice treated with Aβ25–35, and it was shown that repeated cilostazol administration protected against memory impairments in these mice by preventing Aβ aggregation and the accumulation of oxidative stressors.104
On the basis of preliminary data indicating that rolipram reverses Aβ42-induced impairment of LTP in the CA1 hippocampal region, the effects of this drug were also tested in a mouse model of AD. Activation of the cAMP/CREB pathway induced by PDE4 inhibition promoted a long lasting reversal of memory impairments in APP/PS1 mice. The compound led to synaptic remodeling, but that had no effect on Aβ production or deposition.103,106
The PDE5 inhibitor sildenafil exerts potent anti-AD effects when administered at advanced stages of the disease in different mouse models of AD, resulting in the reversal of cognitive impairment.30−32 However, the underlying mechanism of action remains unclear, with several studies reporting conflicting findings. In APP/PS1 AD mice, decreased Aβ levels were described after 3 weeks of sildenafil treatment, while no changes in amyloid burden were reported in the J20 mouse model.30−32 These differences may reflect the mouse model used and/or the dose or duration of treatment. Another PDE5 inhibitor, tadalafil, was also reported recently to improve spatial memory in the J20 mouse model of AD, providing a greater beneficial effect than that observed with comparable doses of sildenafil.32 The longer half-life and proven safety of tadalafil when administered chronically for the treatment of erectile dysfunction and pulmonary arterial hypertension107,108 suggest that this drug may be a promising candidate for AD pharmacotherapy.
In conclusion, PDE inhibitors have demonstrated promising effects in animal models of AD, which will soon be investigated in clinical trials. The proven safety of several PDE inhibitors already in clinical use (often administered chronically) has created high expectations regarding their use for the treatment of AD.
5. PDE Inhibition and Cognitive Enhancement in Humans
A pioneering double-blind crossover study of 10 neurologically healthy young male volunteers investigated the effects of an acute oral dose of 100 mg sildenafil on auditory attention and word recognition tasks, as determined by behavioral pattern analysis and electrophysiological recordings.109 Sildenafil-treated individuals exhibited even-related potentials in auditory tasks, indicating an enhanced ability to focus attention and select relevant target stimuli. In the memory task, the CNS effects of sildenafil were reflected by a reduction in negativity, a neurophysiological index of the detection of a change in the acoustic environment that can be elicited in the absence of focused attention.110 Taken together, these results provided evidence that PDE5 inhibitors can regulate cognition events in the human CNS.
Various PDE5 inhibitors are currently marketed for the treatment of erectile dysfunction and of pulmonary arterial hypertension, the most common of which are sildenafil, tadalafil, and vardenafil. These drugs are safe even when administered as prolonged treatments.111 PDE inhibitors are also used off-label to treat altitude sickness. The Wilderness Medical Society112 recommends tadalafil (10 mg twice a day) or sildenafil (50 mg every 8 h) to prevent high altitude pulmonary edema. The safety of PDE5 inhibitors and their demonstrated beneficial effects in AD animal models prompted a study in patients diagnosed with erectile dysfunction whose cognitive status was tested before and after treatment with udenafil, a recently developed PDE5 inhibitor.113 Udenafil is marketed as Zydena by the Korean Dong-A pharmaceutical company, but it has not yet been approved by the United States Federal Drug Administration or by the European Medicines Agency. Zydena’s market is thus Asia-Pacific and Russia.
In 27 patients with erectile dysfunction due to psychogenic, organic, or mixed causes, and with average illness duration of 33 months, erectile dysfunction was scored in parallel with cognitive function. The Seoul verbal learning test, and the Korean versions of the mini-mental state examination and the frontal assessment battery, were performed before and 2 months after drug administration. Although the patients were not fully diagnosed from a neurological point of view, no neurological or neuropsychiatric pathologies were detected among comorbid conditions, which included diabetes, hypertension, and/or hyperlipidemia. Average mini-mental and frontal assessment scores increased significantly after udenafil administration (p < 0.001). By contrast, the improvement in the Seoul learning test was not significant (p > 0.05), probably due to the already high proficiency of the subjects and the absence of any underlying episodic-memory- or learning-related pathologies. Although this was a nonrandomized study of a small number of patients, and there was no placebo group, the improvement in cognitive scores observed after PDE inhibitor administration paves the way for further randomized and placebo-controlled clinical trials to study the effects of udenafil, and other PDE5 inhibitors approved for use in humans, in patients with cognitive deficits.
As mentioned above, two phase 2 clinical trials have been carried out to determine the effects of a PDE4 inhibitor (MK 0952; ClinicalTrials.gov identifier: NCT00362024) and a PDE9 inhibitor (PF-04447943; ClinicalTrials.gov identifier: NCT00930059) in AD, although no results have yet been disclosed. However, a recent pilot study reported that the PDE 3 inhibitor cilostazol prevents cognitive decline in patients with AD. (114) A phase 4 clinical trial is currently recruiting participants to analyze the effects of cilostazol in patients with mild to moderate AD who have been prescribed donepezil for subcortical white matter hyperintensities (WMHI) (ClinicalTrials.gov identifier: NCT01409564, phase 4). The next decade will be crucial to determine the efficacy of PDE inhibitors for cognitive enhancement and/or neuroprotective therapies in humans.
6. PDE Inhibitors in Alzheimer’s Disease: Mechanism of Action
Almost all PDE isoforms have been implicated in mood and/or cognitive disorders,115 and inhibitors of all isoforms have been patented as potential AD pharmacotherapies (Patent: US 20120076732). Increases in cAMP and/or cGMP levels represent the most reliable end points of the activity of PDE inhibitors. In principle, the effects of PDE inhibitors on cognition or the pathophysiological mechanisms of AD are mediated by increases in the levels of these cyclic nucleotides in the endothelial cells of brain vessels and/or neurons. For example, PDE5 inhibitors trigger vasodilation, and PDE5A mRNA and protein is present in human middle cerebral arteries. (116) As PDE5 is critical to control the diameter of vessels and its inhibition promotes vasodilation, these data suggest that some of the actions of PDE5 inhibitors may be a consequence of increased cerebral blood flow. However, neither cerebral excitability nor hemodynamic responses are affected by the PDE5 inhibitor sildenafil in healthy subjects.117 Moreover, no associations between cerebrovascular risk factors and perfusion scores have been reported for sildenafil, except in stroke patients. Interestingly, in such stroke patients, there were significantly more brain areas with diminished (not augmented) perfusion after sildenafil administration,118 which may be due to the central actions of PDE inhibitors inhibiting the enzyme in peripheral cells. Indeed, a recent study reported that anandamide, an endogenous cannabinoid, suppresses pain initiation via a peripheral mechanism.119 Also, the antidepressant efficacy of ginseng saponins appears to be attributable to its peripheral anti-inflammatory activity.120 A recent study reported that PDE5 inhibitors improve novel object recognition in rats with cognitive deficits induced by scopolamine, or by an N-methyl-d-aspartate (NMDA) receptor antagonist that specifically disrupts short-term memory (STM) and attention processes.121 Based on these findings, the cerebrovasculature was proposed as a locus for the peripheral effect of PDE5 inhibition in the object recognition test.
While the peripheral activity of PDE inhibitors may have an impact on the CNS, many of these compounds cross the blood-brain barrier and reach concentrations high enough to inhibit PDEs in neurons and glial cells. At doses normally administered in animal models of AD, tadalafil may be present in low micromolar concentrations in the brain, sufficient to inhibit both PDE5 and PDE11.32 In another transgenic rodent line, the Flinders sensitive rat model of depression, sildenafil administration results in hippocampal and cortical concentrations of 146 and 185 nM, sufficient to inhibit PDE5.122 Thus, the main mechanism of action of PDE inhibitors probably involves increased levels of cAMP and/or cGMP in cells within the CNS. In addition to replenishing the low levels of cyclic nucleotides present in the aged brain,49,50,55 this effect leads to sustained and/or increased activation of signaling pathways that impact neuroprotection/neurodegeneration processes.
CREB is a cellular transcription factor that binds to specific DNA sequences known as cAMP response elements (CRE), thereby modifying the transcription of downstream genes involved in neural plasticity and neuroprotection.123 Rolipram inhibits the high-affinity, cAMP-specific PDE4, and it was the first PDE inhibitor tested in an animal model of AD, improving synaptic and cognitive functions in the double transgenic APP/PS1.103 Further studies with rolipram124 and cilostazol, a PDE3 inhibitor,104,105 linked their efficacy to the pCREB-mediated reversal of amyloid-beta-induced cognitive deficits. Furthermore, chronic rolipram administration reverses Aβ-induced alterations in pCREB expression in the hippocampus of rats that received intracerebroventricular injections of Aβ peptides, which in turn induced memory deficits.124
Within cells, cGMP and cAMP act through different pathways to induce CREB phosphorylation during LTP induction.125 Specifically, the effects of cGMP and PKG appear to be indirectly due to increases in intracellular Ca2+ levels. Thus, cGMP activates PKG and ADP-ribosyl cyclase, stimulating the production of cADP-ribose, which acts synergistically with cytoplasmic Ca2+ to further promote Ca2+ release from a variety of sources.126−130 When the Ca2+ signal is sufficiently large, it triggers CREB phosphorylation and induces of LTP in parallel with PKA. Therefore, cGMP mimics the beneficial effects of cAMP on pCREB, although it may reverse cognitive impairment via other more specific pathways.
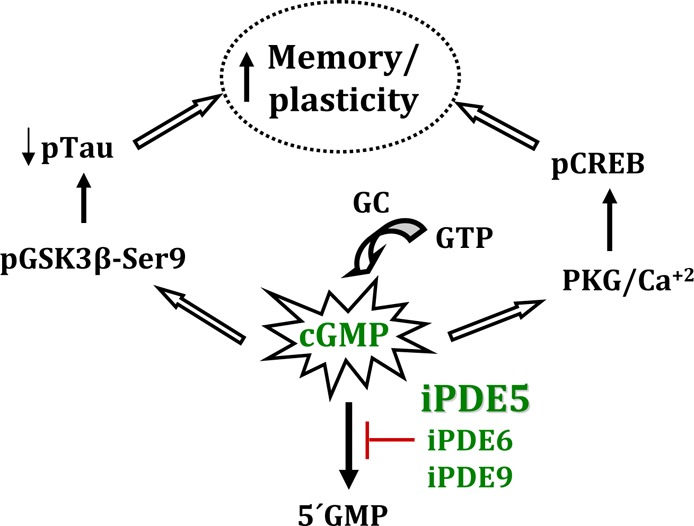
PDE5 is 100-fold more selective for cGMP than cAMP: the Km for cGMP is in the low micromolar range, while that of cAMP is ∼300 μM. Sildenafil inhibits PDE5 and to a lesser extent PDE6 and PDE1,7 and its inhibition of PDE5 counteracts the negative effects of Aβ on CREB phosphorylation.31 Pathways specific to cGMP have been characterized extensively, particularly since the discovery of NO as a neuroregulatory molecule. Indeed, nitric oxide (NO) activity is primarily mediated via increases in cellular cGMP levels.131 Two cGMP-dependent kinases are major downstream effectors of many signaling events of the NO/cGMP pathway. (132) The canonical NO/cGMP/cGK pathway modulates long-term changes in synaptic activity in the hippocampus, amygdala, cerebellum, and other brain regions, and it contributes to distinct forms of learning and memory, such as fear conditioning, motor adaptation, and object recognition. At the molecular level, cGMP-dependent kinase isoforms appear to mediate the effects of cGMP on presynaptic transmitter release and postsynaptic functions, and they have also been proposed to modulate cytoskeletal organization, vesicle and AMPA receptor trafficking, and gene expression through the phosphorylation of various neural substrates, including VASP, RhoA, RGS2, hSERT, GluR1, G-substrate, and DARPP-32. (97) In AD mouse models, cGMP-dependent inactivation of GSK3β by PDE5 inhibitors leads to a decrease in hyperphosphorylated tau in the brain areas involved in learning and memory.30,32 As tau phosphorylation better correlates with cognitive impairment in AD than amyloid burden,133 increased hippocampal pAkt and pGSK3β-Ser9 levels have been proposed to underlie the improvements in learning and memory consolidation observed in sildenafil- and tadalafil-treated AD mice.30,32 Similar activation of the Akt pathway by tadalafil has been proposed to increase cell survival in different peripheral tissues.134,135 This association between NO-cGMP signaling and tau phosphorylation via pGSK3β activity is supported by the existence of a signaling cascade that links cGMP activation with GSK3 inhibition.136
7. Chemical Probes as Pharmacological Tools
Drug discovery approaches in AD research are now focusing on aspects of AD pathology other than the amyloid pathology. A promising approach is to reduce aberrant tau phosphorylation, which may be achieved via the NO-cGMP-related pathway. In fact, it is predicted that increases in cerebral cGMP rather than cAMP will be more beneficial for the treatment of cognitive impairment. Alternative approaches can also increase cAMP, such as drugs that target G-protein-coupled receptors. The best option in PDE-related AD therapy is therefore to target enzymes whose substrate specificity for cGMP is comparable to or greater than that for cAMP (e.g., PDEs 1C, 2A, 5, 6, 9, and 11). Many of these PDEs are present in the CNS (see section 2), and they therefore constitute potential targets for anti-AD drugs. However, there are exceptions that include PDE6, whose expression appears to be restricted to the retina and pineal gland, and PDE11, that has yet to be reported in the human CNS. Finally, PDE4 and PDE10A should also be considered as potential targets for cognition enhancement due to their specific localization in the CNS, the availability of optimal inhibitors, and the efficacy of these compounds in models of memory impairment (Table 1).
Table 1. Summary of PDEs and Their Corresponding Pharmacological Tools, Which Are Critical for Target and Hypothesis Validation.
| target | expressed in brain | substrate | pharmacological tool | cognitive benefit in ad model |
|---|---|---|---|---|
| PDE1B | yes | cAMP/cGMP | 1 | yes |
| PDE2A | yes | cAMP/cGMP | 2 | yes |
| PDE4 | yes | cAMP | 3, 4, 5, 6 | yes (only 3) |
| PDE5 | yes | cGMP | 7, 8, 9 | yes |
| PDE9 | yes | cGMP | 10 | yes |
| PDE10 | yes | cAMP/cGMP | 11 | yes (but not using 11) |
One advantage of using inhibitors of dual-specificity PDEs is that this can promote increases of both cGMP and cAMP in neurons. The ideal inhibitor would be one that increases the levels of both cyclic nucleotides but that primarily targets cGMP. Concomitant increases of cGMP and cAMP are nowadays seen not as detrimental but beneficial to improve cognition deficits. Finally, it is important to consider the safety profile of PDE5 inhibitors in humans, which is extremely favorable, particularly in the case of tadalafil (Cialis) that is approved for both acute and chronic erectile dysfunction, and for arterial pulmonary hypertension. Tadalafil is administered at doses of 2.5 or 5 mg/day for the treatment of chronic erectile dysfunction and up to 40 mg/day for arterial pulmonary hypertension (marketed as Adcirca).137,138 Therefore, safety associated to prolonged treatments of tadalafil is quite notable. In summary, targeting PDEs 1B/C, 2A 5 or 9 represents a promising approach for the chronic treatment of cognitive disorders.
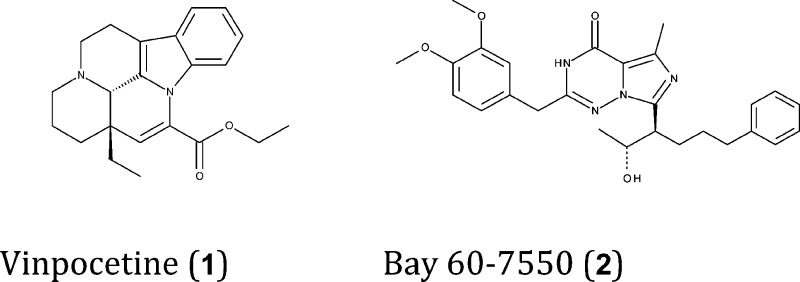
Currently, there are more than 125 entries publicly available in the protein data bank,139 halo- and apo-forms, covering all known PDE isoforms (except PDE11) and some variants. Together with ligand-based chemogenomics approaches, structural information can be used to attain off-target selectivity against other isoforms. For example, PDE1B is expressed at high levels in the brain, particularly in the striatum and the dentate gyrus of the hippocampus,140 and its inhibition by vinpocetine (1) induces memory-enhancing effects.51 Accordingly, structural information pertaining to this isoform (PDB: 1TAZ)139 may help medicinal chemists to design and synthesize novel, and more selective, PDE1B ligands.
PDE2A is also expressed in regions associated with cognitive function, including the cortex, striatum, hippocampus, amygdala, and habenula.141 According to the biochemical and electrophysiological effects of the isoform-selective inhibitor Bay 60-7550 (2), this PDE variant mediates LTP induction by regulating cGMP levels.19 Moreover, as described above, Bay 60-7550 has cognition enhancing properties in animal models. PDE2 has slightly higher affinity for cGMP than cAMP, although it may metabolize both cyclic nucleotides in a tissue-dependent manner. Indeed, the endogenous substrate of this enzyme may vary across tissues in function of its subcellular localization and/or splice variant expression. Interestingly, most studies of the role of PDE2A in brain tissue point to its modulation of cGMP levels.141
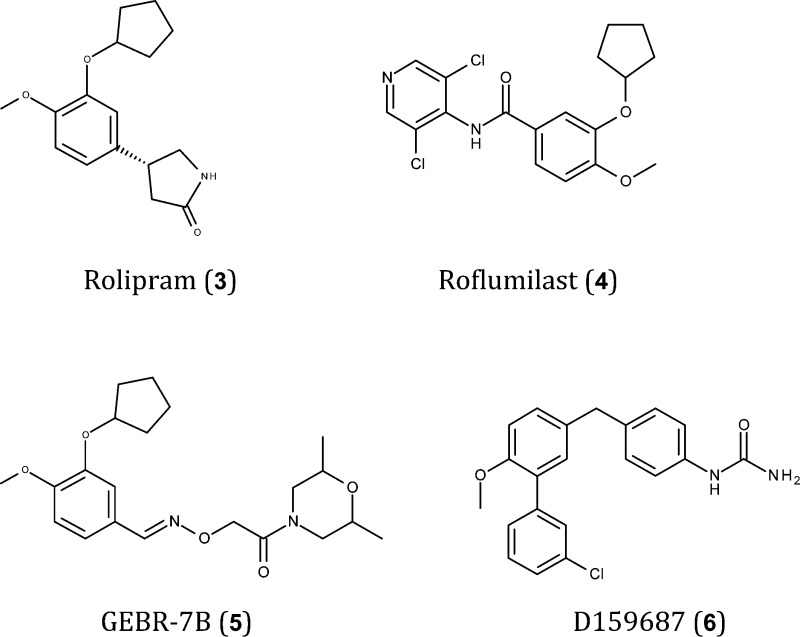
The PDE4 subfamily of PDEs is quite complex, comprising over 20 splice variants, which permits a variety of regional and subcellular localizations.7 Among the different isoforms, PDE4A and B are expressed at high levels in the frontal and temporal cortex, and in the hippocampus of humans. (57) Preclinical studies with a selective and brain-penetrant PDE4 inhibitor, rolipram (3), identified this isoform as a promising target for the treatment of the cognitive dysfunction associated with AD.142 However, serious side effects associated with PDE4 inhibitors have hampered their development. Nevertheless, in 2011, the FDA approved the first and only selective PDE4 inhibitor, roflumilast (4), to reduce the exacerbation and worsening of symptoms associated with severe chronic obstructive pulmonary disease (COPD). Given the potential risks identified in clinical trials, roflumilast was approved with a medication guide informing patients of side effects.143 A recent report highlighted the severity of this drug’s side effects and determined that its benefits are minimal at best: “even a small increase in adverse effects would tilt the risk/benefit ratio against the use of roflumilast”.144 The development of PDE4 inhibitors lacking side effects is critical for therapeutic applications that require chronic treatment, such as AD. In addition to the selective PDE4D inhibitor GEBR-7B (5) (see section 2),145 DeCode Genetics has used structural information (PDB: 3IAD)139 to develop a series of PDE4 allosteric modulators that serve as a molecular glue to close a regulatory domain, in this case acting as a “lid” over the PDE4 catalytic site.146 These compounds do not completely abolish PDE4 enzymatic activity in cellular and in vivo models. Interestingly, the occurrence of emesis, a dose-limiting side effect of existing active site-directed PDE4 inhibitors, is also significantly reduced by D159687 (6).146 This strategy of PDE4 modulation may therefore lead to the development of pharmacotherapeutic compounds in the near future that will ultimately become available to the patient.
PDE5 mRNA has been detected in the cortex and hippocampus of both mice and primates, confirming an important role of this enzyme in neural activities related to cognition.32 The availability of selective, safe, and brain-penetrant PDE5 inhibitors has been crucial to carry out studies in AD animal models to assess cognitive improvements and the biochemical markers underlying such improvement. Vardenafil (7),46,121 sildenafil (8),30,31 and, more recently, tadalafil (9)32 have all been used as pharmacological tools, confirming their efficacy in AD mouse models. Given that (i) PDE5 mRNA is expressed in the cortex and hippocampus;32 (ii) PDE5 inhibitors cross the blood-brain barrier;32 (iii) PDE5 inhibitors, particularly tadalafil, are safe; and (iv) these drugs exhibit favorable pharmacokinetic parameters for these already marketed drugs, PDE5 inhibitors should soon enter into clinical trials for the treatment of mild cognitive impairment and/or AD. The characteristics of tadalafil are more suited for the chronic treatment of mild cognitive impairment than those of vardenafil or sildenafil (see Table 2). Although its logBB ratio is very modest, −0.8932 (close to the lowest accepted ratio for a CNS drug, −1147), tadalafil administration produces a sufficiently high CNS concentration to significantly inhibit PDE5. A comprehensive analysis from a multivariate drug discovery perspective also leads tadalafil to become a more than acceptable candidate for reverting cognitive impairments.
Table 2. Biochemical and Pharmacokinetic Data for Marketed Selective PDE5 Inhibitors113 a.
| compd |
|||
|---|---|---|---|
| 7 | 8 | 9 | |
| half-life (h) | 4–5 | ∼4 | 17.5 |
| bioavailability | ∼15% | ∼40% | N.D. |
| Cmax (approx, nM) | 36b | 950c | 771b |
| logBB32,151 | –1.4d | –1.3d | –1d, −0.89e |
| IC50 (nM)152 | 0.14 | 3.5 | 6.7 |
| selectivity152 f | 4-fold vs PDE6 | 10-fold vs PDE6 | 5-fold vs PDE11 |
The half-life, bioavailability, and Cmax data were obtained from official information provided by the relevant pharmaceutical companies (7,1488,149 and 9(150)).
Single oral dose (20 mg).
Single oral dose (100 mg).
Determined in rats.
Determined in mice.
Selectivity with respect to the second isoform for which the compound exerts significant inhibition.
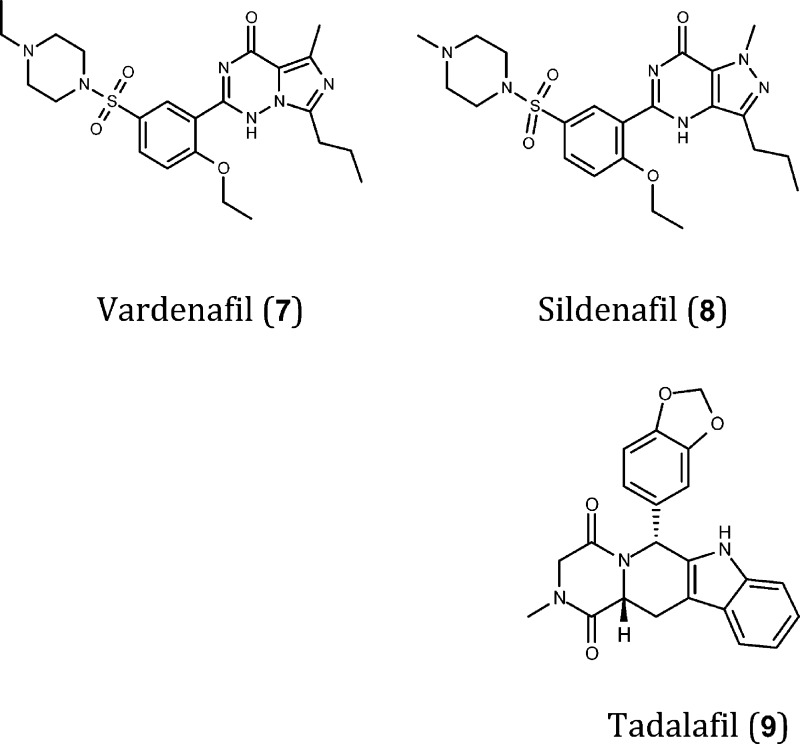
Due to its elevated expression in the brain35 and the established role of cGMP in the regulation of synaptic function, PDE9 is a potential target to treat memory deficits associated with aging and neurodegenerative disorders such as AD.9,17,36 Pfizer has developed PF-4447943 (10), a potent, selective, and brain-penetrant PDE9A inhibitor (logBB = −0.4, IC50 = 12 nM) that increases brain levels of cGMP in vivo88 and that is 78 times more selective for PDE9A than for other PDE family members (tested on all isoforms and some splice variants, 17 in total).153 The in vivo effects of PDE9A inhibitors suggest a role for this enzyme in the regulation of monoaminergic circuits associated with sensory processing and memory. PDE9A activity regulates neuronal cGMP signaling downstream of multiple neurotransmitter systems, and thus, its inhibition may have beneficial effects in the treatment of psychiatric and neurodegenerative diseases.153 However, the lack of results from a 2010 phase 2 clinical trial of subjects with mild to moderate AD154 casts doubts on the utility of PDE9A inhibitors for the treatment of AD.
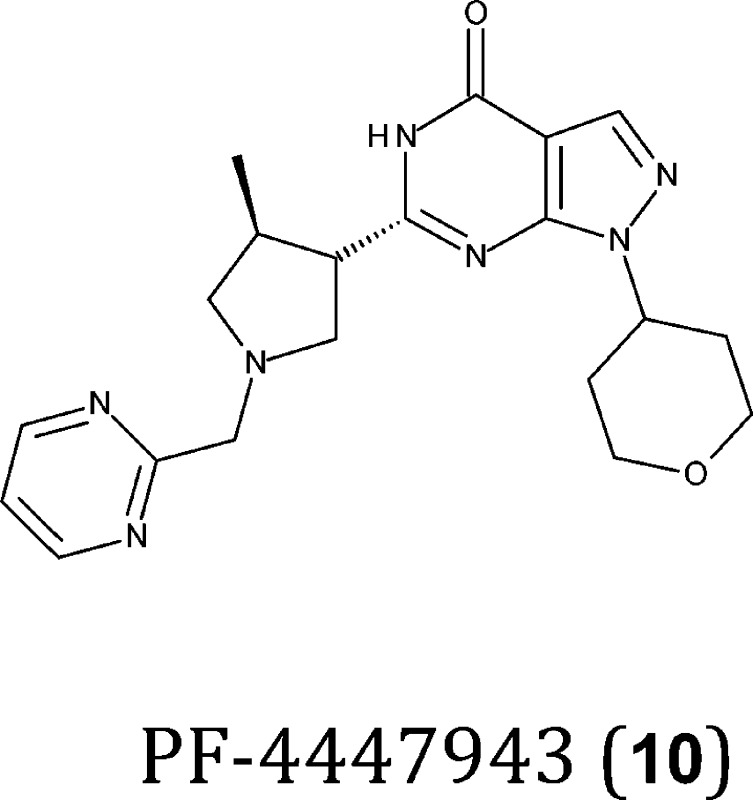
Inhibitors of PDE10A may regulate cyclic nucleotide signaling in the corticostriatothalamic circuit. However the extra-striatal expression of PDE10A37,40 suggests that these inhibitors may also be used to treat cognitive deficits associated with schizophrenia.155,156 The PDE10A inhibitor TP-10 (11) is active in a variety of preclinical models that suggest it may be efficacious in the treatment of schizophrenia.44 Similar effects have been reported for papaverine (12), another PDE10A inhibitor that also improves novel object recognition in rats.157 Although both compounds increase cGMP and cAMP levels, and CREB phosphorylation, a beneficial effect on cognition has only been described for papaverine, which appears to be a positive “side-effect”.44,157 These differential effects are poorly understood and may reflect the specific localization of PDE10A in CNS cells. Defining the specific localization of PDE variants in the brain will be fundamental to identify and select PDEs involved in cognition. A novel pharmacological tool compound, PF-02545920 (13), with optimal activity (IC50 = 0.37 nM) and selectivity (>1000 times more selective for PDE10A) that crosses the BBB (logBB is −0.06),158 is currently being studied for the treatment of schizophrenia in phase 2 clinical trials.154 This drug may be used to selectively increase the levels of cyclic nucleotides in brain areas where PDE10A resides, thereby allowing a careful evaluation of its functional role and its therapeutic value with respect to other PDEs.
8. Future Directions: A Medicinal Chemistry Perspective
The development of AD is preceded by a subtle cognitive decline that affects the individual’s ability to perform their daily activities. Reversal of this decline represents an important unmet medical need, in which PDE inhibition may play a fundamental role.
A considerable number of research studies have focused on the role of PDE inhibition in restoring memory function. Given the profile of currently available PDE5 inhibitors, both in terms of pharmacokinetics and safety, and their demonstrated efficacy in vivo in animal models, clinical trials should be performed to determine their potential in AD patients. The cost-benefit risk underlying such an approach could be assumed by public resources; perhaps, a drug for mild cognitive impairment is already available in the market. It should be noted that PDE5 inhibitors fulfill a number of the new criteria desired in an anti-AD drug: they do not act via anti-amyloid mechanisms, they exhibit good blood-brain barrier penetration, they have long half-lives, and they increase cGMP levels, which in turn decrease GSK3β activity and p-tau levels.
In addition to testing PDE5 inhibitors in clinical trials, several issues must be addressed to facilitate the identification and validation of the most promising PDE isoforms to combat cognitive dysfunction. In particular, the precise localization of distinct PDEs in the brain must be determined, and a greater understanding of the localization of PDEs in different parts of the neuron is required. The development of radiolabeled analogues for PET imaging may also add additional value to current pharmacological tool compounds. Finally, the inhibitors of choice should act on the PDEs specifically located in regions directly involved in cognition, and potential side effects must be taken into account.
Author Contributions
A.G.-O., M.C.-T., J.O., and R.F. researched the scientific literature, wrote sections of the manuscript and prepared diagrams and figures and contributed equally to this work. C.G.-B. contributed in the sections related to PDE5 inhibitors
We thank the Foundation for Applied Medical Research (FIMA) of the University of Navarra for financial support.
The authors declare no competing financial interest.
Author Contributions
§ These authors contributed equally to this work.
References
- Impey S.; Mark M.; Villacres E. C.; Poser S.; Chavkin C.; Storm D. R. (1996) Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 16, 973–982. [DOI] [PubMed] [Google Scholar]
- Lu Y. F.; Kandel E. R.; Hawkins R. D. (1999) Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurosci. 19, 10250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T. (1997) Regulation of gene expression and its role in long-term memory and synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A 94, 4239–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J. C.; Tully T. (1996) CREB and the formation of long-term memory. Curr. Opin. Neurobiol. 6, 264–268. [DOI] [PubMed] [Google Scholar]
- Tully T.; Bourtchouladze R.; Scott R.; Tallman J. (2003) Targeting the CREB pathway for memory enhancers. Nat. Rev. Drug Discovery 2, 267–277. [DOI] [PubMed] [Google Scholar]
- Ran I.; Laplante I.; Lacaille J. C. (2012) CREB-dependent transcriptional control and quantal changes in persistent long-term potentiation in hippocampal interneurons. J. Neurosci. 32, 6335–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. T.; Beavo J. A. (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58, 488–520. [DOI] [PubMed] [Google Scholar]
- Lakics V.; Karran E. H.; Boess F. G. (2010) Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59, 367–374. [DOI] [PubMed] [Google Scholar]
- Reneerkens O. A.; Rutten K.; Steinbusch H. W.; Blokland A.; Prickaerts J. (2009) Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology (Berlin, Ger.) 202, 419–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Albergine M.; Rybalkin S. D.; Rybalkina I. G.; Feil R.; Wolfsgruber W.; Hofmann F.; Beavo J. A. (2003) Individual cerebellar Purkinje cells express different cGMP phosphodiesterases (PDEs): in vivo phosphorylation of cGMP-specific PDE (PDE5) as an indicator of cGMP-dependent protein kinase (PKG) activation. J. Neurosci. 23, 6452–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger G. B.; Rodgers L.; Riggs M. (1994) Differential CNS expression of alternative mRNA isoforms of the mammalian genes encoding cAMP-specific phosphodiesterases. Gene 149, 237–244. [DOI] [PubMed] [Google Scholar]
- Sahu M.; Litvin D. G.; Sahu A. (2011) Phosphodiesterase-3B is expressed in proopiomelanocortin and neuropeptide Y neurons in the mouse hypothalamus. Neurosci. Lett. 505, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Wolda S. L.; Frazier A. L.; Florio V. A.; Martins T. J.; Snyder P. B.; Harris E. A.; McCaw K. N.; Farrell C. A.; Steiner B.; Bentley J. K.; Beavo J. A.; Ferguson K.; Gelinas R. (1997) Identification and characterisation of a human calmodulin-stimulated phosphodiesterase PDE1B1. Cell. Signalling 9, 519–529. [DOI] [PubMed] [Google Scholar]
- Deshmukh R.; Sharma V.; Mehan S.; Sharma N.; Bedi K. L. (2009) Amelioration of intracerebroventricular streptozotocin induced cognitive dysfunction and oxidative stress by vinpocetine – a PDE1 inhibitor. Eur. J. Pharmacol. 620, 49–56. [DOI] [PubMed] [Google Scholar]
- Krahe T. E.; Wang W.; Medina A. E. (2009) Phosphodiesterase inhibition increases CREB phosphorylation and restores orientation selectivity in a model of fetal alcohol spectrum disorders. PLoS One 4, e6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repaske D. R.; Corbin J. G.; Conti M.; Goy M. F. (1993) A cyclic GMP-stimulated cyclic nucleotide phosphodiesterase gene is highly expressed in the limbic system of the rat brain. Neuroscience 56, 673–686. [DOI] [PubMed] [Google Scholar]
- Reyes-Irisarri E.; Markerink-Van Ittersum M.; Mengod G.; de Vente J. (2007) Expression of the cGMP-specific phosphodiesterases 2 and 9 in normal and Alzheimer’s disease human brains. Eur. J. Neurosci. 25, 3332–3338. [DOI] [PubMed] [Google Scholar]
- Van Staveren W. C.; Steinbusch H. W.; Markerink-Van Ittersum M.; Repaske D. R.; Goy M. F.; Kotera J.; Omori K.; Beavo J. A.; De Vente J. (2003) mRNA expression patterns of the cGMP-hydrolyzing phosphodiesterases types 2, 5, and 9 during development of the rat brain. J. Comp. Neurol. 467, 566–580. [DOI] [PubMed] [Google Scholar]
- Boess F. G.; Hendrix M.; van der Staay F. J.; Erb C.; Schreiber R.; van Staveren W.; de Vente J.; Prickaerts J.; Blokland A.; Koenig G. (2004) Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 47, 1081–1092. [DOI] [PubMed] [Google Scholar]
- Rodefer J. S.; Saland S. K.; Eckrich S. J. (2012) Selective phosphodiesterase inhibitors improve performance on the ED/ID cognitive task in rats. Neuropharmacology 62, 1182–1190. [DOI] [PubMed] [Google Scholar]
- Cherry J. A.; Davis R. L. (1999) Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J. Comp. Neurol. 407, 287–301. [PubMed] [Google Scholar]
- Cho C. H.; Cho D. H.; Seo M. R.; Juhnn Y. S. (2000) Differential changes in the expression of cyclic nucleotide phosphodiesterase isoforms in rat brains by chronic treatment with electroconvulsive shock. Exp. Mol. Med. 32, 110–114. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Imaizumi M.; D’Sa C.; Zoghbi S. S.; Crescenzo M. S.; Hong J.; Musachio J. L.; Gee A. D.; Seidel J.; Green M. V.; Pike V. W.; Duman R. S.; Innis R. B. (2007) In vivo and in vitro measurement of brain phosphodiesterase 4 in rats after antidepressant administration. Synapse 61, 78–86. [DOI] [PubMed] [Google Scholar]
- McPhee I.; Cochran S.; Houslay M. D. (2001) The novel long PDE4A10 cyclic AMP phosphodiesterase shows a pattern of expression within brain that is distinct from the long PDE4A5 and short PDE4A1 isoforms. Cell. Signalling 13, 911–918. [DOI] [PubMed] [Google Scholar]
- Conti M.; Richter W.; Mehats C.; Livera G.; Park J. Y.; Jin C. (2003) Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J. Biol. Chem. 278, 5493–5496. [DOI] [PubMed] [Google Scholar]
- Rose G. M.; Hopper A.; De Vivo M.; Tehim A. (2005) Phosphodiesterase inhibitors for cognitive enhancement. Curr Pharm Des 11, 3329–3334. [DOI] [PubMed] [Google Scholar]
- Loughney K.; Hill T. R.; Florio V. A.; Uher L.; Rosman G. J.; Wolda S. L.; Jones B. A.; Howard M. L.; McAllister-Lucas L. M.; Sonnenburg W. K.; Francis S. H.; Corbin J. D.; Beavo J. A.; Ferguson K. (1998) Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3′,5′-cyclic nucleotide phosphodiesterase. Gene 216, 139–147. [DOI] [PubMed] [Google Scholar]
- Prickaerts J.; Sik A.; van Staveren W. C.; Koopmans G.; Steinbusch H. W.; van der Staay F. J.; de Vente J.; Blokland A. (2004) Phosphodiesterase type 5 inhibition improves early memory consolidation of object information. Neurochem. Int. 45, 915–928. [DOI] [PubMed] [Google Scholar]
- Prickaerts J.; Steinbusch H. W.; Smits J. F.; de Vente J. (1997) Possible role of nitric oxide-cyclic GMP pathway in object recognition memory: effects of 7-nitroindazole and zaprinast. Eur. J. Pharmacol. 337, 125–136. [DOI] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M.; Hervias I.; Ricobaraza A.; Puerta E.; Perez-Roldan J. M.; Garcia-Barroso C.; Franco R.; Aguirre N.; Garcia-Osta A. (2011) Sildenafil restores cognitive function without affecting Ass burden in an Alzheimer’s disease mouse model. Br. J. Pharmacol. 164, 2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D.; Staniszewski A.; Deng S. X.; Privitera L.; Leznik E.; Liu S.; Zhang H.; Feng Y.; Palmeri A.; Landry D. W.; Arancio O. (2009) Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J. Neurosci. 29, 8075–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Barroso C.; Ricobaraza A.; Pascual-Lucasa M.; Unceta N.; Rico A. J.; Goicolea M.; Sallés J.; Lanciego J.; Oyarzabal J.; Franco R.; Cuadrado-Tejedor M.; García-Osta A. (2012) Tadalafil crosses the blood brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 64, 114–123. [DOI] [PubMed] [Google Scholar]
- Miro X.; Perez-Torres S.; Palacios J. M.; Puigdomenech P.; Mengod G. (2001) Differential distribution of cAMP-specific phosphodiesterase 7A mRNA in rat brain and peripheral organs. Synapse 40, 201–214. [DOI] [PubMed] [Google Scholar]
- Paterniti I.; Mazzon E.; Gil C.; Impellizzeri D.; Palomo V.; Redondo M.; Perez D. I.; Esposito E.; Martinez A.; Cuzzocrea S. (2011) PDE 7 inhibitors: new potential drugs for the therapy of spinal cord injury. PLoS One 6, e15937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Staveren W. C.; Glick J.; Markerink-van Ittersum M.; Shimizu M.; Beavo J. A.; Steinbusch H. W.; de Vente J. (2002) Cloning and localization of the cGMP-specific phosphodiesterase type 9 in the rat brain. J. Neurocytol. 31, 729–741. [DOI] [PubMed] [Google Scholar]
- Van der Staay F. J.; Rutten K.; Barfacker L.; Devry J.; Erb C.; Heckroth H.; Karthaus D.; Tersteegen A.; van Kampen M.; Blokland A.; Prickaerts J.; Reymann K. G.; Schroder U. H.; Hendrix M. (2008) The novel selective PDE9 inhibitor BAY 73–6691 improves learning and memory in rodents. Neuropharmacology 55, 908–918. [DOI] [PubMed] [Google Scholar]
- Fujishige K.; Kotera J.; Omori K. (1999) Striatum- and testis-specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur. J. Biochem. 266, 1118–1127. [DOI] [PubMed] [Google Scholar]
- Loughney K.; Snyder P. B.; Uher L.; Rosman G. J.; Ferguson K.; Florio V. A. (1999) Isolation and characterization of PDE10A, a novel human 3′,5′-cyclic nucleotide phosphodiesterase. Gene 234, 109–117. [DOI] [PubMed] [Google Scholar]
- Soderling S. H.; Bayuga S. J.; Beavo J. A. (1999) Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc. Natl. Acad. Sci. U.S.A. 96, 7071–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger T. F.; Bartlett B.; Coskran T. M.; Culp J. S.; James L. C.; Krull D. L.; Lanfear J.; Ryan A. M.; Schmidt C. J.; Strick C. A.; Varghese A. H.; Williams R. D.; Wylie P. G.; Menniti F. S. (2003) Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 985, 113–126. [DOI] [PubMed] [Google Scholar]
- Rodefer J. S.; Murphy E. R.; Baxter M. G. (2005) PDE10A inhibition reverses subchronic PCP-induced deficits in attentional set-shifting in rats. Eur. J. Neurosci. 21, 1070–1076. [DOI] [PubMed] [Google Scholar]
- Hebb A. L.; Robertson H. A.; Denovan-Wright E. M. (2004) Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience 123, 967–981. [DOI] [PubMed] [Google Scholar]
- O’Connor V.; Genin A.; Davis S.; Karishma K. K.; Doyere V.; De Zeeuw C. I.; Sanger G.; Hunt S. P.; Richter-Levin G.; Mallet J.; Laroche S.; Bliss T. V.; French P. J. (2004) Differential amplification of intron-containing transcripts reveals long term potentiation-associated up-regulation of specific Pde10A phosphodiesterase splice variants. J. Biol. Chem. 279, 15841–15849. [DOI] [PubMed] [Google Scholar]
- Schmidt C. J.; Chapin D. S.; Cianfrogna J.; Corman M. L.; Hajos M.; Harms J. F.; Hoffman W. E.; Lebel L. A.; McCarthy S. A.; Nelson F. R.; Proulx-LaFrance C.; Majchrzak M. J.; Ramirez A. D.; Schmidt K.; Seymour P. A.; Siuciak J. A.; Tingley F. D. 3rd; Williams R. D.; Verhoest P. R.; Menniti F. S. (2008) Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J. Pharmacol. Exp. Ther. 325, 681–690. [DOI] [PubMed] [Google Scholar]
- Kelly M. P.; Logue S. F.; Brennan J.; Day J. P.; Lakkaraju S.; Jiang L.; Zhong X.; Tam M.; Sukoff Rizzo S. J.; Platt B. J.; Dwyer J. M.; Neal S.; Pulito V. L.; Agostino M. J.; Grauer S. M.; Navarra R. L.; Kelley C.; Comery T. A.; Murrills R. J.; Houslay M. D.; Brandon N. J. (2011) Phosphodiesterase 11A in brain is enriched in ventral hippocampus and deletion causes psychiatric disease-related phenotypes. Proc. Natl. Acad. Sci. U.S.A. 107, 8457–8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutten K.; Prickaerts J.; Hendrix M.; van der Staay F. J.; Sik A.; Blokland A. (2007) Time-dependent involvement of cAMP and cGMP in consolidation of object memory: studies using selective phosphodiesterase type 2, 4 and 5 inhibitors. Eur. J. Pharmacol. 558, 107–112. [DOI] [PubMed] [Google Scholar]
- Abel T.; Nguyen P. V.; Barad M.; Deuel T. A.; Kandel E. R.; Bourtchouladze R. (1997) Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88, 615–626. [DOI] [PubMed] [Google Scholar]
- Bernabeu R.; Bevilaqua L.; Ardenghi P.; Bromberg E.; Schmitz P.; Bianchin M.; Izquierdo I.; Medina J. H. (1997) Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc. Natl. Acad. Sci. U.S.A. 94, 7041–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalimoniuk M.; Strosznajder J. B. (1998) Aging modulates nitric oxide synthesis and cGMP levels in hippocampus and cerebellum. Effects of amyloid beta peptide. Mol. Chem. Neuropathol. 35, 77–95. [DOI] [PubMed] [Google Scholar]
- Domek-Lopacinska K. U.; Strosznajder J. B. (2010) Cyclic GMP and nitric oxide synthase in aging and Alzheimer’s disease. Mol. Neurobiol. 41, 129–137. [DOI] [PubMed] [Google Scholar]
- Molnar P.; Gaal L. (1992) Effect of different subtypes of cognition enhancers on long-term potentiation in the rat dentate gyrus in vivo. Eur. J. Pharmacol. 215, 17–22. [DOI] [PubMed] [Google Scholar]
- DeNoble V. J. (1987) Vinpocetine enhances retrieval of a step-through passive avoidance response in rats. Pharmacol., Biochem. Behav. 26, 183–186. [DOI] [PubMed] [Google Scholar]
- Hindmarch I.; Fuchs H. H.; Erzigkeit H. (1991) Efficacy and tolerance of vinpocetine in ambulant patients suffering from mild to moderate organic psychosyndromes. Int. Clin. Psychopharmacol. 6, 31–43. [DOI] [PubMed] [Google Scholar]
- Medina A. E.; Krahe T. E.; Ramoa A. S. (2006) Restoration of neuronal plasticity by a phosphodiesterase type 1 inhibitor in a model of fetal alcohol exposure. J. Neurosci. 26, 1057–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domek-Lopacinska K.; Strosznajder J. B. (2008) The effect of selective inhibition of cyclic GMP hydrolyzing phosphodiesterases 2 and 5 on learning and memory processes and nitric oxide synthase activity in brain during aging. Brain Res. 1216, 68–77. [DOI] [PubMed] [Google Scholar]
- van Donkelaar E. L.; Rutten K.; Blokland A.; Akkerman S.; Steinbusch H. W.; Prickaerts J. (2008) Phosphodiesterase 2 and 5 inhibition attenuates the object memory deficit induced by acute tryptophan depletion. Eur. J. Pharmacol. 600, 98–104. [DOI] [PubMed] [Google Scholar]
- Perez-Torres S.; Miro X.; Palacios J. M.; Cortes R.; Puigdomenech P.; Mengod G. (2000) Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and[3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J. Chem. Neuroanat. 20, 349–174. [DOI] [PubMed] [Google Scholar]
- Rutten K.; Basile J. L.; Prickaerts J.; Blokland A.; Vivian J. A. (2008) Selective PDE inhibitors rolipram and sildenafil improve object retrieval performance in adult cynomolgus macaques. Psychopharmacology (Berlin, Ger.) 196, 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. T.; Crissman A. M.; Dorairaj N. R.; Chandler L. J.; O’Donnell J. M. (2000) Inhibition of cyclic AMP phosphodiesterase (PDE4) reverses memory deficits associated with NMDA receptor antagonism. Neuropsychopharmacology 23, 198–204. [DOI] [PubMed] [Google Scholar]
- Li Y. F.; Cheng Y. F.; Huang Y.; Conti M.; Wilson S. P.; O’Donnell J. M.; Zhang H. T. (2011) Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J. Neurosci. 31, 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuciak J. A.; McCarthy S. A.; Chapin D. S.; Martin A. N. (2008) Behavioral and neurochemical characterization of mice deficient in the phosphodiesterase-4B (PDE4B) enzyme. Psychopharmacology (Berlin, Ger.) 197, 115–126. [DOI] [PubMed] [Google Scholar]
- Rutten K.; Wallace T. L.; Works M.; Prickaerts J.; Blokland A.; Novak T. J.; Santarelli L.; Misner D. L. (2011) Enhanced long-term depression and impaired reversal learning in phosphodiesterase 4B-knockout (PDE4B–/−) mice. Neuropharmacology 61, 138–147. [DOI] [PubMed] [Google Scholar]
- Barad M.; Bourtchouladze R.; Winder D. G.; Golan H.; Kandel E. (1998) Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc. Natl. Acad. Sci. U.S.A. 95, 15020–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutten K.; Prickaerts J.; Blokland A. (2006) Rolipram reverses scopolamine-induced and time-dependent memory deficits in object recognition by different mechanisms of action. Neurobiol. Learn. Mem. 85, 132–138. [DOI] [PubMed] [Google Scholar]
- Zhang H. T.; Huang Y.; Suvarna N. U.; Deng C.; Crissman A. M.; Hopper A. T.; De Vivo M.; Rose G. M.; O’Donnell J. M. (2005) Effects of the novel PDE4 inhibitors MEM1018 and MEM1091 on memory in the radial-arm maze and inhibitory avoidance tests in rats. Psychopharmacology (Berlin, Ger.) 179, 613–619. [DOI] [PubMed] [Google Scholar]
- Ramos B. P.; Birnbaum S. G.; Lindenmayer I.; Newton S. S.; Duman R. S.; Arnsten A. F. (2003) Dysregulation of protein kinase a signaling in the aged prefrontal cortex: new strategy for treating age-related cognitive decline. Neuron 40, 835–45. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R.; Lidge R.; Catapano R.; Stanley J.; Gossweiler S.; Romashko D.; Scott R.; Tully T. (2003) A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl. Acad. Sci. U.S.A. 100, 10518–10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa T.; Mishima K.; Matsumoto Y.; Iwasaki K.; Iwasaki K.; Fujiwara M. (1997) Rolipram and its optical isomers, phosphodiesterase 4 inhibitors, attenuated the scopolamine-induced impairments of learning and memory in rats. Jpn. J. Pharmacol. 75, 275–281. [DOI] [PubMed] [Google Scholar]
- Imanishi T.; Sawa A.; Ichimaru Y.; Miyashiro M.; Kato S.; Yamamoto T.; Ueki S. (1997) Ameliorating effects of rolipram on experimentally induced impairments of learning and memory in rodents. Eur. J. Pharmacol. 321, 273–278. [DOI] [PubMed] [Google Scholar]
- Zhang H. T.; Zhao Y.; Huang Y.; Dorairaj N. R.; Chandler L. J.; O’Donnell J. M. (2004) Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into the CA1 subregion of the rat hippocampus. Neuropsychopharmacology 29, 1432–1439. [DOI] [PubMed] [Google Scholar]
- Zhang H. T.; O’Donnell J. M. (2000) Effects of rolipram on scopolamine-induced impairment of working and reference memory in the radial-arm maze tests in rats. Psychopharmacology (Berlin, Ger.) 150, 311–316. [DOI] [PubMed] [Google Scholar]
- Gallant M.; Aspiotis R.; Day S.; Dias R.; Dube D.; Dube L.; Friesen R. W.; Girard M.; Guay D.; Hamel P.; Huang Z.; Lacombe P.; Laliberte S.; Levesque J. F.; Liu S.; Macdonald D.; Mancini J.; Nicholson D. W.; Styhler A.; Townson K.; Waters K.; Young R. N.; Girard Y. (2010) Discovery of MK-0952, a selective PDE4 inhibitor for the treatment of long-term memory loss and mild cognitive impairment. Bioorg. Med. Chem. Lett. 20, 6387–6393. [DOI] [PubMed] [Google Scholar]
- NCT00362024, C. g. I. (2006) http://clinicaltrials.gov/ct2/show/NCT00362024.
- Robichaud A.; Savoie C.; Stamatiou P. B.; Tattersall F. D.; Chan C. C. (2001) PDE4 inhibitors induce emesis in ferrets via a noradrenergic pathway. Neuropharmacology 40, 262–269. [DOI] [PubMed] [Google Scholar]
- Robichaud A.; Savoie C.; Stamatiou P. B.; Lachance N.; Jolicoeur P.; Rasori R.; Chan C. C. (2002) Assessing the emetic potential of PDE4 inhibitors in rats. Br. J. Pharmacol. 135, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno O.; Fedele E.; Prickaerts J.; Parker L. A.; Canepa E.; Brullo C.; Cavallero A.; Gardella E.; Balbi A.; Domenicotti C.; Bollen E.; Gijselaers H. J.; Vanmierlo T.; Erb K.; Limebeer C. L.; Argellati F.; Marinari U. M.; Pronzato M. A.; Ricciarelli R. (2011) GEBR-7b, a novel PDE4D selective inhibitor that improves memory in rodents at non-emetic doses. Br. J. Pharmacol. 164, 2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina D. (2008) PDE4 inhibitors: current status. Br. J. Pharmacol. 155, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratti C. M.; Boccia M. M. (1999) Effects of sildenafil on long-term retention of an inhibitory avoidance response in mice. Behav. Pharmacol. 10, 731–737. [DOI] [PubMed] [Google Scholar]
- Boccia M. M.; Blake M. G.; Krawczyk M. C.; Baratti C. M. (2011) Sildenafil, a selective phosphodiesterase type 5 inhibitor, enhances memory reconsolidation of an inhibitory avoidance task in mice. Behav. Brain Res. 220, 319–324. [DOI] [PubMed] [Google Scholar]
- Devan B. D.; Bowker J. L.; Duffy K. B.; Bharati I. S.; Jimenez M.; Sierra-Mercado D. Jr.; Nelson C. M.; Spangler E. L.; Ingram D. K. (2006) Phosphodiesterase inhibition by sildenafil citrate attenuates a maze learning impairment in rats induced by nitric oxide synthase inhibition. Psychopharmacology (Berlin, Ger.) 183, 439–445. [DOI] [PubMed] [Google Scholar]
- Erceg S.; Monfort P.; Hernandez-Viadel M.; Rodrigo R.; Montoliu C.; Felipo V. (2005) Oral administration of sildenafil restores learning ability in rats with hyperammonemia and with portacaval shunts. Hepatology 41, 299–306. [DOI] [PubMed] [Google Scholar]
- Prickaerts J.; Sik A.; van der Staay F. J.; de Vente J.; Blokland A. (2005) Dissociable effects of acetylcholinesterase inhibitors and phosphodiesterase type 5 inhibitors on object recognition memory: acquisition versus consolidation. Psychopharmacology (Berlin, Ger.) 177, 381–390. [DOI] [PubMed] [Google Scholar]
- Rutten K.; Vente J. D.; Sik A.; Ittersum M. M.; Prickaerts J.; Blokland A. (2005) The selective PDE5 inhibitor, sildenafil, improves object memory in Swiss mice and increases cGMP levels in hippocampal slices. Behav. Brain Res. 164, 11–16. [DOI] [PubMed] [Google Scholar]
- Hosseini-Sharifabad A.; Ghahremani M. H.; Sabzevari O.; Naghdi N.; Abdollahi M.; Beyer C.; Bollen E.; Prickaerts J.; Roghani A.; Sharifzadeh M. (2012) Effects of protein kinase A and G inhibitors on hippocampal cholinergic markers expressions in rolipram- and sildenafil-induced spatial memory improvement. Pharmacol., Biochem. Behav. 101, 311–319. [DOI] [PubMed] [Google Scholar]
- Devan B. D.; Sierra-Mercado D. Jr.; Jimenez M.; Bowker J. L.; Duffy K. B.; Spangler E. L.; Ingram D. K. (2004) Phosphodiesterase inhibition by sildenafil citrate attenuates the learning impairment induced by blockade of cholinergic muscarinic receptors in rats. Pharmacol., Biochem. Behav. 79, 691–699. [DOI] [PubMed] [Google Scholar]
- Patil C. S.; Singh V. P.; Kulkarni S. K. (2006) Modulatory effect of sildenafil in diabetes and electroconvulsive shock-induced cognitive dysfunction in rats. Pharmacol. Rep. 58, 373–380. [PubMed] [Google Scholar]
- Fisher D. A.; Smith J. F.; Pillar J. S.; St Denis S. H.; Cheng J. B. (1998) Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J. Biol. Chem. 273, 15559–15564. [DOI] [PubMed] [Google Scholar]
- Hutson P. H.; Finger E. N.; Magliaro B. C.; Smith S. M.; Converso A.; Sanderson P. E.; Mullins D.; Hyde L. A.; Eschle B. K.; Turnbull Z.; Sloan H.; Guzzi M.; Zhang X.; Wang A.; Rindgen D.; Mazzola R.; Vivian J. A.; Eddins D.; Uslaner J. M.; Bednar R.; Gambone C.; Le-Mair W.; Marino M. J.; Sachs N.; Xu G.; Parmentier-Batteur S. (2011) The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 (6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-py ran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one) enhances synaptic plasticity and cognitive function in rodents. Neuropharmacology 61, 665–676. [DOI] [PubMed] [Google Scholar]
- Vardigan J. D.; Converso A.; Hutson P. H.; Uslaner J. M. (2011) The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. J. Neurogenet 25, 120–126. [DOI] [PubMed] [Google Scholar]
- Nicholas T; Evans R; S S. (2009) Pf-04447943, a novel PDE9A inhibitor, increases cGMP levels in cerebrospinal fluid: translation from non-clinical species to healthy human volunteers. Alzheimer's Dementia 5, 330–331. [Google Scholar]
- Kleiman R. J.; Kimmel L. H.; Bove S. E.; Lanz T. A.; Harms J. F.; Romegialli A.; Miller K. S.; Willis A.; des Etages S.; Kuhn M.; Schmidt C. J. (2011) Chronic suppression of phosphodiesterase 10A alters striatal expression of genes responsible for neurotransmitter synthesis, neurotransmission, and signaling pathways implicated in Huntington’s disease. J Pharmacol. Exp. Ther. 336, 64–76. [DOI] [PubMed] [Google Scholar]
- Lehericy S.; Gerardin E. (2002) Normal functional imaging of the basal ganglia. Epileptic Disord. 4(Suppl 3), S23–30. [PubMed] [Google Scholar]
- Roussinov K.; Yonkov D. (1976) Effect of imidazole, papaverine and histamine on learning and memory in albino rats. Acta Physiol. Pharmacol. Bulg 2, 49–57. [PubMed] [Google Scholar]
- Siuciak J. A.; McCarthy S. A.; Chapin D. S.; Fujiwara R. A.; James L. C.; Williams R. D.; Stock J. L.; McNeish J. D.; Strick C. A.; Menniti F. S.; Schmidt C. J. (2006) Genetic deletion of the striatum-enriched phosphodiesterase PDE10A: evidence for altered striatal function. Neuropharmacology 51, 374–385. [DOI] [PubMed] [Google Scholar]
- Lione L. A.; Carter R. J.; Hunt M. J.; Bates G. P.; Morton A. J.; Dunnett S. B. (1999) Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. J. Neurosci. 19, 10428–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernabeu R.; Schroder N.; Quevedo J.; Cammarota M.; Izquierdo I.; Medina J. H. (1997) Further evidence for the involvement of a hippocampal cGMP/cGMP-dependent protein kinase cascade in memory consolidation. NeuroReport 8, 2221–2224. [DOI] [PubMed] [Google Scholar]
- Kleppisch T.; Feil R. (2009) cGMP signalling in the mammalian brain: role in synaptic plasticity and behaviour. Handb. Exp. Pharmacol. 549–579. [DOI] [PubMed] [Google Scholar]
- Dundore R. L.; Clas D. M.; Wheeler L. T.; Habeeb P. G.; Bode D. C.; Buchholz R. A.; Silver P. J.; Pagani E. D. (1993) Zaprinast increases cyclic GMP levels in plasma and in aortic tissue of rats. Eur. J. Pharmacol. 249, 293–7. [DOI] [PubMed] [Google Scholar]
- Paterno R.; Faraci F. M.; Heistad D. D. (1996) Role of Ca(2+)-dependent K+ channels in cerebral vasodilatation induced by increases in cyclic GMP and cyclic AMP in the rat. Stroke 27, 1603–1607discussion 1607–1608. [DOI] [PubMed] [Google Scholar]
- Perez-Torres S.; Cortes R.; Tolnay M.; Probst A.; Palacios J. M.; Mengod G. (2003) Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer’s disease brains examined by in situ hybridization. Exp. Neurol. 182, 322–334. [DOI] [PubMed] [Google Scholar]
- McLachlan C. S.; Chen M. L.; Lynex C. N.; Goh D. L.; Brenner S.; Tay S. K. (2007) Changes in PDE4D isoforms in the hippocampus of a patient with advanced Alzheimer disease. Arch. Neurol. 64, 456–457. [DOI] [PubMed] [Google Scholar]
- Thal L. J.; Salmon D. P.; Lasker B.; Bower D.; Klauber M. R. (1989) The safety and lack of efficacy of vinpocetine in Alzheimer’s disease. J. Am. Geriatr. Soc. 37, 515–520. [DOI] [PubMed] [Google Scholar]
- Gong B.; Vitolo O. V.; Trinchese F.; Liu S.; Shelanski M.; Arancio O. (2004) Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Invest. 114, 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu M.; Takiguchi O.; Nishiyama A.; Mori H. (2010) Cilostazol prevents amyloid beta peptide(25–35)-induced memory impairment and oxidative stress in mice. Br. J. Pharmacol. 161, 1899–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. H.; Kim J. H.; Bae S. S.; Hong K. W.; Lee D. S.; Leem J. Y.; Choi B. T.; Shin H. K. (2011) Protective effect of the phosphodiesterase III inhibitor cilostazol on amyloid beta-induced cognitive deficits associated with decreased amyloid beta accumulation. Biochem. Biophys. Res. Commun. 408, 602–608. [DOI] [PubMed] [Google Scholar]
- Smith D. L.; Pozueta J.; Gong B.; Arancio O.; Shelanski M. (2009) Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. U.S.A. 106, 16877–16882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgue S. T.; Patterson B. E.; Bedding A. W.; Payne C. D.; Phillips D. L.; Wrishko R. E.; Mitchell M. I. (2006) Tadalafil pharmacokinetics in healthy subjects. Br. J. Clin. Pharmacol. 61, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S. K.; Patil C. S. (2004) Phosphodiesterase 5 enzyme and its inhibitors: update on pharmacological and therapeutical aspects. Methods Find. Exp. Clin. Pharmacol. 26, 789–799. [DOI] [PubMed] [Google Scholar]
- Schultheiss D.; Muller S. V.; Nager W.; Stief C. G.; Schlote N.; Jonas U.; Asvestis C.; Johannes S.; Munte T. F. (2001) Central effects of sildenafil (Viagra) on auditory selective attention and verbal recognition memory in humans: a study with event-related brain potentials. World J. Urol. 19, 46–50. [DOI] [PubMed] [Google Scholar]
- Naatanen R.; Kujala T.; Kreegipuu K.; Carlson S.; Escera C.; Baldeweg T.; Ponton C. (2011) The mismatch negativity: an index of cognitive decline in neuropsychiatric and neurological diseases and in ageing. Brain 134, 3435–3453. [DOI] [PubMed] [Google Scholar]
- Fusco F.; Razzoli E.; Imbimbo C.; Rossi A.; Verze P.; Mirone V. (2010) A new era in the treatment of erectile dysfunction: chronic phosphodiesterase type 5 inhibition. BJU Int. 105, 1634–1639. [DOI] [PubMed] [Google Scholar]
- Luks A. M.; McIntosh S. E.; Grissom C. K.; Auerbach P. S.; Rodway G. W.; Schoene R. B.; Zafren K.; Hackett P. H. (2010) Wilderness Medical Society consensus guidelines for the prevention and treatment of acute altitude illness. Wilderness Environ. Med. 21, 146–155. [DOI] [PubMed] [Google Scholar]
- Shim Y. S.; Pae C. U.; Kim S. W.; Kim H. W.; Kim J. C.; Koh J. S. (2011) Effects of repeated dosing with Udenafil (Zydena) on cognition, somatization and erection in patients with erectile dysfunction: a pilot study. Int. J. Impotence Res. 23, 109–114. [DOI] [PubMed] [Google Scholar]
- Sakurai H., Hanyu H., Sato T., Kume K., Hirao K., Kanetaka H., and Iwamoto T. Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: A pilot study. . Geriatr. Gerontol. Int. doi: 10.1111/j.1447-0594.2012.00866.x. [DOI] [PubMed]; Epub June, 2012.
- Xu Y.; Zhang H. T.; O’Donnell J. M. (2011) Phosphodiesterases in the central nervous system: implications in mood and cognitive disorders. Handb. Exp. Pharmacol. 447–485. [DOI] [PubMed] [Google Scholar]
- Kruuse C.; Khurana T. S.; Rybalkin S. D.; Birk S.; Engel U.; Edvinsson L.; Olesen J. (2005) Phosphodiesterase 5 and effects of sildenafil on cerebral arteries of man and guinea pig. Eur. J. Pharmacol. 521, 105–114. [DOI] [PubMed] [Google Scholar]
- Kruuse C.; Hansen A. E.; Larsson H. B.; Lauritzen M.; Rostrup E. (2009) Cerebral haemodynamic response or excitability is not affected by sildenafil. J. Cereb. Blood Flow Metab. 29, 830–839. [DOI] [PubMed] [Google Scholar]
- Lorberboym M.; Mena I.; Wainstein J.; Boaz M.; Lampl Y. (2010) The effect of sildenafil citrate (Viagra) on cerebral blood flow in patients with cerebrovascular risk factors. Acta Neurol. Scand. 121, 370–376. [DOI] [PubMed] [Google Scholar]
- Clapper J. R.; Moreno-Sanz G.; Russo R.; Guijarro A.; Vacondio F.; Duranti A.; Tontini A.; Sanchini S.; Sciolino N. R.; Spradley J. M.; Hohmann A. G.; Calignano A.; Mor M.; Tarzia G.; Piomelli D. (2012) Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 13, 1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang A.; Hao H.; Zheng X.; Liang Y.; Xie Y.; Xie T.; Dai C.; Zhao Q.; Wu X.; Xie L.; Wang G. (2011) Peripheral anti-inflammatory effects explain the ginsenosides paradox between poor brain distribution and anti-depression efficacy. J. Neuroinflammation 8, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reneerkens O. A.; Rutten K.; Akkerman S.; Blokland A.; Shaffer C. L.; Menniti F. S.; Steinbusch H. W.; Prickaerts J. (2012) Phosphodiesterase type 5 (PDE5) inhibition improves object recognition memory: Indications for central and peripheral mechanisms. Neurobiol. Learn. Mem. 97, 370–379. [DOI] [PubMed] [Google Scholar]
- Liebenberg N.; Harvey B. H.; Brand L.; Wegener G.; Brink C. B. (2012) Chronic treatment with the phosphodiesterase type 5 inhibitors sildenafil and tadalafil display anxiolytic effects in Flinders Sensitive Line rats. Metab. Brain Dis 27, 337–340. [DOI] [PubMed] [Google Scholar]
- Sakamoto K.; Karelina K.; Obrietan K. (2011) CREB: a multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 116, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. F.; Wang C.; Lin H. B.; Li Y. F.; Huang Y.; Xu J. P.; Zhang H. T. (2010) Inhibition of phosphodiesterase-4 reverses memory deficits produced by Abeta25–35 or Abeta1–40 peptide in rats. Psychopharmacology (Berlin, Ger.) 212, 181–191. [DOI] [PubMed] [Google Scholar]
- Lu Y. F.; Hawkins R. D. (2002) Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurophysiol. 88, 1270–1278. [DOI] [PubMed] [Google Scholar]
- Alford S.; Frenguelli B. G.; Schofield J. G.; Collingridge G. L. (1993) Characterization of Ca2+ signals induced in hippocampal CA1 neurones by the synaptic activation of NMDA receptors. J. Physiol. 469, 693–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss T. V.; Collingridge G. L. (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. [DOI] [PubMed] [Google Scholar]
- Emptage N.; Bliss T. V.; Fine A. (1999) Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22, 115–124. [DOI] [PubMed] [Google Scholar]
- Lee H. C. (1993) Potentiation of calcium- and caffeine-induced calcium release by cyclic ADP-ribose. J. Biol. Chem. 268, 293–299. [PubMed] [Google Scholar]
- Lee H. C.; Aarhus R.; Graeff R. M. (1995) Sensitization of calcium-induced calcium release by cyclic ADP-ribose and calmodulin. J. Biol. Chem. 270, 9060–6066. [DOI] [PubMed] [Google Scholar]
- Gallo E. F.; Iadecola C. (2011) Neuronal nitric oxide contributes to neuroplasticity-associated protein expression through cGMP, protein kinase G, and extracellular signal-regulated kinase. J. Neurosci. 31, 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F.; Bernhard D.; Lukowski R.; Weinmeister P. (2009) cGMP regulated protein kinases (cGK). Handb. Exp. Pharmacol. 137–162. [DOI] [PubMed] [Google Scholar]
- Arriagada P. V.; Growdon J. H.; Hedley-Whyte E. T.; Hyman B. T. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. [DOI] [PubMed] [Google Scholar]
- Ahmad N.; Wang Y.; Ali A. K.; Ashraf M. (2009) Long-acting phosphodiesterase-5 inhibitor, tadalafil, induces sustained cardioprotection against lethal ischemic injury. Am. J. Physiol.: Heart Circ. Physiol. 297, H387–H391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysiak J. J.; Yang S. K.; Klausner A. P.; Son H.; Tuttle J. B.; Steers W. D. (2008) Tadalafil increases Akt and extracellular signal-regulated kinase 1/2 activation, and prevents apoptotic cell death in the penis following denervation. J. Urol. 179, 779–785. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wang Z.; Gu Y.; Feil R.; Hofmann F.; Ma L. (2009) Regulate axon branching by the cyclic GMP pathway via inhibition of glycogen synthase kinase 3 in dorsal root ganglion sensory neurons. J. Neurosci. 29, 1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington S. L. 3rd; Shindel A. W. (2010) A once-daily dose of tadalafil for erectile dysfunction: compliance and efficacy. Drug Des. Dev. Ther. 4, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin Y. D.; White R. J. (2011) Novel therapeutic approaches in pulmonary arterial hypertension: focus on tadalafil. Drugs Today 47, 145–156. [DOI] [PubMed] [Google Scholar]
- Bank R. P. D.www.pdb.org.
- Polli J. W.; Kincaid R. L. (1992) Molecular cloning of DNA encoding a calmodulin-dependent phosphodiesterase enriched in striatum. Proc. Natl. Acad. Sci. U.S.A. 89, 11079–11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C. J. (2010) Phosphodiesterase inhibitors as potential cognition enhancing agents. Curr. Top. Med. Chem. 10, 222–230. [DOI] [PubMed] [Google Scholar]
- Ghavami A.; Hirst W. D.; Novak T. J. (2006) Selective phosphodiesterase (PDE)-4 inhibitors: a novel approach to treating memory deficit?. Drugs R D 7, 63–71. [DOI] [PubMed] [Google Scholar]
- Administration, U. S. F. a. D. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm244989.htm.
- Gupta S. (2012) Side-effects of roflumilast. Lancet 379, 710–711author reply 711–712. [DOI] [PubMed] [Google Scholar]
- Bruno O.; Romussi A.; Spallarossa A.; Brullo C.; Schenone S.; Bondavalli F.; Vanthuyne N.; Roussel C. (2009) New selective phosphodiesterase 4D inhibitors differently acting on long, short, and supershort isoforms. J. Med. Chem. 52, 6546–6557. [DOI] [PubMed] [Google Scholar]
- Burgin A. B.; Magnusson O. T.; Singh J.; Witte P.; Staker B. L.; Bjornsson J. M.; Thorsteinsdottir M.; Hrafnsdottir S.; Hagen T.; Kiselyov A. S.; Stewart L. J.; Gurney M. E. (2010) Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 28, 63–70. [DOI] [PubMed] [Google Scholar]
- Mensch J.; Oyarzabal J.; Mackie C.; Augustijns P. (2009) In vivo, in vitro and in silico methods for small molecule transfer across the BBB. J. Pharm. Sci. 98, 4429–4468. [DOI] [PubMed] [Google Scholar]
- Levitra, www.levitra.com (accessed on 29th June 2012).
- Viagra, www.viagra.com (accessed on 29th June 2012).
- Cialis, www.cialis.com (accessed on 29th June 2012).
- Unceta N.; Echeazarra L.; Montana M.; Salles J.; Gomez-Caballero A.; Goicolea M. A.; Barrio R. J. (2012) Validation of an LC-ESI-MS/MS method for the quantitation of phosphodiesterase-5 inhibitors and their main metabolites in rat serum and brain tissue samples. J. Pharm. Biomed. Anal. 70, 529–533. [DOI] [PubMed] [Google Scholar]
- Gresser U.; Gleiter C. H. (2002) Erectile dysfunction: comparison of efficacy and side effects of the PDE-5 inhibitors sildenafil, vardenafil and tadalafil–review of the literature. Eur. J. Med. Res. 7, 435–446. [PubMed] [Google Scholar]
- Kleiman R. J.; Chapin D. S.; Christoffersen C.; Freeman J.; Fonseca K. R.; Geoghegan K. F.; Grimwood S.; Guanowsky V.; Hajos M.; Harms J. F.; Helal C. J.; Hoffmann W. E.; Kocan G. P.; Majchrzak M. J.; McGinnis D.; McLean S.; Menniti F. S.; Nelson F.; Roof R.; Schmidt A. W.; Seymour P. A.; Stephenson D. T.; Tingley F. D.; Vanase-Frawley M.; Verhoest P. R.; Schmidt C. J. (2012) Phosphodiesterase 9A regulates central cGMP and modulates responses to cholinergic and monoaminergic perturbation in vivo. J. Pharmacol. Exp. Ther. 341, 396–409. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov, http://clinicaltrials.gov/ct2/show/NCT00930059 (accessed on 29th June 2012).
- Harrison P. J. (2004) The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berlin, Ger.) 174, 151–162. [DOI] [PubMed] [Google Scholar]
- Tan H. Y.; Callicott J. H.; Weinberger D. R. (2007) Dysfunctional and compensatory prefrontal cortical systems, genes and the pathogenesis of schizophrenia. Cereb. Cortex 17(Suppl 1), i171–i181. [DOI] [PubMed] [Google Scholar]
- Grauer S. M.; Pulito V. L.; Navarra R. L.; Kelly M. P.; Kelley C.; Graf R.; Langen B.; Logue S.; Brennan J.; Jiang L.; Charych E.; Egerland U.; Liu F.; Marquis K. L.; Malamas M.; Hage T.; Comery T. A.; Brandon N. J. (2009) Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J. Pharmacol. Exp. Ther. 331, 574–590. [DOI] [PubMed] [Google Scholar]
- Verhoest P. R.; Chapin D. S.; Corman M.; Fonseca K.; Harms J. F.; Hou X.; Marr E. S.; Menniti F. S.; Nelson F.; O’Connor R.; Pandit J.; Proulx-Lafrance C.; Schmidt A. W.; Schmidt C. J.; Suiciak J. A.; Liras S. (2009) Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 52, 5188–5196. [DOI] [PubMed] [Google Scholar]