

Background: Sepp1 transports selenium, but its complete role in selenium homeostasis is not known.
Results: Deletion of Sepp1 in hepatocytes increases liver selenium at the expense of other tissues and decreases whole-body selenium by increasing excretion.
Conclusion: Sepp1 production by hepatocytes retains selenium in the organism and distributes it from the liver to peripheral tissues.
Significance: Sepp1 is central to selenium homeostasis.
Keywords: Liver, Metal Homeostasis, Selenium, Selenoprotein, Transport Metals, Selenium Excretion, Selenium Homeostasis, Selenium in the Hepatocyte, Selenium Transport, Selenoprotein P
Abstract
Sepp1 is a widely expressed extracellular protein that in humans and mice contains 10 selenocysteine residues in its primary structure. Extra-hepatic tissues take up plasma Sepp1 for its selenium via apolipoprotein E receptor-2 (apoER2)-mediated endocytosis. The role of Sepp1 in the transport of selenium from liver, a rich source of the element, to peripheral tissues was studied using mice with selective deletion of Sepp1 in hepatocytes (Sepp1c/c/alb-cre+/− mice). Deletion of Sepp1 in hepatocytes lowered plasma Sepp1 concentration to 10% of that in Sepp1c/c mice (controls) and increased urinary selenium excretion, decreasing whole-body and tissue selenium concentrations. Under selenium-deficient conditions, Sepp1c/c/alb-cre+/− mice accumulated selenium in the liver at the expense of extra-hepatic tissues, severely worsening clinical manifestations of dietary selenium deficiency. These findings are consistent with there being competition for metabolically available hepatocyte selenium between the synthesis of selenoproteins and the synthesis of selenium excretory metabolites. In addition, selenium deficiency down-regulated the mRNA of the most abundant hepatic selenoprotein, glutathione peroxidase-1 (Gpx1), to 15% of the selenium-replete value, while reducing Sepp1 mRNA, the most abundant hepatic selenoprotein mRNA, only to 61%. This strongly suggests that Sepp1 synthesis is favored in the liver over Gpx1 synthesis when selenium supply is limited, directing hepatocyte selenium to peripheral tissues in selenium deficiency. We conclude that production of Sepp1 by hepatocytes is central to selenium homeostasis in the organism because it promotes retention of selenium in the body and effects selenium distribution from the liver to extra-hepatic tissues, especially under selenium-deficient conditions.
Introduction
Selenium is an essential micronutrient that functions through selenoproteins (1). Some tissues, e.g. testis, kidney, and bone marrow, synthesize selenoproteins for export and therefore have greater requirements for selenium than other tissues. The brain must have a reliable supply of selenium for its viability.
Because dietary selenium intake varies widely, physiological mechanisms must regulate whole-body selenium and ensure its availability to tissues in amounts consistent with their needs. Intestinal absorption of selenium is not regulated; the selenium content of the body is regulated by hepatic production of methylated selenium compounds that are excreted predominantly in the urine (2).
The major selenium transport form in plasma is Sepp1.3 Sepp1 in mice, rats, and humans consists of an N-terminal domain with one selenocysteine residue in a thioredoxin-like motif and a smaller C-terminal domain containing nine selenocysteine residues (3). A short Sepp1 isoform, consisting only of the N-terminal domain, has been identified (but not quantified) in rat plasma along with longer isoforms that contain up to the full 10 selenocysteine residues (4). Tissues take up “long isoform” Sepp1 from plasma via apoER2-mediated endocytosis and utilize its selenium for synthesis of selenoproteins (5, 6). Outside the blood circulation, kidney proximal convoluted tubule cells take up fragments of Sepp1 from the urinary filtrate via megalin-mediated endocytosis and use the selenium for the synthesis of Gpx3, an extracellular glutathione peroxidase (7, 8).
The liver is the main source of plasma Sepp1 (9, 10), although lesser amounts of Sepp1 mRNA are present in many other tissues, indicating that Sepp1 is widely expressed (11). Deletion of Sepp1 in the mouse has severe consequences; brain and testis become selenium-deficient and neurological dysfunction occurs unless a high selenium diet is fed (12, 13). Deletion of Sepp1 also increases urinary selenium excretion, causing a decrease in whole-body selenium (14).
We postulated that Sepp1 synthesis and excretory metabolite synthesis compete for selenium in the liver and therefore that deletion of Sepp1 makes more selenium available for excretion (14). Such a loss of selenium in the urine might be expected to lead to inability to conserve the element under conditions of selenium deficiency. We were unable to study Sepp1−/− mice fed selenium-deficient diet, however, because feeding that diet leads to neurological dysfunction and death within weeks (15).
To test our hypothesis that Sepp1 synthesis in the liver prevents selenium deficiency by reducing its excretion, we produced mice with Sepp1 deleted only in hepatocytes. We reasoned that it might be possible to feed those mice selenium-deficient diet without provoking neurological injury. That would allow us to study selenium metabolism under deficiency conditions and to determine whether production of Sepp1 in the liver protects against selenium deficiency. The experiments reported here indicate that loss of hepatocyte Sepp1 synthesis has widespread effects on systemic selenium metabolism and worsens dietary selenium deficiency.
MATERIALS AND METHODS
Reagents
The reagents used for construction of the targeting vector for production of mice with a conditional allele of Sepp1 were described previously (16). Oligonucleotides were obtained from core lab facilities at the University of Utah and Vanderbilt University Medical Center (Enzyme Reagents Core Lab). 75Se-Selenite (specific activity >250 Ci/g selenium) was purchased from the University of Missouri Research Reactor Facility, Columbia, MO. NADPH was purchased from United States Biochemical Corp. (Cleveland, OH). Glutathione reductase was purchased from Sigma. Digoxigenin-labeled nucleotides and alkaline phosphatase-conjugated anti-digoxigenin antibody were purchased from Roche Diagnostics. pCR4 Topo plasmid and OneShot Top 10 cells were purchased from Invitrogen. High capacity cDNA reverse transcriptase kit and Power SYBR Green PCR master mix were purchased from Applied Biosystems. RNeasy mini kits were purchased from Qiagen. All other chemicals were of reagent grade.
Construction of Targeting Vector
To construct the targeting vector, we followed a method described previously (16). The construction process is summarized in Fig. 1. We first used recombineering to subclone a 13.1-kb genomic fragment (Fig. 1A) from a BAC clone RP23-41H17, which had been obtained from BACPAC resources. The two oligonucleotides used in this step (WS785 and WS786) are shown in Table 1. The resulting plasmid from this step was named pStartK-Sepp1.
FIGURE 1.

Generation of a conditional knock-out mouse line for Sepp1. To generate a conditional knock-out allele for Sepp1, a genomic fragment (bold black line) containing sequence from 5′-CTGAAGCAACAGCTAAAAGA-3′ to 5′-AACACTCCATGCAAACTACA-3′ of the Sepp1 gene was used for constructing the targeting vector. A, genomic structure of the wild-type (WT) Sepp1 gene, with its five exons shown in gray. The coding exons are gray and outlined in black. The second exon contains the start codon ATG. Sepp1 has 10 selenocysteines, each encoded by UGA in the mRNA. The first in-frame TGA is in the second exon. The remaining nine in-frame TGA codons are in exon 5. The 3′UTR of exon 5, where the two selenocysteine insertion sequence elements are located, is gray and is not outlined in black. B, loxP sites are represented by the open arrowheads in the targeting vector. The 5′ loxP site was inserted into the BglI site between exon 1 and exon 2. The 3′ loxP site and an FRT-flanked neo selection cassette were inserted into the SpeI site after exon 5. The arrows show the location of primers WS1004 and WS1005 used to assess the presence of the conditional allele. C, mating a heterozygous mouse with an Flp deleter mouse will delete the neo sequence between the two FRT sites (represented by the open hexagon), resulting in a clean Sepp1 conditional allele.
TABLE 1.
Targeting vector and other primers
| Primer designation | primer sequence (5′ → 3′) | |
|---|---|---|
| pStartK | WS785 | GTAGTGCTACTTATAAATCCCAACAGAGTCTCTTTTAGCTGTTGCTTCAGCGACTGAATTGGTTCCTTTAAAGCC |
| WS786 | ATGTTTATTGCTCACCTAGGCATTATACTAAACACTCCATGCAAACTACAGCCGCACTCGAGATATCTAGACCCA | |
| Southern blot probe | WS869–5F | GAGATCCCAGTAGGTAGGCGACTTG |
| WS870–5R | CCTGTAACCTTGAGCCAAACTTCCT | |
| PCR genotyping | WS1004 | TCCTAGATTGGCAGAGGATAGAATGAA |
| WS1005 | TCAGAAACACCTTCCAACTGTAATGC | |
| neo deletion | WS1006 | GGAGAGAATGGATAGTTACAATGAAGATACA |
| WS1007 | GGAGCTGCATTTTATTTGTTATCTGGA | |
| RT-PCR | mSepp1F | GGCCGTCTTGTGTATCACCT |
| mSepp1R | TGGTGTTTGTGGTGGCTATG | |
| mGpx1F | ACAGTCCACCGTGTATGCCTTC | |
| mGpx1R | CTCTTCATTCTTGCCATTCTCCTG | |
| mHprtF | TCCTCCTCAGACCGCTTTT | |
| mHprtR | CCTGGTTCATCGCTAATC |
We inserted the 5′ loxP site into the BglI site before the second exon. The 3′ loxP site and an FRT-flanked neo selection cassette were inserted into the SpeI site located 3′ of the Sepp1 coding sequence (Fig. 1B). The resulting plasmid was named pStartK-Sepp1cond. To add a negative selection HSV-tk gene, Gateway recombination was performed to quickly transfer the modified Sepp1 genomic DNA into an HSV-tk containing vector named pWSTK2. The resulting targeting vector was named pWSTK2-Sepp1cond.
Generation of Conditional Allele of Sepp1 in Mice
Standard electroporation of the linearized targeting vector into embryonic stem cells was performed as described (16). Long range PCR and Southern blot analyses were performed to identify correctly targeted embryonic stem cell clones. The 5′ Southern probe template (476 bp) was amplified by PCR from the BAC clone RP23-41H17 with primers WS869-5F and WS870-5R (Table 1). DNA isolated from embryonic stem cells was digested with XbaI and run on a 0.9% agarose gel. Southern blot was done with the 5′ probe. The wild-type band was 9.6 kb, and the targeted mutant band was 6.5 kb. Targeted embryonic stem cells were injected into blastocysts using a standard protocol. Male chimeric mice were bred with C57BL/6 females to obtain the desired Sepp1c allele. The FRT-flanked neo selection cassette can be deleted by crossing with a mouse line that ubiquitously expresses the Flp recombinase or it may undergo self-excision (Fig. 1C). The University of Utah Institutional Animal Care and Use Committee approved animal protocols used to generate the knock-out mouse.
Animal Husbandry
Adult Sepp1c/+ mice (mice heterozygous for the Sepp1 conditional allele) were transferred to the animal facility at Vanderbilt University without having been mated with Flp deleter mice. The mice were housed in plastic cages with aspen shavings or alpha-dri bedding material. The light/dark cycle was 12:12 h. Mice received pelleted diet and tap water ad libitum. Experimental diets were formulated by Harlan-Teklad (Madison, WI) to our specifications (15). The diets were Torula yeast-based and contained supplemental amounts of selenium as sodium selenite. The basal (selenium-deficient) form of this experimental diet was assayed, and it contained 0.006 ± 0.003 mg of selenium/kg (n = 7). Sodium selenite was added to this diet during mixing to give final added selenium concentrations of 0.25 mg/kg (control diet) and 0.15, 1, or 4 mg/kg. The Vanderbilt University Institutional Animal Care and Use Committee approved animal protocols for studies conducted at Vanderbilt.
Pups from heterozygote matings were weaned 21 days after birth and separated by sex. All mice used in experiments reported here were males. The genotypes of all pups were determined by PCR of genomic DNA isolated from ear notches. PCR amplification with WS1004 and WS1005 (Table 1) identified mice that were homozygous for the floxed gene. Pups from these matings were used to establish the Sepp1c/c colony.
Deletion of Hepatic Sepp1
Sepp1c/c mice were mated with albumin-cre recombinase mice (JAX strain B6.Cg-Tg(Alb-cre)21Mgn/J that is congenic with C57BL/6 mice; stock number 003574). The resulting pups were heterozygous for the conditional Sepp1 allele and were genotyped to identify pups carrying the albumin-cre recombinase transgene. Sepp1c/+ male and female mice that carried the albumin-cre transgene were mated to produce Sepp1c/c mice carrying the albumin-cre transgene (Sepp1c/c/alb-cre+/−). After this initial mating, it was verified by PCR amplification of genomic DNA that a majority of the progeny no longer carried the FRT-flanked neo cassette. PCR amplification using oligonucleotides WS1006 and WS1007 gave a 352-bp product indicating that the neo cassette was absent. Male (Sepp1c/c) and female (Sepp1c/c/alb-cre+/−) mice that did not carry the FRT-flanked neo cassette were used as breeders to establish the colony. Male mice lacking hepatic Sepp1 (Sepp1c/c/alb-cre+/− mice) were identified by PCR and selected for experiments.
Whole-body and Tissue Selenium Determination Experiments
At weaning, Sepp1c/c and Sepp1c/c/alb-cre+/− male mice were fed control diet supplemented with 0.25 mg of selenium/kg. Four weeks after weaning, mice were anesthetized with isoflurane, and blood was removed from the inferior vena cava with a syringe and needle. Blood was treated with disodium EDTA (1 mg/ml) to prevent coagulation. An aliquot of whole blood was taken for selenium assay, and the remainder of the blood was centrifuged. Plasma was frozen for assay to determine selenium biomarkers. Liver, kidney, muscle, testis, and brain were harvested and frozen immediately in liquid nitrogen. The carcass was frozen in liquid nitrogen. Plasma, tissues, and carcasses were stored at −80 °C. Whole-body selenium concentration was calculated as the sum of blood, tissue, and carcass selenium contents divided by body weight.
To assess the effect of selenium deficiency on tissue selenium concentrations, weanling male Sepp1c/c and Sepp1c/c/alb-cre+/− mice were fed a basal (selenium-deficient) diet. They were observed daily and weighed bi-weekly. After 12 weeks, the mice were anesthetized with isoflurane, and blood was removed from the inferior vena cava. Blood, tissues, and carcasses were analyzed as in the group fed the 0.25 mg of selenium/kg diet.
To assess the selenium content of the liver in the form of Gpx1, Gpx1−/− and Gpx1+/+ weanling mice congenic with C57BL/6 mice (17) were fed a diet supplemented with 0.25 mg of selenium/kg for 4 weeks and then exsanguinated via the inferior vena cava under isoflurane anesthesia. Tissues, blood, and carcasses were taken for selenium analysis.
Biochemical Measurements
Selenium was measured using a modification of the fluorometric assay of Koh and Benson (18) and Sheehan and Gao (19). The ELISA used to measure plasma Sepp1 detects the N-terminal domain of the protein, providing a measurement of the sum of all isoforms (20). Gpx activity was determined using the coupled method with 0.25 mm hydrogen peroxide as substrate (21).
75Se Labeling of Mouse Plasma
Sepp1c/c/alb-cre+/− and Sepp1c/c mice fed 0.25 mg of selenium/kg diet were injected intraperitoneally with 10 μCi of [75Se]selenite (in 0.15 m NaCl). Four hours after 75Se administration, the mice were anesthetized. Blood was obtained from the inferior vena cava and treated with disodium EDTA to prevent coagulation. Plasma was separated by centrifugation. Plasma (1 μl) was subjected to SDS-PAGE on a 12.5% acrylamide gel. After staining with Coomassie Blue, the gel was dried and exposed to Kodak XAR film.
Metabolism of 75Se after Gavage of 75Se-Labeled Selenite
Sepp1c/c/alb-cre+/− and Sepp1c/c mice were gavaged with 10 μCi of 75Se-labeled selenite in 0.15 m NaCl. They were individually housed in metabolic cages. After 24 h, the mice were removed from the cages, and urine and feces were collected. The mice were anesthetized and exsanguinated by removal of blood from the inferior vena cava. The liver was removed. 75Se was determined in urine and liver using a γ-counter (PerkinElmer Life Sciences model 1480 Wizard 3″ gamma counter, Shelton, CT).
Collection of Spermatozoa
The cauda of each epididymis was minced in PBS. An aliquot of the sperm suspension was added to 4% formaldehyde in 0.1 m phosphate buffer, pH 7.6. Sperm counts were performed in a hemocytometer. When the spermatozoa were counted, each was noted to be either normal or kinked at the midpiece-principal piece junction.
In Situ Hybridization
Nonisotopic in situ hybridization was performed as described previously (6) using digoxigenin-labeled sense and antisense Sepp1 riboprobes and formaldehyde-fixed cryosections of mouse liver. A 641-bp mSEPP1 cDNA was prepared by PCR using MB23A1 plasmid (12) and primers 5′-AGCCAGCTGATACTTGTGTCTTCTGCAGGCAT-3′ and 5′-AAAGGTGCAAGCCTTCACTTGCTGTGGTGT-3′. The PCR product was gel-purified and ligated into the pCR4 Topo plasmid. Following transformation into OneShot Top10 cells, individual clones were analyzed by PCR to identify the orientation of the mSEPP1 cDNA insert. Clones with opposite insert orientation were used to prepare template DNA by PCR, which included the plasmid T7 promoter and the mSepp1 insert. The purified PCR products were used in transcription reactions to prepare sense and antisense digoxigenin-labeled riboprobes as described previously (6).
RT-PCR
Frozen tissue was pulverized under liquid nitrogen and then treated with TRIzol reagent. Total RNA was isolated according to the manufacturer's protocol. RNA was further purified on an RNeasy mini column following the manufacturer's instructions. RNA concentration was determined by measurement of A260. cDNA was prepared using high capacity cDNA reverse transcriptase kit following the manufacturer's instructions. 100 ng of total RNA was used per 20 μl of cDNA synthesis reaction. Quantitative PCR for gene expression was performed using Power SYBR Green PCR master mix with 1 μl of cDNA in a total reaction volume of 20 μl. Gene-specific primers were used at a final concentration of 250 nm. StepOnePlus RT-PCR system and StepOne software Version 2.1 (Applied Biosystems) were used to collect and analyze data. Three replicates of each sample were amplified. Relative quantitation of RNA levels was determined by comparative CT reactions (ΔΔCT analysis). Primers used for amplification of Sepp1, Gpx1, and hypoxanthine-guanine phosphoribosyltransferase are listed in Table 1. Hypoxanthine-guanine phosphoribosyltransferase served as the endogenous control. The target mRNA quantity in each tissue was expressed in arbitrary units (relative quantitation).
Statistics
Statistical comparisons between groups were made on an iMac using Prism 4 for Macintosh Version 4.0c software program (GraphPad Software, Inc.). Tukey's Multiple Comparison Test was applied after analysis by one-way analysis of variance. Where appropriate, Student's t test was used to compare groups. These groups were considered to be significantly different with p < 0.05.
RESULTS
The Hepatocyte Is the Predominant, but Not Exclusive, Source of Plasma Sepp1
Fig. 2 demonstrates that liver has the greatest relative amount of Sepp1 mRNA of the tissues we examined in Sepp1c/c mice. Kidney ranked second with 38% of the liver level. Skeletal muscle, heart, and testis followed with 10, 6, and 6%, respectively. Sepp1 mRNA was present in whole brain, but at less than 2% of the liver level. These results confirm a report by others that Sepp1 is expressed in many tissues (11).
FIGURE 2.

Expression of Sepp1 mRNA in tissues from mice with deletion of hepatocyte Sepp1 (Sepp1c/c/alb-cre+/− mice) and in floxed (Sepp1c/c) controls. Values are means with 1 S.D. indicated by a bracket, n = 3–4. The effect of gene deletion was assessed in each tissue by Student's t test. The only significant difference (p < 0.05) was in liver, in which the deleted value was 2% of the control. RQ, relative quantitation.
Sepp1c/c mice were bred with alb-cre+/− mice to produce Sepp1c/c/alb-cre+/− mice that had Sepp1 deleted in hepatocytes. Liver Sepp1 mRNA fell to 2% in those mice, but Sepp1 mRNA did not change significantly in the other tissues tested (Fig. 2). Those results are consistent with Sepp1c/c/alb-cre+/− mice having Sepp1 deleted in hepatocytes but not in nonhepatocyte liver cells or in extra-hepatic tissues.
Several populations of hepatocytes are present in the liver. Peri-portal hepatocytes are the first to encounter blood from the portal vein and thus receive greater amounts of oxygen and absorbed nutrients than do peri-central hepatocytes. In situ hybridization revealed a strikingly zonal distribution of Sepp1 mRNA in liver with high staining in the peri-portal zone and low to undetectable staining in the peri-central zone (Fig. 3A). No staining was detectable in liver from a Sepp1−/− mouse (Fig. 3B).
FIGURE 3.

In situ hybridization showing Sepp1 mRNA (dark stain) in mouse liver. Portal veins are indicated by filled squares and central veins by asterisks. Mice had been fed a diet supplemented with 0.25 mg of selenium/kg. A depicts liver from a C57BL/6 mouse and B depicts liver from a congenic Sepp1−/− mouse.
Selective deletion of Sepp1 in hepatocytes lowered plasma Sepp1 concentration in mice fed a selenium-adequate diet (supplemented with 0.25 mg of selenium/kg) to 4% of the value in Sepp1c/c mice fed the same diet (Fig. 4A). The dietary selenium concentration that is needed to maximize selenoprotein levels in wild-type mice is 0.10–0.15 mg/kg (22). In a separate experiment (Fig. 4B), increasing dietary selenium supplementation from 0.25 to 1 mg/kg produced a small, but significant, rise in plasma Sepp1 from 4% of the Sepp1c/c value in the experiment depicted in Fig. 4A to 10% of it, indicating that selenium supply to some nonhepatocyte Sepp1-producing tissues was not adequate in Sepp1c/c/alb-cre+/− mice fed the diet supplemented with 0.25 mg of selenium/kg. An additional increase in dietary selenium supplementation to 4 mg/kg did not raise plasma Sepp1 concentration further. These findings indicate that nonhepatocyte sources supply ∼10% of plasma Sepp1 and imply that hepatocyte-produced Sepp1 provides selenium to other tissues that are also sources of plasma Sepp1.
FIGURE 4.

Effect of dietary selenium on plasma selenium biomarkers in Sepp1c/c/alb-cre+/− mice. A depicts plasma selenium biomarkers in Sepp1c/c/alb-cre+/− (n = 5) and Sepp1c/c (n = 4) mice that had been fed a diet supplemented with 0.25 mg of selenium/kg for 4 weeks beginning at weaning. B depicts plasma selenium biomarkers in mice fed diets supplemented with 0.25, 1, and 4 mg of selenium/kg. In that experiment, mice that had been fed 0.25 mg of selenium/kg diet for 1–2 months beginning at weaning were used for the study. One group (n = 4) continued to be fed the same diet, and two other groups (n = 5 in each group) were switched to the 1 and 4 mg/kg diets for 4 weeks before plasma was obtained. Values in both panels are means with 1 S.D. indicated by a bracket. Percentages in A indicate that values were significantly different (p < 0.05) by Student's t test. Asterisks in B indicate that values were different from the preceding value (p < 0.05) by Tukey's multiple comparison test.
The plasma Gpx activity in Sepp1c/c/alb-cre+/− mice fed the diet supplemented with 0.25 mg of selenium/kg was very low (Fig. 4A). Increasing dietary selenium supplementation to 1 mg/kg and then to 4 mg/kg raised plasma Gpx activity progressively (Fig. 4B). These results strongly suggest that non-Sepp1 selenium forms, likely of low molecular weight, can replace Sepp1 as a source of selenium for Gpx3 synthesis when selenium intake is high. Plasma selenium concentrations in Sepp1c/c/alb-cre+/− mice fed different amounts of selenium reflected the levels of the two selenoproteins (Fig. 4B).
The presence of plasma selenoproteins was further evaluated using autoradiography of an SDS-polyacrylamide gel (Fig. 5). The Sepp1c/c lane contained two 75Se bands as has been observed in wild-type mice (23). The darker band at 50 kDa represents Sepp1, and the lighter one at 23 kDa represents Gpx3. A 50-kDa 75Se band was present in the Sepp1c/c/alb-cre+/− lane in Fig. 5, consistent with Sepp1 being present in Sepp1c/c/alb-cre+/− mouse plasma. The migrations of Sepp1 and Gpx3 were similar in Sepp1c/c and Sepp1c/c/alb-cre+/− plasma, although the relative amounts of 75Se in the bands were reversed. Thus, Sepp1 is present in plasma from Sepp1c/c/alb-cre+/− mice, although at a concentration of 10% or less that in Sepp1c/c mice (Fig. 4).
FIGURE 5.
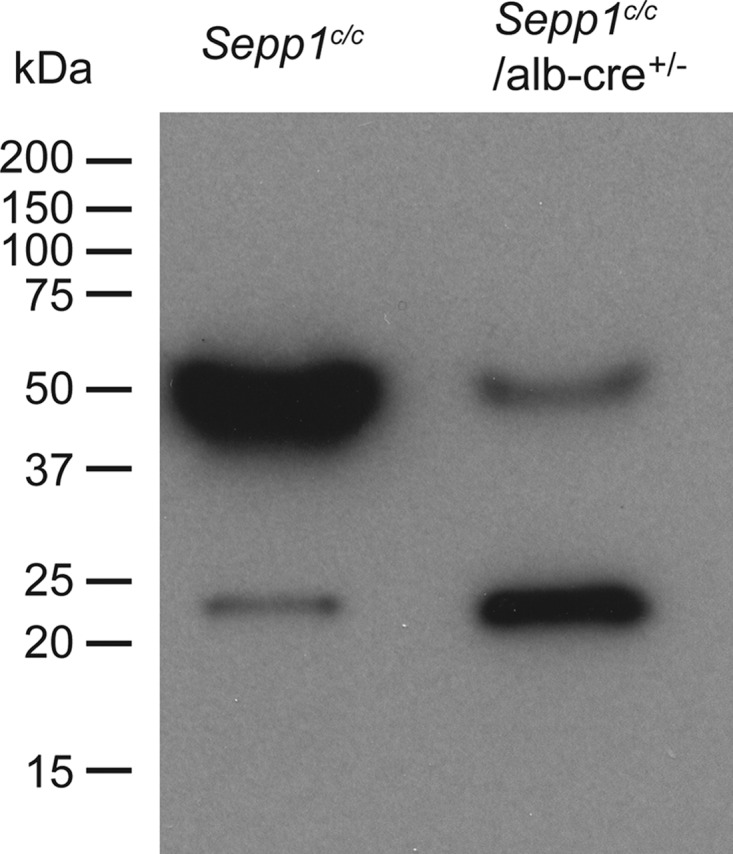
Effect of hepatocyte Sepp1 deletion on the presence of plasma selenoproteins. Mice fed the diet supplemented with 0.25 mg of selenium/kg were injected intraperitoneally with 10 μCi of 75Se-labeled selenite, and plasma was obtained 4 h later. After SDS-PAGE had been performed, the gel was dried and used for autoradiography. Migrations of protein standards are indicated to the left. Sepp1 migrates at 50 kDa and Gpx3 migrates at 23 kDa (25).
Hepatocyte Sepp1 Synthesis Promotes Selenium Retention in the Body and Transfer from the Liver to Extra-hepatic Tissues
Deletion of Sepp1 in the whole mouse increases urinary selenium excretion, thereby decreasing whole-body selenium (14). To explain these findings, we hypothesized that synthesis of Sepp1 competes for metabolically available selenium in hepatocytes with synthesis of excretory selenium metabolites. Thus, deleting hepatocyte Sepp1 should make more selenium available for the excretory pathway. We tested this hypothesis by comparing selenium metabolism in Sepp1c/c/alb-cre+/− and Sepp1c/c mice.
An experiment was designed to determine the fate of newly administered, and thus metabolically available, selenium. Mice fed a selenium-adequate diet (supplemented with 0.15 mg of selenium/kg) received 75Se-labeled selenite by gavage to trace metabolically available selenium. Urine was collected for 24 h, and then the mice were sacrificed. Fig. 6A shows that deletion of hepatocyte Sepp1 more than doubled 75Se excretion in urine. Liver 75Se was 25% higher in the mice with deletion of Sepp1 than in their controls, likely reflecting increased incorporation of metabolically available selenium into liver selenoproteins (see Scheme 1). These results are consistent with there being competition for 75Se between synthesis of Sepp1 and synthesis of excretory metabolites. In addition, they suggest that there is competition for metabolically available selenium between synthesis of Sepp1 and synthesis of liver intracellular selenoproteins.
FIGURE 6.
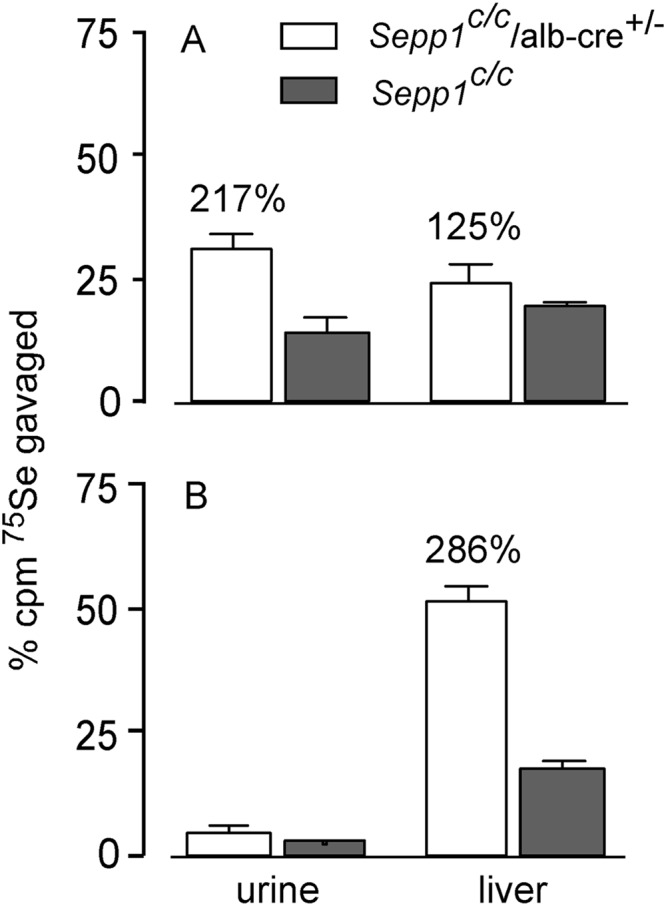
Effect of hepatocyte Sepp1 deletion on urinary excretion and liver incorporation of gavaged 75Se administered as 75Se-labeled selenite. A shows results in mice fed a diet supplemented with 0.15 mg of selenium/kg for 5 weeks beginning at weaning, and B shows results in mice fed a selenium-deficient diet for 20 weeks beginning at weaning. Values are means with 1 S.D. indicated by the bracket, n = 4–6. Percentage differences are given for pairs of values that were significantly different from each other by Student's t test (p < 0.05).
SCHEME 1.

Proposed fate of metabolically available selenium (‘Se’) in the hepatocyte. Hepatocytes have several sources of selenium that are immediately available for further metabolism. Synthesis of sec-tRNA[ser]sec ① competes for selenium with methylation reactions ② that produce excretory metabolites. Synthesis of Sepp1 ③ for export to the plasma competes for sec-tRNA[ser]sec with synthesis of intracellular selenoproteins ④. Liver selenoproteins turn over ⑤ to release selenium. The asterisks on steps ① and ③ indicate the path of selenium favored under conditions of selenium deficiency.
When the same experiment was carried out in selenium-deficient mice, the results were somewhat different (Fig. 6B). Urinary 75Se excretion was very low and not significantly different between mouse groups. These findings are consistent with synthesis of sec-tRNA[ser]sec out-competing synthesis of excretory metabolites for metabolically available selenium under selenium-deficient conditions, even when Sepp1 has been deleted. Liver 75Se was sharply higher in the mice with Sepp1 deleted, likely caused by diversion of sec-tRNA[ser]sec from synthesis of Sepp1 to synthesis of intracellular selenoproteins (Scheme 1). These results indicate that the pathways to synthesis of selenoproteins (sec-tRNA[ser]sec) and to excretory metabolites compete for metabolically available selenium in the hepatocyte and that synthesis of Sepp1 competes with synthesis of other selenoproteins for sec-tRNA[ser]sec.
Gpx1 contains 53% of liver selenium in selenium-replete C57BL/6 mice (Fig. 7A), and its synthesis would therefore be expected to utilize a large fraction of the sec-tRNA[ser]sec produced in that organ. Because synthesis of Gpx1 would compete for sec-tRNA[ser]sec with synthesis of Sepp1, we assessed expression of their mRNAs under selenium-adequate and selenium-deficient conditions in C57BL/6 mice. In confirmation of earlier reports (22), selenium-deficient liver Gpx1 mRNA was only 15% of that in selenium-replete controls, whereas Sepp1 mRNA only fell to 61% under the same conditions (Fig. 7B). These results suggest that utilization of selenium is shifted from synthesis of Gpx1 to synthesis of Sepp1 under selenium-deficient conditions, enhancing the supply of selenium to extra-hepatic tissues when the element is in short supply.
FIGURE 7.

Comparison of Gpx1 and Sepp1 in the liver. A depicts liver and whole-body selenium concentrations of Gpx1−/− and Gpx1+/+ mice fed a diet supplemented with 0.25 mg of selenium/kg for 4 weeks beginning at weaning. B depicts relative Gpx1 and Sepp1 mRNA in livers of mice fed selenium-deficient or selenium-adequate (0.25 mg of selenium/kg) diets for 16 weeks beginning at weaning. Values are means with 1 S.D. indicated by a bracket (n = 5). All pairs were significantly different by Student's t test (p < 0.05).
The effect of hepatocyte Sepp1 deletion on tissue selenium concentrations was determined. Deletion of Sepp1 in hepatocytes decreased whole-body selenium concentration to 58% of the Sepp1c/c value in mice fed our usual selenium-adequate diet supplemented with 0.25 mg of selenium/kg (Fig. 8A), consistent with increased urinary excretion of metabolically available selenium (Fig. 6A). In accord with the decrease in whole-body selenium, its concentrations in tissues were also decreased in Sepp1c/c/alb-cre+/− mice, but to varying degrees. Liver maintained its selenium better than other tissues, probably because deletion of Sepp1 allowed liver intracellular selenoproteins a greater supply of sec-tRNA[ser]sec (Scheme 1). Brain and testis retained selenium better than kidney and muscle, presumably because they have higher expressions of apoER2, which is responsible for endocytosis of plasma Sepp1 (5).
FIGURE 8.

Effect of hepatocyte Sepp1 deletion on tissue and whole-body selenium concentrations in selenium-adequate (A) and selenium-deficient (B) mice. Selenium-adequate mice were fed a diet supplemented with 0.25 mg of selenium/kg for 4 weeks beginning at weaning. Selenium-deficient mice were fed a selenium-deficient diet for 12 weeks beginning at weaning. Values are means with the bracket indicating 1 S.D. (n = 3–4). Values in every pair were significantly different from each other (p < 0.05) by Student's t test. Percentages indicate the differences. Plasma biomarker values of the selenium-adequate animals (A) are shown in Fig. 4A. Plasma Sepp1 was not detectable (<0.5 mg/liter) in any of the four selenium-deficient Sepp1c/c/alb-cre+/− mice (B) and was 2.2 ± 0.1 mg/liter, n = 3, in the Sepp1c/c mice.
Under conditions of selenium deficiency, deletion of hepatocyte Sepp1 lowered whole-body and tissue selenium except for that in liver, which was sharply higher than liver selenium in Sepp1c/c mice (Fig. 8B). Strikingly, the liver contained 53% of whole-body selenium in selenium-deficient Sepp1c/c/alb-cre+/− mice but only 8% of whole-body selenium in selenium-deficient Sepp1c/c mice. Thus, most of the selenium not secreted as Sepp1 in these mice was retained in hepatocyte selenoproteins. These results demonstrate the importance of Sepp1 secretion by the liver in supplying extra-hepatic tissues with selenium and maintaining the metabolic relationships outlined in Scheme 1.
Deletion of Hepatocyte Sepp1 Synthesis Worsens Clinical Signs of Dietary Selenium Deficiency
Because incorporation of metabolically available hepatocyte selenium into Sepp1 is a mechanism for retaining selenium in the body and for distributing it to extra-hepatic tissues (Figs. 6 and 8), deletion of hepatocyte Sepp1 would be expected to exacerbate the signs of dietary selenium deficiency. To evaluate this possibility, Sepp1c/c/alb-cre+/− mice and control Sepp1c/c mice were fed selenium-deficient diet beginning at weaning. Both groups gained weight for 12 weeks, at which time Sepp1c/c/alb-cre+/− mice ceased gaining weight, although Sepp1c/c mice continued to gain (Fig. 9).
FIGURE 9.

Body weights of Sepp1c/c/alb-cre+/− (n = 8) and Sepp1c/c (n = 12) mice fed a selenium-deficient diet for 22 weeks beginning at week 0 (weaning). Values are means with 1 S.D. indicated by a bracket. The horizontal line is above the portion of the weight curves in which the pairs of values differ by Student's t test (p < 0.05).
Selenium deficiency causes azoospermia and structural abnormalities of spermatozoa (24). The sperm count in the epididymal cauda of Sepp1c/c/alb-cre+/− mice fed selenium-deficient diet for 24 weeks was 39% of that in C57BL/6 mice fed the same diet (Table 2). Spermatozoa of selenium-deficient mice become “kinked” at the junction of the mid-piece and principal piece (25). Nearly all the spermatozoa of the selenium-deficient Sepp1c/c/alb-cre+/− mice were kinked in this manner, and many fewer of the spermatozoa of the control mice were so kinked (Fig 10 and Table 2).
TABLE 2.
Sperm counts and morphology in Sepp1c/c/Alb-cre+/− mice fed a selenium-deficient diet for 24 weeks beginning at weaning
| n | Sepp1c/c/Alb-cre+/− micea | C57BL/6 micea | |
|---|---|---|---|
| Sperm count (×106/cauda epididymis) | 5 | 17 ± 6.7b | 43 ± 8.3b |
| Percentage of spermatozoa with kinks | 5 | 95 ± 2.5c | 37 ± 5.5c |
a Values are means ± 1 S.D.
b,c Values with the same superscript are different (p < 0.05) by Student's t test.
FIGURE 10.

Effect of feeding selenium-deficient diet for 24 weeks on the morphology of spermatozoa in the cauda epididymis from C57BL/6 (A) and Sepp1c/c/alb-cre+/− (B) mice. Arrows indicate kinks in spermatozoa at the junction of the midpiece and the principal piece.
The brain is better protected against selenium deficiency than any other tissue (26). However, in Sepp1c/c/alb-cre+/− mice fed a selenium-deficient diet for 12 weeks, brain selenium concentration fell to 37 ± 7 ng/g (n = 4), 48% of the concentration in control Sepp1c/c mice fed the same diet, and 29% of Sepp1c/c mice fed the selenium-adequate diet supplemented with 0.25 mg selenium/kg (Fig. 8B). Thus, deletion of hepatocyte Sepp1 reduced the ability of the brain to maintain its selenium in selenium deficiency.
Beginning 16 weeks after weaning, the Sepp1c/c/alb-cre+/− mice fed selenium-deficient diet began to curl their hind limbs when lifted by the tail (Fig 11A). This finding was described by others in wild-type mice only after they had been fed selenium-deficient diet through three generations (27). In addition, the mice developed wide-based gait in their hind limbs (Fig 11B) beginning 22 weeks after weaning. The hind limbs remained impaired until the observation period was terminated at 80 weeks, but this disability did not worsen significantly and the front limbs remained unaffected. The Sepp1c/c/alb-cre+/− mice fed a selenium-deficient diet for 80 weeks were still able to move about; they did not develop the severe neurological dysfunction observed in Sepp1−/− mice fed a selenium-deficient diet for only 2 weeks (12). Thus, in addition to decreasing the selenium concentration in all tissues except liver, deletion of hepatocyte Sepp1 worsened the clinical signs of selenium deficiency in mice fed a selenium-deficient diet.
FIGURE 11.
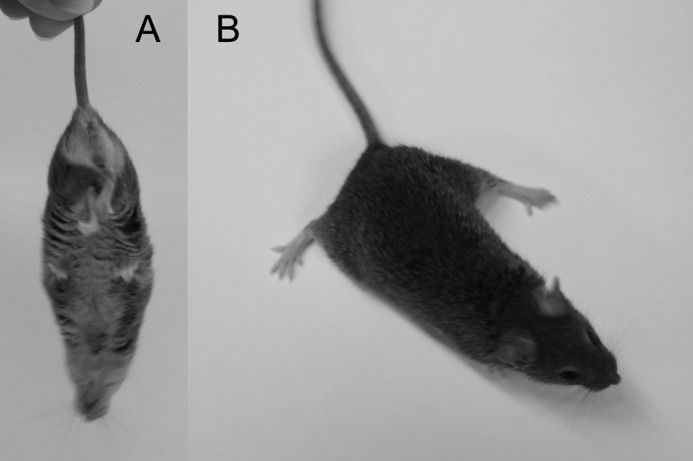
Selenium deficiency affects hind limbs and gait of Sepp1c/c/alb-cre+/− mice. Mice were fed a selenium-deficient diet for 61 weeks beginning at weaning. A shows curling of hind limbs when the mouse was picked up by the tail. B shows splaying of hind limbs but not fore limbs.
DISCUSSION
The results presented here demonstrate that 90% of plasma Sepp1 originates in hepatocytes and that deletion of hepatocyte Sepp1 leads to major changes in whole-body selenium metabolism. Those changes in metabolism impair selenium supply to extra-hepatic tissues and worsen dietary selenium deficiency.
The liver is well suited to regulate whole-body selenium metabolism because it has a greater supply of the element than do other tissues. Portal vein blood transports absorbed selenium directly to the liver in small molecule form (9). Selenomethionine, the major dietary form of selenium, is catabolized in the liver (28, 29), freeing its selenium for further metabolism in the hepatocyte.
Fig. 6 indicates that the liver apportions this rich supply of selenium between selenoprotein synthesis and synthesis of excretory metabolites. This regulation likely occurs at the biochemical level of selenide, the product of selenocysteine catabolism by selenocysteine lyase (28). Selenide can be metabolized to selenophosphate, an immediate precursor of sec-tRNA[ser]sec, by selenophosphate synthetase (30) or it can be methylated to produce excretory metabolites (2, 31).
An early study in rats demonstrated a selenium intake threshold of 0.054 mg of selenium/kg diet below which urinary excretion of metabolically available selenium did not respond to changes in dietary selenium intake (32). However, tissue distribution of selenium and selenoprotein synthesis both responded to dietary selenium levels below that threshold (32, 33). Thus, Fig. 6B and these earlier reports suggest that metabolically available selenium is directed primarily to synthesis of sec-tRNA[ser]sec at very low selenium intakes, and only when intake rises above a “threshold” is selenium also used for the production of excretory metabolites as well. The molecular mechanism of this regulation is not known. However, it promotes selenoprotein synthesis in the hepatocyte and thereby protects the organism against selenium deficiency.
Once sec-tRNA[ser]sec has been produced in the liver, it is used for synthesis of selenoproteins, either Sepp1 for export or selenoproteins that remain in the hepatocyte (Scheme 1). Deletion of Sepp1 in hepatocytes diverted administered 75Se to the intracellular selenoproteins, especially under selenium-deficient conditions (Fig. 6B). As a consequence of this diversion, 53% of whole-body selenium was retained in the livers of selenium-deficient Sepp1c/c/alb-cre+/− mice compared with only 8% in selenium-deficient Sepp1c/c mice that were able to synthesize Sepp1 and thereby export hepatic selenium to other tissues (Fig. 8B). These results demonstrate that the production of Sepp1 by the liver is a mechanism for distributing selenium to extra-hepatic tissues, especially under selenium-deficient conditions.
A further mechanism for ensuring selenium supply to extra-hepatic tissues is the down-regulation of hepatic Gpx1 mRNA under selenium-deficient conditions. Gpx1 contains 53% of hepatic selenium under selenium-adequate conditions (Fig. 7A). In selenium-deficient liver, Gpx1 mRNA fell sharply relative to Sepp1 mRNA (Fig. 7B). A detailed study of most of the mouse liver selenoproteome has shown that in selenium deficiency the mRNAs of three liver selenoproteins (including Gpx1) fall sharply; five (including Sepp1) decrease moderately; and nine remain unchanged (34). These changes in mRNA levels presumably allow a greater amount of the metabolically available selenium in liver to be incorporated into Sepp1 for supply of the element to extra-hepatic tissues while maintaining vital intracellular selenoproteins. Other mechanisms of differentially regulating translation of selenoproteins are under investigation, but no results have yet been presented that are relevant to hepatic Sepp1 production (35). Thus, available results suggest that supply of selenium by the liver to other tissues is regulated at the level of apportionment of sec-tRNA[ser]sec between synthesis of Sepp1 and synthesis of intracellular selenoproteins as well as at the level of competition between selenium excretion and sec-tRNA[ser]sec synthesis.
Sepp1 is not the only transport form of selenium. Sepp1−/− mice are viable when fed a high selenium diet (12). The other plasma selenoprotein, Gpx3, does not appear to transport selenium for specific uptake by cells (5), and mice with both Sepp1 and Gpx3 deleted are viable (23). Thus, the non-Sepp1 transport form appears to be a small molecule.
This small molecule form (or forms) of selenium has been detected but not characterized (9, 36). When compared with the Sepp1-apoER2 pathway, the small molecule pathway appears to lack the ability to distinguish between high need tissues and low need tissues. Moreover, to be effective, the small molecule pathway requires a much higher selenium intake than does the Sepp1-apoER2 pathway (12). Thus, this second tier pathway of selenium transport appears to be nonspecific and to be less effective than the Sepp1-apoER2 pathway.
Until 10 years ago, selenium deficiency could only be produced by feeding selenium-deficient diet. The major clinical sign of selenium deficiency in mice was male infertility caused by abnormal spermatozoa (27). Only when fed a selenium-deficient diet through three generations did mice develop a mild neurological abnormality (27). Thus, wild-type mice tolerate severe nutritional selenium deficiency with relatively mild clinical impairment, aside from male infertility.
Ten years ago, Sepp1−/− mice were produced and were observed to have abnormal spermatozoa and severe neurological injury leading to death when fed a diet considered to be adequate in selenium for wild-type mice (12, 13). Feeding a high selenium diet (1 mg of selenium/kg diet) to Sepp1−/− mice prevented overt neurological injury and allowed a long life span, but it did not completely prevent neuropathological abnormalities (37). Neither did it completely correct the sperm abnormalities (25). We interpret this to indicate that the small molecule selenium could largely replace Sepp1 in supplying the brain with selenium but that it did not meet the apparently higher selenium demands of the testis.
Other groups have studied mice with selective deletion of Trsp (the gene for tRNA[ser]sec) in hepatocytes (10, 38). Those mice were unable to synthesize any selenoproteins in their hepatocytes. Brain selenium concentration was not affected by loss of hepatocyte selenoprotein synthesis. The plasma Sepp1 concentration was shown by Western blot to be sharply decreased, but it was not quantified in those studies. This study demonstrates that up to 10% of plasma Sepp1 is produced by extra-hepatocyte tissues (Fig. 4). Severe selenium deficiency in wild-type mice lowers plasma Sepp1 concentration to less than 10% without producing neurological signs (26). Thus, it is not surprising that mice with deletion of Sepp1 or of Trsp in hepatocytes that are fed selenium-adequate diet maintain their brain selenium (Fig. 8A) and do not develop neurological abnormalities.
When fed a selenium-deficient diet, however, Sepp1c/c/alb-cre+/− mice developed signs of selenium deficiency not seen in selenium-deficient wild-type mice. In addition to developing abnormal spermatozoa, they ceased gaining weight 12 weeks post-weaning (Fig. 9) and began losing strength in their hind limbs at 22 weeks (Fig 10). Plasma Sepp1 was undetectable (<2% of Sepp1 in wild-type selenium-adequate mice) in Sepp1c/c/alb-cre+/− mice fed a selenium-deficient diet for 12 weeks, although it was detectable at 8% in Sepp1c/c mice under the same conditions (Fig. 8 legend). The observation that clinical signs did not worsen further after 22 weeks suggests the establishment of a new selenium steady state at a very low intake (dietary selenium concentration of 0.006 mg/kg). Thus, mice with deletion of hepatic Sepp1 can be used as a model for producing extremely severe dietary selenium deficiency in all tissues with the exception of the liver.
Sepp1 is expressed within the brain by astrocytes and likely by other cells (39, 40). The Sepp1 receptor apoER2 is present on neurons (41), so it is possible that Sepp1 functions to maintain appropriate distribution of selenium among cells in the brain as it does among tissues in the body (Fig. 8). Supporting this possibility is the very low selenium concentration in the brains of selenium-deficient Sepp1c/c/alb-cre+/− mice (37 ng/g) with no overt neurological signs (Fig. 8B). Sepp1−/− mice fed a selenium-deficient diet for 1–2 weeks have essentially the same brain selenium level but have severe neurological abnormalities.4 Thus, in mice with very low brain selenium concentrations, Sepp1 expression in brain protects against brain injury. This strongly suggests that Sepp1 produced in the brain provides selenium to high need brain cells. However, it does not rule out other, perhaps enzymatic, protective functions of Sepp1 in the brain.
In conclusion, this study demonstrates that the process of incorporating metabolically available selenium into Sepp1 in the hepatocyte is central to selenium homeostasis in the mouse. As selenium supply becomes limited, alternative routes of its hepatic metabolism (production of excretory metabolites and of intracellular selenoproteins) diminish, allowing continued Sepp1 synthesis for export into the plasma. The Sepp1 in plasma provides selenium to extra-hepatic tissues via apoER2-mediated endocytosis (5), thus protecting against selenium deficiency in them.
This work was supported, in whole or in part, by National Institutes of Health Grants R37 ES002497, 5P30 DK058404, and 5P30 ES000267.
R. F. Burk, manuscript in preparation.
- Sepp1
- selenoprotein P
- apoER2
- apolipoprotein E receptor-2
- Gpx
- glutathione peroxidase
- FRT
- flippase recognition target
- Flp
- flippase recombination enzyme
- Sepp1c/c
- floxed Sepp1
- Sepp1c/c/alb-cre+/−
- conditional deletion of Sepp1 in hepatocytes
- sec-tRNA[ser]sec
- tRNA for selenocysteine charged with selenocysteine
- trsp
- gene for tRNA[ser]sec.
REFERENCES
- 1. Kryukov G. V., Castellano S., Novoselov S. V., Lobanov A. V., Zehtab O., Guigó R., Gladyshev V. N. (2003) Characterization of mammalian selenoproteomes. Science 300, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 2. Kobayashi Y., Ogra Y., Ishiwata K., Takayama H., Aimi N., Suzuki K. T. (2002) Selenosugars are key and urinary metabolites for selenium excretion within the required to low-toxic range. Proc. Natl. Acad. Sci. U.S.A. 99, 15932–15936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burk R. F., Hill K. E. (2009) Selenoprotein P-expression, functions, and roles in mammals. Biochim. Biophys. Acta 1790, 1441–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma S., Hill K. E., Caprioli R. M., Burk R. F. (2002) Mass spectrometric characterization of full-length rat selenoprotein P and three isoforms shortened at the C terminus. Evidence that three UGA codons in the mRNA open reading frame have alternative functions of specifying selenocysteine insertion or translation termination. J. Biol. Chem. 277, 12749–12754 [DOI] [PubMed] [Google Scholar]
- 5. Kurokawa S., Hill K. E., McDonald W. H., Burk R. F. (2012) Long isoform mouse selenoprotein P (Sepp1) supplies rat myoblast L8 cells with selenium via endocytosis mediated by heparin binding properties and apolipoprotein E receptor-2 (ApoER2). J. Biol. Chem. 287, 28717–28726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olson G. E., Winfrey V. P., Nagdas S. K., Hill K. E., Burk R. F. (2007) Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 282, 12290–12297 [DOI] [PubMed] [Google Scholar]
- 7. Chiu-Ugalde J., Theilig F., Behrends T., Drebes J., Sieland C., Subbarayal P., Köhrle J., Hammes A., Schomburg L., Schweizer U. (2010) Mutation of megalin leads to urinary loss of selenoprotein P and selenium deficiency in serum, liver, kidneys, and brain. Biochem. J. 431, 103–111 [DOI] [PubMed] [Google Scholar]
- 8. Olson G. E., Winfrey V. P., Hill K. E., Burk R. F. (2008) Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J. Biol. Chem. 283, 6854–6860 [DOI] [PubMed] [Google Scholar]
- 9. Kato T., Read R., Rozga J., Burk R. F. (1992) Evidence for intestinal release of absorbed selenium in a form with high hepatic extraction. Am. J. Physiol. 262, G854–G858 [DOI] [PubMed] [Google Scholar]
- 10. Schweizer U., Streckfuss F., Pelt P., Carlson B. A., Hatfield D. L., Köhrle J., Schomburg L. (2005) Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem. J. 386, 221–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann P. R., Höge S. C., Li P. A., Hoffmann F. W., Hashimoto A. C., Berry M. J. (2007) The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. 35, 3963–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Atkins J. F., Gesteland R. F., Burk R. F. (2003) Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646 [DOI] [PubMed] [Google Scholar]
- 13. Schomburg L., Schweizer U., Holtmann B., Flohé L., Sendtner M., Köhrle J. (2003) Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370, 397–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burk R. F., Hill K. E., Motley A. K., Austin L. M., Norsworthy B. K. (2006) Deletion of selenoprotein P upregulates urinary selenium excretion and depresses whole-body selenium content. Biochim. Biophys. Acta 1760, 1789–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Burk R. F. (2004) Neurological dysfunction occurs in mice with targeted deletion of selenoprotein P gene. J. Nutr. 134, 157–161 [DOI] [PubMed] [Google Scholar]
- 16. Wu S., Ying G., Wu Q., Capecchi M. R. (2008) A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nat. Protoc. 3, 1056–1076 [DOI] [PubMed] [Google Scholar]
- 17. Ho Y. S., Magnenat J. L., Bronson R. T., Cao J., Gargano M., Sugawara M., Funk C. D. (1997) Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 272, 16644–16651 [DOI] [PubMed] [Google Scholar]
- 18. Koh T. S., Benson T. H. (1983) Critical re-appraisal of fluorometric method for determination of selenium in biological materials. J. Assoc. Off. Anal. Chem. 66, 918–926 [PubMed] [Google Scholar]
- 19. Sheehan T. M., Gao M. (1990) Simplified fluorometric assay of total selenium in plasma and urine. Clin. Chem. 36, 2124–2126 [PubMed] [Google Scholar]
- 20. Hill K. E., Zhou J., Austin L. M., Motley A. K., Ham A. J., Olson G. E., Atkins J. F., Gesteland R. F., Burk R. F. (2007) The selenium-rich C-terminal domain of mouse selenoprotein P is necessary for supply of selenium to brain and testis but not for maintenance of whole-body selenium. J. Biol. Chem. 282, 10972–10980 [DOI] [PubMed] [Google Scholar]
- 21. Lawrence R. A., Burk R. F. (1976) Glutathione peroxidase activity in selenium-deficient rat liver. Biochem. Biophys. Res. Commun. 71, 952–958 [DOI] [PubMed] [Google Scholar]
- 22. Sunde R. A., Raines A. M. (2011) Selenium regulation of the selenoprotein and nonselenoprotein transcriptomes in rodents. Adv. Nutr. 2, 138–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olson G. E., Whitin J. C., Hill K. E., Winfrey V. P., Motley A. K., Austin L. M., Deal J., Cohen H. J., Burk R. F. (2010) Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol. Renal Physiol. 298, F1244–F1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gunn S. A., Gould T. C., Anderson W. A. (1967) Incorporation of selenium into spermatogenic pathway in mice. Proc. Soc. Exp. Biol. Med. 124, 1260–1263 [DOI] [PubMed] [Google Scholar]
- 25. Olson G. E., Winfrey V. P., Nagdas S. K., Hill K. E., Burk R. F. (2005) Selenoprotein P is required for mouse sperm development. Biol. Reprod. 73, 201–211 [DOI] [PubMed] [Google Scholar]
- 26. Nakayama A., Hill K. E., Austin L. M., Motley A. K., Burk R. F. (2007) All regions of mouse brain are dependent on selenoprotein P for maintenance of selenium. J. Nutr. 137, 690–693 [DOI] [PubMed] [Google Scholar]
- 27. Wallace E., Calvin H., Cooper G. (1983) Progressive defects observed in mouse sperm during the course of three generations of selenium deficiency. Gamete Res. 7, 377–387 [Google Scholar]
- 28. Esaki N., Nakamura T., Tanaka H., Soda K. (1982) Selenocysteine lyase, a novel enzyme that specifically acts on selenocysteine. Mammalian distribution and purification and properties of pig liver enzyme. J. Biol. Chem. 257, 4386–4391 [PubMed] [Google Scholar]
- 29. Okuno T., Ueno H., Nakamuro K. (2006) Cystathionine γ-lyase contributes to selenomethionine detoxification and cytosolic glutathione peroxidase biosynthesis in mouse liver. Biol. Trace Elem. Res. 109, 155–171 [DOI] [PubMed] [Google Scholar]
- 30. Guimarães M. J., Peterson D., Vicari A., Cocks B. G., Copeland N. G., Gilbert D. J., Jenkins N. A., Ferrick D. A., Kastelein R. A., Bazan J. F., Zlotnik A. (1996) Identification of a novel selD homolog from eukaryotes, bacteria, and archaea: Is there an autoregulatory mechanism in selenocysteine metabolism? Proc. Natl. Acad. Sci. U.S.A. 93, 15086–15091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McConnell K. P., Portman O. W. (1952) Excretion of dimethyl selenide by the rat. J. Biol. Chem. 195, 277–282 [PubMed] [Google Scholar]
- 32. Burk R. F., Seely R. J., Kiker K. W. (1973) Selenium: Dietary threshold for urinary excretion in the rat. Proc. Soc. Exp. Biol. Med. 142, 214–216 [DOI] [PubMed] [Google Scholar]
- 33. Yang J. G., Hill K. E., Burk R. F. (1989) Dietary selenium intake controls rat plasma selenoprotein P concentration. J. Nutr. 119, 1010–1012 [DOI] [PubMed] [Google Scholar]
- 34. Sunde R. A., Raines A. M., Barnes K. M., Evenson J. K. (2009) Selenium status highly regulates selenoprotein mRNA levels for only a subset of the selenoproteins in the selenoproteome. Biosci. Rep. 29, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Budiman M. E., Bubenik J. L., Miniard A. C., Middleton L. M., Gerber C. A., Cash A., Driscoll D. M. (2009) Eukaryotic initiation factor 4a3 is a selenium-regulated RNA-binding protein that selectively inhibits selenocysteine incorporation. Mol. Cell 35, 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shiobara Y., Ogra Y., Suzuki K. T. (1999) Speciation of metabolites of selenate in rats by HPLC-ICP-MS. Analyst 124, 1237–1241 [DOI] [PubMed] [Google Scholar]
- 37. Valentine W. M., Abel T. W., Hill K. E., Austin L. M., Burk R. F. (2008) Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. J. Neuropathol. Exp. Neurol. 67, 68–77 [DOI] [PubMed] [Google Scholar]
- 38. Carlson B. A., Novoselov S. V., Kumaraswamy E., Lee B. J., Anver M. R., Gladyshev V. N., Hatfield D. L. (2004) Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J. Biol. Chem. 279, 8011–8017 [DOI] [PubMed] [Google Scholar]
- 39. Yang X., Hill K. E., Maguire M. J., Burk R. F. (2000) Synthesis and secretion of selenoprotein P by cultured rat astrocytes. Biochim. Biophys. Acta 1474, 390–396 [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y., Zhou Y., Schweizer U., Savaskan N. E., Hua D., Kipnis J., Hatfield D. L., Gladyshev V. N. (2008) Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J. Biol. Chem. 283, 2427–2438 [DOI] [PubMed] [Google Scholar]
- 41. Beffert U., Nematollah Farsian F., Masiulis I., Hammer R. E., Yoon S. O., Giehl K. M., Herz J. (2006) ApoE receptor 2 controls neuronal survival in the adult brain. Curr. Biol. 16, 2446–2452 [DOI] [PubMed] [Google Scholar]