

Background: Consecutive proton stimulation reduces ASIC1a peak currents leading to silencing of channels.
Results: Kinetic analysis using a fast perfusion system shows that human ASIC1a has two desensitized states with markedly different stabilities.
Conclusion: High frequency trains of short stimuli prevent desensitization.
Significance: The results predict steady ASIC1a responses to high frequency release of protons as in synaptic transmission.
Keywords: Acid Sensing Ion Channels (ASIC), Kinetics, Patch Clamp, Perfusion, Synaptic Plasticity, Desensitization, High Frequency Stimulation, Rundown
Abstract
ASIC1a is a neuronal sodium channel activated by external H+ ions. To date, all the characterization of ASIC1a has been conducted applying long H+ stimuli lasting several seconds. Such experimental protocols weaken and even silence ASIC1a currents to repetitive stimulation. In this work, we examined ASIC1a currents by methods that use rapid application and removal of H+. We found that brief H+ stimuli, <100 ms, even if applied at high frequency, prevent desensitization thereby generate full and steady peak currents of human ASIC1a. Kinetic analysis of recovery from desensitization of hASIC1a revealed two desensitized states: short- and long-lasting with time constants of τDs ≤0.5 and τDl = 229 s, while in chicken ASIC1a the two desensitized states have similar values τD 4.5 s. It is the large difference in stability of the two desensitized states that makes hASIC1a desensitization more pronounced and complex than in cASIC1a. Furthermore, recovery from desensitization was unrelated to cytosolic variations in pH, ATP, PIP2, or redox state but was dependent on the hydrophobicity of key residues in the first transmembrane segment (TM1). In conclusion, brief H+-stimuli maintain steady the magnitude of peak currents thereby the ASIC1a channel is well poised to partake in high frequency signals in the brain.
Introduction
Activation of the acid sensing ion channel isoform (ASIC1a)2 by consecutive pulses of external H+ leads to progressive attenuation of the peak current in channels from rat, mouse, and human (1–4). This usage-dependent decrease in channel response, previously referred to as rundown, lasts minutes before it fully recovers in both endogenous ASIC1a from mammalian neurons (5) and recombinant channels expressed in heterologous cell lines: CHO (4) and Xenopus laevis oocytes (3). However, not all mammalian ASIC isoforms or orthologs from other species exhibit it (3, 6, 7); the cause of this variability is also not understood. Since ASIC1a is the isoform most abundant and with the broadest expression in neurons of the mammalian CNS, this phenomenon predicts reduction of the ASIC1a response when more than one isolated stimulus activates the channel in the brain. Such behavior would interfere with the functions attributed to ASIC1, namely modulation of synaptic transmission (8), induction of fear responses (9, 10), and even in the transduction of pain in sensory neurons (11, 12).
Most ligand-activated channels in the brain desensitize in the presence of their respective agonists but mechanisms that remove neurotransmitters in the synaptic cleft and around synapses such as degradation, reuptake, and diffusion minimize desensitization by keeping the exposure brief. In the instance of ASICs, pH fluctuations in microdomains of the brain evoked by neuronal activity have been documented (13). However, the intensity and time course of variations in H+ concentration have not been resolved yet, in large extend because of limits of the techniques used to report rapid pH changes in poorly accessible perisynaptic spaces of the intact brain.
The purpose of this work was to investigate hASIC1a responses to high frequency stimulation and to provide a quantitative analysis of the decrease in peak current magnitude upon repetitive stimuli. The results indicate that hASIC1a exhibits slow desensitization and slow recovery. We demonstrate the existence of two desensitized states with markedly different stability, their contribution to the time course of recovery from desensitization, the role of TM1 in stabilizing the long-lasting desensitized state; lastly, we provide a kinetic model that predicts the attenuation of hASIC1a currents upon repetitive stimulation.
EXPERIMENTAL PROCEDURES
Cell Culture
N1E-115 mouse neuroblastoma and HEK-293 cell lines (both from ATCC) were maintained on DMEM supplemented with 10% fetal bovine serum in an incubator set at 37 °C and 5% CO2. Cells were grown on 25 cm2 flasks and were transferred to acid-washed cover slips pre-coated with 0.1% poly-l-lysine for experiments.
cDNAs and Mutagenesis
The coding region of human ASIC1a cDNA (hASIC1a) was cloned from total RNA extracted from HEK-293 cells using reverse transcriptase (SuperScript RT III, Invitrogen) and polymerase chain reaction amplification with primers corresponding to the first 27 (Forward primer: ATGGAACTGAAGGCCGAGGAGGAGGAG) and last 26 base pairs (Reverse primer: GCAGGTAAAGTCCTCGAACGTGCCTC) of the coding sequence of the human gene ACCN2 (NCBI Reference sequence NM 020039.2). The PCR product was ligated to pCDNA3.1/V5 (Invitrogen) plasmid, and the DNA was sequenced from five independent clones. Chicken ASIC1 (cASIC1) was originally cloned in our laboratory (14). Single or multiple point mutations were introduced using Quickchange (Agilent). Chimeras of human and chicken ASIC1 were made by a two-step approach. First, unique restriction sites were introduced in the junctions to swap domains (MfeI or NdeI). Second, the restriction sites were reverted by site-directed mutagenesis using Quickchange (Agilent) to keep the original amino acids. All constructs and mutations were verified by DNA sequencing.
Isolation and Injection of Xenopus Oocytes
Oocytes were harvested from female Xenopus laevis frogs according to procedures approved by the Institutional Animal Care and Use Committee at Yale University. RNAs were synthesized in vitro using T7 RNA polymerase (mMESSAGEmMACHINE, Agilent). 5 ng of cRNA were injected per oocyte in ≤50 nl. Cells were maintained at 16 °C for 24 to 48 h until used for experiments.
Two-electrode Voltage Clamp (TEVC)
Function and expression level of ASIC1a wild type, chimeras, and mutants were evaluated in injected oocytes by the TEVC (oocyte Clamp OC-725-B, PowerLab 4/30 ADInstruments). Whole-cells currents were elicited by changing the perfusate from a solution of pH 7.4 to 6.5 as previously described (Li et al., 2009). Oocytes expressing ≥20μA of current while the membrane potential was clamped at −60 mV were selected for patch clamp studies.
Patch Clamp Recordings
Patch clamp recordings from oocytes were conducted in excised patches in the outside-out configuration. Patch pipettes were pulled from PG150T glass (Warner Instruments) to tip diameter of 2–4 μm after heat polishing. In outside-out patches the pipette solution was (mm): 120 KCl, 5 EDTA, 20 HEPES titrated with KOH to pH 7.4. Experiments that evaluated the effect of internal pH, the pipette's pH solution was either 7.4 or 6.5 buffered with 30 mm HEPES. When indicated the pipette solution was supplemented with either: 50 μm 1,2-dioctanoyl-sn-glycerol-3-phosphate-(1′-myo-inositol-4,5-bisphosphate (C8 PIP2) (Avanti Polar Lipids, Inc.), 5 mm ATP, or 5 mm dithiothreitol (DTT). Bath and activating solutions were (mm): 120 NaCl, 2 mm KCl, 1 mm CaCl2, 20 HEPES buffered to pH 7.4 or 6.5. Recordings were made using an EPC-9 amplifier and the Pulse acquisition program v8.78 (HEKA Electronic). Membrane potential was held at −30 mV unless indicated. Experiments were conducted at room temperature. Measurements of ASIC currents in N1E-115 and HEK 293 cells were conducted in the whole-cell configuration.
Fast Perfusion
Solutions were applied using a Perfusion Fast Step device SF-77B (Warner Instruments) controlled by the data acquisition program Pulse v8.78. In experiments requiring short perfusion times (milliseconds as shown in Fig. 3) two solution streams, pH 7.4 and 6.5, flowed from a pulled theta-glass capillary with a tip diameter of ∼300 μm to a stationary outside-out patch. The flow velocity of the solutions was 75–150 μm/ms, chosen to form a sharp solution interface while minimizing the chance of breaking the patch. Mechanical vibration artifacts were reduced by decreasing the stepper motor drive voltage, and the residual artifacts were removed by subtracting averaged null traces from the trace with channel activity. After each experiment, the rate of the solution switching was measured by monitoring the current steps resulting from changes in the liquid junction potential. In a typical experiment, the time between the 10 and 90% amplitude points of the junction potential change was 1 ms.
FIGURE 3.

Short stimuli prevent hASIC1a desensitization. A, outside-out patches expressing hASIC1a were activated 50 consecutive times with stimuli of pH 6.5 of duration: 20, 100, and 500 ms followed by recovery for 5 s with pH 7.4. Bars indicate amplitude and time scales. B, plot of normalized peak current from 1 up to 100 consecutive stimuli. C, a train of 8 consecutive stimuli of pH 6.5 for 10 ms followed by 200 ms at pH 7.4.
Data Analysis: Kinetics of Desensitization
The first peak current (PC) and subsequent ones were expressed as: PC1, PC2 = 1 − PS1 to PCn = 1 − PS1, where PS is the component of the peak current that remains in the long-lasting desensitized state. The value of the peak current at sweep n is expressed by Equation 1.
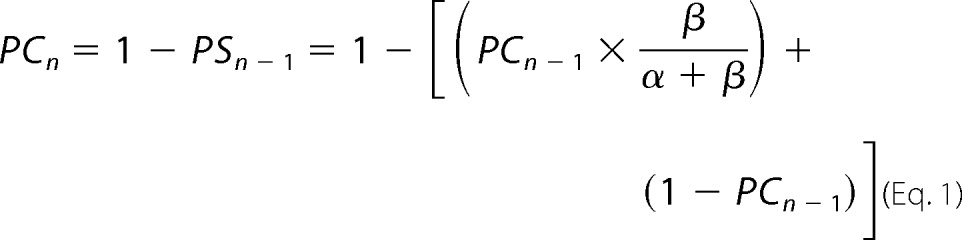 |
Equation 1 has two components in the brackets: the first one reflects the fraction of channels entering the long-lasting desensitized state after each sweep. The second one reflects the current that remains in the long-lasting desensitized state for each recovery time, Tr, expressed in seconds. δ is the rate of the transition from the slow-recovery desensitized state to the open state measured at pH 7.4 (Fig. 5A) (15). All data are shown as mean ± S.D. Statistical significance was determined by the Student's t test and indicates p ≤ 0.01.
FIGURE 5.

Kinetic model of the recovery from desensitization of ASIC1a. A, kinetic scheme describing the recovery from desensitization of mammalian ASIC1a. Squares indicate the minimal number of states: Closed, Open, and two Desensitized States. One short-lasting that recovers rapidly and another long-lasting that recovers slowly. The symbols indicate the rate constants of the transitions. Gray arrows show the dominant direction of transitions at low pH, and black arrows the direction at pH 7.4. B, superimposed consecutive currents of an outside-out patch expressing hASIC activated by the protocol described by the bar: activation with pH 6.0 for 10 s, a duration that allows all open channels to desensitized. pH 7.4 for a Tr of 10 s to recover channels from the desensitized states. The peak current (PC) of the first acid stimulus is the largest as all channels in the patch open. The second peak (PC2) and subsequent ones (PCn) have progressively decreased magnitude as some channels remain in the slow-recovery desensitized state, until a steady state is reached, PC∞. C, plot of the normalized mean values of PC2 for various Tr. When Tr is short, ≤10 s, PC2 value reflects the fraction of channels entering the slow-recovery desensitized state: β/α+β. D, table shows the experimental values of PC∞ for various Tr that were used to calculate δ according to Equation 4. E, graph shows a plot of mean values of PC∞, each symbol is the mean of 3–4 independent experiments. The line is the fit to Equation 4 with calculated δ of 0.00437 ± 0.00156.
RESULTS
Repetitive H+ Activation Reduces ASIC1a Currents of Native and Recombinant Mouse and Human ASIC1a Channels
Native mouse ASIC1a in the neuronal cell line N1E-115 (16) and human ASIC1a in HEK-293 cells (17) were examined using whole cell currents and excised patches. Channels were activated by a solution of pH 6.5 applied for 10 s; this stimulus produced transient inward currents that completely desensitized in 10 s. It was followed by perfusion for 10, 30, or 180 s with pH 7.4 to recover channels from the desensitized state. There are 10 sweeps in each of the representative examples shown for HEK 293 (Fig. 1A) and N1E-115 (Fig. 1B), which were obtained from independent cells. The first sweep is always indicated in black and subsequent ones in gray. The examples show that consecutive stimuli progressively attenuate the peak current and that the attenuation decreases by lengthening the recovery time (Tr): compare peak currents at 10 versus 180 s intervals. These results reproduce rundown of mammalian ASIC1a reported previously by several groups (1–5) and highlight the importance of the recovery time at pH 7.4 to rescuing channels from the desensitized state.
FIGURE 1.

Consecutive stimuli decrease peak currents of native mouse and human ASIC1a. A, representative examples of endogenous human ASIC1a in HEK-293 cells and B, mouse ASIC1a currents from the neuroblastoma cell line N1E-115. Traces represent currents in the whole-cell configuration elicited by a change of pH from 7.4 to 6.0 for 10 s followed by 10, 30, or 180 s of recovery at pH 7.4. Response to first stimulus is indicated in black and subsequent ones in gray. Bars are current amplitude and time scales.
In addition to the desensitization-dependent attenuation of ASIC1a currents in the time range of 10 to 300 s, we also observed a gradual and irreversible loss of channel activity in both whole-cell and excised patch configurations. Typically, after obtaining a high resistance seal a patch was activated by pH 6.5 and returned to pH 7.4 for 20 to 40 min before applying a second pulse of pH 6.5. The peak current of the second pulse was usually smaller, 0.6 to 0.8, than the first one. This much slower form of current loss or actual rundown differs from the one examined in this study and was not pursued here.
Modifications of the Cytosolic Solution Do Not Change Recovery from Desensitization
The results in Fig. 1 show equal magnitude and time course of peak current changes in endogenous and heterologous expressed ASIC1a making it unlikely that an auxiliary protein, with expression restricted to neurons, accounts for this phenomenon. Most likely it arises from an intrinsic property of mammalian ASIC1a channels or from conditions under which measurements are recorded. Chen and Gründer reported that it was caused by acidification of the cytoplasm due to permeation of H+ through rat ASIC1a (3). According to this explanation one may posit that the effect of cytosolic H+ ions decreases ASIC1a activity by screening charges in the channel protein that favor a desensitized conformation. Alternatively, H+ may neutralize negative charges of phospholipids from the inner leaflet of the plasma membrane reducing their apparent concentration in the vicinity of the channel. To test the effect of intracellular pH (pHi), we compared excised patches in the outside-out configuration with pipette solutions of pH 6.5 or 7.4 buffered with 30 mm HEPES to ensure constant pHi even in the event of H+ permeation. Currents were evoked by pH 6.5 for 10 s followed by 3 s of recovery at pH 7.4. Fig. 2A shows the desensitization rate faster at pHi 6.5 than at 7.4, and it follows two exponentials. Approximately half of the current (0.53 ± 0.05) desensitized with a time constant of τ1 0.71 ± 0.1 s and the other half (0.47 ± 0.08) with τ2 0.03 ± 0.004 s. In patches with pHi 7.4 the desensitization had a single time constant of 0.77 ± 0.08 s. These results indicate that internal H+ ions accelerate the process of desensitization but they do not influence its recovery.
FIGURE 2.
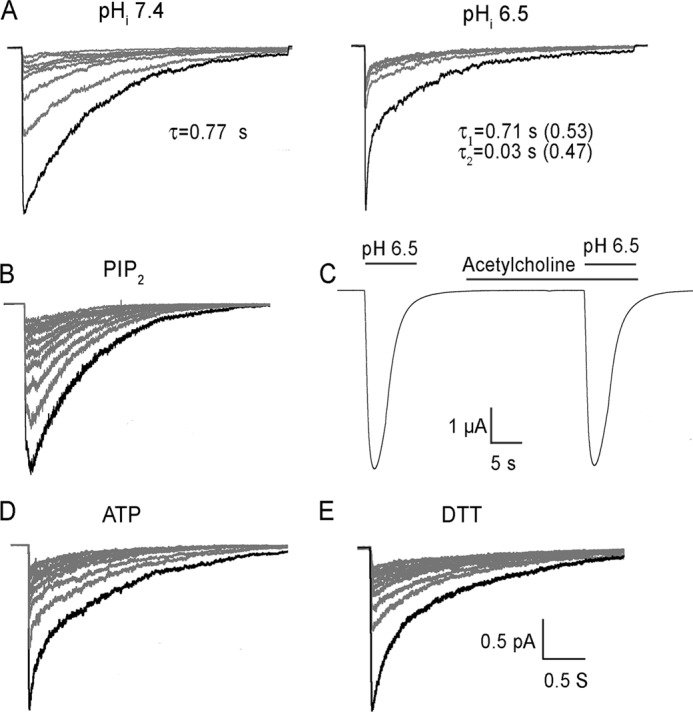
Changes in composition of the internal solution on hASIC1a response to protons. A, effects of acidifying of internal pH from 7.4 to 6.5. Excised patches in the outside-out configuration activated by external perfusion with pH 6.5 followed by 3 s of recovery at pH 7.4. Values of time constants of desensitization τ and fraction of total current (in parenthesis) calculated from fitting the black traces with one or two exponentials are shown. B, representative example of outside-out patch activated by pH 6.5 and 10 s recovery time at pH 7.4. Pipette solution pHi 7.4 supplemented with 100 μm C8-PIP2. C, whole-cell currents from co-expression of ASIC1a and muscarinic acetylcholine receptor (MR). Activations of ASIC1a by pH 6.5 and MR with acetylcholine are indicated by the bars above the current trace. D, outside-out patch activated by pH 6.5, recovery time 5 s. Pipette solution supplemented with 0.5 mm ATP. E, recovery time 5 s. Pipette solution supplemented with 5 mm DTT.
We next examined the possibility of H+-mediated screening of negatively charged phospholipids leading to an apparent depletion of phospahtidylinositol-4,5-bisphosphate, PIP2, which is the most abundant phospholipid in the plasma membrane and has been shown to modulate the activity of channels closely related to ASIC1a, namely ENaC (18, 19). Fig. 2B shows that supplementing the pipette solution with a short-chain PIP2 (50 μm C8-PIP2) was not effective. Furthermore, co-expression and activation of a muscarinic receptor that hydrolysis endogenous PIP2 (20) did not diminish hASIC1a currents (Fig. 2C). Addition of 0.5 mm ATP in the pipette solution also did not alter the decrease of peak currents in consecutive stimuli (Fig. 2D).
Another possible modification is oxidation of intracellular cysteines introduced by washout of cytosolic reducing factors in whole-cell and excised patches. This consideration is underscored by a previous study that reported application of oxidizing agents to the external side of ASIC1a decreased channel currents in mouse neurons while the reducing reagent DTT reverted the effect (21). There are four free cysteines in the intracellular C terminus of hASIC1a and none in the N terminus that could be targeted by oxidation while most cysteines in the extracellular domain form disulfide bridges. We compared the response of hASIC1a in patches containing 0 or 5 mm DTT. Fig. 2E shows no detectable difference in the two conditions.
Since modifications targeted to the channel protein, both endogenous and recombinant, and to the environment immediately surrounding ASIC1a, did not alter the decrease in peak currents induced by repetitive stimuli we conclude that it most likely reflects a structural property intrinsic to the channel protein.
Short Stimuli Applied at High Frequency Maintain Stable hASIC1a Peak Currents
Trains of consecutive stimuli predict progressive attenuation and even suppression of hASIC1a currents. To cope with effect channels must either recover rapidly from desensitization or prevent it. The previous experiments indicate that decrease in peak currents arises from the desensitized state since channels were exposed to pH 6.5 for 10 s that induces complete desensitization every single sweep. However, it remains possible that deactivation, unbinding of H+ from open channels, is enough to induce desensitization. We examined this possibility by activating channels for short periods of time: 20, 100, or 500 ms with pH 6.5, followed by a fixed recovery time with pH 7.4 for 5 s. If opening is enough to induce desensitization, then the magnitude of the peak currents should decrease on consecutive stimulations. Fig. 3A shows that repetitive H+ stimuli shorter than 100 ms did not decrease the peak currents; they remained constant even after 100 sweeps, whereas peak currents progressively decreased after a few stimuli longer than ≥500 ms. Fig. 3B summarizes the relation of peak currents as a function of the stimulus duration. These results demonstrate that hASIC1a desensitizes slowly and that deactivated channels do not lead to desensitization. However, we have not proved yet that currents remain stable under high frequency stimulation, which is the prevalent form of signaling in the CNS. We addressed this issue by decreasing the time of recovery at pH 7.4 from 5 s to 200 ms, the limit speed of our patch perfusion system. As shown in Fig. 3C, channels were activated for 10 ms with pH 6.5 and recovered for 200 ms with pH 7.4; peak currents remained stable over the first 8 consecutive sweeps shown in Fig. 3C. Together these results indicate that the decrease of peak current on consecutive stimuli originates only from the desensitized state and that short stimuli, even if applied at high frequency, keep ASIC1a response stable.
Kinetics of Recovery from Desensitization of hASIC1a
We next examined the kinetics of recovery from desensitization in outside-out patches exposed to pH 6.5 for 10 s followed by recovery times, Tr, of increasing duration, from 0.5 to 180 s. For each recovery time the protocol was repeated until a steady state current was reached. Only patches with initial peak current equal or greater than 500 nA were examined. A single condition was used per patch to avoid accumulation of inactive channels induced by mechanisms acting on a longer time scale. Currents were normalized to facilitate comparison of patches examined at various Tr values, representative examples are shown in Fig. 4A. Steady-state peak currents are plotted as a function of the recovery time in Fig. 4B where the line is the fit to a single exponential with time constant τ of 48 ± 2 s. This time constant however does not represent the life-time of the desensitized state(s) as explained in the analysis below.
FIGURE 4.
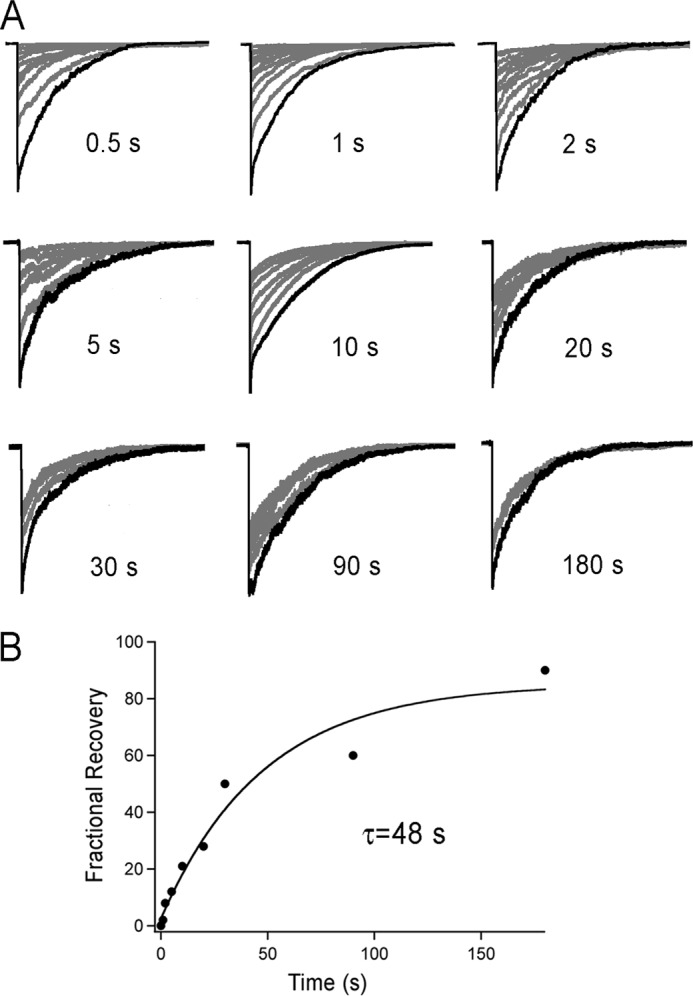
Recovery from desensitization of hASIC1a reflects the presence of more than a single process. A, excised outside-out patches expressing hASIC1 were activated by pH 6.5 for 10 s followed by recovery at pH 7.4 for the indicated time periods. B, plot of the stable peak current after 20 consecutive stimuli. Symbols are the mean of 4 to 7 independent experiments. Line is the fit to a single exponential with calculated time constant of τ = 48 s.
The most parsimonious explanation for the progressive decrease of the magnitude of the peak current (PC) is described in the kinetic scheme shown in Fig. 5A. Here, open channels desensitize to two states with different stability: short-lasting and long-lasting desensitized states. After each stimulus, the PC decreases owing to channels remaining in the long-lasting state, which in turn depends inversely to the length of the recovery time, Tr, as shown in Fig. 5B and described by Equation 1 in “Experimental Procedures.” The Equation 1 can be rewritten as Equation 2.
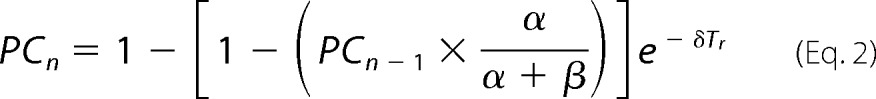 |
When n = 2 and Tr is short, Equation 2 reduces to: PC2 = α/(α+β), where α and β are the rate constants to the two desensitized states, respectively.
A plot of experimentally obtained PC2 values for various Tr is shown in Fig. 5C. Accordingly, PC2 has value of 0.8 indicating that after each sweep ∼80% of open channels enter the short-lasting desensitized state and 20% enter the long-lasting desensitized state. On the other hand, when the peak current reaches steady state after many sweeps, P∞, Equation 2 can be expressed as Equation 3.
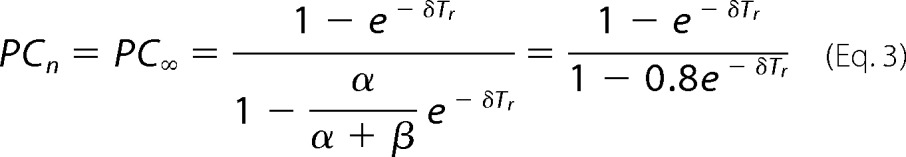 |
Solving Equation 3 for δ gives the expression in Equation 4.
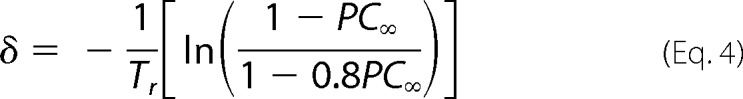 |
Data values for PC∞ obtained at various recovery times are shown in the table in Fig. 5D, and plotted in Fig. 5E. Circles are mean data points and the line is the fit to Equation 4, which yields a calculated value for δ of 0.00437 s−1.
This analysis is consistent with hASIC1a having at least two desensitized states that are distinguishable by their different stabilities. At any H+-activating stimulus, 80% of open channels enter the short-lasting desensitized state whose τDs is estimated to be <0.5 s. The remaining channels enter a stable and long-lasting desensitized state with a τDl of 229 ± 12 s (tDl = 1/δ).
Recovery from Desensitization of Avian ASIC1a
All the ASIC1a channels from mammals studied to date exhibit slow recovery from desensitization but it is not known whether this property is shared by other species. Here we examined chicken ASIC1a (cASIC1a) since it is the only channel of this class with solved atomic structures in several conformational states: open (22), closed (23), and desensitized (23–25).
We used the same protocol as in hASIC1a except that the stimulus duration was reduced from 10 to 5 s since cASIC1a exhibits faster desensitization rate. Fig. 6A shows representative examples of sequential activation at various Tr values, from 0.5 to 30 s. The recovery from desensitization was also dependent on the duration of Tr though it differed markedly from hASIC1a. Here, for all Tr examined a steady state was reached at the second sweep rather than the slow and progressive decrease over many sweeps before reaching steady state observed with hASIC1a currents. However, expansion of the traces at Tr 0.5 s (inset, Fig. 6A) shows that only after several sweeps the steady state was reached, suggesting the presence of a second desensitized state. Thus in cASIC1a the two desensitized states differ only slightly in stability; detection of the difference was barely within the limits of the exchange solution system. A plot of the stabilized peak current as a function of Tr is shown in Fig. 6B where the line is the fit to a single exponential with t of 4.5 ± 0.6 s. This τ value applies for the two desensitized states of cASIC1a.
FIGURE 6.

Recovery from desensitization of avian ASIC1a follows a single exponential. A, excised outside-out patches expressing cASIC1 were activated by pH 6.0 for 5 s followed by recovery at pH 7.4 for the indicated time periods. In black is the first sweep and in gray subsequent 10 sweeps. Bars below current traces indicate amplitude and time scales. B, plot of the stable peak current after 10 consecutive stimuli. Symbols are the mean of 4 to 7 independent experiments. The line is the fit to a single exponential with calculated time constant for recovery from the desensitized state of τD = 4.5 s. The inset shows a magnification of currents elicited after 0.5 s recovery at pH 7.4.
These results show a broad variability in the stability of the two desensitized states among ASIC1 channels. As indicated in the previous section, the τD values of the short- and long-lasting desensitized states of hASIC1 are ∼300-fold different. In contrast, we show here that the τD values of the desensitized states of cASIC1a are both so close to 4.5 s that they are barely distinguishable, given the impression that recovery from desensitization follows a single exponential.
Structural Determinants of Recovery from Desensitization in Human ASIC1a
In a prior study, Chen et al. reported that rat ASIC1b isoform recovers rapidly from desensitization (3). The latter uses different exons of the same ASIC1 gene to encode the first 185 amino acids (26). These observations suggest that some of the elements conferring stability to the desensitized states are located in the first 185 amino acids of the protein. The rat isoforms ASIC1a and ASIC1b differ in 47 out of 185 amino acids interspersed in the whole spliced region whereas cASIC1a has only 30 different amino acids in the same region making easier the identification of important residues. Since ASIC1a and ASIC1b are essentially chimeras, we started with the equivalent chimera between human and chicken ASIC1a (CH1) and constructed additional ones (CH2-CH5) as shown in Fig. 7A. CH3 behaves as hASIC1a while CH1, CH2, and CH4 do not, indicating that TM1 from chicken accelerates recovery from the long-lasting desensitized state of hASIC1. There are eight different residues in human TM1 compared with chicken: A44, L45, L51, V56, L58, C59, E63, and V65. Replacing those positions with residues in cASIC1 TM1 showed that those in the first half of TM1, close to the cytosolic side, had the largest functional effect (CH5), but there was not a single residue that alone reproduced the whole effect of TM1 as indicated by the examples shown in Fig. 7B. To further explore whether hydrophobicity of the side chains was a factor, we made the substitutions A44T and M51T in hASIC1. These mutations delayed even more the recovery, Fig. 7C shows that the steady state current reached at 10 and 30 s recovery times are smaller than in wild type channels. A graph of steady-state currents as a function of Tr is shown in Fig. 7D.
FIGURE 7.

Functional analysis of human and chicken ASIC1a chimeras. A, schematic representation of chimeras: human sequences are indicated in black and chicken ones in gray. Numbers represent the amino acid position in the hASIC1a protein. Presence of rundown in chimeras, similar as in hASIC1a, is indicated by the sign +, absence by −, and intermediate rundown by ±. B, superimposed consecutive currents evoked by pH 6.0 for 5 s and recovery by pH 7.4 for 10 s of human-chicken chimeras (CH) and mutants. C, representative superimposed currents from hASIC1-A44T and hASIC1-L51T recorded at Tr of 10 and 30 s. D, graph of steady-state currents as a function of Tr of mutants A44T and L51T compared with hASIC1 wild type: τA44T = 300 ± 26 and τL51T = 280 ± 39 s.
DISCUSSION
Usage-dependent attenuation is one of the means ion channels use in the CNS for preventing excessive stimulation. It is particularly important for cation channels that drive membrane depolarization and thus facilitate influx of calcium. Multiple mechanisms have been suggested to explain usage-dependent attenuation of the mammalian ASIC1a and several parts of the channel protein have been implicated in this process (3–27). Chen et al. proposed that ASIC1a exhibits rundown but not ASIC1b because only the former permeates H+ and Ca2+, which in turn might interact with the first 19 residues of the amino terminus to induce rundown in ASIC1a. To explain their findings they figured a qualitative scheme containing two desensitized states. However, no further analysis was offered because all the data were collected as whole cell currents from oocytes and at a single recovery time (Tr of 60 s). Kinetic analysis can be obtained only using rapid perfusion methods that allow fast deliver and removal of H+ i.e. patches perfused with rapid solution-exchange systems. The reason is that with slowly rising concentrations of agonist, channel activity is highly desynchronized since some channels are opening or desensitizing while others are still waiting to bind agonist. The response to a rapid step into high agonist concentration provides a better macroscopic representation of intrinsic channel kinetics. The time constants of the ensemble response still do not correspond to single microscopic transitions rates, but depend on both forward and backward rates of the relevant transitions. Thus, even with rapid agonist application, additional methods are necessary to determine microscopic rate constants from macroscopic data. In our study accurate estimates of microscopic parameters were obtained by fitting a single model to multiple Tr values, from 0.5 to 180 s.
Our results indicate that ASIC1 channels, from different isoforms and different species, exhibit a broad range of peak current attenuation as a consequence of desensitization and that the marked differences are due to variations in stability of two desensitized states. When the stability of the desensitized states differs markedly, as in mammalian ASIC1a: τDs <0.5 s and τDl 229 ± 12 s, the channel exhibits progressive attenuation of peak currents upon consecutive stimuli (Fig. 4A). The longer the recovery period, Tr > τDl, the fewer desensitized channels since they have more time to leave the long-lasting desensitized state. When both desensitized states have similar stability, as in the case of cASIC1a: τDs∼τDl = 4.5 ± 0.6 s (Fig. 6A), the decay of the currents follows a single exponential. In this instance, peak currents decrease and reach steady value as soon as the second stimulus. Since all desensitized channels recovered at similar rates, a single exponential describes the kinetics of recovery. In the event of an ASIC1 channel with very short and similar τD values for the two desensitized states, there would be minimal attenuation of peak currents at any Tr thereby no rundown. Our analysis therefore can be applied to any ASIC from any species to describe the kinetics of recovery from desensitization and predict the degree of recovery.
We also demonstrate that stability of the desensitized states is intrinsic to the channel protein and independent of modifications in the composition of the cytosol as previously proposed. In addition, it is not the amino terminus but TM1 what influences the kinetics of recovery from desensitization. Specifically, residues in the initial segment of TM1, close to the cytosolic side, change the rate constants of the desensitized states. It has become clear that the desensitized states identified here by functional means have structural counterparts as indicated by three independent crystal structures of cASIC1 in the putative desensitized state(s). The first structure was published by Jasti et al. (25) and showed an asymmetric nonconductive pore; the second one published by Gonzales et al. (24) showed a symmetric crossing of three TM2 occluding the pore. Most recently, a third structure by Dawson et al. (23) has been crystallized at pH 5.5 in a complex with the inhibitory toxin PcTx, which induces desensitization specifically of ASIC1a channels (28, 29). Thus, the structural data give credence to the existence of several desensitized states, two of which we were able to identify functionally because of their quite different stabilities. Presently, the data do not allow yet assigning the slow- or the fast-recovery desensitized states to any of the three structures already available. However, additional crystal structures in the open conformation by Baconguis and Gouaux show that the initial segment of TM1 rotates during gating shifting interactions of the side chains between the lipid bilayer and residues of TM2 from the adjacent subunit, including the interaction of V46 in TM1 with I446 in TM2 of cASIC1 (22). Therefore, changes in the amino acid composition in the initial segment of TM1 are poised to alter kinetics of rundown as shown here.
We also demonstrate that unbinding of protons from open channels does not lead to desensitization. This assertion is based on the finding that short stimuli, <100 ms, keep the magnitude of peak currents constant, thus repetitive stimulation of short duration does not induce desensitization. Even if the recovery interval, Tr, shortens to deliver high frequency stimulation, peak currents remain stable. Together these experiments demonstrate that hASIC1a sustains steady responses to stimuli delivered at high frequency. The data also support the notion that long periods of acidification, even if followed by recovery to baseline pH, attenuate or abolish hASIC1a signals by retaining channels in the desensitized state.
Although our analysis predicts the behavior of ASIC1a to various durations and frequencies of H+-mediated stimulation, crucial questions of how much the local pH fluctuates in microdomains of the CNS and the time course of the pH changes remain mostly unanswered. Despite uncertainties, the transient changes in local pH required to activate ASIC1a are within physiological boundaries. For instance, a small spherical compartment in the brain of 100 nm diameter with limited diffusion and low buffering capacity compared with plasma, could reversible change pH from 7.3 to 6.5 by addition or removal of only 0.8 free H+ ions, which would be sufficient for ASIC1a activation.
From a solely practical point, desensitization interferes with measurements of ASIC1a when more than one stimulus is applied experimentally. A way to avoid this confounding effect is to activate ASIC1a briefly, ≤100 ms, which prevents channels entering the desensitized state. Alternatively, if experiments are conducted with recombinant mammalian channels and using slow perfusion systems as in oocytes, the substitution of a few residues in TM1, abrogates most of the slow recovery from desensitization.
In summary, we provide a formal explanation for attenuation of peak currents upon consecutive activation of ASIC1a based on two desensitized states with different stabilities, and predict stable response of hASIC1a to short and high frequency stimuli as those induced by the release of H+ through synaptic transmission in the CNS.
Footnotes
- ASIC1a
- acid-sensing ion channel-1a
- TEVC
- two-electrode voltage clamp
- PC
- peak current.
REFERENCES
- 1. Neaga E., Amuzescu B., Dinu C., Macri B., Pena F., Flonta M. L. (2005) Extracellular trypsin increases ASIC1a selectivity for monovalent versus divalent cations. J. Neurosci. Methods 144, 241–248 [DOI] [PubMed] [Google Scholar]
- 2. Gitterman D. P., Wilson J., Randall A. D. (2005) Functional properties and pharmacological inhibition of ASIC channels in the human SJ-RH30 skeletal muscle cell line. J. Physiol. 562, 759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen X., Gründer S. (2007) Permeating protons contribute to tachyphylaxis of the acid-sensing ion channel (ASIC) 1a. J. Physiol. 579, 657–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kusama N., Harding A. M., Benson C. J. (2010) Extracellular chloride modulates the desensitization kinetics of acid-sensing ion channel 1a (ASIC1a). J. Biol. Chem. 285, 17425–17431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weng J. Y., Lin Y. C., Lien C. C. (2010) Cell type-specific expression of acid-sensing ion channels in hippocampal interneurons. J. Neurosci. 30, 6548–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li T., Yang Y., Canessa C. M. (2010) Two residues in the extracellular domain convert a nonfunctional ASIC1 into a proton-activated channel. Am. J. Physiol. Cell Physiol. 299, C66–C73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li T., Yang Y., Canessa C. M. (2010) Asn415 in the β11-β12 linker decreases proton-dependent desensitization of ASIC1. J. Biol. Chem. 285, 31285–31291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wemmie J. A., Chen J., Askwith C. C., Hruska-Hageman A. M., Price M. P., Nolan B. C., Yoder P. G., Lamani E., Hoshi T., Freeman J. H., Jr., Welsh M. J. (2002) The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34, 463–477 [DOI] [PubMed] [Google Scholar]
- 9. Wemmie J. A., Askwith C. C., Lamani E., Cassell M. D., Freeman J. H., Jr., Welsh M. J., (2003) Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J. Neurosci. 23, 5496–5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wemmie J. A., Coryell M. W., Askwith C. C., Lamani E., Leonard A. S., Sigmund C. D., Welsh M. J. (2004) Overexpression of acid-sensing ion channel 1a in transgenic mice increases acquired fear-related behavior. Proc. Natl. Acad. Sci. U.S.A. 101, 3621–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deval E., Gasull X., Noël J., Salinas M., Baron A., Diochot S., Lingueglia E. (2010) Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol. Ther. 128, 549–558 [DOI] [PubMed] [Google Scholar]
- 12. Bohlen C. J., Chesler A. T., Sharif-Naeini R., Medzihradszky K. F., Zhou S., King D., Sánchez E. E., Burlingame A. L., Basbaum A. I., Julius D. (2011) A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 479, 410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaila K., Chesler M. (1998) pH and Brain Function, Wiley-Liss, Inc [Google Scholar]
- 14. Coric T., Zheng D., Gerstein M., Canessa C. M. (2005) Proton sensitivity of ASIC1 appeared with the rise of fishes by changes of residues in the region that follows TM1 in the ectodomain of the channel. J. Physiol. 568, 725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Colquhoun D., Hawkes A. (1995) G. Single-Channel Recording, 2nd Ed., Plenum, NY [Google Scholar]
- 16. Arteaga M. F., Coric T., Straub C., Canessa C. M. (2008) A brain-specific SGK1 splice isoform regulates expression of ASIC1 in neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gunthorpe M. J., Smith G. D., Davis J. B., Randall A. D. (2001) Characterisation of a human acid-sensing ion channel (hASIC1a) endogenously expressed in HEK293 cells. Pflügers Arch. 442, 668–674 [DOI] [PubMed] [Google Scholar]
- 18. Yue G., Malik B., Yue G., Eaton D. C. (2002) Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J. Biol. Chem. 277, 11965–11969 [DOI] [PubMed] [Google Scholar]
- 19. Pochynyuk O., Tong Q., Medina J., Vandewalle A., Staruschenko A., Bugaj V., Stockand J. D. (2007) Molecular determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. J. Gen. Physiol. 30, 399–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Falkenburger B. H., Jensen J. B., Hille B. (2010) Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 135, 99–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chu X. P., Close N., Saugstad J. A., Xiong Z. G. (2006) ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J. Neurosci. 26, 5329–5339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baconguis I., Gouaux E. (2012) Structural plasticity and dynamic selectivity of acid sensing ion channel-toxin complexes. Nature 489, 400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dawson R. J., Benz J., Stohler P., Tetaz T., Joseph C., Huber S., Schmid G., Hügin D., Pflimlin P., Trube G., Rudolph M. G., Hennig M., Ruf A. (2012) Structure of the Acid-sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat. Commun. doi: 10.1038/ncomms1917 [DOI] [PubMed] [Google Scholar]
- 24. Gonzales E. B., Kawate T., Gouaux E. (2009) Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 460, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jasti J., Furukawa H., Gonzales E. B., Gouaux E. (2007) Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449, 316–323 [DOI] [PubMed] [Google Scholar]
- 26. Chen C. C., England S., Akopian A. N., Wood J. N. (1998) A sensory neuron-specific, proton-gated ion channel. Proc. Natl. Acad. Sci. U.S.A. 95, 10240–10245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang W. Z., Chu X. P., Li M. H., Seeds J., Simon R. P., Xiong Z. G. (2006) Modulation of acid-sensing ion channel currents, acid-induced increase of intracellular Ca2+, and acidosis-mediated neuronal injury by intracellular pH. J. Biol. Chem. 281, 29369–29378 [DOI] [PubMed] [Google Scholar]
- 28. Escoubas P., De Weille J. R., Lecoq A., Diochot S., Waldmann R., Champigny G., Moinier D., Ménez A., Lazdunski M. (2000) Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J. Biol. Chem. 275, 25116–25121 [DOI] [PubMed] [Google Scholar]
- 29. Chen X., Kalbacher H., Gründer S. (2006) Interaction of acid-sensing ion channel (ASIC) 1 with the tarantula toxin psalmotoxin 1 is state dependent. J. Gen. Physiol. 127, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]