

Abstract
BACKGROUND AND PURPOSE
Noscapine is a promising anti-tumour agent. The purpose of the present study was to describe the metabolic map and investigate the bioactivation of noscapine.
EXPERIMENTAL APPROACH
Ultra-performance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight mass spectrometry-based metabolomics was used to analyse the in vitro incubation mixtures, urine and faeces samples from mice treated with noscapine. Recombinant drug-metabolizing enzymes were employed to identify those involved in noscapine metabolism. Hepatic GSH levels and serum biochemistry were also carried out to determine reactive metabolites of noscapine.
KEY RESULTS
Several novel phase I metabolites of noscapine were detected after oral gavage of mice, including an N-demethylated metabolite, two hydroxylated metabolites, one metabolite undergoing both demethylation and cleavage of the methylenedioxy group and a bis-demethylated metabolite. Additionally, several novel glucuronides were detected, and their structures were elucidated through MS/MS fragmentology. Recombinant enzymes screening showed the involvement of several cytochromes P450, flavin-containing mono-oxygenase 1 and the UDP-glucuronosyltransferases UGT1A1, UGT1A3, UGT1A9 and UGT2B7, in noscapine metabolism. In vitro glutathione trapping revealed the existence of an ortho-quinone reactive intermediate formed through further oxidation of a catechol metabolite. However, this bioactivation process of noscapine does not occur in vivo. Similar to this result, altered glutathione levels in liver and serum biochemistry revealed no evidence of hepatic damage, thus indicating that, at least in mice, noscapine does not induce hepatotoxicity through bioactivation.
CONCLUSIONS AND IMPLICATIONS
A comprehensive metabolic map and bioactivation evaluation provides important information for the development of noscapine as an anti-tumour drug.
Keywords: noscapine, metabolic map, bioactivation
Introduction
Bioactivation through drug metabolism is one of the mechanisms giving rise to drug toxicity and clinical drug–drug interactions (DDI) (Walsh and Miwa, 2011). To date, many structural alerts (toxicophores) have been summarized based on retrospective analyses of structure–toxicity relationships, and medicinal chemists try to avoid these functional groups in drug design (Kalgutkar and Solia, 2005; Baillie, 2006; Fang et al., 2011). A methylenedioxyphenyl group has been widely accepted as an important structural alert, and many methylenedioxyphenyl-containing compounds were demonstrated to undergo bioactivation to induce clinical toxicity or DDI. For example, the formation of 3,4-dihydroxymetamphetamine ortho-quinone (HHMA-o-quinone) and 3,4-dihydroxyamphetamine ortho-quinone (HHA-o-quinone) via bioactivation of 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy) plays an important role in the toxic effects of MDMA (Tucker et al., 1994; Antolino-Lobo et al., 2010).
Noscapine, a phthalideisoquinoline alkaloid isolated from opium, has been clinically used as an efficient cough suppressant (Empey et al., 1979). In recent years, the inhibitory potential of noscapine towards the growth of various tumours has been demonstrated using in vitro and in vivo methods, including the tumour types lymphoma, breast cancer, melanoma, ovarian carcinoma, glioblastoma, colon cancer and non–small cell lung cancer (Ye et al., 1998; Mahmoudian and Rahimi-Moghaddam, 2009). Based on these studies, noscapine is undergoing phase I/II clinical trials for anti-cancer therapy. New therapeutic regimens are being developed, including noscapine–doxorubicin and noscapine–cisplatin combination chemotherapy regimens (Chougule et al., 2011a,b). Additionally, medicinal chemists are making efforts to modify the structure of noscapine in an attempt to synthesize better derivatives of noscapine (Naik et al., 2011; Santoshi et al., 2011).
To date, the safety of noscapine is still controversial. Although there is general agreement that the clinical application of noscapine is safe, some concerns on its possible adverse side effects have been raised. Some studies suggest that noscapine is potentially carcinogenic (Porter et al., 1992; Schuler et al., 1999). For example, oral administration of 200 mg·kg−1 body weight noscapine to rats can decrease GSH contents and enhance lipid peroxidation (Aneja et al., 2004). In previous studies, a link between time-dependent inhibition of cytochromes P450, CYP2C9 and CYP3A, and the observed clinical noscapine–warfarin interaction has been firmly established (Fang et al., 2010). Noscapine undergoes extensive ‘first pass’ metabolism mainly by C–C cleavage, O-demethylation and cleavage of methylenedioxy group in rats, rabbits and humans (Tsunoda and Yoshimura, 1981). Of these biotransformation pathways, cleavage of the methylenedioxy group is of interest due to its potential to induce the formation of quinone reactive metabolites.
Ultra performance chromatography electrospray ionization quadrupole time-of-flight mass spectrometry (UPLC-ESI-QTOFMS)-based metabolomics has been applied to determine the metabolic behaviour of many xenobiotics, including thioTEPA (Li et al., 2011b) and arecoline (Giri et al., 2006). Additionally, this method has proven to be an efficient method to determine the reactive metabolites formed through bioactivation of xenobiotics (Li et al., 2011a). The aims of the present study were (i) to elucidate the metabolic pathway of noscapine in the mouse, a useful animal model for anti-tumour studies of noscapine; (ii) to identify the drug-metabolizing enzymes (DMEs) involved in noscapine metabolism; and (iii) to evaluate the bioactivation of noscapine.
Methods
Chemicals and reagents
Noscapine, alamethicin, NADPH, uridine 5′-diphosphoglucuronic acid trisodium salt (UDPGA) were purchased from Sigma-Aldrich (St. Louis, MO). All other reagents were of the highest grade commercially available.
In vitro phase I metabolism of noscapine
Livers from five untreated 6- to 8-week-old age male C57BL/6 mice were homogenized, and mouse liver microsomes (MLM) were prepared as previously described (Fang et al., 2010). A pool was prepared by mixing equal volumes of each liver microsomal preparation (n= 5), with identical protein contents per sample. Human liver microsomes (HLM) from a pool of 50 donors were purchased from BD Gentest Corp (Woburn, MA). The incubation system (200 µL) contains 50 mM Tris–HCl buffer solution (pH = 7.4), 0.5 mg·mL−1 HLM or MLM, 2 mM MgCl2, 100 µM noscapine and 1 mM freshly prepared NADPH. After 1 h incubation at 37°C, the reaction was stopped using 200 µL cold 50% aqueous acetonitrile containing 5 µM chlorpropamide. After centrifugation at 14 000×g for 20 min, a 5 µL aliquot of the supernatant was injected into a UPLC-ESI-QTOFMS. The in vitro incubation system for recombinant phase I enzymes was similar to the liver microsomes incubation system. The recombinant enzymes used – CYP1A1, CYP1A2, CYP2C8, CYP2C9, CYP2C9*2, CYP2C9*3, CYP2D6, CYP2A6, CYP2B6, CYP3A5, CYP3A4, CYP3A7, CYP2C19, CYP2E1, CYP4A11, CYP4F12, FMO-1, FMO-3 and FMO-5 – were produced in baculovirus, and purchased from BD Gentest. CYPs (2 pmol) and 5 µg FMOs were incubated with 100 µM of noscapine. The reaction time was 30 min, and metabolites were analysed using UPLC-ESI-QTOFMS.
Co-activation of phase I enzymes and UGTs in liver microsomes
A dual-activity incubation system was used (Yan and Caldwell, 2003) with an incubation system (200 µL) containing 50 mM Tris–HCl buffer solution (pH 7.4), 0.5 mg·mL−1 HLM or MLM, 2 mM MgCl2, 100 µM noscapine, 1 mM freshly prepared NADPH and UDPGA, and 25 µg·mL−1 alamethicin. After 1 h incubation at 37°C, the reaction was stopped using 200 µL cold 50% aqueous acetonitrile containing 5 µM chlorpropamide. After centrifugation at 14 000×g for 20 min, a 5 µL aliquot of the supernatant was injected into UPLC-ESI-QTOFMS. The dual-activity incubation system was also employed to screen the hepatic UGT isoforms involved in the glucuronidation elimination of noscapine's phase I metabolites. The recombinant UGTs UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B10, UGT2B15, and UGT2B17 were obtained from BD Gentest. To maximize the quantity of phase I metabolites formed, the phase I enzymes exhibiting relatively high catalytic activity were used. To study the UGT forms involved in generation of the C1 metabolite, 0.5 pmol CYP2C9*2 was used. CYP3A5 was employed to investigate the UGT forms involved in formation of the glucuronides C3 and C4 using 5 µg of each recombinant UGT.
In vivo metabolism of noscapine
Mice are frequently used for evaluating the anti-cancer activity of noscapine (Jackson et al., 2008; Liu et al., 2011). However, the metabolism of noscapine in mice remains unclear. In this experiment, two strains of mice (male Sv/129 mice and C57BL/6, 6 to 8 weeks old) were selected for urine metabolite profiling. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Handling was in accordance with an animal study protocol approved by the National Cancer Institute Animal Care and Use Committee. The mice were maintained under a standard 12 h light, 12 h dark cycle with water and chow provided ad libitum. Noscapine dissolved in corn oil was administered by oral gavage at a dose of 300 mg·kg−1 BW. For Sv/129 mice, four mice were used to collect the urine before and after administration of noscapine (24 h). For C57BL/6 mice, eight mice were used, including three control and five noscapine-treated mice. Control mice were treated with corn oil alone. Urine and faeces samples (24 h) were collected using metabolic cages (Metabowls, Jencons Scientific USA, Bridgeville, PA), and blood samples were collected in BD microtainer serum separator tubes (Franklin Lakes, NJ) by retro-orbital bleeding at 24 h. After centrifugation for 15 min at 8000×g, serum was obtained for clinical biochemistry analysis. Twenty-four hours after treatment, the livers of C57BL/6 were also collected for the following analysis of GSH levels and GSH adducts. Urine samples were prepared through mixing the 20 µL urine with 180 µL 50% aqueous acetonitrile containing 5 µM chlorpropamide. After centrifugation at 14 000×g for 20 min, a 5 µL aliquot of the supernatants was injected into a Waters UPLC–ESI-QTOFMS system (Waters Corporation, Milford, MA). Faeces homogenates in 50% aqueous acetonitrile containing 5 µM chlorpropamide were prepared. After centrifugation at 14 000×g for 20 min, a 5 µL aliquot of each supernatants was injected into a Waters UPLC–ESI-QTOFMS system (Waters Corporation).
Evaluation of bioactivation of noscapine
GSH was used as a trapping agent to confirm the existence of reactive metabolites. The incubation was conducted in 50 mM Tris–HCl buffer solution (pH 7.4), containing 100 µM noscapine, 2.5 mM GSH, 1 mM NADPH and 0.5 mg·mL−1 HLM or MLM in a final volume of 200 µL. The reaction was initiated by the addition of NADPH and continued for 1 h. The reaction was stopped using 200 µL cold 50% aqueous acetonitrile containing 5 µM chlorpropamide. After centrifugation at 14 000×g for 20 min, a 5 µL aliquot of the supernatant was injected into UPLC-ESI-QTOFMS. The incubation for screening for enzymes involved in the bioactivation of noscapine was carried out as described above for the microsomal incubation system. Detection of GSH adducts in liver was carried out as previously described (Li et al., 2011a). In brief, the livers were homogenized in water (100 mg liver in 400 µL H2O), and then 200 µL acetonitrile was added to 200 µL of liver homogenates. After 20 min centrifugation at 14 000×g, a 5 µL aliquot of the supernatant was analysed.
UPLC-ESI-QTOFMS and Triple quadrupole MS analysis
An Acquity C18 BEH UPLC column (Waters Corporation) was employed to separate components in urine and microsomal incubation samples. The mobile phase consisted of water containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). The following gradient condition was used: 100% A for 0.5 min, increased to 100% B over the next 7.5 min and returned to 100% A in last 2 min. The flow rate of the mobile phase was set 0.5 mL·min−1. Data were collected in both positive ion mode on a Waters Q-TOF Premier mass spectrometer, which was operated in full-scan mode at 50–850 m/z. Nitrogen was used as both cone gas (50 L·h−1) and desolvation gas (600 L·h−1). Source temperature and desolvation temperature were set at 120°C and 350°C, respectively. The capillary voltage and cone voltage were 3000 and 20 V, respectively. The structures of metabolites were elucidated by tandem MS fragmentography with collision energies ranging from 15 to 40 eV. To determine the GSH adducts produced by recombinant enzymes and liver extracts, an acquity UPLC system coupled with a XEVO triple-quadrupole tandem mass spectrometer (Waters Corporation) was used. Multiple reaction monitoring (MRM) was employed, and the MRM transition 707 → 384 was used. An Acquity UPLC BEH C18 column (1.7 µm, 2.1 × 50 mm, Waters Corporation) was used to separate the samples. The mobile phase was comprised of 0.1% aqueous formic acid (A) and acetonitrile containing 0.1% formic acid (B). The gradient elution was performed over 6 min at a flow rate of 0.3 mL using 1–99% B in 4 min, holding at 99% B up to 5.0 min, bringing back to 1% at 5.5 min and holding at 1% until the end. Column temperature was maintained at 40°C throughout the run.
Multivariate data analysis (MDA)
MarkerLynx software (Waters Corporation) was utilized to deconvolute the chromatographic and mass spectrometric data. A multivariate data matrix containing information on sample identity, ion identity (Rt and m/z), and ion abundance was generated through centroiding, deisotoping, filtering, peak recognition and integration. The data matrix was further analysed using SIMCA-P+ 12.0 software (Umetrics, Kinnelon, NJ). OPLS was adopted to analyse the data to identify the major latent variables in the data matrix. Potential metabolites were identified by analysing the ions contributing to the separation of sample groups in the loading scatter plots.
Serum clinical biochemistry
The catalytic activities of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in serum were determined using Catachem VetSpec™ Kit (Catachem Inc, Bridgeport, CT) according to the manufacturer's instructions. Briefly, 1 µL serum was mixed with 200 µL AST assay buffer (Catachem) in a 96-well microplate, and the oxidation of NADH to NAD+ was monitored at 340 nm for 5 min. Alkaline phosphatase (ALP) analysis was carried out to monitor the catalytic conversion of p-nitrophenyl phosphate (PNPP) to p-nitrophenol at 414 nm using Catachem VetSpec™ Kit (Catachem Inc).
Measurement of glutathione (GSH) levels in liver
The Glutathione Assay Kit obtained from Sigma-Aldrich was used to measure the total liver glutathione. The procedure was in accordance with the manufacturer's instructions. In brief, the kinetic assay at 412 nm was used to determine GSH levels in which the catalytic amounts of GSH caused a continuous reduction of 5,5-dithiobis (2-nitrobenzoic acid) (DTNB) to TNB.
Statistical analysis
The experimental data are presented as mean ± SD. Statistical analysis was carried out using Student's independent t-test.
Results
In vitro phase I metabolism of noscapine in HLMs and MLMs
Microsomal incubations with noscapine were carried out, and the metabolites were analysed by metabolomics using UPLC-ESI-QTOFMS. The data were deconvoluted and analysed by OPLS to filter out variations not directly related to the response. Multivariate data analysis using OPLS scores revealed two clusters corresponding to metabolites from the in vitro MLM incubation samples in the presence and absence of noscapine (Figure 1A). The loading plot (Figure 1B) revealed 10 major ions contributing to the group separation, as listed in Table 1. Ions I–X were proposed to be the parent compound and phase I metabolites of noscapine based on the trend plots (Figure S1). The chemical structures of these metabolites were elucidated on the basis of MS/MS fragmentation patterns (Table 2). The presence of unchanged noscapine (ion I) was determined from the occurrence of an ion of [M + H]+= 414.154 m/z, which gave a good match for C22H23NO7 with a mass error of 2.7 ppm. The main MS/MS fragmentation ions of noscapine were 353, 220 and 205. The MS/MS spectrum of noscapine and the fragmentation scheme are given in Table 2. A metabolite (ion II) produced through cleavage of the methylenedioxy group eluted at 2.96 min and yielded an ion of [M + H]+= 402.157 m/z, which gave a mass error of 5 ppm with the matched molecular formula C21H23NO7. The MS/MS fragmentation ions 208 and 193 correspond to the loss of 12 from noscapine's fragmentation ions 220 and 205, which further supported cleavage of the methylenedioxy group.
Figure 1.
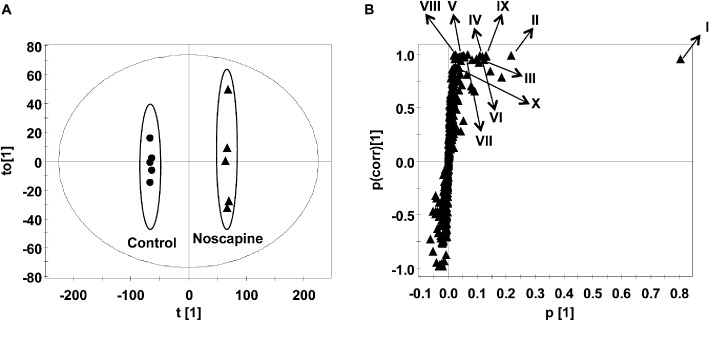
Identification of phase I metabolites in MLM incubation system. (A) Scores plot of an OPLS model from MLM incubation mixture without and with noscapine. (B) OPLS loading S-plot; noscapine and its phase I metabolites are labelled in the S-plot (I, II, III, IV, V, VI, VII, VIII, IX and X). The p(corr)[1] values represent the interclass difference, and the p[1] values represent the relevant abundance of ions.
Table 1.
Metabolites detected in mice urine dosed with noscapine (300 mg·kg−1 body weight)
| Symbol | Rt (min) | Observed m/z | Formula | Mass error (ppm) | Identity |
|---|---|---|---|---|---|
| I | 4.59 | 414.1542 | C22H23NO7[H+] | −2.7 | Noscapine |
| II | 2.96 | 402.1573 | C21H23NO7[H+] | 5.0 | Cleavage of methylenedioxy |
| III | 3.83 | 400.1394 | C21H21NO7[H+] | −0.5 | Demethylated noscapine |
| IV | 4.68 | 400.1411 | C21H21NO7[H+] | 3.7 | Demethylated noscapine |
| V | 3.36 | 400.1367 | C21H21NO7[H+] | −7.2 | Demethylated noscapine |
| VI | 4.50 | 430.1523 | C22H23NO8[H+] | 4.9 | Hydroxylated noscapine |
| VII | 3.97 | 400.1400 | C21H21NO7[H+] | 1.0 | Demethylated noscapine |
| VIII | 3.75 | 195.0658 | C10H10O4[H+] | 0.5 | Meconine |
| IX | 2.25 | 220.0960 | C12H13NO3[H+] | 5.9 | Cotarnine |
| X | 4.17 | 430.1508 | C22H23NO8[H+] | 1.4 | Hydroxylated noscapine |
| C1 | 2.56 | 578.1837 | C27H31NO13[H+] | −6.4 | Mono-glucuronidation product of cleavage of methylenedioxy |
| C2 | 2.26 | 578.1875 | C27H31NO13[H+] | −0.2 | Mono-glucuronidation product of cleavage of methylenedioxy |
| C3 | 2.78 | 576.1740 | C27H29NO13[H+] | 4.0 | Demethylated noscapine glucuronide |
| C4 | 3.16 | 576.1750 | C27H29NO13[H+] | 5.7 | Demethylated noscapine glucuronide |
| C5 | 3.66 | 606.1866 | C28H31NO14[H+] | 7.1 | Hydroxylated noscapine glucuronide |
| C6 | 4.35 | 606.1804 | C28H31NO14[H+] | −3.1 | Hydroxylated noscapine glucuronide |
| C7 | 3.13 | 388.1422 | C20H21NO7[H+] | 6.7 | Cleavage of methylenedioxy and demethylated noscapine |
| C8 | 2.87 | 562.1576 | C26H27NO13[H+] | 2.7 | Bisdemethylated noscapine glucuronide |
| C9 | 4.04 | 386.1240 | C20H19NO7[H+] | 0.0 | Bisdemethylated noscapine |
| C10 | 2.21 | 564.1691 | C26H29NO13[H+] | −4.6 | Cleavage of methylenedioxy and demethylated noscapine glucuronide |
| C11 | 2.73 | 564.1697 | C26H29NO13[H+] | −3.5 | Cleavage of methylenedioxy and demethylated noscapine glucuronide |
Table 2.
MS/MS fragments and proposed structures of metabolites of noscapine
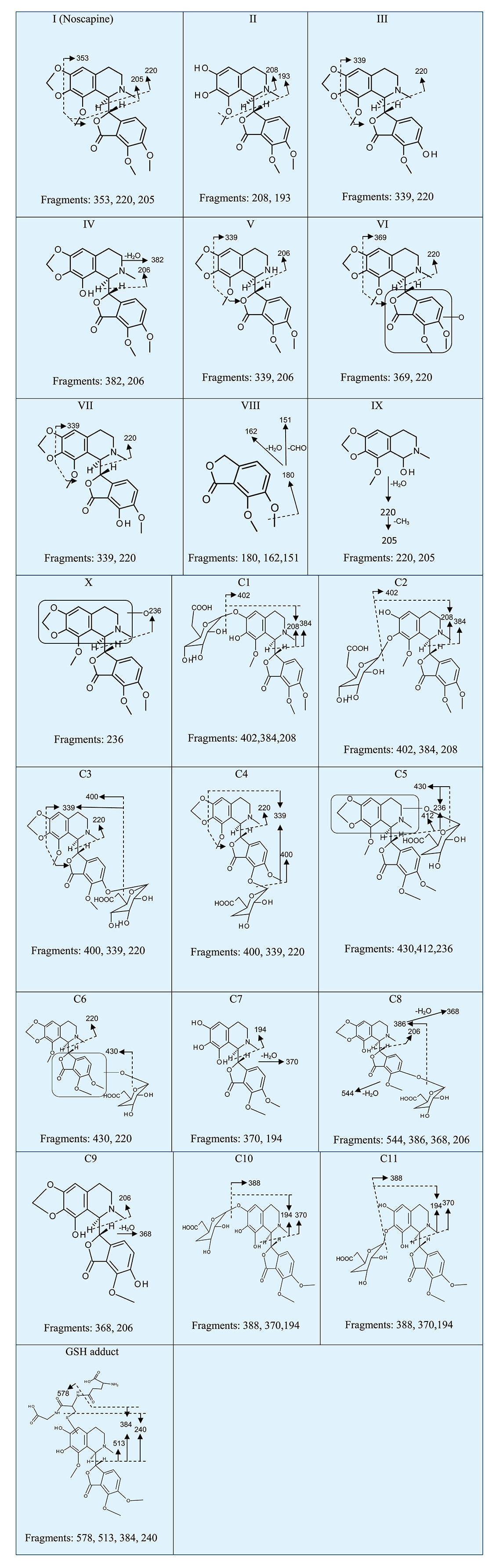 |
Four demethylated metabolites (ions III, IV, V, VII) of noscapine were identified at retention times of 3.83 min, 4.68 min, 3.36 min and 3.97 min, which all gave a match for C21H21NO7, with the mass errors of 0.5, 3.7, 7.2 and 1.0 ppm, respectively. By a comparison of MS/MS fragmentations between noscapine and demethylated metabolites (Table 2), other metabolites were identified to be O-demethylated noscapine, and one novel metabolite was elucidated to be N-demethylated noscapine.
Ions VI (retention time = 4.5 min) and X (retention time = 4.17 min) showed the accurate molecular weights 430.1523 and 430.1508, matching the molecular formula C22H23NO8. One fragment ion, 220, obtained from ion VI, was also detected in the fragments of the parent compound, indicating a hydroxylation site located in the lower fraction of noscapine, as shown in Figure 2 and Table 2. Similarly, one fragment ion 236 from ion X suggested the addition of 16 in the fragment ion 220 of the parent compound, indicating the location of hydroxylation site in the upper part of noscapine (Figure 2, Table 2). These two phase I metabolites have not previously been reported. The accurate molecular weight of metabolite VIII was 195.0658, which gave a match for C10H10O4. The fragments of this metabolite contain 180, 162 and 151. This metabolite was proposed to be meconine according to previous literature (Tsunoda and Yoshimura, 1981), and the fragment ions corresponding to the loss of CH3, CH3+ H2O, and CH3+ CHO, respectively.
Figure 2.
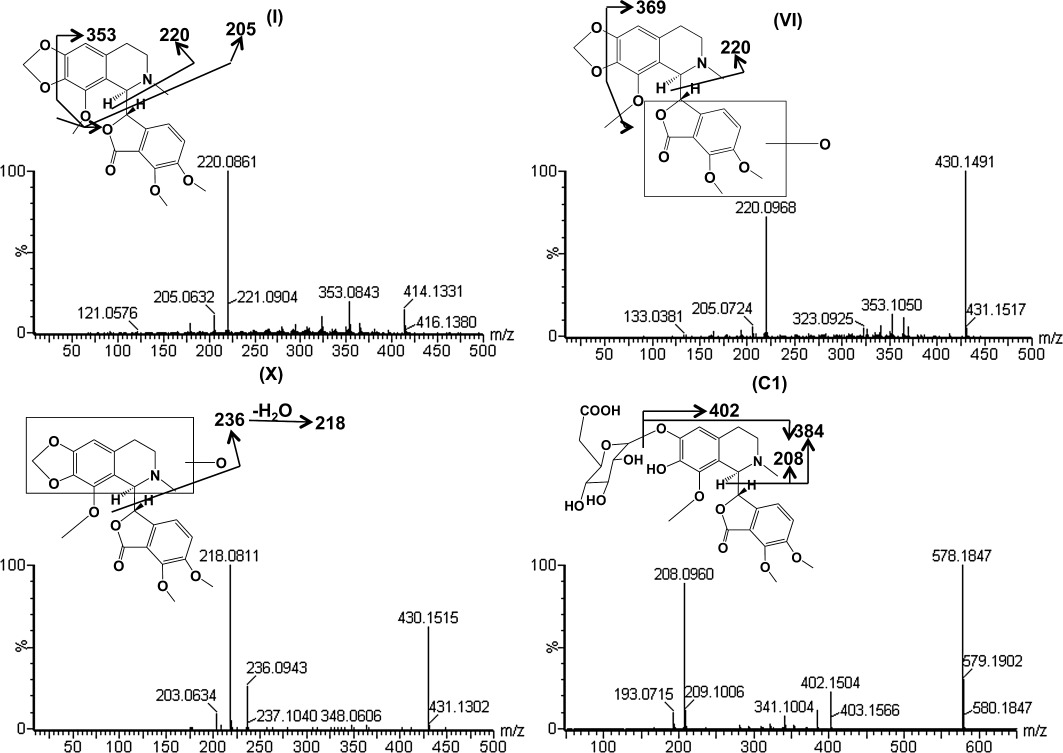
Tandem MS spectrum and proposed chemical structures of noscapine (I) and its representative metabolites (VI, X, C1).
The metabolite IX showed the ion [M + H]+= 220.0960, which gave a match for C12H13NO3. As previously reported (Tsunoda and Yoshimura, 1981), the mass spectrum of metabolite cotarnine didn't give the m/z 238, which is the ion of parent compound, and instead gave the major fragment ion 220 (loss of H2O). So metabolite IX was proposed to be cotarnine. All phase I metabolites were also produced by HLM with the comparative metabolism noted between MLM and HLM (Figure S3A). At the same microsomal protein concentration, the relative abundance of metabolites II, III, V, VI, VII and IX in MLM is higher than HLM. The production of IV and X was very similar between HLM and MLM. The formation of metabolite VIII was greater in HLM than in MLM.
Investigation of noscapine's metabolic behaviour in mice
As shown in the scores scatter plot (Figure 3A), urine samples from the noscapine-treated Sv/129 mice were significantly separated from the control group. All the phase I metabolites identified using this in vitro system were found in the loading plots (Figure 3B). Additionally, 11 more metabolites (C1–C11) were detected; trend plots are shown in Figure S2.
Figure 3.
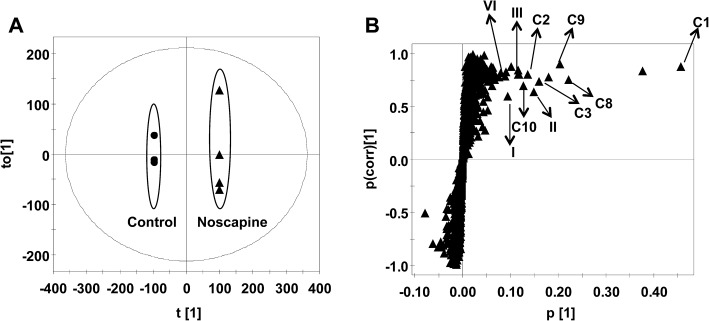
Identification of noscapine metabolites in male Sv/129 mice after oral gavage of noscapine (300 mg·kg−1 body weight). Scores plot of an OPLS model (A) and OPLS loading S-plot (B) of urine ions from control and noscapine-treated mice. Noscapine and some metabolites are labelled in the S-plot. The p(corr)[1] values represent the interclass difference, and the p[1] values represent the relevant abundance of ions.
Metabolite C1 and C2 exhibited a molecular ion at m/z 578.1837 and 578.1875, which matched C27H31NO13. The molecular ion yielded three fragment ions at m/z 402, 384 and 208 in the MS/MS experiment (Figure 2 and Table 2). The fragment ion 402 occurs from the loss of one molecule of glucuronic acid. The fragmentation mechanism for formation of the fragments 384 and 208 was similar with metabolite II. Based on this information, metabolites C1 and C2 were proposed to be the O-glucuronide of metabolite II at the catechol site of metabolite II.
The metabolites C3 (Rt= 2.78 min) and C4 (Rt= 3.16 min) yielded the same molecular formula C27H29NO13. The base peak at m/z 400 was formed through the elimination of 176 Da (one molecule of glucuronic acid) from the molecular ions. MS/MS fragments 339 and 220 were similar to the fragments from metabolites III and VII. Based on this date, these two metabolites were likely to be the glucuronides of demethylated noscapine (III and VII).
The metabolites C5 (Rt= 3.66 min, m/z= 606.1866) and C6 (Rt= 4.35 min, m/z= 606.1804) gave the match for C28H31NO14, with the mass error of 7.1 and 3.1 ppm, respectively. According to the MS/MS fragments and fragmentation mechanism shown in Table 2, these two metabolites were glucuronides of the hydroxylated noscapine X and VI.
The metabolite C7 exhibited the molecular ion at m/z 388.1422, which gave a match for C20H21NO7. Based on the MS/MS fragments and fragmentation mechanism shown in Table 2, this metabolite was suggested to be a metabolite of noscapine that underwent both the cleavage of methylenedioxy group and demethylation.
Metabolite C8 (Rt= 2.87 min, m/z= 562.1576) gave a match for the molecular formula C26H27NO13, which corresponded to the addition of glucuronic acid to metabolite C9 (glucuronide of bis-demethylated noscapine). The MS/MS fragments and proposed structure of C8 is given in Table 2.
The metabolite C9 (Rt= 4.04 min, m/z= 386.1240), matching for the molecular formula C20H19NO7, was proposed to be bis-demethylated noscapine based on the MS/MS fragmentation mechanism shown in Table 2.
Metabolites C10 (Rt= 2.21 min, m/z= 564.1691) and C11 (Rt= 2.73 min, m/z= 564.1697) gave a match for the molecular formula C26H19NO13. The fragmentation mechanism shown in Table 2 suggested the addition of one molecule of glucuronic acid to the catechol sites of metabolite C7. All these metabolites found in Sv/129 mice could also be detected in C57BL/6 mice. The relevant abundance of these metabolites in urine of these two kinds of mice is shown in Figure S3B.
The parent compound noscapine and its metabolites II, III, IX, C1, C2, C3, C7, C8 and C9 can be detected in faeces samples.
Screening the DMEs involved in the metabolism of noscapine
The identity of the human phase I and phase II enzymes involved in the metabolism of noscapine were investigated using recombinant human enzymes. The main phase I enzymes involved in the metabolism of noscapine include three allelic types of CYP2C9 (CYP2C9, CYP2C9*2 and CYP2C9*3) that exhibited highest catalytic activity towards the formation of metabolite II (Figure 4). For formation of metabolite III, CYP3A5, CYP1A1/2 and CYP2C19 showed higher catalytic activity than other CYP isoforms. For metabolite IV, CYP3A4/5 and corresponding fetal CYP3A7 showed higher catalytic activity. FMO-1 played a key role in the formation of metabolite VI. For metabolite VII, CYP3A5 was the major enzyme involved. For C–C cleavage reaction of noscapine to form the metabolite VIII and IX, many phase I enzyme isoforms were involved. CYP3A4/5 were the major enzymes involved in the formation of X metabolite.
Figure 4.
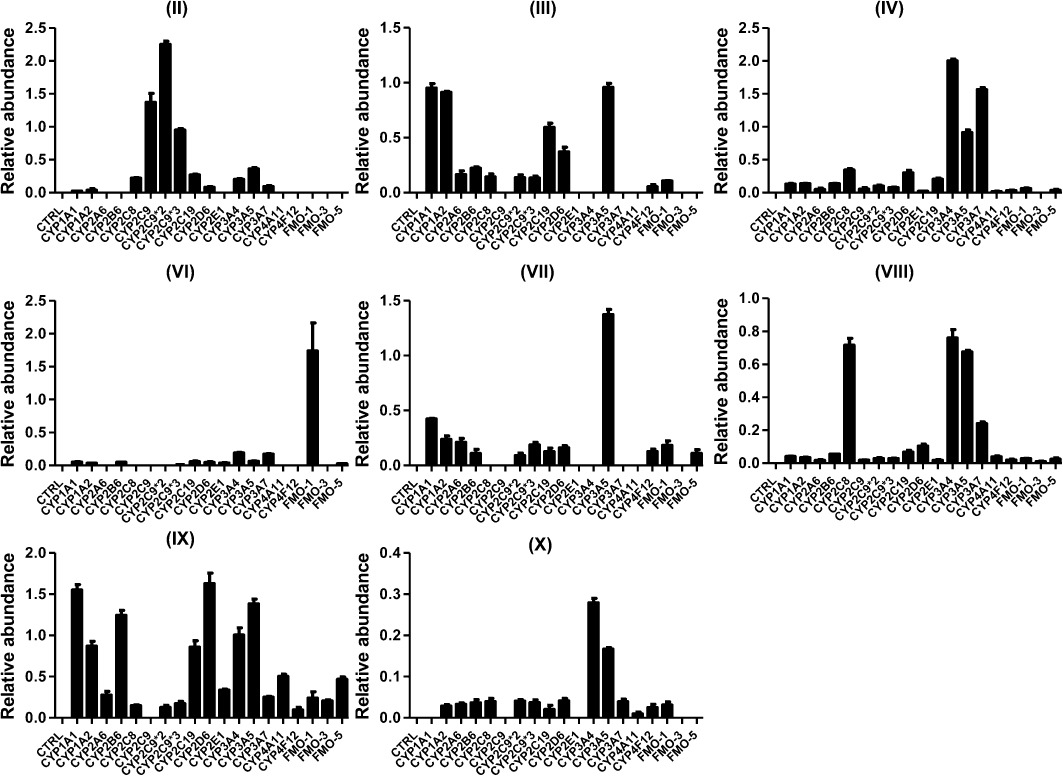
In vitro recombinant enzymes screening for identification of the phase I drug-metabolizing enzymes. The incubation system (200 µL) contained 50 mM Tris–HCl buffer solution (pH = 7.4), 2 pmol CYP isoforms or 5 µg FMO isoforms, 2 mM MgCl2, 100 µM noscapine and 1 mM freshly prepared NADPH. The relative abundance (peak area ratio of metabolites/internal standard) is displayed. The experiments were performed in triplicate, and the results are given as mean ± SD.
The ‘dual-activity’ incubation system was used to form glucuronides, and only glucuronides C1, C3 and C4 could be detected using this ‘dual-activity’ incubation system. As shown in Figure 5, screening of recombinant UGTs yielded the following results: (i) UGT2B7 was mainly involved in the formation of C1; (ii) UGT1A3 was mainly involved in the formation of C3; (iii) UGT1A1, UGT1A3 and UGT1A9 were involved in the formation of C4.
Figure 5.
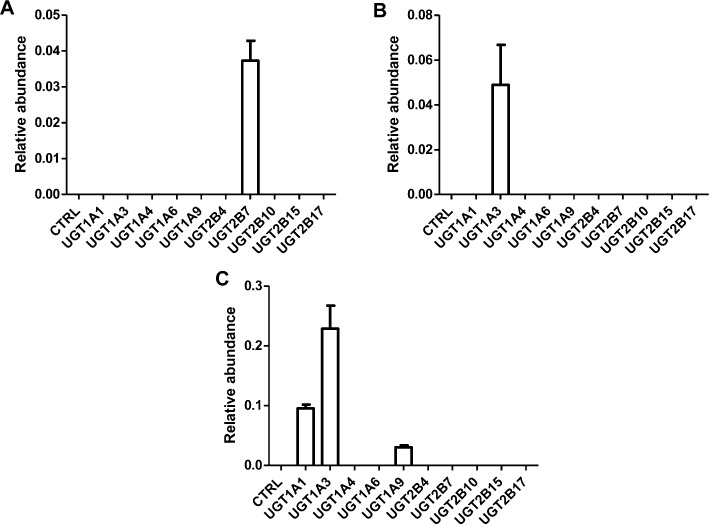
In vitro recombinant enzymes screening for identification of UGT forms involved in noscapine metabolism. The ‘dual-activity’ incubation system (200 µL) contains 50 mM Tris–HCl buffer solution (pH = 7.4), 5 µg of each UGT form, phase I enzyme, 2 mM MgCl2, 100 µM noscapine, 1 mM freshly prepared NADPH and UDPGA, and 25 µg·mL−1 alamethicin. To study the UGT forms involved in C1 formation, 0.5 pmol CYP2C9*2 was used. CYP3A5 (0.5 pmol) was employed to investigate the UGTs involved in the formation of glucuronide C3 and C4. The relative abundance (peak area ration of metabolites/internal standard) is given. The experiments were performed in triplicate, and the results are given as mean ± SD.
Evaluation of noscapine's bioactivation
For the HLM incubation system, OPLS-DA analysis revealed different clustering among the three groups (+NADPH+GSH group, +NADPH-GSH group, +GSH-NADPH groups) (Figure 6A). A GSH adduct of noscapine with the molecular ion m/z= 707.2222 was found, and the MS/MS fragmentation is displayed in Figure 6B. The formation of this adduct was HLM-dependent, NADPH-dependent and GSH-dependent (Figure 6C). The GSH adduct gave the MS/MS fragment ions of 578, 513, 384 and 240 (Table 2). The fragmentation mechanism is given in Figure 6B and Table 2. The fragment ion at m/z 578 was consistent with the elimination of a pyroglutamate residue (–129) of GSH. The fragment ion at m/z 384 was consistent with the elimination of a pyroglutamate residue (–129) of GSH in the fragment ion 513. The fragment ion at m/z 240 was formed via cleavage of the sulfur–carbon bond of the glutathionyl moiety in the fragment ion 513. This adduct was also identified in the MLM incubation system (data not shown). A further experiment was carried out to identify the human phase I enzymes involved in the bioactivation of noscapine using the MRM method with the transition 707 → 384. The results showed that CYP2C9, CYP2C9*2, CYP2C9*3, CYP1A2, CYP2D6, CYP3A5, CYP1A1, CYP2C8, CYP3A7, CYP3A4 and CYP2C19 were involved in the bioactivation of noscapine (Figure 7A). The GSH adduct of noscapine was not detected in urine, faeces and liver extracts of noscapine-treated C57BL/6 mice.
Figure 6.
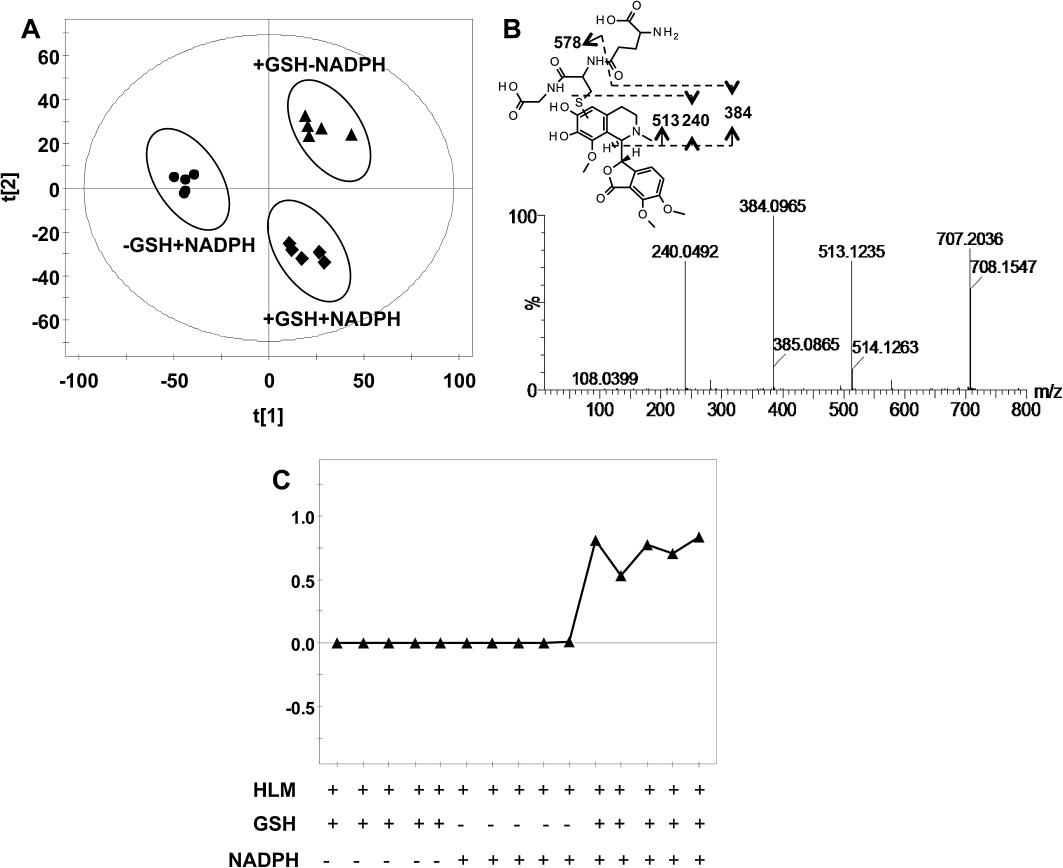
Metabolomic analysis of GSH adducts in the HLM incubation system. (A) Scores scatter plot of OPLS-DA model to characterize three mice groups (+NADPH+GSH, +NADPH-GSH, -NADPH+GSH). The t[1] and t[2] values represent the score of each sample in principle component 1 and 2, respectively. (B) The MS/MS fragments and proposed structure of the GSH adduct. (C) Trend plots of GSH adduct formation.
Figure 7.
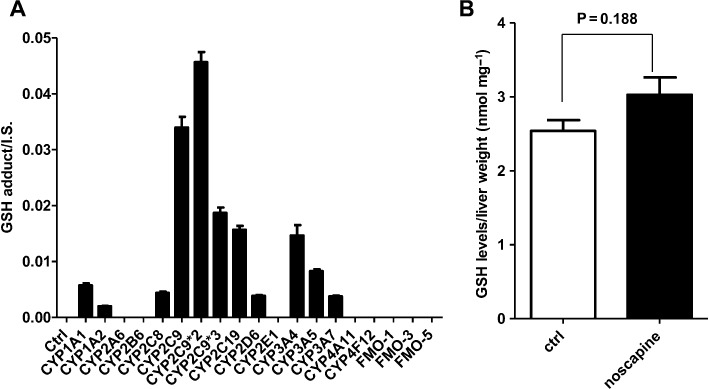
MRM monitoring of the GSH adduct of noscapine produced by recombinant enzymes (A), and GSH determination (B) in liver extracts from control (n= 3) and noscapine-treated (n= 5) C57BL/6 mice. For GSH determination, the results are given as mean ± SD.
The determination of total GSH level in liver and serum chemistry
Compared with the control group, the GSH level in liver obtained from noscapine-treated C57BL/6 mice did not show significant alterations (Figure 7B). There was no also significant difference in ALT, ALP and AST activity between the control group and the noscapine treated group (Figure S4).
Discussion and conclusions
Drug metabolism plays a key role in determining the efficiency and safety of drugs (Lin and Lu, 1997). On the one hand, drugs undergo extensive first-pass metabolism elimination to form numerous stable metabolites, most of which are pharmacologically inactive, which may result in significant alteration of drugs' therapeutic potential. On the other hand, in some cases, metabolism can catalyse the transformation of drugs to reactive metabolites that can induce adverse effects. Mice are a commonly used model to determine anti-tumour activity. The anti-tumour activities of noscapine towards multiple tumours have been demonstrated using mouse models, including melanoma (Ke et al., 2000) and T cell lymphoma (Landen et al., 2002). Additionally, compared with low antitussive doses, relatively high anti-tumour doses of noscapine might have unfavourable pharmacokinetic and metabolic behaviour. In a previous study, the pharmacokinetics and bioavailability of noscapine in mice was carried out to determine its anti-tumour potential (Aneja et al., 2007). However, detailed metabolic information was not given in this study. Therefore, the first task of the present study was to analyse the metabolism of noscapine at a standard anti-tumour dose (300 mg·kg−1 body weight) in mice (Landen et al., 2004), in order to facilitate a mechanistic bridge between the metabolism and pharmacological activity of the drug.
Metabolomics, a powerful tool for elucidation of the metabolic pathway of xenobiotics, was applied to the present study of noscapine. In addition to the phase I metabolites found in rats, rabbits and humans dosed with noscapine, several novel phase I metabolites were detected in mice, including a N-demethylated metabolite (V), two hydroxylated metabolites (VI and X), one metabolite undergoing both demethylation and cleavage of methylenedioxy group (C7), and one bisdemethylated metabolite (C9). In previous studies (Tsunoda and Yoshimura, 1981), the levels of phase I metabolites significantly increased after hydroxylation of urine of rats, human and rabbits using β-glucuronidase, suggesting the existence of glucuronide as one of these phase I metabolites. In the present study, numerous corresponding glucuronides of phase I metabolites were discovered in the urine of Sv/129 mice and C57BL/6 mice treated with noscapine, and the structures of these glucuronides were elucidated using MS/MS fragmentation patterns.
Identification of the enzymes involved in noscapine metabolism is an important task for deciphering the metabolic pathway, which facilitates the understanding of an individual's response to drugs and drug–drug interactions. In the present study, the phase I and phase II enzymes involved in the metabolism of noscapine were identified. Earlier studies demonstrated that noscapine exhibits competitive and non-competitive inhibition towards CYP3A4/5 and CYP2C9 (Fang et al., 2010), indicating the potential importance of these two P450s in noscapine's metabolism. The present study showed that CYP3A4/5 and CYP2C9 exhibited high catalytic activity towards the formation of many of noscapine's metabolites. CYP3A7, the predominant P450 expressed in the human fetal liver, accounts for 87–100% of total fetal hepatic CYP3A content (Pang et al., 2012). Similar to the CYP3A4/5 results, CYP3A7 plays an important role in the metabolism of noscapine, which provides useful information for fetal exposure of noscapine. These results also support the drug–drug interactions noted when noscapine was used in combination with other anti-tumour drugs (e.g. doxorubicin, paclitaxel, etc.). The formation of aglycone metabolites of doxorubicin are produced by CYP3A (Choi et al., 2011), and pregnane X receptor (PXR)-induced expression of CYP3A4 is significantly associated with lower doxorubin clearance (Sandanaraj et al., 2008). The 3′-p-hydroxypaclitaxel reaction was mainly catalysed by CYP3A4, and thus substrates and potent inhibitors of CYP3A4 might interfere with its pharmacokinetic behaviour (Cresteil et al., 1994). Given that noscapine is good substrate and strong inhibitor of CYP3A, drug–drug interactions between noscapine and these anti-tumour drugs could be expected, and thus further clinical monitoring is needed. Additionally, gender and age differences should be considered when attempting to explain the metabolism and pharmacokinetic behaviour of noscapine. It is generally regarded that women exhibit higher CYP3A activity (Scandlyn et al., 2008). These factors might result in individual differences in noscapine's metabolism. Due to the absence of standards for the phase I metabolites of noscapine, the ‘dual-activity’ incubation system was used to investigate the UGTs involved in its metabolism. Using this ‘dual-activity’ incubation system, three glucuronides (C1, C3, C4) were detected. UGT2B7 was involved in the glucuronidation of catechol sites of II, consistent with a previous report that UGT2B7 plays a key role in the glucuronidation of catechol compounds (Cheng et al., 1998). UGT1A1, UGT1A3 and UGT1A9 were involved in the glucuronidation of phenol hydroxyl group of noscapine's demethylated metabolites. It should be noted that mice might have different UGT forms than humans (Buckley and Klaassen, 2007). Therefore, although the same glucuronides also exist in mice, the UGTs involved in glucuronidation in mice might be different. It should be noted that the formation of meconine and cotarnine are not equimolar in recombinant enzymes and mice, as revealed in a previous study (Tsunoda and Yoshimura, 1981).
A GSH adduct of an ortho-quinone reactive intermediate of noscapine was also detected using the liver microsomal incubation system supplemented with NADPH and GSH, revealing that the same enzymes producing the demethylenated metabolite (II) are involved in the formation of the reactive intermediate of noscapine. Ortho-quinones cause toxicity via covalent interactions with biomacromolecules, depletion of GSH, and oxidative stress due to redox cycling with the semiquinones/catechol forms (Monks et al., 1992; Bolton et al., 2000; Monks and Jones, 2002; Jian et al., 2009). Due to the potential risks resulting from noscapine's bioactivation, in vivo studies were carried out. The GSH adduct was not detected in either liver, urine or faeces obtained from the noscapine-treated mice. Additionally, the N-acetyl-L-cysteine (NAC) conjugates (mercapturic acid conjugates), produced through a series of biotransformation steps of GSH adducts mediated by multiple enzymes including γ-glutamylcysteine transpeptidase, dipeptidase and N-acetyl transferase (Jian et al., 2009), were also not detected. Therefore, the bioactivation of noscapine might not or only weakly occur (below detection limits) in vivo. In agreement with this result, hepatic GSH levels were not significantly changed after treatment with noscapine. Additionally, analysis of ALT, AST and ALP levels did not reveal significant liver toxicity after noscapine treatment. For longer-term toxicity, the experiment carried out by Ke et al. (2000) demonstrated that noscarpine, administered to C57BL/6 mice for three months, did not induce any obvious specific toxicity towards normal organs, including liver. The discrepancy between the in vitro bioactivation potential and in vivo results might be explained by the following: (i) Noscapine can inactivate the enzyme(s) catalysing its bioactivation (mainly CYP2C9). With the enzymes inactivated, the amount of noscapine undergoing metabolism at its methylenedioxyphenyl group to the catechol and ortho-quinone would be lower. This explanation may also apply for another methylenedioxyphenyl-containing compound paroxetine for which a disconnection between safety and bioactivation potential exists (Zhao et al., 2007). (ii) The glucuronidation pathway might effectively compete with the bioactivation pathway for the catechol metabolite of noscapine (II).
In conclusion, the present results provide a more comprehensive analysis of the metabolism of noscapine (Figure 8), including deciphering the detailed metabolism map and evaluation of bioactivation potential. These results provide important information for the development of noscapine for anti-tumour therapy.
Figure 8.
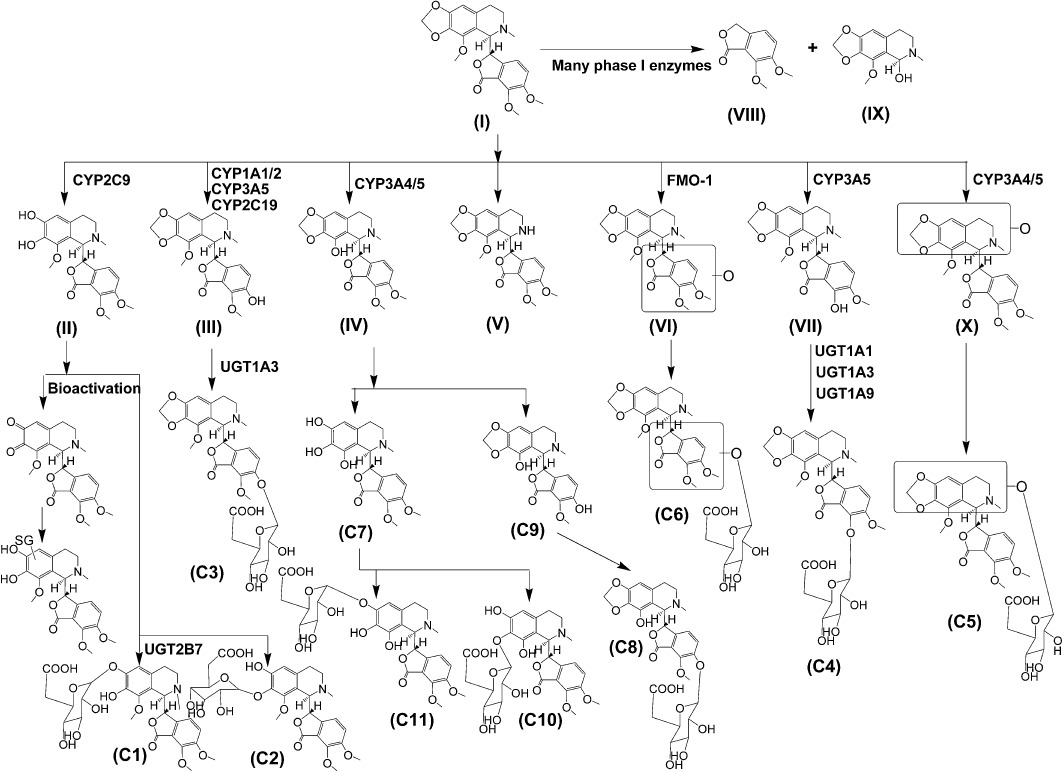
Metabolic map of noscapine.
Glossary
- CYP
cytochrome P450
- DME
drug-metabolizing enzymes
- FMO
flavin mono-oxygenase
- GSH
glutathione
- HLM
human liver microsome
- MLM
mice liver microsome
- UDPGA
uridine 5′-diphosphoglucuronic acid trisodium salt
- UGT
UDP-glucuronosyltransferase
- UPLC-ESI-QTOFMS
ultra performance chromatography electrospray ionization quadrupole time-of-flight MS
Statement of conflicts of interests
There is no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Trend plots of phase I metabolites of noscapine in MLMs with and without noscapine. Metabolites codes are the same with those in Figure 1.
Figure S2 Trend plots of metabolites C1–C11 found in male 129 mice after oral gavage of 300mg·kg−1 BW.
Figure S3 The comparison of relative quantitation of metabolites. (A) The comparison of relative quantitative of phase I metabolites between MLM and HLM incubation system. (B) The comparison of relative quantitative of metabolites in the urine between male Sv/129 mice and C57BL/6 mice.
Figure S4 Determination of ALT (A), AST (B) and ALP (C) activity in serum obtained from control (n = 3) and noscapine-treated (n = 5) C57BL/6 mice. The results were given as Mean ± SD.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aneja R, Katyal A, Chandra R. Modulatory influence of noscapine on the ethanol-altered hepatic biotransformation system enzymes, glutathione content and lipid peroxidation in vivo in rats. Eur J Drug Metab Pharmacokinet. 2004;29:157–162. doi: 10.1007/BF03190592. [DOI] [PubMed] [Google Scholar]
- Aneja R, Dhiman N, Idnani J, Awasthi A, Arora SK, Chandra R, et al. Preclinical pharmacokinetics and bioavailability of noscapine, a tubulin-binding anticancer agent. Cancer Chemother Pharmacol. 2007;60:831–839. doi: 10.1007/s00280-007-0430-y. [DOI] [PubMed] [Google Scholar]
- Antolino-Lobo I, Meulenbelt J, Nijmeijer SM, Scherpenisse P, van den Berg M, van Duursen MBM. Differential roles of phase I and phase II enzymes in 3,4-methylendioxymethamphetamine-induced cytotoxicity. Drug Metab Dispos. 2010;38:1105–1112. doi: 10.1124/dmd.110.032359. [DOI] [PubMed] [Google Scholar]
- Baillie TA. Future of toxicology-metabolic activation and drug design: challenges and opportunities in chemical toxicology. Chem Res Toxicol. 2006;19:889–893. doi: 10.1021/tx060062o. [DOI] [PubMed] [Google Scholar]
- Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Roles of quinines in toxicity. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- Buckley DB, Klaassen CD. Tissue- and Gender-specific mRNA expression of UDP-glucuronosyltransferases (UGTs) in mice. Drug Metab Dispos. 2007;35:121–127. doi: 10.1124/dmd.106.012070. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Rios GR, King CD, Coffman BL, Green MD, Mojarrabi B, et al. Glucuronidation of catechol estrogens by expressed human UDP-glucuronosyltransferases (UGTs) 1A1, 1A3, and 2B7. Toxicol Sci. 1998;45:52–57. doi: 10.1006/toxs.1998.2494. [DOI] [PubMed] [Google Scholar]
- Choi JS, Piao YJ, Kang KW. Effects of quercetin on the bioavailability of doxorubicin in rats: role of CYP3A4 and P-gp inhibition by quercetin. Arch Pharm Res. 2011;34:607–613. doi: 10.1007/s12272-011-0411-x. [DOI] [PubMed] [Google Scholar]
- Chougule M, Patel AR, Jackson T, Singh M. Antitumor activity of noscapine in combination with doxorubicin in triple negative breast cancer. PLoS ONE. 2011a;6:e17733. doi: 10.1371/journal.pone.0017733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chougule M, Patel AR, Sachdeva P, Jackson T, Singh M. Anticancer activity of noscapine, an opioid alkaloid in combination with cisplatin in human non-small cell lung cancer. Lung Cancer. 2011b;71:271–282. doi: 10.1016/j.lungcan.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresteil T, Monsarrat B, Alvinerie P, Treluyer JM, Vieira I, Wright M. Taxol metabolism by human liver microsomes: identification of cytochrome P450 isozymes involved in its biotransformation. Cancer Res. 1994;54:386–392. [PubMed] [Google Scholar]
- Empey DW, Laitinen LA, Young GA, Bye CE, Hughes DTD. Comparison of the antitussive effects of codeine phosphate 20 mg, dextromethorphan 30 mg and noscapine 30 mg using citric acid-induced cough in normal subjects. Eur J Clin Pharmacol. 1979;16:393–397. doi: 10.1007/BF00568199. [DOI] [PubMed] [Google Scholar]
- Fang ZZ, Zhang YY, Ge GB, Huo H, Liang SC, Yang L. Time-dependent inhibition (TDI) of CYP3A4 and CYP2C9 by noscapine potentially explains clinical noscapine-warfarin interaction. Br J Clin Pharmacol. 2010;69:193–199. doi: 10.1111/j.1365-2125.2009.03572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang ZZ, Zhang YY, Wang XL, Cao YF, Huo H, Yang L. Bioactivation of herbal constituents: simple alerts in the complex system. Expert Opin Drug Metab Toxicol. 2011;7:989–1007. doi: 10.1517/17425255.2011.586335. [DOI] [PubMed] [Google Scholar]
- Giri S, Idle JR, Chen C, Zabriskie TM, Krausz KW, Gonzalez FJ. A metabolomic approach to the metabolism of the areca nut alkaloids arecoline and arecaidine in the mouse. Chem Res Toxicol. 2006;19:818–827. doi: 10.1021/tx0600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson T, Chougule MB, Ichite N, Patlolla RR, Singh M. Antitumor activity of noscapine in human non-small cell lung cancer xenograft model. Cancer Chemother Pharmacol. 2008;63:117–126. doi: 10.1007/s00280-008-0720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian W, Yao M, Zhang D, Zhu M. Rapid detection and characterization of in vitro and urinary N-acetyl-L-cysteine conjugates using quadrupole-linear ion trap mass spectrometry and polarity switching. Chem Res Toxicol. 2009;22:1246–1255. doi: 10.1021/tx900035j. [DOI] [PubMed] [Google Scholar]
- Kalgutkar AS, Solia JR. Minimising the potential for metabolic activation in drug discovery. Expert Opin Drug Metab Toxicol. 2005;1:91–142. doi: 10.1517/17425255.1.1.91. [DOI] [PubMed] [Google Scholar]
- Ke Y, Ye K, Grossniklaus HE, Archer DR, Joshi HC, Kapp JA. Noscapine inhibits tumor growth with little toxicity to normal tissues or inhibition of immune responses. Cancer Immunol Immunother. 2000;49:217–225. doi: 10.1007/s002620000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen JW, Lang R, McMahon SJ, Rusan NM, Yvon AM, Adams AW, et al. Noscapine alters microtubule dynamics in living cells and inhibits the progression of melanoma. Cancer Res. 2002;62:4109–4114. [PubMed] [Google Scholar]
- Landen JW, Hau V, Wang M, Davis T, Ciliax B, Wainer BH, et al. Noscapine crosses the blood-brain barrier and inhibits glioblastoma growth. Clin Cancer Res. 2004;10:5187–5201. doi: 10.1158/1078-0432.CCR-04-0360. [DOI] [PubMed] [Google Scholar]
- Li F, Lu J, Ma X. Profiling the reactive metabolites of xenobiotics using metabolomic technologies. Chem Res Toxicol. 2011a;24:744–751. doi: 10.1021/tx200033v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Patterson AD, Hofer CC, Krausz KW, Gonzalez FJ, Idle JR. A comprehensive understanding of thioTEPA metabolism in the mouse using UPLC-ESI-QTOFMS-based metabolomics. Biochem Pharmacol. 2011b;81:1043–1053. doi: 10.1016/j.bcp.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Lu AYH. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev. 1997;49:403–449. [PubMed] [Google Scholar]
- Liu M, Luo XJ, Liao F, Lei XF, Dong WG. Noscapine induces mitochondria-mediated apoptosis in gastric cancer cells in vitro and in vivo. Cancer Chemother Pharmacol. 2011;67:605–612. doi: 10.1007/s00280-010-1356-3. [DOI] [PubMed] [Google Scholar]
- Mahmoudian M, Rahimi-Moghaddam P. The anti-cancer activity of noscapine: a review. Recent Pat Anticancer Drug Discov. 2009;4:92–97. doi: 10.2174/157489209787002524. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks TJ, Jones DC. The metabolism and toxicity of quinines, quinonimines, quinine methides, and quinine-thioethers. Curr Drug Metab. 2002;3:425–438. doi: 10.2174/1389200023337388. [DOI] [PubMed] [Google Scholar]
- Monks TJ, Hanzlik RP, Cohen GM, Ross D, Graham DG. Quinone chemistry and toxicity. Toxicol Appl Pharmacol. 1992;112:2–16. doi: 10.1016/0041-008x(92)90273-u. [DOI] [PubMed] [Google Scholar]
- Naik PK, Chatterji BP, Vangapandu SN, Aneja R, Chandra R, Kanteveri S, et al. Rational design, synthesis and biological evaluations of amino-noscapine: a high affinity tubulin-binding noscapinoid. J Comput Aided Mol Des. 2011;25:443–454. doi: 10.1007/s10822-011-9430-4. [DOI] [PubMed] [Google Scholar]
- Pang XY, Cheng J, Kim JH, Matsubara T, Krausz KW, Gonzalez FJ. Expression and regulation of human fetal-specific CYP3A7 in mice. Endocrinology. 2012;153:1453–1463. doi: 10.1210/en.2011-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter R, Parry EM, Parry JM. Morphological transformation of an established-hamster dermal cell with the anti-tussive agent noscapine. Mutagenesis. 1992;7:205–209. doi: 10.1093/mutage/7.3.205. [DOI] [PubMed] [Google Scholar]
- Sandanaraj E, Lal S, Selvarajan V, Ooi LL, Wong ZW, Wong NS, et al. PXR pharmacogenetics: association of haplotypes with hepatic CYP3A4 and ABCB1 messenger RNA expression and doxorubicin clearance in asian breast cancer patients. Clin Cancer Res. 2008;14:7116–7126. doi: 10.1158/1078-0432.CCR-08-0411. [DOI] [PubMed] [Google Scholar]
- Santoshi S, Naik PK, Joshi HC. Rational design of novel anti-microtubule agent (9-azido-noscapine) from quantitative structure activity relationship (QSAR) evaluation of noscapinoids. J Biomol Screen. 2011;16:1047–1058. doi: 10.1177/1087057111418654. [DOI] [PubMed] [Google Scholar]
- Scandlyn MJ, Stuart EC, Rosengren RJ. Sex-specific differences in CYP450 isoforms in humans. Expert Opin Drug Metab Toxicol. 2008;4:413–424. doi: 10.1517/17425255.4.4.413. [DOI] [PubMed] [Google Scholar]
- Schuler M, Muehlbauer P, Guzzie P, Eastmond DA. Noscapine hydrochloride disrupts the mitotic spindle in mammalian cells and induces aneuploidy as well as polyploidy in cultured human lymphocytes. Mutagenesis. 1999;14:51–56. doi: 10.1093/mutage/14.1.51. [DOI] [PubMed] [Google Scholar]
- Tsunoda N, Yoshimura H. Metabolic fate of noscapine. III. Further studies on identification and determination of the metabolites. Xenobiotica. 1981;11:23–32. doi: 10.3109/00498258109045268. [DOI] [PubMed] [Google Scholar]
- Tucker GT, Lennard MS, Ellis SW, Woods HF, Cho AK, Lin LY, et al. The demethylenation of methylenedioxymethamphetamine (‘ecstasy’) by debrisoquine hydroxylase (CYP2D6) Biochem Pharmacol. 1994;47:1151–1156. doi: 10.1016/0006-2952(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Walsh JS, Miwa GT. Bioactivation of drugs: risk and drug design. Annu Rev Pharmacol Toxicol. 2011;51:145–167. doi: 10.1146/annurev-pharmtox-010510-100514. [DOI] [PubMed] [Google Scholar]
- Yan Z, Caldwell GW. Metabolic assessment in liver microsomes by co-activating cytochrome P450s and UDP-glucuronosyltransferases. Eur J Drug Metab Pharmacokinet. 2003;28:223–232. doi: 10.1007/BF03190489. [DOI] [PubMed] [Google Scholar]
- Ye KQ, Ke Y, Keshava N, Shanks J, Kapp JA, Tekmal RR, et al. Opium alkaloid noscapine is an antitumor agent that arrests metaphase and induces apoptosis in dividing cells. Proc Natl Acad Sci U S A. 1998;95:1601–1606. doi: 10.1073/pnas.95.4.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao SX, Dalvie DK, Kelly JM, Soglia JR, Frederick KS, Smith EB, et al. NADPH-dependent covalent binding of [3H]paroxetine to human liver microsomes and S-9 fractions: identification of an electrophilic quinine metabolite of paroxetine. Chem Res Toxicol. 2007;20:1649–1657. doi: 10.1021/tx700132x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.