

Abstract
BACKGROUND AND PURPOSE
Antidepressants are known to interact with the opioid system through mechanisms not completely understood. We previously reported that tricyclic antidepressants act as agonists at distinct opioid receptors. Here, we investigated the effect of the atypical antidepressant mianserin at cloned and native opioid receptors.
EXPERIMENTAL APPROACH
Effects of mianserin were examined in CHO cells transfected with human opioid receptors, C6 glioma cells and rat brain membranes by the use of radioligand binding and functional assays including the stimulation of [35S]GTPγS binding and MAPK phosphorylation.
KEY RESULTS
Mianserin displayed 12- and 18-fold higher affinity for κ- than µ- and δ-opioid receptors respectively. In [35S]GTPγS assays, mianserin selectively activated κ-opioid receptors. The agonist activity was antagonized by the selective κ-opioid blocker nor-binaltorphimine (nor-BNI). The mianserin analogue mirtazapine also displayed κ-opioid agonist activity. Mianserin and mirtazapine increased ERK1/2 phosphorylation in CHO cells expressing κ-opioid receptors and C6 cells, and these effects were antagonized by nor-BNI. In rat striatum and nucleus accumbens, mianserin stimulated [35S]GTPγS binding in a nor-BNI-sensitive manner with maximal effects lower than those of the full κ-opioid agonists (–)-U50,488 and dynorphin A. When combined, mianserin antagonized the effects of the full κ-opioid receptor agonists in [35S]GTPγS assays and reduced the stimulation of p38 MAPK and ERK1/2 phosphorylation by dynorphin A.
CONCLUSIONS AND IMPLICATIONS
In different cell systems, mianserin directly activates κ-opioid receptors, displaying partial agonist activity at brain receptors. Thus, this property appears to be a common feature of different classes of antidepressants.
Keywords: atypical antidepressants, cloned and native κ-opioid receptor, radioligand binding assays, [35S]GTPγS binding assay, MAPK phosphorylation, CHO cells, rat C6 glioma cells, mouse primary neurons
Introduction
Antidepressants, and in particular tricyclic antidepressants (TCAs), have long been known to interact with the opioid system. A number of studies have shown that the administration of opioid receptor antagonists reverses the anti-nociceptive effects of TCAs (Biegon and Samuel, 1980; Gray et al., 1998; Marchand et al., 2003; Ozturk et al., 2006; Benbouzid et al., 2008a,b), and that TCAs potentiate morphine-induced analgesia in both animals and humans (Mico et al., 2006). Moreover, in animal behavioural tests predictive of antidepressant activity in humans, such as the forced swimming and learned helplessness tests, the antidepressant action of TCAs has been found to be antagonized by blockade of opioid receptors (Devoize et al., 1982; Tejedor-Real et al., 1995; Besson et al., 1999). Although these studies support the involvement of the opioid system in the therapeutic activity of TCAs, how these drugs act on opioid neurotransmission has not been completely elucidated.
We have recently reported that a number of TCAs bind to and activate distinct opioid receptors with a preference for either δ- or κ-opioid receptor subtypes (Onali et al., 2010; receptor nomenclature follows Alexander et al., 2011). For instance, amoxapine displayed higher potency and efficacy at δ-opioid receptors, whereas amitriptyline, nortriptyline, desipramine and imipramine showed higher agonist activity at κ-opioid receptors. At the µ-opioid receptor, these drugs had low affinity and no significant agonist activity. From a pharmacodynamic point of view, the agonist activity at opioid receptors appears to be a unique property. In fact, these drugs, besides blocking monoamine transporters, have generally been found to behave as antagonists of neurotransmitter receptors (Baldessarini, 2006). As the receptor stimulation occurred at concentrations compatible with the brain levels reached by these drugs, we proposed that the direct agonist activity at opioid receptors could contribute to the analgesic and antidepressant actions of TCAs. However, it remains to be examined whether this property is typical of TCAs or shared by other structurally different antidepressants.
Mianserin is a tetracyclic compound that is approved for the treatment of major depression in several countries. A number of studies have indicated that mianserin differs from TCAs not only chemically but also in its pharmacological profile. In fact, unlike several TCAs, mianserin does not inhibit neuronal 5-HT reuptake and in the brain is only weakly active in blocking noradrenaline uptake (Marshall, 1983). Moreover, mianserin shows low affinity for cholinergic muscarinic receptors, but it has been found to block with high-affinity 5-HT2 receptors, histamine H1 receptors and α2-adrenoceptors (Peroutka and Snyder, 1981; Richelson and Nelson, 1984). Indeed, the antidepressant effect of mianserin is considered to mainly derive from the blockade of pre-synaptic, auto- and hetero-α2-adrenoceptors, with the consequent increase of noradrenergic and 5-hydroxytryptaminergic neurotransmission (Marshall, 1983; Pinder, 1985). However, like TCAs, mianserin has been reported to produce analgesia that was blocked by opioid antagonists, indicating that also this antidepressant acts through the opioid system (Reichenberg et al., 1985; Schreiber et al., 1998). Moreover, mianserin has been shown to either potentiate or antagonize the analgesic effect of the selective κ-opioid receptor agonist U-50 488 (Ho and Takemori, 1989).
In the present study, we investigated the actions of mianserin on opioid receptors by using both heterologous and homologous expression systems. We also examined the effects of mirtazapine, as this newer antidepressant is structurally and pharmacologically similar to mianserin (Croom et al., 2009).
Part of this study has been presented in an abstract form (Olianas et al., 2011).
Methods
Cell culture
CHO-K1 cells (American Type Culture Collection, Manassas, VA, USA) were grown as a monolayer culture in tissue culture flasks that were incubated at 37°C in a humidified atmosphere (5% CO2) in Ham's F12 medium (Invitrogen, Carlsbad, CA) containing l-glutamine and sodium bicarbonate and supplemented with 10% fetal calf serum (FCS; Invitrogen), 0.5% penicillin/streptomycin. CHO-K1 cells stably expressing the human δ-opioid receptor (CHO/DOP), κ-opioid (CHO/KOP), µ-opioid receptor-1 (CHO/MOP) were generated as previously described (Olianas et al., 2006).
C6 rat glioma cells (European Collection of Cell Cultures, Porton Down, Salisbury, UK) were grown in Ham's F12 supplemented with 2 mM l-glutamine, 0.5% penicillin/streptomycin and 10% FCS in a humidified 95% air and 5% CO2 at 37°C.
Primary cultures of mouse neurons
All animal care and experimental procedures were in accordance with the European Communities Council Directive of November 24 1986 (86/609/EEC) and with the principles of Laboratory Animal Care in Italy (D.L. 116/92). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). The total of 44 animals were used in the experiments described here.
CD-1 mice were obtained from Harlan (Udine, Italy). One-day-old mice of both sexes were used to prepare primary cultures of neuronal cells of mouse striatum and hippocampus. The animals were anaesthetized by hypothermia and killed by decapitation. The dorsal and ventral striatum and the dorsal hippocampus were dissected from coronal brain slices, and the tissue fragments were incubated with 0.2% trypsin (type IX from porcine pancreas; Sigma Aldrich, St. Louis, MO, USA) for 30 min at 30°C. Thereafter, 1% soya bean trypsin inhibitor and 0.1 mg·mL−1 DNase type I (Sigma Aldrich) were added, and the incubation was continued for additional 5 min. Following trituration, the cell suspension was layered on the top of a 4% BSA solution in complete Neurobasal medium and centrifuged at 1200×g for 10 min. The cells were re-suspended in complete Neurobasal medium and plated on either glass coverslips (Bellco Brand, Electron Microscopy Sciences, Fort Washington, PA, USA) or six-well plates pre-coated with 0.01% L-poly-lysine (Sigma Aldrich) at the density of 1 × 105 cells and 1 × 106 per well respectively. Cultures were placed in a humidified incubator and maintained at 37°C in 5% CO2. Cells were used 8–10 days after plating.
Cell membrane preparation
Cells were washed with ice-cold PBS (pH 7.4), scraped into an ice-cold buffer containing 10 mM HEPES/NaOH (pH 7.4) and 1 mM EDTA and lysed with a Dounce tissue grinder. The cell lysate was centrifuged at 1000×g for 2 min at 4°C. The supernatant was collected and centrifuged at 32 000×g for 20 min at 4°C. The pellet was re-suspended in homogenization buffer at a protein concentration of 1.0–1.5 mg·mL−1 and stored in aliquots at −80°C.
Cell treatments and cell extracts preparation
CHO/KOP cells and C6 glioma cells were serum-starved for 24 h or 48 h, respectively, whereas primary neuronal cultures were incubated in the absence of B 27 supplement for 24 h. The cells were then treated with the test agents as indicated in the legends to figures, washed with ice-cold PBS and lysed by scraping into PBS containing 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 2 mM EDTA, 2 mM EGTA, 4 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 10 mM sodium fluoride, 20 nM okadaic acid, 0.1% phosphatase inhibitor cocktail 1, 1% protease inhibitor cocktail and 1 mM PMSF. The samples were stored at −80°C. Aliquots of cell extracts were taken for protein determination.
Protein content was determined by the method of Bradford (1976) using BSA as a standard.
Dissection of brain regions and membrane preparation
Male Sprague–Dawley rats (200–300 g) (Harlan) were used. Animals were maintained in a 12 h light/dark cycle with food and water ad libitum. The dorsal striatum and nucleus accumbens were rapidly microdissected from 300 µm brain coronal slices as previously described (Onali and Olianas, 2002). Freshly dissected tissue samples were homogenized in an ice-cold buffer containing 10 mM HEPES-NaOH, 1 mM EDTA (pH 7.40) using a Teflon-glass tissue grinder. The homogenate was centrifuged at 27 000× g for 20 min at 4°C. The pellet was re-suspended in the same buffer at a protein concentration of 0.8-1.0 mg·mL−1 and stored at −80°C for binding assays.
[35S]GTPγS binding assay
The binding of [35S]GTPγS was assayed in a reaction mixture (final volume 100 µL) containing 25 mM HEPES/NaOH (pH 7.4), 10 mM MgCl2, 1 mM EDTA, 150 mM KCl and 1.0 nM [35S]GTPγS. The GDP concentration was optimized for each receptor system and was 30 µM for CHO/KOP and CHO/DOP, 10 µM for CHO/MOP and 50 µM for rat brain membranes. Membranes (2–4 µg of protein) were pre-incubated for 20 min at 30°C with the test compounds. For each compound, control samples received an equal volume (10 µL) of vehicle. The reaction was started by the addition of [35S]GTPγS and continued for 40 min at 30°C. The incubation was terminated by the addition of 5 mL of ice-cold buffer containing 10 mM HEPES/NaOH (pH 7.4) and 1.0 mM MgCl2, immediately followed by rapid filtration on glass fibre filters (Whatman GF/C). The filters were washed twice with 5 mL of buffer, and the radioactivity trapped was determined by liquid scintillation spectrometry. Non-specific binding was determined in the presence of 50 µM GTPγS. Assays were performed in duplicate.
Receptor binding assays
In CHO cell membranes, receptor binding assays were carried out by using [3H]diprenorphine to label κ- and µ-opioid receptors and [3H]naltrindole (NTI) to label δ-opioid receptors and by incubating the membrane preparations (15–40 µg of protein) at 30°C for 120 min in a buffer containing 25 mM HEPES/NaOH (pH 7.4), 10 mM MgCl2, 1 mM EDTA and 150 mM KCl. For saturation binding assays, the concentrations of [3H]diprenorphine ranged from 40 pM to 3 nM. For competition binding assays, the concentration of either [3H]diprenorphine or [3H]NTI was 0.20 nM. Non-specific binding was determined in the presence of 10 µM naloxone and corresponded to 4–12% and 12–30% of total [3H]diprenorphine and [3H]NTI binding respectively. Triplicate determinations were made for each experiment.
[3H]U-69 593 binding was performed in membranes prepared from rat brain tissue containing both dorsal striatum and nucleus accumbens. Tissue samples were homogenized in 50 mM Tris–HCl (pH 7.4) containing 0.1 mM PMSF, centrifuged at 27 000×g for 20 min at 4°C and frozen at −80°C. Thereafter, the tissue preparation was thawed and diluted in 50 mM Tris/HCl (pH 7.4), centrifuged as above and re-suspended in the same buffer. Aliquots containing ∼300 µg of protein were incubated with 4 nM [3H]U-69 593 at 25°C for 1 h in the presence of the indicated concentrations of mianserin. Non-specific binding was determined in the presence of 10 µM naloxone and corresponded to 25% of total radioligand binding. Saturation binding assays were performed using 3H]U-69 593 concentrations ranging from 0.5 to 25 nM. The estimated KD of [3H]U-69 593 was 1.2 nM. Triplicate determinations were made for each experiment.
Reactions were terminated by filtration through Whatman GF/C filters pre-soaked with 0.1% polyethylenimine, which were washed three times with 5 mL of ice-cold buffer containing 10 mM HEPES/NaOH (pH 7.4) and 1 mM MgCl2. The radioactivity trapped was determined by liquid scintillation spectrometry.
Western blot analysis
Aliquots of cell extracts containing equal amount of protein were subjected to SDS-PAGE, and the proteins were electrophoretically transferred to PVDF membranes (Hybond-P, Amersham Biosciences, Piscataway, NJ, USA). Non-specific binding sites were blocked by incubation in 20 mM Tris–HCl, 137 mM NaCl and 0.1% Tween-20 (pH 7.6) (TBS-T buffer) containing 5% BSA for 1 h. After washing with TBS-T buffer, the membranes were incubated overnight at 4°C with one of the following primary antibodies: rabbit polyclonal anti-phospho-ERK1 (Thr202/Tyr204)/ERK2 (Thr185/Tyr187) (pERK1/2) (1:20 000) (Neuromics, Northfield, MN); rabbit polyclonal anti-ERK1/2 (1:2000), anti-phospho-CREB (Ser133) (1:1000), anti-phospho-p38 MAPK (Thr180/Tyr182) (1:1000), mouse monoclonal anti-p38 MAPK (1:1000) and rabbit monoclonal anti-CREB (1:1000) (Cell Signaling Technology, Danvers, MA, USA). The membranes were then incubated with a HRP-conjugated secondary antibody (1:10 000), and immunoreactive bands were detected by using ECL Plus and ECL Hyperfilm (Amersham). The size of the immunoreactive bands was determined by using molecular weight standards detected with a specific antibody suitable for the ECL system (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were stripped of antibodies with Western-Reprobe reagent (Calbiochem, La Jolla, CA, USA) and re-probed with rabbit polyclonal anti-actin (1:3000) (Sigma Aldrich) to control for sample loading. Band densities were determined by densitometric analysis using Image Scanner III (GE Healthcare, Milan, Italy) and NIH ImageJ software (US National Institutes of Health, Bethesda, MA, USA). The optical density of phosphoprotein bands was normalized to the density of the corresponding total protein to yield the relative optical density value.
Statistical analysis
Results are reported as mean ± SEM. Data from concentration–response curves were analysed by the program Graph Pad Prism (San Diego, CA, USA), which yielded agonist concentration producing half-maximal effect (EC50 values) and maximal effects (Emax). Saturation binding data were analysed by the non-linear curve fitting program LIGAND, which provided ligand dissociation constant (KD) and maximal binding capacity (Bmax). Antagonist potencies were analysed by nonlinear regression analysis. When increasing concentrations of antagonists in the presence of a fixed concentration of agonist were examined, the antagonist inhibitory constant (Ki) was calculated according to the equation: Ki = IC50/1 + (A/EC50), where IC50 is the antagonist concentration producing half-maximal inhibition, A is the agonist concentration and EC50 is the agonist EC50 value. The pA2 of mianserin was calculated from Schild plots, where the ratios −1 (DR-1) of the EC50 values of the agonist in the absence and in the presence of mianserin was plotted as a function of the mianserin concentration. When the effect of a fixed mianserin concentration in the presence of increasing agonist concentrations was examined, the Ki value was calculated according to the equation: EC50b = EC50a (1 + I/Ki), where EC50a and EC50b are agonist EC50a values in the absence and in the presence of mianserin, respectively; and I is the concentration of mianserin. Statistical analysis was performed by either Student's t-test when comparing two groups or one-way anova followed by Newman–Keuls post hoc tests when comparing more than two groups.
Materials
[35S]GTPγS (1306 Ci mmol−1), [15,16-3H]diprenorphine (53 Ci mmol−1), [3H]U-69 593 (43 Ci mmol−1) and [5′,7′-3H]naltrindole ([3H]NTI) (20 Ci mmol−1) were obtained from Perkin Elmer (Boston, MA, USA). GTPγS was from Boehringer (Mannheim, Germany). (–)-U-50 488 hydrochloride, nor-binaltorphimine dihydrochloride (nor-BNI), CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2) and NTI hydrochloride were from Tocris Cookson Ltd (Avonmouth, UK). (2-D-penicillamine, 5-D-penicillamine)-enkephalin (DPDPE) was purchased from Bachem AG (Bubendorf, Switzerland). Mianserin hydrochloride, mirtazapine hydrochloride, (D-Ala2-N-methyl-Phe-Gly-ol5)-enkephalin (DAMGO), dynorphin A 1–13, Pertussis toxin (PTX) and the other reagents were from Sigma Aldrich.
Results
Mianserin binding to opioid receptor subtypes
In radioligand binding assays carried out with membranes of CHO cells expressing the human opioid receptors, mianserin caused a concentration-dependent displacement of [3H]diprenorphine specifically bound to κ- and µ-opioid receptors and [3H]NTI bound to δ-opioid receptors (Figure 1A). Estimation of the corresponding Ki values indicated that mianserin was about 12- and 18-fold more potent in displacing the radioligand bound to the κ-opioid receptor (Ki = 1.7 ± 0.3 µM) than the µ-opioid receptor-1 (Ki = 21 ± 1.2 µM) and δ-opioid receptors (Ki = 30.2 ± 1.9 µM). In CHO/KOP cell membranes, Scatchard analysis of saturation binding data indicated that mianserin (10 µM) increased the KD value of [3H]diprenorphine from 160 ± 20 to 870 ± 50 pM (P < 0.05, n = 3) without significantly changing the Bmax value (1450 ± 60 and 1590 ± 70 fmol·mg−1 protein for control and mianserin respectively, n = 3) (Figure 1B).
Figure 1.
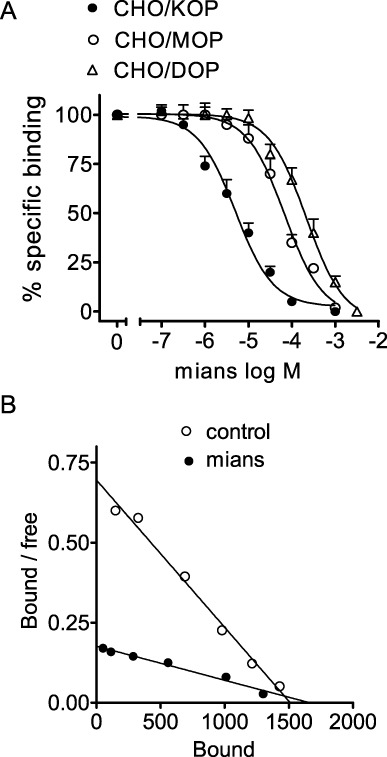
Mianserin displays higher affinity for κ- than µ- and δ-opioid receptors. (A) Radioligand binding assays were performed in membranes of CHO cells stably transfected with human κ- (CHO/KOP), µ- (CHO/MOP) and δ- (CHO/DOP) opioid receptors by using 0.2 nM [3H]diprenorphine (CHO/KOP and CHO/MOP) or 0.2 nM [3H]NTI (CHO/DOP). Non-specific binding was determined in the presence of 10 µM naloxone. Values are the mean ± SEM of three experiments. (B) Scatchard plot of [3H]diprenorphine saturation binding carried out in CHO/KOP membranes in the presence of either vehicle (control) or 10 µM mianserin (mians). Values are the mean of three experiments.
Stimulation of [35S]GTPγS binding in CHO cells transfected with human opioid receptors
In functional assays, mianserin stimulated [35S]GTPγS binding to CHO/KOP membranes in a concentration-dependent manner (Figure 2A). The estimated EC50 value was 0.53 ± 0.05 µM (n = 3), whereas the Emax value corresponded to 2.5-fold increase of control activity (P < 0.001, n = 4). No significant stimulatory effects were observed in either CHO/MOP and CHO/DOP cells or in untransfected CHO/K1 cells. To gain information on the relative intrinsic activity, the stimulatory effect of mianserin on [35S]GTPγS binding was compared with that of the full κ-opioid receptor agonist (–)-U-50 488. In these experiments, the effect of mirtazapine was also investigated. In agreement with previous data (Onali et al., 2010), (–)-U-50 488 potently stimulated [35S]GTPγS binding with an EC50 value of 0.66 ± 0.04 nM (n = 5) and an Emax value (2.3-fold increase of control value, n = 5), which was similar to that of mianserin (Figure 2B). Mirtazapine was effective in stimulating [35S]GTPγS binding almost as much as mianserin, but with a lower potency (EC50 = 7.2 ± 0.8 µM, n = 3). The stimulatory effects of both mianserin and mirtazapine were effectively blocked by the selective κ-opioid receptor antagonist nor-BNI (100 nM) (Figure 2C). The nor-BNI antagonism of mianserin (30 µM)-stimulated [35S]GTPγS binding was concentration-dependent with an estimated Ki value of 39 ± 5 pM (Figure 2D).
Figure 2.
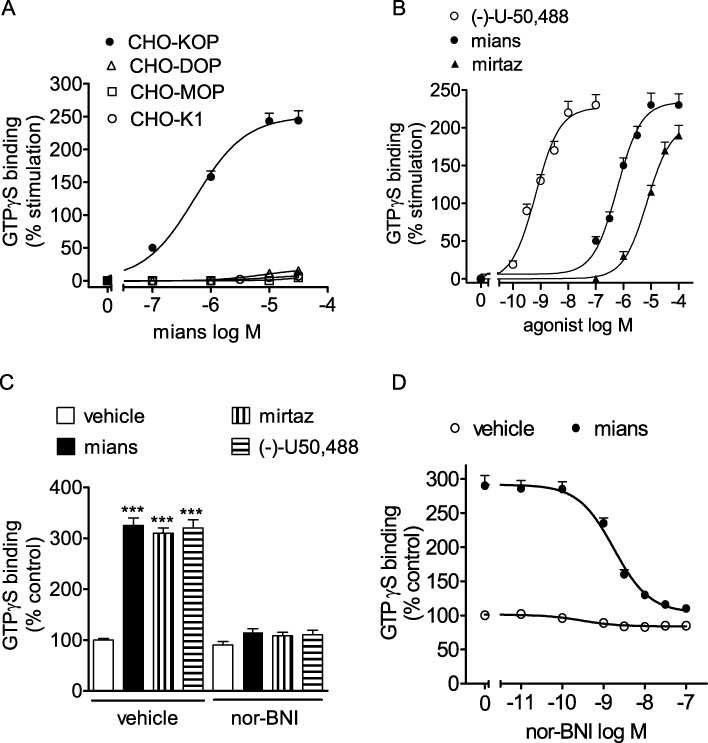
Stimulation of [35S]GTPγS binding by mianserin and mirtazapine in CHO cells. (A) Membranes of CHO cells untransfected (CHO/K1) or stably transfected with the different opioid receptor subtypes were incubated with 1.0 nM [35S]GTPγS in the presence of the indicated concentrations of mianserin (mians). Values are the mean ± SEM of three experiments. (B) Stimulation of [35S]GTPγS binding by mianserin, mirtazapine (mirtaz) and the full κ receptor agonist (–)-U-50 488 in CHO/KOP membranes. Values are the mean ± SEM of three to five experiments. (C) Antagonism of [35S]GTPγS binding stimulation by the κ receptor antagonist nor-BNI. CHO/KOP cell membranes were incubated with either vehicle, 100 nM (–)-U50,488, 30 µM mianserin or 50 µM mirtazapine in the presence of either vehicle or 100 nM nor-BNI. Values are the mean ± SEM of three experiments. ***P < 0.001 significantly different from control (vehicle + vehicle) by anova. (D) Concentration-dependent-inhibition of mianserin-stimulated [35S]GTPγS binding by nor-BNI. CHO/KOP cell membranes were incubated with either vehicle or 30 µM mianserin in the presence of the indicated concentrations of nor-BNI. Values are the mean ± SEM of three experiments.
Stimulation of ERK1/2 phosphorylation in CHO/KOP and C6 glioma cells
In a variety of cell types, ERK1/2, which belong to the MAPK family, have been shown to be critical down-stream components of opioid receptor-mediated regulation of cell proliferation and differentiation (Tegeder and Geisslinger, 2004). Previous studies have shown that ERK1/2 activity is critical for the action of antidepressants (Duman et al., 2007). As shown in Figure 3A, exposure of CHO/KOP cells to mianserin (1 µM) induced a rapid increase in dual ERK1/2 phosphorylation, which peaked at 10 min and then declined, although remaining above control up to 120 min. The stimulation of ERK1/2 phosphorylation by mianserin was concentration-dependent with an EC50 value of 1.5 ± 0.3 µM and Emax corresponding to about 10-fold increase of control value (P < 0.001, n = 5) (Figure 3B). For comparison, the effect of the naturally occurring κ-opioid agonist dynorphin A 1–13 (dyn A) was examined. Dyn A stimulated ERK1/2 phosphorylation by about 15-fold with an EC50 value of 20 ± 3 pM (P < 0.001, n = 3) (Figure 3C). The stimulation of ERK1/2 phosphorylation elicited by mianserin (1 µM) was significantly attenuated by nor-BNI (100 nM), which per se had no effect (Figure 3D). Mirtazapine (3 µM) also significantly increased phospho-ERK1/2 levels, and the effect was effectively antagonized by nor-BNI (Figure 3D).
Figure 3.
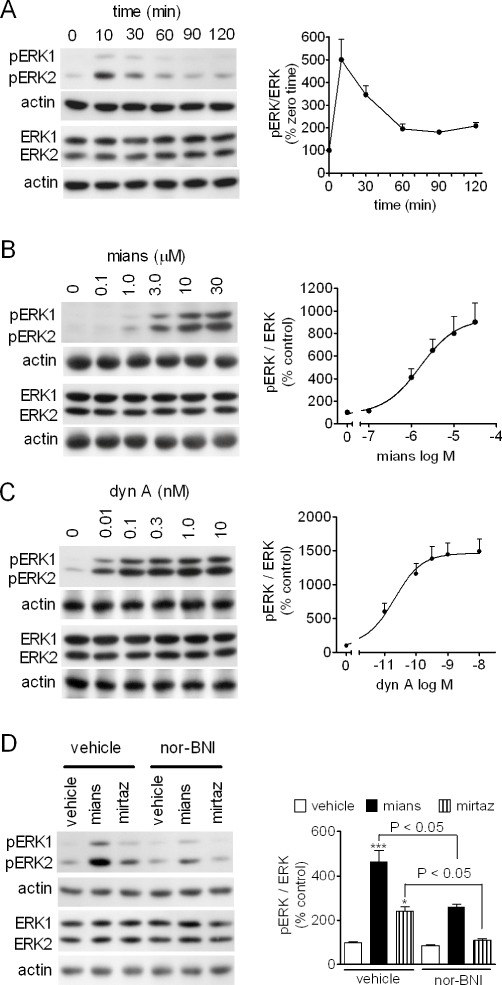
Stimulation of ERK1/2 phosphorylation by mianserin and mirtazapine in CHO/KOP cells. (A) Cells were incubated for the indicated time periods in the presence of 1 µM mianserin. Cell extracts were analysed for ERK1/2 phosphorylation (pERK1 and pERK2), total ERK1/2 and actin levels by Western blot. Densitometric ratios of pERK1/2 normalized for total ERK1/2 are expressed as percent of zero time values and are the mean ± SEM of four experiments. (B and C) CHO/KOP cells were incubated for 10 min in the presence of the indicated concentrations of either mianserin (mians) (B) or dynorphin A 1–13 (dyn A) (C). Values are the mean ± SEM of five and three experiments respectively. (D) CHO/KOP cells were pre-incubated in the presence of either vehicle or 100 nM nor-BNI for 10 min and then exposed to either vehicle, 1 µM mianserin or 3 µM mirtazapine (mirtaz) for 10 min. Values are the mean ± SEM of four experiments. ***P < 0.001, *P < 0.05 significantly different from control (vehicle + vehicle) by anova.
C6 glioma cells are known to endogenously express κ-opioid receptors coupled to stimulation of ERK1/2 phosphorylation (Bohn et al., 2000), and we have previously reported that, in these cells, TCAs activate ERK1/2 through a mechanism that involves κ-opioid receptors (Onali et al., 2010). As shown in Figure 4A, in C6 glioma cells mianserin induced a concentration-dependent stimulation of ERK1/2 phosphorylation with an EC50 value of 1.6 ± 0.3 µM and Emax corresponding to fivefold increase of control value (P < 0.001, n = 4). The stimulatory effects elicited by low concentrations of mianserin (0.3 and 0.5 µM) were completely blocked by nor-BNI (100 nM) (Figure 4B). Like mianserin, mirtazapine (3 µM) stimulated ERK1/2 phosphorylation, and this effect was suppressed by nor-BNI (Figure 4C). Moreover, the stimulatory effects of both mianserin (10 µM) and mirtazapine (10 µM) were completely prevented in cells pre-treated with PTX, which uncouples the G-proteins of the Gi and Go family from the receptors (Figure 4D).
Figure 4.
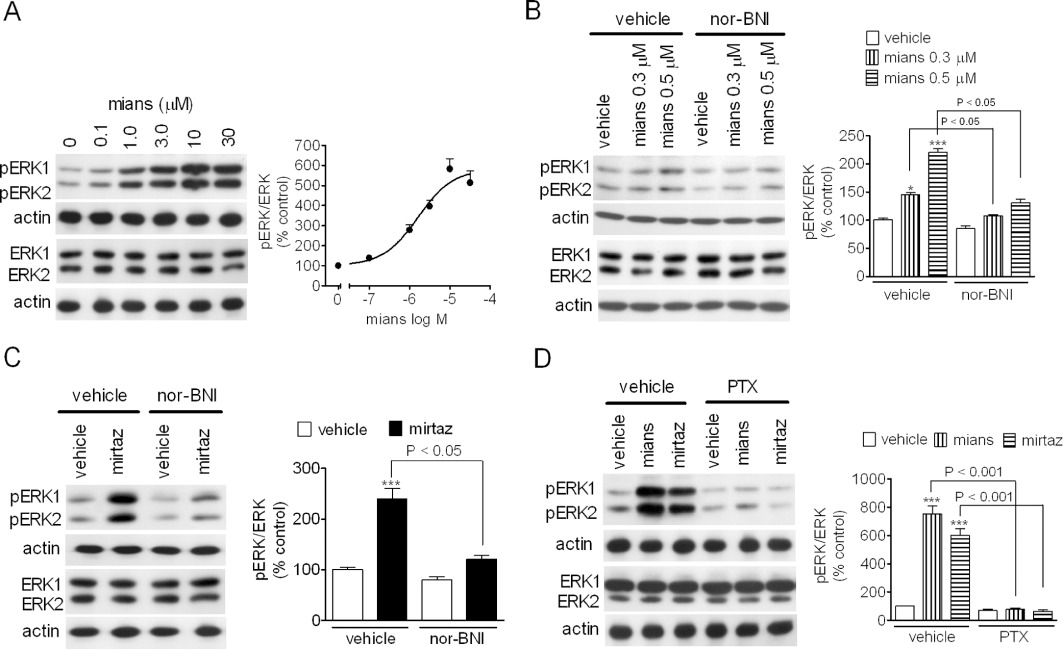
Stimulation of ERK1/2 phosphorylation by mianserin and mirtazapine in C6 glioma cells. (A) Cells were incubated for 15 min in the presence of the indicated mianserin (mians) concentrations. Values are the mean ± SEM of four experiments. (B and C) C6 glioma cells were pre-incubated for 10 min with either vehicle or 100 nM nor-BNI and then exposed to either vehicle, the indicated concentrations of mianserin (B) or 3 µM mirtazapine (mirtaz; C). Values are the mean ± SEM of four experiments. ***P < 0.001, *P < 0.05 significantly different from control by anova. (D) PTX prevents the stimulation of ERK1/2 phosphorylation by mianserin and mirtazapine. C6 glioma cells were pre-incubated for 24 h with either vehicle or 100 ng·mL−1 PTX and then exposed for 15 min to either 10 µM mianserin or 10 µM mirtazapine. Values are the mean ± SEM of three experiments. ***P < 0.001 significantly different from control (vehicle + vehicle) by anova.
Stimulation of CREB phosphorylation
CREB is a well-characterized stimulus-dependent transcription factor that is activated by phosphorylation on Ser133 by cAMP-dependent protein kinase and other protein kinases pathways, including that involving ERK1/2 (Lonze and Ginty, 2002). CREB up-regulation has been observed following antidepressant treatment in animals and humans and changes in CREB expression and/or activity has been associated with antidepressant-like behaviours (Blendy, 2006). In CHO/KOP cells, mianserin (1 µM) induced a time-dependent increase of phospho-CREB, which reached a peak at 30 min (about twofold increase of control levels) and then slowly declined returning at control levels at 120 min (Figure 5A). Mianserin significantly increased phospho-CREB also in C6 cells, and in either these cells or CHO/KOP cells, the stimulatory effect was blocked by nor-BNI (100 nM) (Figure 5B and C). Like mianserin, mirtazapine (3 µM) was effective in increasing phospho-CREB in both cell systems in a nor-BNI-sensitive manner (Figure 5B and C).
Figure 5.
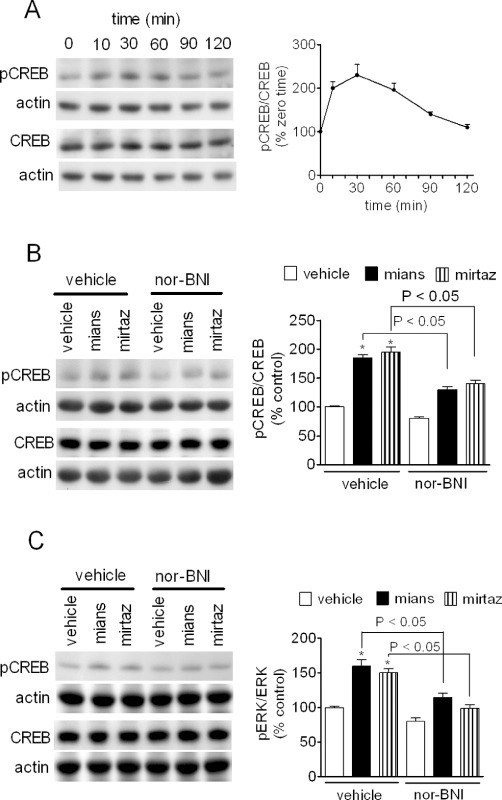
Stimulation of CREB phosphorylation by mianserin and mirtazapine. (A) CHO/KOP cells were incubated in the presence of 1 µM mianserin for the indicated time periods. Cell extracts were then analysed for phospho-Ser133-CREB, total CREB and actin levels by Western blot. Values are the mean ± SEM of three experiments. (B and C) CHO/KOP (B) and C6 glioma (C) cells were pre-incubated for 10 min in the presence of either vehicle or 100 nM nor-BNI. Cells were then exposed for 20 min to either vehicle, 1 µM mianserin (mians) or 3 µM mirtazapine (mirtaz). Densitometric values are the mean ± SEM of four experiments. *P < 0.05 significantly different from control by anova.
Selective activation of κ-opioid receptors in rat striatum and nucleus accumbens
To investigate whether mianserin acted on κ-opioid receptors expressed in the brain, radioligand binding studies using the selective κ-opioid receptor ligand [3H]U-69 593 were first conducted in membranes derived from rat dorsal and ventral striatum. Mianserin completely displaced [3H]U-69 593 specific binding with a Ki value of 1.7 ± 0.3 µM (n = 3) (Figure 6A). In membranes of individually dissected rat striatum and nucleus accumbens, mianserin elicited a concentration-dependent stimulation of [35S]GTPγS binding with EC50 values of 0.70 ± 0.05 and 0.39 ± 0.06 µM, respectively, and Emax values corresponding to 13 ± 2% (P < 0.05, n = 5) and 22 ± 3% increase of control activity (P < 0.05, n = 4) respectively (Figure 6B). In the same membrane preparations, (–)-U-50 488 increased [35S]GTPγS binding by 20 ± 2% (P < 0.05, n = 4) and 29 ± 3%, respectively (P < 0.05, n = 4), with EC50 of 7.4 ± 0.5 and 4.0 ± 0.3 nM respectively (Figure 6B). In nucleus accumbens, the stimulation of [35S]GTPγS binding by mianserin (30 µM) was completely blocked by nor-BNI (1 nM) but was not affected by either the selective µ-opioid receptor antagonist CTAP (10 nM) or the selective δ-opioid antagonist NTI (1nM) (Figure 6C).
Figure 6.
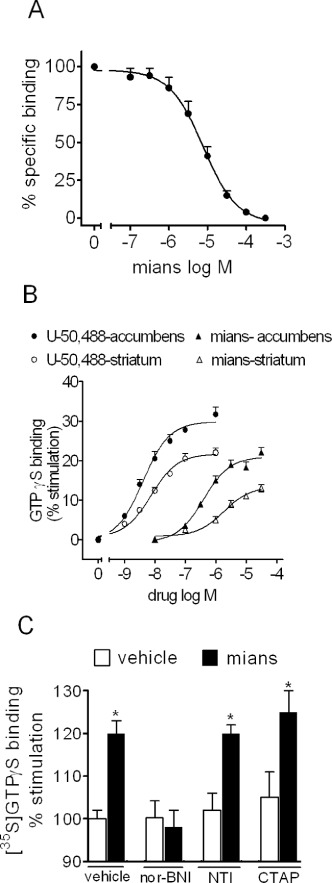
Selective activation of κ-opioid receptors by mianserin in rat striatum and nucleus accumbens. (A) Mianserin (mians) displaces the specific binding of the selective κ-opioid receptor ligand [3H]U-69 593 in membranes of rat striatum and nucleus accumbens. Tissue membranes were incubated with 4 nM [3H]U-69 593 in the presence of the indicated concentrations of mianserin for 60 min at 25°C. Values are the mean ± SEM of three experiments. (B) Stimulation of [35S]GTPγS binding by (–)-U-50 488 and mianserin in membranes of individually dissected rat striatum and nucleus accumbens. Tissue membranes were incubated in the presence of the indicated concentrations of the test compounds for 20 min at 30°C. Thereafter, 1 nM [35S]GTPγS was added and the incubation continued for 20 min. Values are the mean ± SEM of four to five experiments. (C) Selective antagonism of mianserin stimulation of [35S]GTPγS binding by nor-BNI in rat nucleus accumbens. Tissue membranes were incubated in the presence of either vehicle or 30 µM mianserin with either vehicle, 1 nM nor-BNI, the selective δ-opioid receptor antagonist NTI (1 nM) or the µ-opioid antagonist CTAP (10 nM). Values are the mean ± SEM of three experiments. *P < 0.05 significantly different from the corresponding control by Student's t-test.
Effects of mianserin in combination with opioid receptor agonists
In rat striatal membranes, the curve of [35S]GTPγS binding stimulation by dynorphin A (EC50 = 3.9 ± 0.5 nM) was progressively shifted to the right in the presence of increasing concentrations of mianserin (Figure 7A). Schild analysis of mianserin antagonism yielded a pA2 value of 5.9 ± 0.2 (n = 4). Similarly, mianserin (10 µM) caused a rightward shift in (–)-U-50 488 concentration–response curve with an estimated Ki of 1.5 ± 0.2 µM (n = 4) (Figure 7B). Conversely, mianserin (10 µM) failed to affect the stimulation of [35S]GTPγS binding elicited by either the selective δ-opioid receptor agonist DPDPE (Figure 7C) or the selective µ-opioid receptor agonist DAMGO (Figure 7D).
Figure 7.
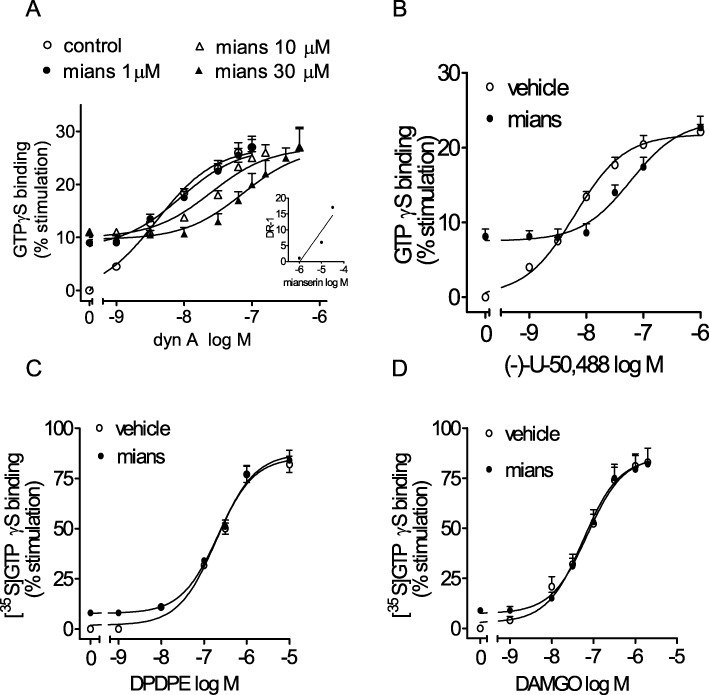
Partial agonist activity of mianserin at κ-opioid receptors in rat striatum. (A) Antagonism of dynorphin A 1–13 (dyn A)-stimulated [35S]GTPγS binding by mianserin. Tissue membranes were incubated with increasing concentrations of dyn A without and with the indicated concentrations of mianserin (mians). The inset shows the Schild plot of mianserin antagonism. Values are the mean ± SEM of four experiments. (B–D) Mianserin antagonizes the stimulation of [35S]GTPγS binding by (–) U-50 488 but not the δ-opioid agonist DPDPE and the µ-opioid agonist DAMGO. Tissue membranes were incubated in the presence of the indicated concentrations of the opioid agonists without and with 10 µM mianserin. Values are the mean ± SEM of three to four experiments.
Effects of mianserin on p38 MAPK and ERK1/2 phosphorylation in primary cultures of mouse neurons
Like ERK1/2, the p38 MAPK is activated by dual phosphorylation on Thr and Tyr residues by the upstream kinases MEK 3 and 6. A variety of cellular stressors activate this kinase and p38 MAPK is activated by κ-opioid receptor agonists in both neurons and astrocytes (Bruchas et al., 2006). We found that in primary cultures of mouse striatal neurons, exposure to dynorphin A (30 nM) increased p38 MAPK phosphorylation by about twofold (P < 0.05, n = 4) (Figure 8A). Addition of mianserin (10 µM) caused a modest increase of p38 phosphorylation and, when combined with dynorphin A, inhibited the stimulatory effect of the full κ-opioid agonist (Figure 8A). In primary cultures of mouse hippocampal neurons, dynorphin A (30 nM) increased phospho-p38 MAPK levels by about threefold (P < 0.001, n = 4) (Figure 8B), and this response was antagonized in the presence of mianserin (10 µM), which per se caused only a small stimulatory effect (Figure 8B).
Figure 8.
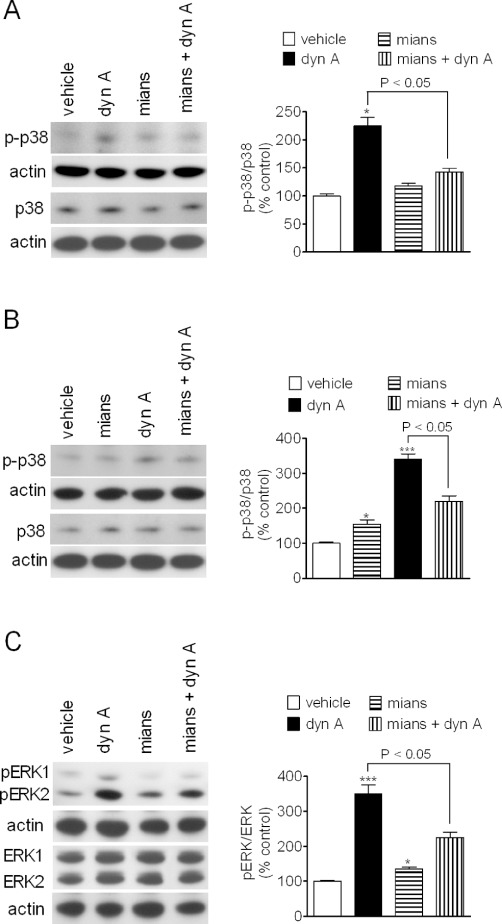
Antagonism of dynorphin A stimulated p38 MAP kinase and ERK1/2 phosphorylation by mianserin in mouse primary neurons. Primary cultures of mouse striatal (A) and hippocampal (B) neurons grown for 8–10 days were treated with either vehicle or 10 µM mianserin for 10 min and then exposed to either vehicle or 30 nM dynorphin A (dyn A) for 15 min. Cell extracts were analysed for phospho-p38 MAPK (p-p38), total p38 MAPK (p38) and actin levels by Western blot. Values are the mean ± SEM of four experiments. ***P < 0.001, *P < 0.05 significantly different from control by anova. (C) Primary cultures of mouse striatal neurons were treated as indicated above and cell extracts were analysed for phospho-ERK1/2, total ERK1/2 and actin levels. Values are the mean ± SEM of four experiments. ***P < 0.001 significantly different from control by anova.
We next examined the effect of mianserin on κ-opioid receptor-stimulated ERK1/2 phosphorylation in primary cultures of striatal neurons. In these cells, dyn A (30 nM) increased phospho-ERK1/2 by 3.5-fold (P < 0.001, n = 4). As observed for p38 MAPK responses, mianserin (10 µM) induced a slight increase in phospho-ERK1/2 levels (35 ± 8%, P < 0.05, n = 4) but significantly attenuated the stimulatory effect of dynorphin A (Figure 8C).
Discussion
In the present study, we report for the first time that the tetracyclic atypical antidepressant mianserin behaves as an agonist at both recombinant and native κ-opioid receptors. The κ-opioid selectivity of mianserin was first documented by studies conducted in CHO cells stably expressing the different opioid receptor subtypes. Thus, in radioligand competition experiments, mianserin affinity for κ-opioid receptors was 18- and 12-fold higher than that for δ- and µ-opioid receptors respectively. As expected for a competitive type of ligand–receptor interaction, Scatchard analysis of [3H]diprenorphine binding to κ-opioid receptors showed that mianserin decreased the affinity of the radioligand without significantly affecting the Bmax value. Functional studies using the stimulation of GTPγS binding in cell membranes as readout showed that the antidepressant selectively activated the κ-opioid receptors, being without significant effect at µ- and δ-opioid receptors. The mianserin agonist activity was blocked by nor-BNI with a potency (Ki = 39 pM) consistent with the antagonist affinity for the κ-opioid receptor (Metcalf and Coop, 2005). Moreover, in CHO/KOP cells and C6 glioma cells, nor-BNI effectively antagonized the mianserin-induced stimulation of ERK1/2 and CREB phosphorylation, indicating the involvement of κ-opioid receptors also in functional responses measured in intact cell preparations. Interestingly, mirtazapine, which is close to mianserin both structurally and pharmacologically (Croom et al., 2009), was found to behave similarly to mianserin, being able to elicit nor-BNI-sensitive responses in both CHO/KOP cells and C6 glioma cells. Thus, the agonist activity at κ-opioid receptors appears to be an additional property shared by the two antidepressants.
The ability of mianserin to selectively interact with κ-opioid receptors was also observed in rat striatum and nucleus accumbens, two brain areas known to express all three opioid receptor subtypes (Mansour et al., 1995). In rat striatum, mianserin displaced the binding of the selective κ-agonist [3H]U-69 593 with an affinity similar to that displayed in CHO/KOP cells. Mianserin has previously been reported to antagonize opiate binding in extracts of whole rat brain, although with a potency (IC50 = 88 µM) much lower than that observed in the present study (Isenberg and Cicero, 1984). The reason for this discrepancy may be due to the fact that in the latter study the non-selective radioligand [3H]naltrexone was employed. The lower mianserin affinity could therefore have been due to [3H]naltrexone binding to the highly abundant µ- and δ-opioid receptors, for which mianserin displays low affinity.
Mianserin was found to be more effective in stimulating GTPγS binding in nucleus accumbens than striatum, consistent with the higher density of κ-opioid receptors in ventral than in dorsal, striatum (Mansour et al., 1995). However, in contrast to the responses observed in CHO cells overexpressing the κ-opioid receptor, where mianserin was as effective as (–)-U-50 488, the maximal stimulation by mianserin of GTPγS binding in the brain areas was lower than that elicited by full κ-agonists. When combined with either dynorphin A or (–)-U-50 488, mianserin antagonized the stimulation of GTPγS binding elicited by the highly efficacious κ-agonists with a potency equal to its affinity for κ-opioid receptors. Moreover, in primary cultures of mouse striatal and hippocampal neurons, mianserin weakly enhanced the phosphorylation of p38 MAPK but effectively antagonized the stimulation elicited by dynorphin A. Similar results were obtained by examining dynorphin A-induced stimulation of phospho-ERK1/2 in striatal neurons. These data indicate that at brain κ-opioid receptors, mianserin behaves as a partial agonist. In this respect, its activity is similar to that of TCAs, which have also been found to act as partial κ-agonists in the brain (Onali et al., 2010).
A critical issue concerning the κ-opioid agonist activity of mianserin is whether the observed receptor affinity is comparable with the tissue concentrations reached by the drug following therapeutic doses. In patients with depression, therapeutic mianserin plasma concentrations have been reported to range from 0.1 to 0.3 µM (Otani et al., 1993). In mice, either acute or chronic administration of mianserin yielded brain concentrations that were at least 10-fold higher than the plasma concentrations (Altamura et al., 1987). In rats, a chronic continuous infusion of mianserin produced serum concentrations of 118 ng·mL−1, a value close to the therapeutic plasma concentrations, and whole brain concentrations of 1520 ng·g−1, with a brain/serum ratio of 13.6 (Kurata and Kurachi, 1989). Thus, although the mianserin concentrations in the biophase are not known, the affinity of mianserin for κ-opioid receptors (∼1.5 µM) observed in the present study is compatible with the concentrations that can be reached by the drug in the brain. Nonetheless, in vivo functional studies are required to investigate whether mianserin is capable of affecting brain κ-opioid receptor signalling following acute and chronic administration.
Previous studies have reported that mianserin binds to 5-HT2 receptors and α2-adrenoceptors with low nanomolar affinities (Peroutka and Snyder, 1981; Richelson and Nelson, 1984), whereas we found that the drug displays an affinity for κ-opioid receptors in the low micromolar range. This difference suggests that the activity of mianserin would be much more potent at receptor sites other than k-opioid receptors. However, in different studies using either brain synaptosomes or tissue slices, micromolar concentrations of mianserin were used to increase noradrenaline release via blockade of pre-synaptic α2-adrenoceptors (Schoemaker et al., 1981; Raiteri et al., 1983; Rose et al., 1984), a major mechanism thought to mediate the antidepressant action of the drug (Marshall, 1983; Pinder, 1985). Similarly, electrophysiological studies in guinea pig caecum found that mianserin blocked pre-synaptic and post-synaptic α2-adrenoceptors with micromolar potencies (Tokimasa et al., 1987). On the other hand, in rat vas deferens, mianserin was reported to competitively antagonize the pre-synaptic action of the α2-adrenoceptor agonist clonidine with a pA2 value of 7.3 (Doxey et al., 1978). Thus, it appears that, at least in some functional assays, mianserin acted as α2-adrenoceptor antagonist at concentrations comparable with those required for activating κ-opioid receptors.
The relevance of the κ-opioid agonist activity for the pharmacological actions of mianserin remains to be determined. Nonetheless, some potential implications can be considered. Behavioural studies in mice have reported that mianserin produced analgesic effects in the hotplate test, and this effect was blocked not only by naloxone but also nor-BNI and β-funaltrexamine, a µ-opioid antagonist, but not NTI (Schreiber et al., 1998). The anti-nociception induced by mirtazapine in mice was also reported to be prevented by nor-BNI (Schreiber et al., 2002). In addition to δ-opioid receptors, κ-opioid receptors have been found to be involved in the attenuation of neuropathic allodynia by chronic TCA treatment in mice (Benbouzid et al., 2008a). The precise mechanisms underlying the participation of the endogenous opioid system in the analgesic actions of mianserin and mirtazapine have not been elucidated. For antidepressants in general, indirect effects on the opioid system mediated by monoamine transmission have been hypothesized (Mico et al., 2006). However, as suggested by the present data, it is possible that, in addition to these mechanisms, a direct agonist activity at κ-opioid receptors may contribute to the analgesic effects of mianserin and mirtazapine. It is important to mention that mianserin has been reported to either antagonize or potentiate the analgesic effect of U-50 488 (Ho and Takemori, 1989; Schreiber et al., 1998). The antagonist effects of mianserin have been attributed to blockade of 5-HT neurotransmission (Ho and Takemori, 1989), which has been shown to be required for opioid-induced analgesia (Zhao et al., 2007). On the other hand, the mechanisms underlying the potentiating effect of mianserin on U-50 488-induced analgesia have not been elucidated. The present data showing that mianserin antagonized U-50 488 indicated that a synergistic interaction at the κ-opioid receptor is unlikely to be involved.
Besides controlling pain, κ-opioid receptors have been shown to modulate mood and anxiety, although with controversial results. In rats, the administration of low doses of κ-opioid receptor agonists was found to produce anxiolytic-like effects in the elevated plus-maze test (Privette and Terrian, 1995). An anxiolytic-like behaviour was also observed following U-69 593 in the infralimbic cortex of mice (Wall and Messier, 2000). In mice, the modulation of anxiety-related behaviour by κ-opioid receptors was found to be dependent on social status, as U-50 488 had no effect on winners but had anxiolytic-like effects in repeatedly defeated subjects (Kudryavtseva et al., 2004). On the other hand, a large body of evidence indicates that κ-opioid receptor activation produces depressive-like behaviour and anxiety. Thus, the administration of κ-opioid agonists has been reported to induce dysphoric and psychotomimetic effect in humans (Pfeiffer et al., 1986) and pro-depressive-like and anxiogenic effects in rodents (Bals-Kubik et al., 1993; Narita et al., 2006). Moreover, several studies have shown that blockade of central κ-opioid receptors has anxiolytic and antidepressant-like effects (Mague et al., 2003; Bruijnzeel, 2009; Carlezon et al., 2009). Similarly, prodynorphin gene disruption produced less anxiety and pro-depressive signs in mice with C57BL/6N genetic background (McLaughlin et al., 2003; Wittmann et al., 2009) but an increase in anxiety in mice with C57BL/6J background (Bilkei-Gorzo et al., 2008). Mice lacking the κ-opioid receptor gene exhibited either no change in anxiety-like behaviour (Filliol et al., 2000) or increased depressive-like behaviour (McLaughlin et al., 2003) likely depending on the test conditions. Recently, it has been shown that behavioural stressors can lead to activation of the endogenous κ-opioid system (Land et al., 2008), and that dynorphin-stimulated p-38 MAPK signalling plays a critical role in stress-induced impairment of 5-HT transmission and dysphoria (Bruchas et al., 2007; 2011). Thus, the majority of the studies using either pharmacological or genetic approaches indicate that an increased activity of the κ-opioid receptor system mediates pro-depressive and anxiogenic effects. Therefore, the finding that mianserin activates κ-opioid receptors seems to oppose the possibility that this action may participate in the therapeutic effects of the drug. On the other hand, it should be considered that mianserin acts as a partial agonist at brain κ-opioid receptors, causing an attenuation of receptor stimulation by highly efficacious agonists. Thus, it is possible that mianserin may inhibit the increased κ-opioid receptor activation elicited by endogenous agonists released under stress conditions. This hypothesis remains to be verified in vivo by conducting specific behavioural tests.
In conclusion, the present study shows that the atypical antidepressant mianserin exerts direct agonist activity at κ-opioid receptors. As TCAs have previously been found to behave similarly, the study supports the idea that this opioid receptor agonism may be a common property of different classes of antidepressant drugs.
Acknowledgments
This work was supported by MIUR and Regione Autonoma della Sardegna, PO Sardegna FSE 2007-13, L.R. 7/2007 ‘Promozione della Ricerca Scientifica e dell' Innovazione Tecnologica in Sardegna’ with a post-doctoral fellowship to SD.
Glossary
- CHO/KOP
/DOP and /MOP, CHO cells stably expressing the human κ-, δ- and µ-opioid receptor-1, respectively
- CTAP
(D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2)
- DAMGO
(D-Ala2-N-methyl-Phe-Gly-ol5)-enkephalin
- DPDPE
[D-Pen(2,5)]-enkephalin
- ECL
enhanced chemiluminescence
- FCS
fetal calf serum
- nor-BNI
nor-binaltorphimine
- NTI
naltrindole
- PTX
Pertussis toxin
- TCA
tricyclic antidepressant
Conflicts of interest
The authors disclose no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamura AC, De Novellis F, Mauri MC, Gomeni R. Plasma and brain pharmacokinetics of mianserin after single and multiple dosing in mice. Prog Neuropsychopharmacol Biol Psychiatry. 1987;11:23–33. doi: 10.1016/0278-5846(87)90028-5. [DOI] [PubMed] [Google Scholar]
- Baldessarini RJ. Drug therapy of depression and anxiety disorders. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 11th edn. New York, NY: McGraw-Hill Book Companies; 2006. pp. 429–459. [Google Scholar]
- Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place-preference paradigm in rats. J Pharmacol Exp Ther. 1993;264:489–495. [PubMed] [Google Scholar]
- Benbouzid M, Choucair-Jaafar N, Yalem I, Waltisperger E, Muller A, Freund-Mercier MJ, et al. Chronic, but not acute, tricyclic antidepressant treatment alleviates neuropathic allodynia after sciatic nerve cuffing in mice. Eur J Pain. 2008a;12:1008–1017. doi: 10.1016/j.ejpain.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Benbouzid M, Gaveriaux-Ruff C, Yalcin I, Waltisperger E, Tessier L-H, Muller A, et al. Delta-opioid receptors are critical for tricyclic antidepressant treatment of neuropathic allodynia. Biol Psychiatry. 2008b;63:633–636. doi: 10.1016/j.biopsych.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Besson A, Privat AM, Eschalier A, Fialip J. Dopaminergic and opioidergic mediations of tricyclic antidepressants in the learned helplessness paradigm. Pharmacol Biochem Behav. 1999;64:541–548. doi: 10.1016/s0091-3057(99)00102-1. [DOI] [PubMed] [Google Scholar]
- Biegon A, Samuel D. Interaction of tricyclic antidepressants with opiate receptors. Biochem Pharmacol. 1980;29:460–462. doi: 10.1016/0006-2952(80)90531-6. [DOI] [PubMed] [Google Scholar]
- Bilkei-Gorzo A, Racz I, Michel K, Mauer D, Zimmer A, Klingmuller D, et al. Control of hormonal stress reactivity by the endogenous opioid system. Psychoneuroendocrinology. 2008;33:425–436. doi: 10.1016/j.psyneuen.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006;59:1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Belcheva MM, Coscia CJ. Mitogenic signaling via endogenous κ-opioid receptors in C6 glioma cells: evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J Neurochem. 2000;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, et al. Stress-induced p38 mitogen-activated protein kinase activation mediates κ-opioid-dependent dysphoria. J Neurosci. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, et al. Selective p38α MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;11:383–385. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW. kappa-Opioid receptor signaling and brain reward function. Brain Res Rev. 2009;62:127–146. doi: 10.1016/j.brainresrev.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Beguin C, Knoll AT, Cohen BM. Kappa-opioid ligands in the study and treatment of mood disorders. Pharmacol Ther. 2009;123:334–343. doi: 10.1016/j.pharmthera.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croom KF, Perry CM, Plosker GL. Mirtazapine. A review of its use in major depression and other psychiatric disorders. CNS Drugs. 2009;23:427–452. doi: 10.2165/00023210-200923050-00006. [DOI] [PubMed] [Google Scholar]
- Devoize JL, Rigal F, Eschalier A, Trolese JF. Naloxone inhibits clomipramine in mouse forced swimming test. Eur J Pharmacol. 1982;78:229–231. doi: 10.1016/0014-2999(82)90241-2. [DOI] [PubMed] [Google Scholar]
- Doxey JC, Everitt J, Metcalf G. Mianserin- An analysis of its peripheral autonomic actions. Eur J Pharmacol. 1978;51:1–10. doi: 10.1016/0014-2999(78)90055-9. [DOI] [PubMed] [Google Scholar]
- Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS. A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry. 2007;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Gray AM, Spencer PSJ, Sewell RDE. The involvement of the opioidergic system in the antinociceptive mechanisms of action of antidepressant compounds. Br J Pharmacol. 1998;124:669–674. doi: 10.1038/sj.bjp.0701882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BY, Takemori AE. Serotonergic involvement in the antinociceptive action of and the development of tolerance to the kappa-opioid receptor agonist, U-50,488H. J Pharmacol Exp Ther. 1989;250:508–514. [PubMed] [Google Scholar]
- Isenberg KE, Cicero TJ. Possible involvement of opiate receptors in the pharmacological profiles of antidepressant compounds. Eur J Pharmacol. 1984;103:57–63. doi: 10.1016/0014-2999(84)90189-4. [DOI] [PubMed] [Google Scholar]
- Kudryavtseva NN, Gerrits MAFM, Avgustinovich DF, Tenditnik MV, Van Ree JM. Modulation of anxiety-related behaviors by µ- and κ-opioid receptor agonists depends on the social status of mice. Peptides. 2004;25:1355–1363. doi: 10.1016/j.peptides.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Kurata K, Kurachi M. Heterogeneous distribution of mianserin in rat brain during chronic continuous infusion. Pharmacology. 1989;39:285–290. doi: 10.1159/000138611. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, et al. Antidepressant-like effects of κ-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- Marchand F, Ardid D, Chapuy E, Alloui A, Jourdan D, Eschalier A. Evidence for an involvement of supraspinal δ- and spinal µ-opioid receptors in the antihyperalgesic effect of chronically administered clomipramine in mononeuropathic rats. J Pharmacol Exp Ther. 2003;307:268–274. doi: 10.1124/jpet.103.052613. [DOI] [PubMed] [Google Scholar]
- Marshall RJ. The pharmacology of mianserin – An update. Br J Clin Pharmacol. 1983;15:263S–268S. doi: 10.1111/j.1365-2125.1983.tb05874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf MD, Coop A. Kappa-opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mico JA, Ardid D, Berrocoso E, Eschalier A. Antidepressants and pain. Trends Pharmacol Sci. 2006;27:348–354. doi: 10.1016/j.tips.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Narita M, Kaneko C, Miyoshi K, Nagumo Y, Kuzumaki N, Nakajima M, et al. Chronic pain induces anxiety with concomitant changes in opioidergic function in the amygdala. Neuropsychopharmacology. 2006;31:739–750. doi: 10.1038/sj.npp.1300858. [DOI] [PubMed] [Google Scholar]
- Olianas MC, Concas D, Onali P. Agonist activity of naloxone benzoylhydrazone at recombinant and native opioid receptors. Br J Pharmacol. 2006;147:360–370. doi: 10.1038/sj.bjp.0706601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olianas MC, Dedoni S, Onali P. 2011. Antidepressants and opioid receptors: evidence that mianserin acts a san agonist at the kappa receptor subtype. Anxiety and Depression. 21st Neuropharmacology Conference Abstr. p.37.
- Onali P, Olianas MC. Muscarinic M4 receptor inhibition of dopamine D1-like receptor signalling in rat nucleus accumbens. Eur J Pharmacol. 2002;448:105–111. doi: 10.1016/s0014-2999(02)01910-6. [DOI] [PubMed] [Google Scholar]
- Onali P, Dedoni S, Olianas MC. Direct agonist activity of tricyclic antidepressants at distinct opioid receptor subtypes. J Pharmacol Exp Ther. 2010;332:255–265. doi: 10.1124/jpet.109.159939. [DOI] [PubMed] [Google Scholar]
- Otani K, Sasa H, Kaneko S, Kondo T, Fukushima Y. Steady-state plasma concentrations of mianserin and its major active metabolita, desmethylmianserin. Ther Drug Monit. 1993;15:113–117. doi: 10.1097/00007691-199304000-00008. [DOI] [PubMed] [Google Scholar]
- Ozturk Y, Aydin S, Beis R, Herekman-Demir T. The involvement of endogenous opioid mechanisms in the antinociceptive effects induced by antidepressant drugs, desipramine and trimipramine. Pharmacol Biochem Behav. 2006;83:592–597. doi: 10.1016/j.pbb.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Peroutka S, Snyder SH. 3H]Mianserin: differential labelling of serotonin2 and histamine1 receptors in rat brain. J Pharmacol Exp Ther. 1981;216:142–148. [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pinder RM. Adrenoreceptor interactions of the enantiomers and metabolites of mianserin: are they responsible for the antidepressant effect ? Acta Psychiatr Scand. 1985;72(Suppl. 320):1–9. doi: 10.1111/j.1600-0447.1985.tb08068.x. [DOI] [PubMed] [Google Scholar]
- Privette TH, Terrian DM. Kappa opioid agonists produce anxiolytic-like behaviour on the elevated plus-maze. Psychopharmacology. 1995;118:444–450. doi: 10.1007/BF02245945. [DOI] [PubMed] [Google Scholar]
- Raiteri M, Maura G, Gemignani A, Pittaluga A. Differential blockade by (-)mianserin of the alpha2-adrenoceptors mediating inhibition of noradrenaline and serotonin release from rat brain synaptosomes. Naunyn Schmiedebergs Arch Pharmacol. 1983;322:180–182. doi: 10.1007/BF00512394. [DOI] [PubMed] [Google Scholar]
- Reichenberg K, Gaillard-Plaza G, Montastruc JL. Influence of naloxone on the antinociceptive effects of some antidepressant drugs. Arch Int Pharmacodyn Ther. 1985;275:78–85. [PubMed] [Google Scholar]
- Richelson E, Nelson A. Antagonism by antidepressants of neurotransmitter receptors of normal human brain in vitro. J Pharmacol Exp Ther. 1984;230:94–102. [PubMed] [Google Scholar]
- Rose A, McCulloch MW, Sarantos-Laska C, Rand MJ. Effects of mianserin on noradrenergic mechanisms. J Psychiatr Res. 1984;18:79–88. doi: 10.1016/0022-3956(84)90049-9. [DOI] [PubMed] [Google Scholar]
- Schoemaker H, Berendsen HHG, Stevens HJT, Nickolson VJ. Differences in presynaptic α-blockade, noradrenaline uptake inhibition, and potential antidepressant activity between (+)- and (-)mianserin. Psychopharmacology. 1981;74:137–142. doi: 10.1007/BF00432680. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Backer MM, Kaufman JP, Pick CG. Interaction between the tetracyclic antidepressant mianserin HCl and opioid receptors. Eur Neuropsychopharmacol. 1998;8:297–302. doi: 10.1016/s0924-977x(97)00088-6. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Rigai T, Katz Y, Pick CG. The antinociceptive effect of mirtazapine in mice is mediated through serotonergic, noradrenergic and opioid mechanisms. Brain Res Bull. 2002;28:601–605. doi: 10.1016/s0361-9230(02)00825-0. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Geisslinger G. Opioids as modulators of cell death and survival- Unraveling mechanisms and revealing new indications. Pharmacol Rev. 2004;56:351–369. doi: 10.1124/pr.56.3.2. [DOI] [PubMed] [Google Scholar]
- Tejedor-Real P, Mico JA, Maldonado R, Roques BP, Gibert-Rahola J. Implication of endogenous opioid system in the learned helplessness model of depression. Pharmacol Biochem Behav. 1995;52:145–152. doi: 10.1016/0091-3057(95)00067-7. [DOI] [PubMed] [Google Scholar]
- Tokimasa T, Ariyoshi M, Akasu T. Mianserin blocks α2 adrenoceptors in submucous neurones of the guinea-pig caecum. Eur J Pharmacol. 1987;143:243–250. doi: 10.1016/0014-2999(87)90539-5. [DOI] [PubMed] [Google Scholar]
- Wall PM, Messier C. U-69,593 microinjection in the infralimbic cortex reduces anxiety and enhances spontaneous alternation memory in mice. Brain Res. 2000;856:259–280. doi: 10.1016/s0006-8993(99)01990-3. [DOI] [PubMed] [Google Scholar]
- Wittmann W, Schunk E, Rosskothen I, Gaburro S, Singewald N, Herzog H, et al. Prodynorphin-derived peptides are critical modulators of anxiety and regulate neurochemistry and corticosterone. Neuropsychopharmacology. 2009;34:775–785. doi: 10.1038/npp.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z-Q, Gao Y-J, Sun Y-G, Zhao C-S, Gereau IVRW, Chen Z-F. Central serotonergic neurons are differentially required for opioid analgesia but not for morphine tolerance or morphine reward. Proc Natl Acad Sci USA. 2007;104:14519–14524. doi: 10.1073/pnas.0705740104. [DOI] [PMC free article] [PubMed] [Google Scholar]