

Abstract
Possible prevention of the effects of prenatal alcohol exposure has been investigated using peptides that were previously shown to be involved in neuroprotection both in vitro and in vivo. I focused in this study on investigating the neuro-protective effects of one of these peptides with regard to the determination of the downstream signaling pathways involved in neuroprotection. This peptide with the sequence NAPVSIPQ, known as NAP, a fragment of activity-dependent neuroprotective protein, demonstrated a potent protective effect against oxidative stress associated with alcohol exposure. On embryonic day 7 (E7), weight-matched C57BL/6 pregnant females were assigned the following groups: (1) Ethanol liquid diet group (ALC) 25% (4.49%, v/v) ethano-derived calories, (2) Pair-fed (PF) control group (3) Chow control group, (4) treatment groups with alcohol alongside i.p. injections of d-NAP (ALC/d-NAP, 20 or 30 μg/20 g body weight), (5) PF/d-NAP control group. On E13, fetal brains were collected and assayed for TdT-mediated dUTP nick end labeling (TUNEL) staining, caspase-3 colorimetric assay and ELISA for cytochrome c detection. My results show that NAP significantly prevented alcohol-induced weight reduction of the fetal brain. Apoptosis was determined by TUNEL staining; NAP administration significantly prevented alcohol-induced increases in TUNEL-positive cells in primordium cingulate cortex and basal ganglia eminence. The investigation of downstream signaling pathways involving NAP neuroprotection revealed that this peptide significantly prevented alcohol-induced increase in the concentrations of caspase-3 in E13 fetal brains. Moreover, ELISA for cytochrome c shows that NAP significantly prevented both alcohol-induced increases in the level of cytosolic cytochrome c and alcohol-induced decreases in the level of mitochondrial cytochrome c. These data provide an understanding of NAP intracellular target, and the downstream mechanisms of action that will pave a path toward potential therapeutics against alcohol intoxication during prenatal stages.
Keywords: neuroprotection, fetal alcohol syndrome, cytochrome c, mitochondria, caspase-3, TUNEL
Brain growth restriction is a hallmark of fetal alcohol exposure (FAE) and fetal alcohol syndrome (FAS) models (Roebuck et al., 1998). Alcohol exposure impedes cerebrum and cerebellum growth (Bauer-Moffett and Altman, 1975, 1977; Kornguth et al., 1979; Samson and Diaz, 1981; Sulik et al., 1981; Barron et al., 1988; Bonthius and West, 1990; Sari and Gozes, 2006). Our and other studies have demonstrated that the effects of prenatal alcohol exposure depend on the dose and timing of alcohol treatment (Webster et al., 1983; Sari and Gozes, 2006). During the period between embryonic day 7 (E7) and E14, the developing brain exhibited the highest susceptibility to alcohol exposure. Deficits in fetal brain growth might be induced possibly through an apoptotic mechanism (Ikonomidou et al., 2000). Mitochondrial activation of apoptotic pathways is one of the major players in ethanol-mediated neuronal death. Although studies have demonstrated that prenatal alcohol exposure induces mitochondrial dysfunction, little is known about its signaling pathways. The mitochondrion is a target organelle in ethanol-induced organ-system toxicity (Hoek et al., 2002). Prenatal alcohol exposure–induced mitochondrial dysfunction includes decreased mitochondrial glutathione concentration, decreased activities of respiratory chain complex IV and ATP synthase, and increased mitochondrial permeability transition (Ramachandran et al., 2001; Spong et al., 2001; Xu et al., 2005a; Green et al., 2006). We have recently found that prenatal alcohol exposure induced decreases in ATP synthase, ADP/ATP translocase, NADH dehydrogenase, ubiquinol-cytochrome-c reductase and prohibitin at E13 (Sari and Mechref, 2008). Together, these findings indicate that mitochondrial dysfunction is a key factor in alcohol-induced apoptosis.
E13 age was chosen as an end-point of alcohol exposure because this developmental age holds several interests in my ongoing research projects in FAE model. Most of the developmental regulatory proteins are highly expressed at this age of development and altered as a consequence of alcohol exposure (Sari and Gozes, 2006; Sari and Mechref, 2008). In addition, at E13, the neural tube has undergone five major divisions to form a fetal brain composed of telencephalon, diencephalon, midbrain, hind-brain and spinal cord. Moreover, at this developmental stage, the 5-HT neurotransmitter system has formed and initiated its differentiation. Our previous study reported that prenatal alcohol exposure induces neural tube defects early at E13 (Zhou et al., 2003) and similarly alters early 5-HT neurons [for review see ref. (Sari and Gozes, 2006)].
Studies investigating alcohol exposure during development in animal models may provide important information for the identification of possible mechanisms of neuroprotection and allow for the development of intervention procedures, which may protect or attenuate the deleterious effects of alcohol exposure during pregnancy. Several studies have investigated the possible prevention of the effects of prenatal alcohol exposure by the treatment of pregnant mice with peptides that have been shown to be involved in neuroprotection (Spong et al., 2001; Poggi et al., 2003; Brenneman et al., 2004; Zhou et al., 2004; Sari and Gozes, 2006). Among these peptides, SALLRSIPA, known as SAL or ADNF-9, is derived from activity-dependent neurotrophic factor (ADNF) (Brenneman and Gozes, 1996; Brenneman et al., 1998). Another peptide with the sequence NAPVSIPQ peptide, termed NAP, derived from activity-dependent neuroprotective protein (ADNP) (Bassan et al., 1999; Zamostiano et al., 2001), demonstrated a potent protective effect against oxidative stress.
In the present study, using our established FAE model in C57BL/6, we tested the neuroprotective effects of NAP at early stage of development E13. The ADNP is released from astroglia (Bassan et al., 1999) and found to be regulated by vasoactive intestinal peptide, VIP (Furman et al., 2004). High ADNP mRNA expression was found in the hippocampus, cerebral cortex and cerebellum in the mouse (Bassan et al., 1999) and in the human brain (Zamostiano et al., 2001), indicating a potential role in the central nervous system (Gozes et al., 1999). It has been further shown that knocking out the ADNP gene is lethal to the embryo and the ADNP-deficient embryos die at the time of neural tube closure (Pinhasov et al., 2003).
Our and other studies have focused on the role of NAP in neuroprotection against prenatal alcohol exposure [for review see ref. (Sari and Gozes, 2006)]. A recent study demonstrated a significant decrease in the ADNP mRNA in the cerebral cortex of 7-day-old pups that were exposed prenatally to alcohol (Pascual and Guerri, 2007). Using an established model for FAS, it has been demonstrated at National Institutes of Health–National Institute of Child Health and Human Development (NIH-NICHD) that pretreatment with NAP prevented the fetal death and abnormalities induced by prenatal alcohol exposure (Spong et al., 2001). Importantly, the effects of enantiomer conformation (l- and d-forms) of this peptide against the insult of alcohol exposure was also investigated in FAS model (Brenneman et al., 2004). The d-form of NAP resulted in a potent effect with no loss of protective response regardless of increases in the doses.
We have demonstrated previously the effects of d-NAP for its protective properties against fetal alcohol–related brain growth restriction in FAE model in C57BL/6 mice (Sari et al., 2001, 2003; Sari and Zhou, 2004; Zhou et al., 2004; Sari and Gozes, 2006). Administration of d-NAP alongside alcohol exposure, from E7 to E15, antagonized the effects of alcohol on brain weight, the size of several forebrain regions, and neural tube development [recently reviewed by us (Sari and Gozes, 2006)]. In the present study, using a similar liquid diet drinking paradigm, we investigated the cellular mechanism of NAP in neuroprotection with more focus on the apoptotic mechanisms that may involve extrinsic and intrinsic mitochondrial signaling pathways at E13.
EXPERIMENTAL PROCEDURES
Synthesis of d-NAP peptide
The peptide d-NAP (NAPVSIPQ, all amino acids are d form) was synthesized in-house at Indiana University using a standard solid-phase Fmoc chemistry on a ABI 433 peptide synthesizer. The peptide was de-protected/cleaved from the resin in TFA cocktail and purified by HPLC. The purity of the final compound was >98% as identified with analytical HPLC. The mass of d-NAP was determined to be m/z 825.3[M+H]+ (calc.825.4) by ESI-MS.
Animals
Mice C57BL/6 were used in these studies. Both male and female mice were obtained at 6–7 weeks of age from Harlan Laboratory at Indianapolis, IN, USA. All mice were housed in the departmental animal colony in a vivarium with a controlled climate (temperature 22 °C, 30% humidity) with a 12-h light/dark cycle. Animals used in these procedures were maintained in facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All experimental procedures were approved by the Institutional Animal Care and Use Committee of Indiana University Bloomington and are in accordance with the guidelines of the Institutional Animal Care and Use Committee of the NIH, and the Guide for the Care and Use of Laboratory Animals. Pregnants mice were euthanized by CO2 followed by physical translocation. This method is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. In addition the number of animals used in this study was minimized according to our previous studies and power analyses.
Breeding and treatment procedure
Female mice were placed into male home cages for 2 h and were then checked for a sperm plug by vaginal smear immediately afterward. When the plugs were positive, we designated this time point as E0. On E7, weight-matched pregnant females were assigned to the following groups: (1) Ethanol liquid diet group (ALC, n=7) fed with chocolate Sustacal (supplemented with vitamins and minerals, Bio-Serv, Frenchtown, NJ, USA) liquid diet 25% (4.49%, v/v) ethanol derived calories (EDC), (2) Pair-fed control group (PF, pair-fed to ethanol-fed group, n=7) fed with a maltose–dextrin solution which was made isocaloric to the dose of ethanol used, (3) Chow group fed with mouse chow pellets and water (Chow, n=5), (4) treatment group, i.p. injections of d-NAP alongside alcohol exposure (ALC/NAP20, 20 μg/20 g body weight, n=6), (5) treatment group, i.p. injections of d-NAP alongside alcohol exposure (ALC/NAP30, 30 μg/20 g body weight, n=5), and (6) PF/NAP control group (d-NAP i.p. injected at a dose of 30 μg/20 g body weight, n=5). NAP was administered once a day from E7 through E13. The PF, PF/NAP, and Chow served as control groups. We have generated fetal brains from 35 pregnant mice (Chow, PF, PF/NAP, ALC, ALC/NAP20 and ALC/NAP30) at E13.
I used my established liquid diet mixed with alcohol in FAE model that mimics moderate alcohol drinking [for review see (Sari and Gozes, 2006)]. As published by Middaugh and colleagues (Middaugh et al., 1988; Middaugh and Boggan, 1995), the fortified liquid diet contained 237 ml of chocolate-flavored Sustacal (CVS Pharmacy), 1.44 g Vitamin Diet Fortification Mixture and 1.2 g Salt Mixture XIV. For the ethanol diet, 15.3 ml (4.49% v/v; 25% EDC) of 95% ethanol was added to the fortified Sustacal formula with water that was added to make 320 ml of diet with 1 cal/ml (ethanol). For the isocaloric control diet, 20.2 maltose dextrin was added to the fortified Sustacal formula with water that was added to bring it to 1 cal/ml (Middaugh et al., 1988; Middaugh and Boggan, 1995). One day before treatment, the ALC, ALC/NAP20 and ALC/NAP30 dams and also the PF and PF/NAP dams were adapted to the PF liquid diet, the dams were weighed, the volume of liquid diet consumed during the previous 24 h was recorded from 30-ml graduated screw-cap tubes, and freshly prepared diet was provided. The PF or PF/NAP subjects had limited access to the PF liquid diet each day to match the amount of calories given to ALC, ALC/NAP20 and ALC/NAP30 subjects.
Maternal blood alcohol levels
Maternal blood alcohol levels were tested in a separate group of C57BL/6 dams with the 25% (4.49%, v/v) EDC diet (Sari and Zhou, 2004; Sari and Gozes, 2006). Pregnant mice were given the same feeding protocol as the other experimental dams (EDC diet provided on E7 at 700 h), and two 50-μl tail blood samples were obtained (at 09:00 and 11:00 h in a reverse dark cycle) at E8 and E11. The blood samples were collected in heparinized capillary tubes and centrifuged, and 5-μl plasma samples were analyzed for alcohol concentrations using the Analox Alcohol Analyzer, calibrated with a 100 mg/dl ethanol standard. Blood alcohol concentrations (BACs) were evaluated at 2 and 4 h on E8 and E11. The BACs were consistently higher at 2 h exposure. The average peaks obtained in the 25% EDC group at 2 h were ~40 mg/dl on E8 and ~55 mg/dl on E11.
Fetal brains
Pregnant mice were deeply anesthetized with CO2 procedure followed by physical translocation at E13 and the fetuses were removed and further dissected from the base of the primordium olfactory bulb to the base of the metencephalon. From the same dam, a group of fetal brains was frozen and stored at −70 °C until used for chemical assays and the other group was fixed in 4% paraformaldehyde for TdT-mediated dUTP nick end labeling (TUNEL)–positive cell detection and fetal brain weight determination.
Protein and concentration of active caspase-3 detections
For tissue extracts, frozen fetal brain from E13 Chow, PF, PF/NAP, ALC, ALC/NAP20, and ALC/NAP30 was ground to a powder with a pestle. The powdered tissue was mixed with TNE buffer (10 mM Tris, pH 7.4; 0.15 M NaCl; 1 mM EDTA) supplemented with protease inhibitor cocktail (Sigma, St Louis, ME, USA) under continuous grinding until the suspension was homogeneous. The suspension was centrifuged at 14,000 rpm for 10 min at 4 °C. The supernatant was collected and frozen at −70 °C until a later date for detection of protein or active caspase-3 concentration. The total protein estimation in each sample was evaluated with the Bio-Rad (Hercules, CA, USA) protein assay.
The concentrations of active caspase-3 were determined using caspase-3 colorimetric assay kit (Assay Designs, Inc, Ann Arbor, MI, USA). The kit involves the conversion of a specific chromogenic substrate for caspase-3 followed by the colorimetric detection of the colored product of a reaction that absorbs visible light at 405 nm. The samples in variant dilutions, standards, p-nitroaniline calibrator (pNA), and blank controls were plated in duplicate in 96 microplates. The blank control was a mixture of active caspase-3 reaction buffer and caspase-3 substrate. The conversion of substrate into the colored product was measured after a 3-h incubation at 37 °C, and the reaction was stopped by a 1 N solution of hydrochloric acid. The multiple samples, standards, pNA, and blank controls were read rapidly by an absorbance reader (SUNRISE, Phoenix Research Products, Phoenix, CA, USA). The average net optical density (OD) for each standard and sample was calculated by subtracting the average blank OD from the average OD for each standard and sample. The activity measurements were quantitated by comparisons of the ODs obtained with standards or with the pNA. The concentration of active caspase-3 in the samples was then determined by interpolation of the average net OD for each standard versus the actual concentration of active caspase-3 substrate for the standards. The concentrations of active caspase-3 in the samples for all groups were expressed as units per milligram of total protein tissue. The colorimetric assay was repeated twice for confirmation of the quantitative analyses.
Analysis of cytosolic and mitochondrial fractions of cytochrome c
Frozen brains were homogenized with digitonin (0.05%) in a lysis buffer (250 mM sucrose, 20 mM Hepes, 10 mM KCl, 5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, protease cocktail inhibitor 1:100). The homogenates were then centrifuged for 12 min (12,000 rpm, 4 °C). The supernatant (cytosolic fraction) was removed and stored at −70 °C. The pellet was resuspended in a second lysis buffer for 30 min (133 mM NaCl, 50 mM Tris pH 8.0, SDS 0.1% [w/v], sodium deoxycholate 0.5% [w/v], Igepal CA630 1.0% [v/v] and protease cocktail inhibitor 1:100). The mixture was then centrifuged and the supernatant (mitochondrial fraction) was collected and stored at −70 °C until ELISA (Chemicon International, Temecula, CA, USA) testing for cytochrome c fractions. Cytosolic and mitochondrial fractions were assayed for ELISA for determination of cytochrome c in nanograms as detailed in the manufacturer protocol. In brief, an anti–cytochrome c monoclonal coating antibody was absorbed onto a 96 microtiter plate. Cytochrome c from samples or standards was incubated to the absorbed antibody for 2 h at room temperature (RT). After incubation, unbound anti–cytochrome c was removed with wash buffer and a biotin-conjugated monoclonal anti–cytochrome c for 2 h at RT. Following incubation, unbound biotin-conjugated anti–cytochrome c was removed by several steps of washes with wash buffer. Streptavidin-HRP was incubated for 1 h at RT. After washes, a substrate solution reactive with HRP was added to each well for 5 min at RT. The enzymatic reaction was stopped with stop solution and the absorbance was read immediately on a spectrophotometer at 450 nm. Total protein concentration in the cytosolic or mitochondrial fraction in each sample was evaluated with the Bio-Rad protein assay. We then determined the average nanograms cytochrome c in cytosolic and/or mitochondrial fractions per milligram of protein tissue. This assay was repeated twice for confirmation of the quantitative analyses.
TUNEL immunostaining reaction for cell death detection and counting
Cell death was determined using TUNEL reaction. Apoptosis and cell-mediated cytotoxicity are characterized by a fragmentation of the genomic DNA. The many breakpoints can be visualized with the TUNEL reaction. To keep consistent conditions of immunostaining for both the experimental and the control groups, we embedded the fetal brains from ALC, ALC/NAP, PF/NAP, PF, and Chow groups in gelatin. All fetal brains were aligned at the same level in gelatin, and serial 50-μm coronal sections were then cut using a vibratome apparatus. Fetal brain sections were treated with proteinase K (10–20 μg/ml) for 5 min at 37 °C, rinsed with PBS three times for 5 min and then incubated with 3% H2O2 in methanol for 10 min at RT. The sections were again rinsed with PBS three times for 5 min and then incubated in a permeabilization solution (0.1% TX-100 in 0.1% sodium citrate) for 2 min at 4 °C. After the sections were rinsed twice in PBS for 5 min, they were incubated with a TUNEL reaction mixture (50 μl from bottle 1 and 450 μl from bottle 2, Roche Pharmaceuticals, Inc., Indianapolis, IN, USA) for 1 h at 37 °C. The control was prepared by incubation in solution from bottle 2 only. The sections were rinsed three times for 5 min with PBS and incubated in converter-POD for 30 min at 37 °C. After the sections were rinsed with TBS, they were incubated in 0.05% 3′-3′-diaminobenzidine tetrahydrochloride and 0.003% H2O2 in TBS to reveal the peroxidase activity.
TUNEL-positive cells were counted in the primordium cingulate cortex and basal ganglia eminence of E13 fetal brains from PF, ALC and ALC/NAP20 groups by an experimenter who was blind to the treatment groups. The penetration of TUNEL-staining through a thickness of 50 μm was verified at 100x magnification. The expected shrinkage of a 50 μm-thick section in the z plane was averaged to approximately 14 μm. The number of sections in the selected brain regions for TUNEL-positive cell counting was also considered and controlled in this study to avoid the bias of any missing sections from PF, ALC, and ALC/NAP20 groups. The entire population of TUNEL-positive cells was counted manually in every other section of the primordium cingulated cortex and basal ganglia eminence, and this overcomes the bias of over-counting the number of TUNEL-positive cells in adjacent sections.
Statistical analyses
Statistical analysis was performed using Prizm software. The data collected in this study was analyzed statistically using one-way analysis of variance (ANOVA) and Newman-Keuls multiple comparison test between control groups and treatment groups. All tests of significance were performed at P<0.05.
RESULTS
Protective effects of NAP administration on fetal brain weight against the insult of alcohol exposure
Morphological observations of fetal brains show smaller size of forebrain primordium and midbrain primordium in ALC fetal brain as compared with PF/NAP and ALC/NAP fetal brains (Fig. 1a, b, c). The one-way ANOVA demonstrated a significant difference between groups (P=0.0001). Post hoc comparison using Newman-Keuls test shows significant reduction in fetal brain weights in the ALC group as compared with control groups (Chow, PF and PF/NAP) (P<0.001) (Fig. 1d). Administration of NAP at two different doses (20 or 30 μg/20 g body weight i.p.) alongside prenatal alcohol exposure prevented such weight reduction and stabilized brain weights comparable approximately to control groups (Chow, PF and PF/NAP). There were significant differences between ALC/NAP treatment groups (ALC/NAP20 and ALC/NAP30) and the ALC group (P<0.001 and P<0.01, respectively). However, there were no significant differences between Chow, PF, PF/NAP, ALC/NAP20, and ALC/NAP30 groups (P>0.05).
Fig. 1.
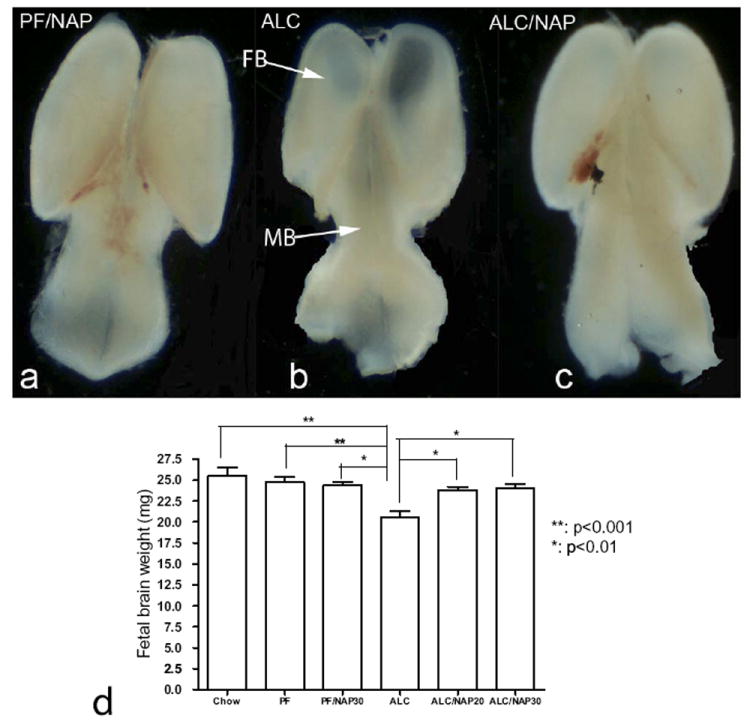
Protective effect of NAP on fetal brain weight at E13 in mice exposed prenatally to alcohol. Reduction of the sizes of the forebrain primordium (FB) and midbrain primordium (MB) can be observed in ALC (b) fetal brain as compared with PF/NAP (a) and ALC/NAP (c) fetal brains as indicated by arrows. (d) Statistical analyses demonstrated that prenatal alcohol exposure induced significant fetal brain reduction as compared with Chow and PF control groups (P<0.001). NAP treatment prevented the deficit in fetal brain weight found in the ALC alone group at both doses, 20 and 30 μg/20 g body weight (P<0.01). No significant differences were found between Chow, PF, PF/NAP, ALC/NAP20 and ALC/NAP30 groups. Values shown as means±SEM. Chow, n=5; PF, n=7; PF/NAP, n=5; ALC, n=7; ALC/NAP20, n=5;and ALC/NAP30, n=5.
Neuroprotective effect of NAP administration against prenatal alcohol-induced apoptosis
In order to determine whether NAP plays a role in prevention of alcohol-induced apoptosis, we used TUNEL-staining to evaluate cell death. Among the fetal brain regions that were analyzed anatomically and statistically are primordium cingulated cortex and basal ganglia eminence. These fetal brain regions have been well studied previously by us in an FAE model [for review see (Sari and Gozes, 2006)]. In primordium cingulated cortex, we found that prenatal alcohol exposure induced significant increases in TUNEL-positive cells in ALC group (Fig. 2b) as compared with control group PF (Fig. 2a). NAP administration alongside alcohol exposure prevented the alcohol-induced cell death in primordium cingulated cortex (Fig. 2c) as compared with ALC group. Statistical analyses of cell counts show significant increase in the TUNEL-positive cells in primordium cingulated cortex of the ALC group as compared with the PF group (P<0.01) (Fig. 2d). Importantly, NAP (ALC/NAP20) significantly prevented alcohol-induced increases in the number of TUNEL-positive cells as compared with the ALC group (P<0.01). No significance difference was found between PF and ALC/NAP20 groups (P>0.05). In basal ganglia eminence, alcohol exposure induced increases in TUNEL-positive cells (Fig. 3b) as compared with the PF control group (Fig. 3a). Administration of NAP peptide alongside alcohol exposure decreases the number of TUNEL-positive cells in basal ganglia eminence (Fig. 3c) when compared with ALC groups. Statistical analyses of cell counts show a significant increase in the number of TUNEL-positive cells in basal ganglia eminence of the ALC group as compared with the PF group (P<0.001) (Fig. 3d). Importantly, NAP (ALC/NAP20) significantly prevented alcohol-induced increases in the number of TUNEL-positive cells as compared with the ALC group (P<0.001). No significance difference was found between PF and ALC/NAP groups (P>0.05).
Fig. 2.
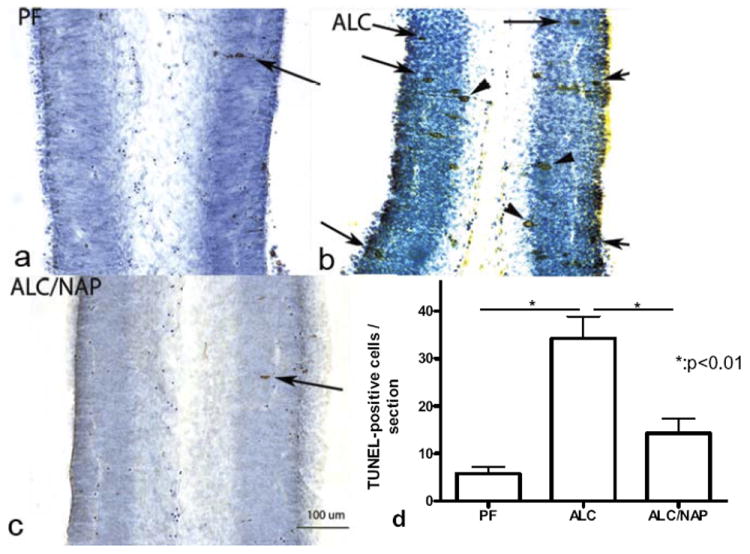
Prenatal alcohol exposure increases the number of TUNEL positive cells in primordium cingulate cortex (a–c) at E13. Administration of NAP alongside alcohol exposure prevented alcohol-induced increases in TUNEL-positive cells. Arrowheads indicate cell undergoing apoptosis as indicated by cell processes, however, arrows indicate the final stage of cell death. Scale bars=100 μm. The one-way ANOVA demonstrated a significant difference between groups (P=0.0016). Increases in TUNEL-positive cells were found in the ALC group as compared with the PF group (P<0.01) (d). Importantly, NAP administration alongside alcohol exposure prevented a significantly alcohol-induced increase in TUNEL-positive cells (P<0.01) (d). Values shown as means±SEM. N=5 for each group (PF, ALC, and ALC/NAP).
Fig. 3.
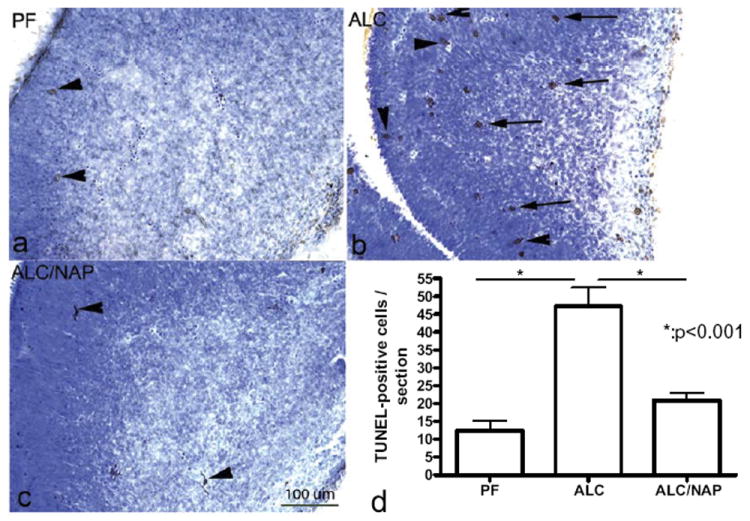
Prenatal alcohol exposure increases the number of TUNEL positive cells in basal ganglia eminence of E13 fetal brains (b) as compared with control group (a). Administration of NAP alongside alcohol exposure prevented alcohol-induced increases in TUNEL-positive cells (c). Scale bar=200 μm. The one-way ANOVA demonstrated a significant difference between groups (P=0.0005). Increases in TUNEL-positive cells were found in the ALC group as compared with the PF group (P<0.001) (d). Importantly, NAP administration alongside alcohol exposure prevented a significantly alcohol-induced increase in TUNEL-positive cells (P<0.001) (d). Values shown as means±SEM. N=5 for each group (PF, ALC, and ALC/NAP).
Neuroprotective effect of NAP administration against prenatal alcohol-induced caspase-3 activation
Caspase-3 colorimetric assay was used in this experiment to determine the concentration of active caspase-3 in the E13 fetal brain in order to determine if the neuroprotective effect of NAP is mediated through caspase-3 downstream apoptotic pathway. Caspase-3 is a cysteine protease that plays a critical role in the induction of apoptosis. Activation of caspase-3 indicates that cells entered an apoptotic pathway. Using a colorimetric assay, we demonstrated a significant difference between groups in the concentration of caspase-3 activity as shown with one-way ANOVA (P=0.0039). Post hoc comparison shows a significant difference in the concentration of active caspase-3 in the fetal brains between the ALC group and control groups Chow (P<0.05) and PF (P<0.01) (Fig. 4). Moreover, administration of NAP at the two different doses (20 and 30 μg/dam/day) significantly prevented alcohol-induced increases in the concentration of active caspase-3 (P<0.05). No significant difference was found in the concentration of active caspase-3 in the fetal brain between NAP treatment groups (ALC/NAP20 and ALC/NAP30) and control groups (Chow and PF) (Fig. 4).
Fig. 4.
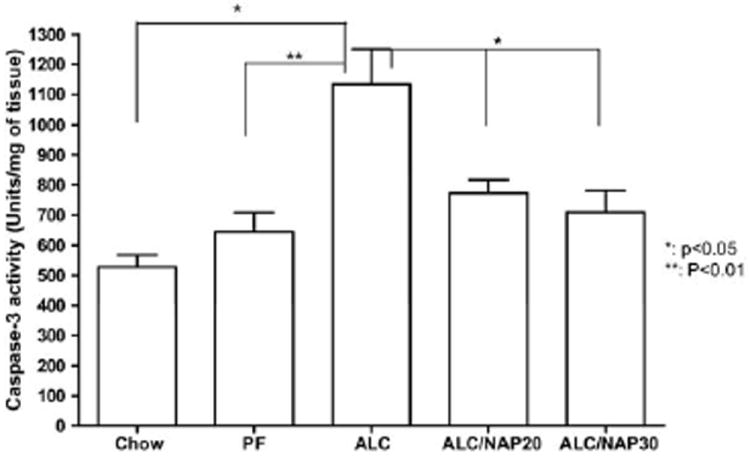
Neuroprotective effect of NAP against the insult of prenatal alcohol exposure is mediated through caspase-3 activation as tested by caspase-3 colorimetric assay in E13 fetal brains. The assay was repeated twice for quantitative analysis. Prenatal alcohol exposure induced significant increase in the concentrations of active caspase-3 as compared with Chow and PF control groups (P<0.05 and P<0.01, respectively). NAP administration at 20 or 30 μg/20 g body weight prevented significantly the effect of alcohol-induced increases in the concentrations of active caspase-3 (P<0.05). There were no significant difference between control groups and NAP treatment groups. Values shown as means±SEM. Chow, n=5; PF, n=7; ALC, n=7; ALC/NAP20, n=5; and ALC/NAP30, n=5.
Neuroprotective effect of NAP administration against prenatal alcohol-induced increases in the level of cytosolic cytochrome c and decreases in the level of mitochondrial cytochrome c
We next tested whether cytosolic cytochrome c release is correlated with changes in the concentration of active caspase-3 and whether the neuroprotective effects of NAP are mediated through the downstream signaling pathways that involved cytosolic cytochrome c. The one-way ANOVA demonstrated a significant difference between groups (P=0.0082). Post hoc comparison shows that prenatal alcohol exposure (ALC group) induced significant increases in the level of cytosolic cytochrome c as compared with PF (P<0.01) and PF/NAP (P<0.01) control groups (Fig. 5a). Simultaneous NAP administration at two different doses (20 and 30 μg/20 g body weight) alongside prenatal alcohol exposure significantly prevented this increase of the level of cytosolic cytochrome c (ALC/NAP20 and ALC/NAP30) as compared with the ALC group (P<0.05) (Fig. 5a). No significant differences were found in the levels of cytosolic cytochrome c in fetal brain between NAP treatment groups (ALC/NAP20 and ALC/NAP30) and control groups (PF and PF/NAP) (Fig. 5a).
Fig. 5.
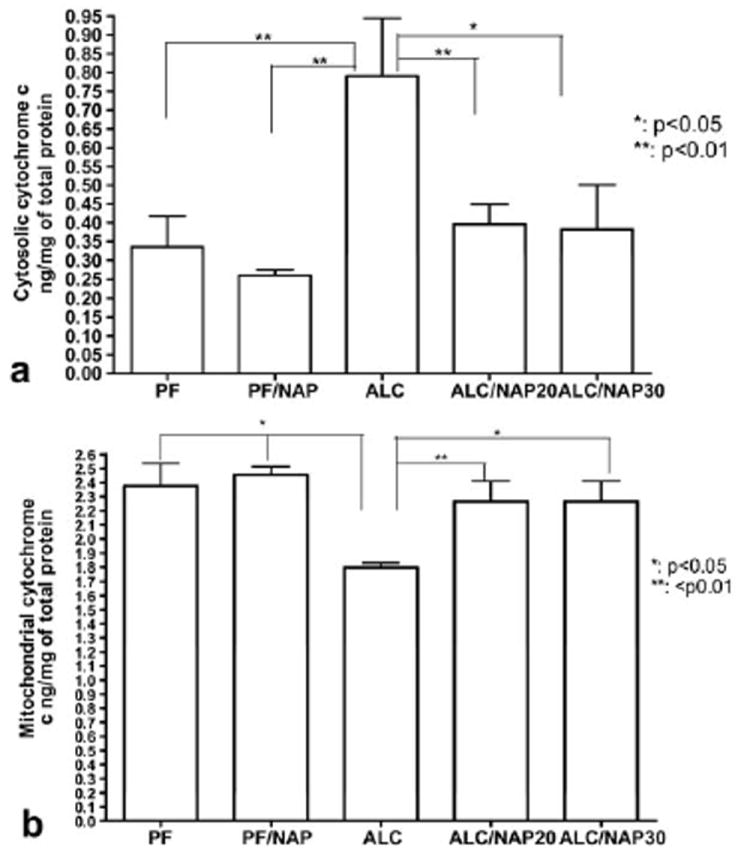
Neuroprotective effect of NAP against the insult of prenatal alcohol exposure is mediated through cytochrome c as tested by ELISA assay in E13 fetal brains. ELISA for determination of the levels of cytosolic and/or mitochondrial cytochrome c was repeated twice for quantitative analysis. (a) Prenatal alcohol exposure induced a significant increase in the concentrations of cytosolic cytochrome c as compared with PF (P<0.01) and PF/NAP (P<0.01) control groups. NAP administration at 20 or 30 μg/dam/day prevented significantly the increases in the levels of cytosolic cytochrome c as compared with the ALC group (P<0.01 and P<0.05, respectively). (b) Prenatal alcohol exposure induced significant decreases in the levels of mitochondrial cytochrome c as compared with PF and PF/NAP control groups (P<0.05). NAP administration at 20 and 30 μg/20 g body weight prevented the reduction in mitochondrial cytochrome c in both ALC/NAP20 (P<0.01) and ALC/NAP30 (P<0.05) as compared with the ALC group. There were no significant differences between control groups and NAP treatment groups. Values shown as means±SEM. N=5 for each group (PF, PF/NAP, ALC, ALC/NAP20, and ALC/NAP30).
We next determine whether changes in the cytosolic cytochrome c are correlated with changes in the levels of mitochondrial cytochrome c. The one-way ANOVA demonstrated a significant difference between groups (P=0.0157). Prenatal alcohol exposure (ALC group) induced significant decreases in mitochondrial cytochrome c as compared with PF and PF/NAP control groups (P<0.05) (Fig. 5b). Importantly, administration of NAP at two different doses (20 and 30 μg/20 g body weight) alongside prenatal alcohol exposure prevented alcohol-induced reduction in mitochondrial cytochrome c in both ALC/NAP20 and ALC/NAP30 as compared with ALC (P<0.01 and P<0.05, respectively) (Fig. 5b). No significant differences were found between ALC/NAP20 and ALC/NAP30, and PF, PF/NAP control groups (P>0.05).
DISCUSSION
We report here that prenatal alcohol exposure induced apoptosis through cytochrome c and caspase-3 downstream signaling pathways. Importantly, NAP administration reversed alcohol-induced increases in the levels of cytosolic cytochrome c and caspase-3; NAP administration also prevented alcohol-induced decreases in the levels of mitochondrial cytochrome c. In addition, NAP prevented alcohol-induced increases in TUNEL-positive cells, which is correlated with down-regulation of downstream signaling pathways.
Regarding the downstream signaling pathways involved in alcohol-induced apoptosis, my findings are in accordance with others that demonstrated that prenatal alcohol exposure (from E17 to E18) caused an increase in mitochondrial permeability transition, which is accompanied by an elevation of cytochrome c and apoptosis-inducing factor in mitochondria of E19 fetal rat brain (Ramachandran et al., 2001). Moreover, acute administration of alcohol on postnatal day 4 leads to apoptotic Purkinje cells in early generated cerebellar lobules (Light et al., 2002). These apoptotic neurons show release of cytochrome c from mitochondria. Cytochrome c release from mitochondria is a critical event in the intrinsic pathway of apoptosis that is suggested to be activated by alcohol exposure. The dissociation of cytochrome c from the inner mitochondrial membrane is the first step in the mitochondrial-directed apoptotic pathway. This is correlated to my findings which suggest that alcohol exposure causes a decrease in the level of mitochondrial cytochrome c. In addition, we have found recently that prenatal alcohol exposure induced significant down-regulation of some of the inner mitochondrial proteins including ADP/ATP translocase, ATP synthase subunit alpha and ubiquinol-cytochrome-c reductase in E13 fetal brain exposed prenatally to alcohol (Sari and Mechref, 2008). Our findings validate previous work which utilizes a different model of alcohol exposure (Xu et al., 2005a,b). These studies demonstrated that intragastric ethanol exposure from days E6 to E15 decreased the activities of respiratory chain complex I and IV and ATP synthase in the fetal cerebral mitochondria of mice. Together, these findings suggest that alcohol-induced apoptosis may be mediated through mitochondrial dysfunction.
The release of cytochrome c from mitochondria into the cytoplasm activates caspases by binding to apoptotic protease activating factor 1 (Apaf-1), inducing it to associate with pro-caspase-9, thereby triggering caspase-9 activation and initiating the proteolytic cascade that culminates in apoptosis (Liu et al., 1996; Zou et al., 1997; Green and Reed, 1998). One of these caspases is caspase-3 which plays a critical role in normal brain development and regulates neuronal apoptosis (Kuida et al., 1996; Keramaris et al., 2000). The regulation of survival/apoptosis during brain development is controlled by the Bcl-2 family which is located at the mitochondrial outer membrane (Cory and Adams, 2002). These Bcl-2 family proteins have been implicated in alcohol-induced apoptosis in the neonatal developing brain (Moore et al., 1999; Heaton et al., 2003; Young et al., 2003; Ge et al., 2004). Exposure of alcohol at postnatal day 4 alters the transcription and expression of pro- and anti-apoptotic Bcl-2 family members (Moore et al., 1999; Heaton et al., 2003). Using proteomics assay, we have recently demonstrated that prenatal alcohol exposure reduced the expression of Bcl-2 protein in E13 fetal brain (Sari and Mechref, 2008).
There are other potential mechanisms of action of alcohol exposure; alcohol may alter the Ca2+ handling in developing neurons. This results in an overload of cytosolic Ca2+ along with an excess of mitochondrial Ca2+ storage, which leads to cytochrome c release into the cytosol inducing apoptosis through activation of caspase cascade pathways (Susin et al., 1996; Leist and Nicotera, 1998; Thornberry and Lazebnik, 1998). The caspases convey the apoptotic signal in a proteolytic cascade, with caspases cleaving and activating other caspases that subsequently degrade cellular targets, ultimately leading to cell death. Alcohol may induce mitochondrial stress through disruption of Ca2+ homeostasis, the release of cytochrome c occurred from mitochondria, which then interacts with Apaf-1, causing self-cleavage and activation of caspase-9. The effector caspases: caspase-3, -6 and -7, are downstream of the activator caspases and act to cleave various cellular targets of neuronal death (Ashkenazi and Dixit, 1998). In addition to the present data related to E13 stage, we also have shown in a previous study using a similar model of FAE that prenatal alcohol exposure from E7–E18 induced significant increases in the concentration of active caspase-3 in the brainstem at E18 (Sari and Zhou, 2004).
In focus of the preventive effect of NAP against the insult of prenatal alcohol exposure, studies conducted at the NIH-NICHD and my laboratory have investigated the protective effects of this peptide using fetal brain morphological (at mid-age of development E15) and fetal death analyses (late age of development E18). Spong et al. (2001) originally demonstrated that single administration (i.p.) of NAP alongside single (i.p.) injection of alcohol at E8 prevented alcohol-induced fetal death, microencephaly and brain weight reduction at E18. We have previously shown that NAP administration alongside prenatal alcohol exposure antagonized the effects of alcohol-induced reduction in the brain weight and the volume of forebrain regions such as septal primordium, hippocampal primordium, amygdalar primordium, rostral and caudal ganglion eminences and diencephalon primordium (Sari and Gozes, 2006). In addition, a neuroprotective effect against prenatal alcohol exposure has also been found in the primordium cingulate and frontal cortices. The cortical plate and intermediate cortical layer in the primordium frontal cortex have also been protected with NAP against prenatal alcohol exposure from E7–E15 (Sari and Gozes, 2006). In the present study I found similar effect on fetal brain weight at E13. NAP at different doses (20 or 30 μg/20 g body weight) prevented alcohol-induced reduction in fetal brain weight at E13; there were no dose-dependent effects. These findings confirm previous findings from ours and others demonstrating that NAP prevented alcohol-induced microencephaly and growth restriction (Spong et al., 2001; Zhou et al., 2004; Sari and Gozes, 2006). Importantly, NAP administration prevents alcohol-induced increases in TUNEL-positive cells in two selected fetal brain regions such as primordium cingulated cortex and basal ganglia eminence regions. A number of studies have shown that prenatal alcohol exposure induced alterations in the primordium cerebral cortex, ganglion eminence, and also midline subcortical nuclei (thalamus and septal primordium), and corpus callosum (Clarren et al., 1978; Peiffer et al., 1979; Riley et al., 1995; Mattson et al., 1996; Roebuck et al., 1998; Sowell et al., 2001). The disruption of the organization between the primordium cortices and basal ganglia eminence regions by prenatal alcohol exposure was closely related to deficits in motor skills, visual–spatial skills, memory, and learning found in children who were born from mothers who were drinking alcohol during their pregnancy (Streissguth, 1986; Mattson et al., 1996; Olson et al., 1998).
Although the molecular mechanisms of action of NAP against the insult of alcohol exposure are not fully clear; my current findings and findings from others indicate that the neuroprotective effect of NAP against the insult of prenatal alcohol exposure involves mitochondria. A previous study demonstrated that NAP prevented alcohol-induced increases in the oxidized mitochondrial glutathione levels (Spong et al., 2001). I report here that NAP prevented alcohol-induced increases in the levels of cytosolic cytochrome c and decreases in the levels of mitochondrial cytochrome c. Interestingly, the downstream signaling pathway, caspase-3, was also found to be involved in this process involving NAP neuroprotection. Together, these findings suggest that NAP prevented alcohol-induced apoptosis through downstream signaling pathways that involve mitochondria.
There is a possibility that NAP involves other signaling pathways; for example, in a model of oxidative stress (H2O2), NAP reduces the level of the pro-apoptotic protein p53 following H2O2 exposure in neuronal-like cells (Gozes and Divinski, 2004; Divinski et al., 2006; Steingart and Gozes, 2006). These findings suggest that the neuroprotective effects of NAP might also be mediated through p53 signaling pathway. Since alcohol is considered an inductor of oxidative stress, it is possible that the mechanism underlying neuroprotection against alcohol-induced cell death may act through the p53 pathway. This pathway may not interact with the mitochondrial intrinsic pathway unless a dual action of NAP is involved in this mechanism of neuroprotection against the insult of prenatal alcohol exposure. Further studies using a model of FAE or FAS are warranted to investigate the upstream signaling pathways that are involved in NAP neuroprotection to determine whether this effect of NAP against the insult of ethanol exposure is mediated through intrinsic mitochondrial or extrinsic pathways or through both pathways.
CONCLUSION
In conclusion, I report in this present study that induced cell death that is caused by prenatal alcohol exposure is mediated in part through mitochondrial dysfunction. Increases of cytosolic cytochrome c levels as a result of prenatal alcohol exposure were directly linked with an increase of caspase-3 activation. Importantly, administration of NAP simultaneously with ethanol exposure reversed ethanol-induced changes in cytochrome c and caspase-3 levels and consequently prevented ethanol-induced increases in cell death. This derived peptide from ADNP is considered as a potential therapeutic drug for the treatment against oxidative stress associated with ethanol exposure.
Acknowledgments
The author would like to thank NIH-NIAAA for their full support in this project, grant number R21AA016115. The author also would like to acknowledge Metabolic and Cytomic Initiative (METACyt) funded by a Lilly endowment for providing the resources and equipment to accomplish this work. The author would like to thank Eta Isiorho for her technical assistance in TUNEL detection assay and ELISA and animal care. The author would like to thank also undergraduate students, Brandi Emerick, Ali Nosher and Aus Aburashed who have helped in the breeding and feeding the pregnant mice with liquid diet paradigm. The author would like to thank Jason Weedman for editing this manuscript. Finally, the author would like also to thank Faye Caylor for her administrative assistance.
Abbreviations
- ADNP
activity-dependent neuroprotective protein
- ALC
ethanol liquid diet group
- ANOVA
analysis of variance
- Apaf-1
apoptotic protease activating factor 1
- BAC
blood alcohol concentration
- E
embryonic day
- EDC
ethanol derived calories
- EDTA
ethyl-enediaminetetraacetic acid
- FAE
fetal alcohol exposure
- FAS
fetal alcohol syndrome
- NIH-NICHD
National Institutes of Health–National Institute of Child Health and Human Development
- OD
optical density
- PF
pair-fed control group
- RT
room temperature
- TUNEL
TdT-mediated dUTP nick end labeling
References
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Barron S, Gagnon WA, Mattson SN, Kotch LE, Meyer LS, Riley EP. The effects of prenatal alcohol exposure on odor associative learning in rats. Neurotoxicol Teratol. 1988;10:333–339. doi: 10.1016/0892-0362(88)90036-0. [DOI] [PubMed] [Google Scholar]
- Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, Bassan H, Blat C, Gibney G, Glazner G, Brenneman DE, Gozes I. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem. 1999;72:1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x. [DOI] [PubMed] [Google Scholar]
- Bauer-Moffett C, Altman J. Ethanol-induced reductions in cerebellar growth of infant rats. Exp Neurol. 1975;48:378–382. doi: 10.1016/0014-4886(75)90164-8. [DOI] [PubMed] [Google Scholar]
- Bauer-Moffett C, Altman J. The effect of ethanol chronically administered to preweanling rats on cerebellar development: a morphological study. Brain Res. 1977;119:249–268. doi: 10.1016/0006-8993(77)90310-9. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Alcohol-induced neuronal loss in developing rats: increased brain damage with binge exposure. Alcohol Clin Exp Res. 1990;14:107–118. doi: 10.1111/j.1530-0277.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Gozes I. A femtomolar-acting neuroprotective peptide. J Clin Invest. 1996;97:2299–2307. doi: 10.1172/JCI118672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Hauser J, Neale E, Rubinraut S, Fridkin M, Davidson A, Gozes I. Activity-dependent neurotrophic factor: structure-activity relationships of femtomolar-acting peptides. J Pharmacol Exp Ther. 1998;285:619–627. [PubMed] [Google Scholar]
- Brenneman DE, Spong CY, Hauser JM, Abebe D, Pinhasov A, Golian T, Gozes I. Protective peptides that are orally active and mechanistically nonchiral. J Pharmacol Exp Ther. 2004;309:1190–1197. doi: 10.1124/jpet.103.063891. [DOI] [PubMed] [Google Scholar]
- Clarren SK, Alvord EC, Jr, Sumi SM, Streissguth AP, Smith DW. Brain malformations related to prenatal exposure to ethanol. J Pediatr. 1978;92:64–67. doi: 10.1016/s0022-3476(78)80072-9. [DOI] [PubMed] [Google Scholar]
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Divinski I, Holtser-Cochav M, Vulih-Schultzman I, Steingart RA, Gozes I. Peptide neuroprotection through specific interaction with brain tubulin. J Neurochem. 2006;98:973–984. doi: 10.1111/j.1471-4159.2006.03936.x. [DOI] [PubMed] [Google Scholar]
- Furman S, Steingart RA, Mandel S, Hauser JM, Brenneman DE, Gozes I. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 2004;1:193–199. doi: 10.1017/S1740925X05000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Belcher SM, Pierce DR, Light KE. Altered expression of Bcl2, Bad and Bax mRNA occurs in the rat cerebellum within hours after ethanol exposure on postnatal day 4 but not on postnatal day 9. Brain Res Mol Brain Res. 2004;129:124–134. doi: 10.1016/j.molbrainres.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Gozes I, Bassan M, Zamostiano R, Pinhasov A, Davidson A, Giladi E, Perl O, Glazner GW, Brenneman DE. A novel signaling molecule for neuropeptide action: activity-dependent neuroprotective protein. Ann N Y Acad Sci. 1999;897:125–135. doi: 10.1111/j.1749-6632.1999.tb07884.x. [DOI] [PubMed] [Google Scholar]
- Gozes I, Divinski I. The femtomolar-acting NAP interacts with microtubules: Novel aspects of astrocyte protection. J Alzheimers Dis. 2004;6:S37–S41. doi: 10.3233/jad-2004-6s605. [DOI] [PubMed] [Google Scholar]
- Green CR, Watts LT, Kobus SM, Henderson GI, Reynolds JN, Brien JF. Effects of chronic prenatal ethanol exposure on mitochondrial glutathione and 8-iso-prostaglandin F2alpha concentrations in the hippocampus of the perinatal guinea pig. Reprod Fertil Dev. 2006;18:517–524. doi: 10.1071/rd05128. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Moore DB, Paiva M, Madorsky I, Mayer J, Shaw G. The role of neurotrophic factors, apoptosis-related proteins, and endogenous antioxidants in the differential temporal vulnerability of neonatal cerebellum to ethanol. Alcohol Clin Exp Res. 2003;27:657–669. doi: 10.1097/01.ALC.0000060527.55252.71. [DOI] [PubMed] [Google Scholar]
- Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Keramaris E, Stefanis L, MacLaurin J, Harada N, Takaku K, Ishikawa T, Taketo MM, Robertson GS, Nicholson DW, Slack RS, Park DS. Involvement of caspase 3 in apoptotic death of cortical neurons evoked by DNA damage. Mol Cell Neurosci. 2000;15:368–379. doi: 10.1006/mcne.2000.0838. [DOI] [PubMed] [Google Scholar]
- Kornguth SE, Rutledge JJ, Sunderland E, Siegel F, Carlson I, Smollens J, Juhl U, Young B. Impeded cerebellar development and reduced serum thyroxine levels associated with fetal alcohol intoxication. Brain Res. 1979;177:347–360. doi: 10.1016/0006-8993(79)90785-6. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Leist M, Nicotera P. Apoptosis, excitotoxicity, and neuropathology. Exp Cell Res. 1998;239:183–201. doi: 10.1006/excr.1997.4026. [DOI] [PubMed] [Google Scholar]
- Light KE, Belcher SM, Pierce DR. Time course and manner of Purkinje neuron death following a single ethanol exposure on postnatal day 4 in the developing rat. Neuroscience. 2002;114:327–337. doi: 10.1016/s0306-4522(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Riley EP, Delis DC, Stern C, Jones KL. Verbal learning and memory in children with fetal alcohol syndrome. Alcohol Clin Exp Res. 1996;20:810–816. doi: 10.1111/j.1530-0277.1996.tb05256.x. [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Boggan WO. Perinatal maternal ethanol effects on pregnant mice and on offspring viability and growth: influences of exposure time and weaning diet. Alcohol Clin Exp Res. 1995;19:1351–1358. doi: 10.1111/j.1530-0277.1995.tb01624.x. [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Randall CL, Favara JP. Prenatal ethanol exposure in C57 mice: effects on pregnancy and offspring development. Neurotoxicol Teratol. 1988;10:175–180. doi: 10.1016/0892-0362(88)90082-7. [DOI] [PubMed] [Google Scholar]
- Moore DB, Walker DW, Heaton MB. Neonatal ethanol exposure alters bcl-2 family mRNA levels in the rat cerebellar vermis. Alcohol Clin Exp Res. 1999;23:1251–1261. doi: 10.1111/j.1530-0277.1999.tb04286.x. [DOI] [PubMed] [Google Scholar]
- Olson HC, Feldman JJ, Streissguth AP, Sampson PD, Bookstein FL. Neuropsychological deficits in adolescents with fetal alcohol syndrome: clinical findings. Alcohol Clin Exp Res. 1998;22:1998–2012. [PubMed] [Google Scholar]
- Pascual M, Guerri C. The peptide NAP promotes neuronal growth and differentiation through extracellular signal-regulated protein kinase and Akt pathways, and protects neurons co-cultured with astrocytes damaged by ethanol. J Neurochem. 2007;103:557–568. doi: 10.1111/j.1471-4159.2007.04761.x. [DOI] [PubMed] [Google Scholar]
- Peiffer J, Majewski F, Fischbach H, Bierich JR, Volk B. Alcohol embryo- and fetopathy. Neuropathology of 3 children and 3 fetuses J Neurol Sci. 1979;41:125–137. doi: 10.1016/0022-510x(79)90033-9. [DOI] [PubMed] [Google Scholar]
- Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, Servoss SJ, Brenneman DE, Gozes I. Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res. 2003;144:83–90. doi: 10.1016/s0165-3806(03)00162-7. [DOI] [PubMed] [Google Scholar]
- Poggi SH, Goodwin K, Hill JM, Brenneman DE, Tendi E, Schinelli S, Abebe D, Spong CY. The role of activity-dependent neuro-protective protein in a mouse model of fetal alcohol syndrome. Am J Obstet Gynecol. 2003;189:790–793. doi: 10.1067/s0002-9378(03)00834-2. [DOI] [PubMed] [Google Scholar]
- Ramachandran V, Perez A, Chen J, Senthil D, Schenker S, Henderson GI. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4-hydroxynonenal. Alcohol Clin Exp Res. 2001;25:862–871. [PubMed] [Google Scholar]
- Riley EP, Mattson SN, Sowell ER, Jernigan TL, Sobel DF, Jones KL. Abnormalities of the corpus callosum in children prenatally exposed to alcohol. Alcohol Clin Exp Res. 1995;19:1198–1202. doi: 10.1111/j.1530-0277.1995.tb01600.x. [DOI] [PubMed] [Google Scholar]
- Roebuck TM, Mattson SN, Riley EP. A review of the neuroanatomical findings in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res. 1998;22:339–344. doi: 10.1111/j.1530-0277.1998.tb03658.x. [DOI] [PubMed] [Google Scholar]
- Samson HH, Diaz J. Altered development of brain by neonatal ethanol exposure: zinc levels during and after exposure. Alcohol Clin Exp Res. 1981;5:563–569. doi: 10.1111/j.1530-0277.1981.tb05362.x. [DOI] [PubMed] [Google Scholar]
- Sari Y, Gozes I. Brain deficits associated with fetal alcohol exposure may be protected, in part, by peptides derived from activity-dependent neurotrophic factor and activity-dependent neuroprotective protein. Brain Res Brain Res Rev. 2006;52:107–118. doi: 10.1016/j.brainresrev.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Sari Y, Mechref Y. Differential expression of proteins in fetal brains of alcohol-treated prenatally C57BL/6 mice: a proteomic investigation. Soc Neurosc. 2008 doi: 10.1002/elps.200900385. 663.1/II26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y, Powrozek T, Spong CY, Zhou FC. Activity-dependent neurotrophic factor protects serotonin neurons against prenatal ethanol exposure in midgestation brain. Soc Neurosci. 2003;157(5):419.411. [Google Scholar]
- Sari Y, Powrozek T, Zhou FC. Alcohol deters the outgrowth of serotonergic neurons at midgestation. J Biomed Sci. 2001;8:119–125. doi: 10.1007/BF02255980. [DOI] [PubMed] [Google Scholar]
- Sari Y, Zhou FC. Prenatal alcohol exposure causes long-term serotonin neuron deficit in mice. Alcohol Clin Exp Res. 2004;28:941–948. doi: 10.1097/01.alc.0000128228.08472.39. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Mattson SN, Thompson PM, Jernigan TL, Riley EP, Toga AW. Mapping callosal morphology and cognitive correlates: effects of heavy prenatal alcohol exposure. Neurology. 2001;57:235–244. doi: 10.1212/wnl.57.2.235. [DOI] [PubMed] [Google Scholar]
- Spong CY, Abebe DT, Gozes I, Brenneman DE, Hill JM. Prevention of fetal demise and growth restriction in a mouse model of fetal alcohol syndrome. J Pharmacol Exp Ther. 2001;297:774–779. [PubMed] [Google Scholar]
- Steingart RA, Gozes I. Recombinant activity-dependent neuro-protective protein protects cells against oxidative stress. Mol Cell Endocrinol. 2006;252:148–153. doi: 10.1016/j.mce.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Streissguth AP. The behavioral teratology of alcohol: Performance, behavioral, and intellectual deficits in prenatally exposed children. In: Lisberger SG, Zhong W, editors. Alcohol and brain development. New York: Oxford University; 1986. pp. 3–44. [Google Scholar]
- Sulik KK, Johnston MC, Webb MA. Fetal alcohol syndrome: embryogenesis in a mouse model. Science. 1981;214:936–938. doi: 10.1126/science.6795717. [DOI] [PubMed] [Google Scholar]
- Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Webster WS, Walsh DA, McEwen SE, Lipson AH. Some teratogenic properties of ethanol and acetaldehyde in C57BL/6J mice: implications for the study of the fetal alcohol syndrome. Teratology. 1983;27:231–243. doi: 10.1002/tera.1420270211. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu P, Li Y. Impaired development of mitochondria plays a role in the central nervous system defects of fetal alcohol syndrome. Birth Defects Res A Clin Mol Teratol. 2005a;73:83–91. doi: 10.1002/bdra.20110. [DOI] [PubMed] [Google Scholar]
- Xu YJ, Zhang GQ, Li Y. In utero ethanol exposure alters the mitochondrial protein expression of fetal mouse cerebrum. Beijing Da Xue Xue Bao. 2005b;37:346–350. [PubMed] [Google Scholar]
- Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, Holtzman DM, Roth KA, Olney JW. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003;10:1148–1155. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]
- Zamostiano R, Pinhasov A, Gelber E, Steingart RA, Seroussi E, Giladi E, Bassan M, Wollman Y, Eyre HJ, Mulley JC, Brenneman DE, Gozes I. Cloning and characterization of the human activity-dependent neuroprotective protein. J Biol Chem. 2001;276:708–714. doi: 10.1074/jbc.M007416200. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Sari Y, Powrozek T, Goodlett CR, Li TK. Moderate alcohol exposure compromises neural tube midline development in prenatal brain. Brain Res Dev Brain Res. 2003;144:43–55. doi: 10.1016/s0165-3806(03)00158-5. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Sari Y, Powrozek TA, Spong CY. A neuroprotective peptide antagonizes fetal alcohol exposure-compromised brain growth. J Mol Neurosci. 2004;24:189–199. doi: 10.1385/JMN:24:2:189. [DOI] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]