

Abstract
Background
Certain bacteria from the genus Streptomyces are currently used as biological control agents against plant pathogenic fungi. Hydrolytic enzymes that degrade fungal cell wall components, such as chitinases, are suggested as one possible mechanism in biocontrol interactions. Adaptive evolution of chitinases are previously reported for plant chitinases involved in defence against fungal pathogens, and in fungal chitinases involved in fungal-fungal interactions. In this study we investigated the molecular evolution of chitinase chiJ in the bacterial genus Streptomyces. In addition, as chiJ orthologs are previously reported in certain fungal species as a result from horizontal gene transfer, we conducted a comparative study of differences in evolutionary patterns between bacterial and fungal taxa.
Findings
ChiJ contained three sites evolving under strong positive selection and four groups of co-evolving sites. Regions of high amino acid diversity were predicted to be surface-exposed and associated with coil regions that connect certain α-helices and β-strands in the family 18 chitinase TIM barrel structure, but not associated with the catalytic cleft. The comparative study with fungal ChiJ orthologs identified three regions that display signs of type 1 functional divergence, where unique adaptations in the bacterial and fungal taxa are driven by positive selection.
Conclusions
The identified surface-exposed regions of chitinase ChiJ where sequence diversification is driven by positive selection may putatively be related to functional divergence between bacterial and fungal orthologs. These results show that ChiJ orthologs have evolved under different selective constraints following the horizontal gene transfer event.
Keywords: Chitinase, Functional divergence, Homology modelling, Horizontal gene transfer, Hypocrea, Protein evolution, Streptomyces, Trichoderma
Findings
Background
Chitin is an insoluble linear β-1,4-linked polymer of N-acetylglucosamine which is an essential structural component of fungal cell walls. Chitinases (EC 3.2.1.14) are hydrolytic enzymes that catalyse the degradation of chitin and are classified into glycoside hydrolase families 18 and 19, based on amino acid sequence similarity [1,2]. These two families possess different three-dimensional structural folds as well as modes of action. Family 18 chitinases is based on a triosephosphate isomerase (TIM) barrel structure and use a substrate-assisted double-displacement with net retention of the configuration of the anomeric carbon during the catalysis [3].
Biological control of pathogenic fungi causing diseases on important crop plants is an attractive alternative to the extensive use of chemical fungicides in modern agriculture, which may have negative environmental impacts and result in evolution of fungicide resistance in pathogen populations. Certain bacteria from the genus Streptomyces and fungi from the genera Hypocrea (anamorph Trichoderma) and Bionectria (anamorph Clonostachys) are currently used as biological control agents. The biocontrol ability of these microbes are suggested to be the result of several mechanisms that act synergistically, including direct antagonism by toxins and cell wall degrading enzymes (including chitinases), induced resistance in plants, or competition for nutrients and space [4].
Streptomyces bacteria are well known for their production of secreted hydrolytic enzymes and secondary metabolites [5], illustrating their ecological niche as ubiquitous soil bacteria. The genetically best studied species is S. coelicolor which is reported to possess 12 different chitinase genes, ten family 18 members (chiA, B, C, D, E, H, I, J, K, L) and two family 19 members (chiF, G) [6]. The ten family 18 chitinases can be phylogenetically divided into subfamilies A, B and C, and subgroups II, IVa and VI [6,7]. Seven genes, chiA, B, C, D, F, I and J, are reported to be induced to varying degrees in the presence of colloidal chitin [6,8], while studies of enzymatic activity are reported for S. coelicolor ChiA, C, D, F and G [6,9]. Recently, chiJ is shown to have an ortholog in Hypocrea species (chi18-15/chit36) and in Bionectria ochroleuca (ech37), acquired through horizontal gene transfer (HGT) [7,10,11].
Certain Hypocrea and Bionectria species are mycoparasitic, i.e. they have the ability to parasitize other fungi and use them as a source of nutrients. Studies of gene family evolution show that the mycoparasitic lifestyle has resulted in selection for increased number of cell wall degrading chitinases in Hypocrea[10]. In addition, certain Hypocrea chitinases evolve rapidly in a manner consistent with co-evolutionary interactions between parasite and host [10], similar to certain plant chitinases involved in defence against fungal pathogens [12,13].
In this study we investigate the molecular evolution of Streptomyces chitinase ChiJ, under the hypothesis that its involvement in interactions with soil fungi have resulted in sequence diversification between taxa. In addition, the HGT event presents an opportunity for a comparative investigation of the molecular fate of ChiJ orthologs evolving in bacterial and fungal species, respectively. We hypothesize that bacterial and fungal ChiJ orthologs evolve differently due to the fundamentally different organismal environments.
Materials and methods
Identification of genes and phylogenetic analysis
Streptomyces genome sequences available at the National Center for Biotechnological Information (NCBI) were screened for the presence of chitinase chiJ [GenBank:NP_626743], DNA recombinase A recA [GenBank:NP_629894] and DNA polymerase III subunit beta dnaN [GenBank:NP_628065] using BLAST [14]. GenBank [15] sequences were screened with ChiJ using BLAST. Sequences were aligned with Clustal X [16]. Phylogenetic analysis was performed using maximum likelihood methods implemented in PhyML-aLRT ver. 2.4.5 [17,18]. The JTT amino acid substitution model [19] was used, the proportion of invariable sites was set to 0, one category of substitution rate was used and gaps were treated as unknown characters. The starting tree to be refined by the maximum likelihood algorithm was a distance-based BIONJ tree estimated by the program [17]. Statistical support for phylogenetic grouping was assessed by approximate likelihood-ratio tests based on a Shimodaira-Hasegawa-like procedure [18] and by bootstrap analysis (1000 resamplings).
Homology modelling
Similar sequences were located by BLAST in the protein entries of GenBank and aligned using Clustal W [20]. Similar chitinase catalytic module structures (Additional file 1) were obtained from the Protein Data Bank (PDB [21]), then superimposed and compared with the program O [22]. Multiple sequence alignments were used to identify the best pair-wise alignment of the Streptomyces sp. Mg1 [GenBank:ZP_04999219] enzyme with that of Lactococcus lactis subsp. lactis. This pair-wise alignment was the basis of creating a homology model, with PDB entry [PDB:3IAN] (L. lactis subsp. lactis) as the template in the program SOD [23]. The model was adjusted in O, using rotamers that would improve packing in the interior of the protein. The model is available upon request from the authors. The figure was prepared using O, MOLSCRIPT [24] and Molray [25].
Reverse conservation analysis
Amino acid diversity in sequence alignments were analysed using Reverse Conservation Analysis (RCA). Amino acid sequences were aligned by Clustal X [16] and amino acid conservation at each position was analysed by RCA analysis as described by Lee (2008) [26]. In short, Rate4Site (Version 2.01) was used to calculate the degree of conservation (S score) for each amino acid position using the empirical Bayesian method [27,28]. A sliding window-average (n = 7) S score was plotted (W mean score) and significant peaks were defined by intensity (I) values of 0.5, as recommended by Lee (2008) [26].
Codon-based analyses
The rate of non-synonymous (dN) and synonymous (dS) substitutions at each codon, and identification of sites evolving under positive or negative selection, was determined using random effects maximum likelihood models (REL) [29] implemented in the HyPhy software package [30] through the Datamonkey webserver [31]. The optimal nucleotide substitution model was estimated for each gene separately [32] and included the following modifications to the general reversible nucleotide model [29] for chiJ: C↔G: R1, C↔T: RAC, G↔T: RAT; for recA: A↔C: R1, A↔T: R1, C↔G: RCG, C↔T: RCT, G↔T: RGT, and for dnaN: A↔C: R1, A↔T: R1, C↔G: RCG, G↔T: R1. A Bayes factor value ≥ 50 (default) was used as an indication of strong positive selection at a site, while values between 10 and 49 were considered to indicate weak support of positive selection [29].
Identification of co-evolving sites was done using the Spidermonkey/BGM program [33] implemented in the HyPhy software package [30] through the Datamonkey webserver [31]. The same nucleotide substitution models as in REL analysis was used, global dN/dS values were estimated by the program, ambiguous characters were averaged, a two-parent, directed network was used and sites were selected based on non-synonymous branch counts (threshold ≥ 3). A posterior probability value ≥ 0.5 (default) was used as the definition of association between sites.
Results
Identification of chiJ orthologs
The translated chiJ gene from S. coelicolor was used as the template for screening Streptomyces genome sequences available at NCBI. ChiJ orthologs were present in ten species, S. avermitilis, S. clavuligerus, S. ghanaensis, S. griseus, S. lividans, S. sp. AA4, S. sp. ACTE, S. sp. C, S. sp. e14 and S. sp. Mg1, in addition to S. coelicolor. With the exception of S. sp. Mg1 and S. sp. C that only contained one additional similar gene (E-value ≤ 1e-5), the other nine species contained two additional similar genes (E-value ≤ 1e-5). A phylogenetic analysis of translated amino acid sequences confirmed that the identified proteins represented the three Streptomyces chitinase paralogs ChiA, ChiB and ChiJ in the IVa group [6,7], and confirmed the orthologous status of the identified chiJ sequences (Figure 1). The translated S. coelicolor ChiJ protein was 358 amino acids long and consisted of a signal peptide for secretion and a family 18 chitinase catalytic module, but no additional substrate-binding modules [34]. Streptomyces recA and dnaN were present in the selected bacterial genomes, and were retrieved for comparative purposes.
Figure 1.

Phylogenetic relationships of Streptomyces ChiJ and related proteins. Phylogenetic analyses were performed using maximum likelihood methods as implemented in PhyML-aLRT, based on an alignment of chitinase amino acid sequences. Branch support values (approximate likelihood-ratio test probabilities (> 0.95) / bootstrap proportions (> 70%)) are associated with nodes. The bar marker indicate the number of amino acid substitutions per 100 amino acids. Streptomyces ChiA, ChiB, ChiJ and fungal ChiJ orthologs are indicated. Sequence identifiers include protein name or locus ID.
Homology modelling of ChiJ
Homology modelling revealed that the Streptomyces sp. Mg1 chitinase had a TIM barrel fold (Figure 2) with six insertions and three deletions compared to the chitinase structure from L. lactis subsp. lactis. In comparison to the hevamine chitinase from rubber tree [35] the catalytic cleft of ChiJ was more open at the substrate entry side of the cleft. Catalytic proton donor (E171) and the residues assisting in the catalysis (D169 and Y251) were conserved. Residues along the cleft that may play important roles in substrate binding were also conserved. The homology model of ChiJ displayed structural similarities to retaining endochitinases [36].
Figure 2.
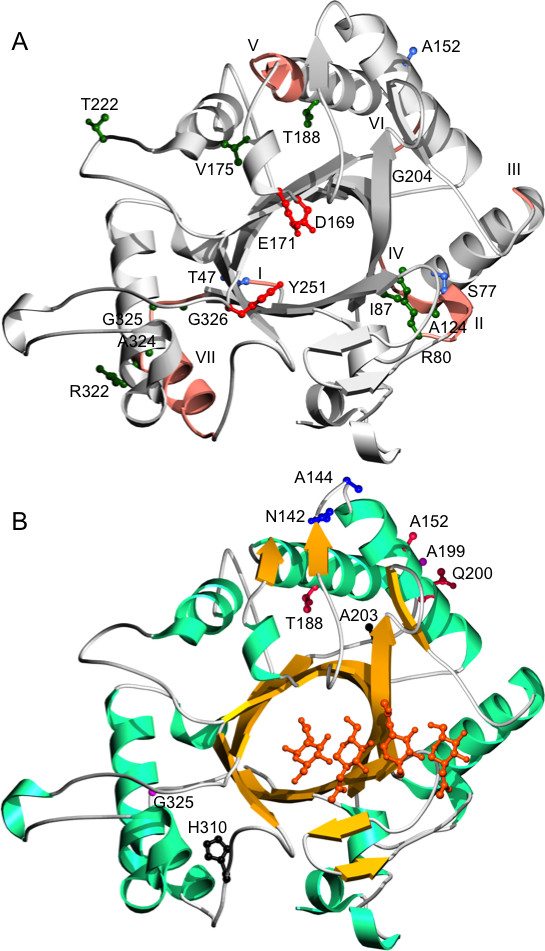
Homology model of Streptomyces sp. Mg1 ChiJ chitinase. A. Catalytically important residues are marked in red. Amino acids under strong and weak positive selection are shown in royal blue and dark green respectively. Variable regions from reverse conservation analysis are coloured in salmon and marked I-VII (I = 44–47, II = 75–80, III = 110–111, IV = 123–127, V = 178–185, VI = 200–203, VII = 312–327). B. Secondary structure elements of the enzyme are coloured differently. Side chains of co-evolving amino acids are shown in balls and sticks. Co-evolving groups of residues are illustrated in the same colour. Chito-tetraose is modelled to show the substrate-binding cleft.
Distribution of amino acid diversity
Patterns of amino acid diversity can provide important information about the type of selective constraints that act on different parts of an enzyme. In distantly related orthologous sequences only functionally important regions are expected to be conserved between species, while other regions are expected to have a low degree of conservation due to accumulation of neutral mutations. In the case of closely related orthologs, such as ChiJ in the current work, a high degree of conservation is expected for most positions as the limited time since speciation has not allowed sufficient sequence diversification by genetic drift. However, as outlined by Lee (2008) [26], residues important for enzyme properties may be expected to display higher diversity than other positions in closely related orthologs due to selection for modified enzymatic properties between species.
Therefore, amino acid diversity in a ChiJ alignment (Additional file 2) was analysed by RCA. The first 36 amino acids, including the signal peptide, were excluded from the analysis as this part could not be reliably aligned. High amino acid diversity was distributed amongst seven regions, labelled I through VII, with W mean scores above the 0.5 standard deviation threshold from the RCA analysis (Figure 3). All predicted residues important for catalysis and substrate-binding (Additional file 3) were localised in conserved regions with low W mean scores (Figure 3). Structural positions of the identified regions were visualized on the ribbon cartoon of the homology model of S. sp. Mg1 ChiJ (Figure 2A). S. sp. Mg1 ChiJ was used as a reference sequence for residue positions throughout this paper. All seven regions with high amino acid diversity (W score ≥ 0.5) were predicted to be surface-exposed, but neither forms a part of the catalytic cleft (Figure 2A). A closer examination of the S scores for individual residue positions revealed that all seven regions of high amino acid diversity were associated with coil regions that connect certain α-helices and β-strands in the family 18 chitinase TIM barrel fold structure (Figure 3). More specifically, highly divergent residue positions (S scores ≥ 0.5) in region I were located in a coil region placed on the N-terminal side of a β-strand at residue positions 50–56. Regions II, III, V, VI and VII contained highly divergent residue positions located in coil regions adjacent to α-helices (pos. 77–79, 111–125, 183–198, 316–323) (Figure 2). Region IV was associated with a α-helix / β-strand junction (Figure 3).
Figure 3.

Reverse conservation analysis of ChiJ orthologs. Amino acid conservation was estimated using Rate4Site, based on a Clustal X alignment of ChiJ Streptomyces orthologs, and plotted as W mean scores in arbitrary units (solid line). Amino acid conservation was estimated in a similar way for fungal orthologs in a previous work [10], and included in the figure (dashed line). Horizontal line indicates a 0.5 standard deviation cut-off. The x-axis represent residue position, asterisks (*) indicate positions of predicted catalytic residues, diamonds (◊) indicate predicted substrate-interacting residues, boxed P indicate residues under strong (Bayes factor ≥ 50) positive selection, P indicate residues under weak (Bayes factor 10–49) positive selection, boxed C interconnected by horizontal lines indicate co-evolving residue groups and vertical dashed lines indicate identical residues. The position of highly variable regions successfully visualised by homology modelling are indicated (I through VII). Magnifications illustrate residue S score distribution of the selected region. The position of a highly variable region in the fungal orthologs is indicated by A.
Evolutionary patterns among sites
High sequence diversity in certain regions and sites between closely related orthologs may be the result of either low selective constraint, i.e. mutations do not alter enzyme function in a significant manner, or to positive selection where certain mutations provide a selective advantage that improve enzyme function. The ratio between non-synonymous and synonymous substitutions is expected to approach 1 in the case of low selective constraints and be > 1 in the case of positive selection.
Therefore, the rate of dN and dS at each codon and identification of sites evolving under positive or negative selection was determined using REL. The first 117 bp of chiJ was removed from the analysis as this part could not be reliably aligned. REL fits both dN and dS substitution rates into three discrete rate classes, yielding a total of nine different rate classes of dN/dS. For chiJ, one rate class was estimated to have a dN/dS value above 1 (dN (0.37) / dS (0.22) = 1.68). Three sites evolved under strong positive selection (Bayes factor ≥ 50) and an additional eleven sites evolved under weak positive selection (Bayes factor 10 – 49) (Table 1). From these fourteen sites, eleven were associated with coil-regions exhibiting high amino acid diversity from the RCA analysis (Figure 3). In addition, site I87 was predicted to be located in a β-strand but not surface-exposed. Both A152 and T222 were predicted to be surface-exposed but located in a α-helix and a coil region, respectively (Figure 2A).
Table 1.
Positively selected sites in ChiJ
| Amino acid position | Posterior probability | Bayes factor |
|---|---|---|
| T47 |
0.77 |
68 |
| S77 |
0.73 |
53 |
| R80 |
0.66 |
39 |
| I87 |
0.34 |
10 |
| A124 |
0.69 |
46 |
| A152 |
0.91 |
197 |
| V175 |
0.68 |
43 |
| T188 |
0.65 |
38 |
| G204 |
0.40 |
13 |
| T222 |
0.63 |
34 |
| R322 |
0.55 |
25 |
| A324 |
0.45 |
16 |
| G325 |
0.61 |
31 |
| G326 | 0.44 | 16 |
Site positions are given in reference to S. sp. Mg1 ChiJ. Posterior probabilities and Bayes factors are determined using random effects likelihood method.
For comparative purposes, REL analysis was performed on sequence alignments of two Streptomyces genes whose functions were assumed to be independent from chitin degradation or microbial interactions, recA and dnaN. No rate class was estimated to have a dN/dS value above 1 for either recA or dnaN.
Co-evolution between sites
Amino acid residues in an enzyme can interact functionally to perform a specific function, or structurally to stabilize protein structures. Co-evolution between sites can result from selection for mutations that compensate for other, slightly deleterious mutations, to preserve protein function or structure. Identification of co-evolving sites in a protein may thus provide information about the boundaries of functional or structural domains. Functional domains can be especially difficult to define as functionality may depend on residues that are distributed over the whole protein.
Therefore, co-evolution between sites in chiJ was studied using evolutionary-network models implemented in Spidermonkey/BGM. Five interacting pairs of sites were identified (Table 2), forming four groups of co-evolving sites (Figure 3). Two co-evolving groups contained residues located in coil-regions VI and VII (Figure 3), located distantly to the catalytic cleft of the homology model of ChiJ (Figure 2A and B). Another group of three co-evolving sites connected region VI with site A152, evolving under strong positive selection (Figure 3). Finally, the co-evolving N142 – A144 pair was predicted to be physically close to site A152 and regions V and VI in the homology model (Figure 2A and B). In three cases, co-evolving sites were also evolving under positive selection (Figure 3).
Table 2.
Co-evolving sites in ChiJ
| Amino acid position 1 | Amino acid position 2 | Posterior probability |
|---|---|---|
| N142 |
A144 |
0.85 |
| A152 |
Q200 |
0.59 |
| T188 |
Q200 |
0.55 |
| A199 |
G325 |
0.58 |
| A203 | H310 | 0.68 |
Site positions are given in reference to S. sp. Mg1 ChiJ. Posterior probabilities for site 1 and site 2 to be conditionally dependent, determined by Spidermonkey/BGM, is given.
Comparison between chiJ and fungal orthologs
Fungal species of the genera Hypocrea and B. ochroleuca were previously shown to possess an ortholog to chiJ (chi18-15/chit36 and ech37 respectively), acquired through HGT from its actinobacterial origin [7,10,11]. An alignment of ChiJ (excluding the first 36 amino acids) from S. coelicolor and S. sp. Mg1, chi18-15 from H. jecorina, CHIT36 from H. atroviridis and Ech37 from B. ochroleuca shows that no insertions or deletions have occurred since the HGT event (data not shown). However, when comparing regions exhibiting high amino acid diversity from the RCA analysis in the current paper with a previous analysis on the fungal (Hypocrea) ChiJ orthologs [10], several differences were observed. ChiJ regions I and IV both exhibited high amino acid variation between Streptomyces species and contained rapidly evolving sites, while these regions were highly conserved in the fungal counterparts (Figure 3). The same trend was observed for ChiJ regions VI and VII (Figure 3). On the other hand, one region (region A) in the fungal chi18-15/CHIT36 protein alignment was highly variable between fungal species at the amino acid level, but conserved between bacterial ChiJ orthologs (Figure 3).
Discussion
Adaptive evolution of chitinases have previously been shown for plant chitinases involved in defence against fungal pathogens [12,13], and in fungal chitinases involved in mycoparasitic interactions [10]. The common theme between these examples is that parasite – host interaction seem to exert selection for rapid modification of the involved enzymes in an arms race fashion. Enzyme modification may thus partly account for variation in virulence or disease resistance in the parasite and host, respectively.
The biological function of ChiJ is suggested to include degradation of exogenous chitin for nutrient acquisition and aggressive interactions with soil fungi. Evolution of ChiJ may be influenced by the presence of several additional chitinases in certain Streptomyces species [6]. Conservation of gene duplications may be favoured if high levels of a specific chitinase is needed. However, the substantial divergence between ChiA, ChiB and ChiJ that is evident from our phylogenetic analysis suggest that these paralogous proteins represent isozymes with different functional properties that may act synergistically in chitin degradation. Presence of a fungal chiJ ortholog in both Hypocrea spp. and in B. ochroleuca suggests that the HGT event preceded the split of these species. These species belong to the same order (Hypocreales) but different families, so the HGT event cannot be considered to be recent. B. ochroleuca and certain Hypocrea species are mycoparasitic, and thus Ech37/CHIT36 are suggested to participate in degradation of the cell wall of antagonistic fungal species [36,37]. Expression data show that ech37 in B. ochroleuca is indeed induced during interaction with Botryotinia fuckeliana on strawberry leaves [37], while chit36 in T. asperellum is induced during plate confrontation with Rhizoctonia solani[38].
No region of high amino acid diversity or sites showing signs of adaptive evolution constitute parts of the catalytic cleft of ChiJ. This suggests that adaptation for changes in substrate-specificity between different Streptomyces species is not the primary selection force for ChiJ evolution. This conclusion is further supported by the localization of all residues predicted to be important for catalysis and substrate-binding to conserved regions. In addition, Hypocrea spp. Chi18-15/CHIT36 interspecific sequence variability is also reported to localize to other parts of the enzyme than the catalytic cleft [10]. This may indicate that the primary substrate of ChiJ and Chi18-15/CHIT36 are of similar composition, presumably fungal cell wall material.
Instead, regions with high amino acid diversity and sites under positive selection in ChiJ coincided with coil regions that connect certain α-helices and β-strands in the family 18 chitinase TIM barrel structure. This result is not surprising as the basic architecture of the TIM barrel is highly conserved between species, while surface loops may vary considerably due to low selective constraints which results in accumulation of neutral mutations. However, some of these surface loops will also be determinants of enzyme properties and interactions with other proteins, and may thus be targets for diversifying selection. Therefore, a major challenge in studies of enzyme evolution is to determine if the observed sequence diversity between orthologs is due to low selective constraints or due to positive selection. Identification of positive selection in a gene strongly implies that these changes are the result from diversifying selection and that the involved sites are connected to protein function.
In the current work, regions I, II, IV, V, VI and VII all contain sites evolving under positive selection. Hence, the most valid interpretation is that the high amino acid diversity between Streptomyces species in these regions is the results from diversifying selection and may influence functional properties of ChiJ. The localization of the variable parts to certain coil-regions predicts changes in enzyme architecture between species. Another observation is that two pairs of co-evolving sites are located on opposite sides of the enzyme, indicating that they participate in the same functional domain, i.e. slightly deleterious mutations in the substrate side of ChiJ may be compensated by other mutations on the products side, or vice versa. The surface-exposure of all seven regions of high amino acid diversity may suggest that interactions with environmental factors are the cause of the observed interspecific variability. No single factor can be identified at this stage, but may include interactions with chitinase inhibitors that are produced from soil fungi [39], plants [40], or from Streptomyces itself [41]. ChiJ may also interact and evolve together with chitin-binding proteins that enhance the efficiency of the degradation process [42,43]. A comparison of the enzymatic activities of S. coelicolor ChiA, ChiC, ChiD and ChiF showed that both pH optima and hydrolytic activity against soluble or crystalline chitin differed substantially between the enzymes [6]. Sites evolving under positive selection in ChiJ are candidates for targeted mutagenesis to elucidate mutational effects on enzyme properties.
The comparison between ChiJ and its fungal orthologs identified two regions that display high amino acid diversity in combination with sites evolving under positive selection in Streptomyces but not in fungi (regions I and IV), and one region of high sequence diversity in the fungal orthologs only (region A). This difference in amino acid conservation/diversity patterns is a hallmark of selection for type 1 functional divergence [44]; if the observed diversity in these regions is due to low selective constraints we would expect a similar level of diversity in both bacterial and fungal orthologs. Regions I and IV may thus represent areas on the ChiJ surface that contain structural features that specifically interact with environmental factors, such as inhibitors or chitin-binding helper proteins, present in the ecological niche of Streptomyces but not Hypocrea. Similarly, region A with high amino acid diversity between fungal orthologs may represent adaptations after the HGT event towards fungal preferences. In fact, this particular region was previously identified in Hypocrea chi18-15/CHIT36 to contain sites evolving under positive selection [10]. Furthermore, that study also shows discrete differences in amino acid conservation pattern between Hypocrea clades in this region; species in clades Rufa and Pachybasioides display high interspecific amino acid diversity in this region compared to a conserved interspecific pattern in other clades [10].
In summary, we show that several surface-exposed regions of Streptomyces chitinase ChiJ evolves rapidly and that this divergence is driven by selection for adaptive modifications. This shows that ChiJ function, whether it be interactions with soil fungi or nutrient acquisition, is important in the ecological niche of Streptomyces spp. We also identify three regions that display signs of type 1 functional divergence, where unique adaptations in the bacterial and fungal taxa are driven by positive selection. These results are noteworthy as it shows that different regions of the ChiJ orthologs have evolved under different selective constraints following the HGT event. The identified regions may thus influence functional properties of ChiJ.
Availability of supporting data
The data set supporting the results of this article is included within the article (and its additional files).
Abbreviations
HGT: Horizontal gene transfer; NCBI: National center for biotechnological information; PDB: Protein data bank; RCA: Reverse conservation analysis; REL: Random effects maximum likelihood; TIM: Triosephosphate isomerase.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WU carried out homology modelling studies and helped to draft the manuscript. MK conceived of the study, carried out studies of molecular evolution and wrote the manuscript. Both authors read and approved the final manuscript.
Supplementary Material
Protein structures used for structural alignment.
Alignment of Streptomyces sp.ChiJ sequences.
Predicted catalytic and substrate binding residues in ChiJ.
Contributor Information
Wimal Ubhayasekera, Email: wimal@xray.bmc.uu.se.
Magnus Karlsson, Email: magnus.karlsson@slu.se.
Acknowledgements
Contributions to Streptomyces genome sequence data to NCBI are gratefully acknowledged. This work was supported by grant CTS 08:189 from the Carl Trygger Foundation (MK), by the Royal Swedish Academy of Sciences (MK) and by DANSCATT (WU). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Henrissat B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1991;280:309–316. doi: 10.1042/bj2800309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B, Bairoch A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1993;293:781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tews I, van Scheltinga ACT, Perrakis A, Wilson KS, Dijkstra BW. Substrate-assisted catalysis unifies two families of chitinolytic enzymes. J Am Chem Soc. 1997;119:7954–7959. doi: 10.1021/ja970674i. [DOI] [Google Scholar]
- Harman GE, Howell CR, Viterbo A, Chet I, Lorito M. Trichoderma species - Opportunistic, avirulent plant symbionts. Nature Rev Microbiol. 2004;2:43–56. doi: 10.1038/nrmicro797. [DOI] [PubMed] [Google Scholar]
- Hodgson DA. Primary metabolism and its control in Streptomycetes: A most unusual group of bacteria. Adv Microb Physiol. 2000;42:47–238. doi: 10.1016/s0065-2911(00)42003-5. [DOI] [PubMed] [Google Scholar]
- Kawase T, Yokokawa S, Saito A, Fujii T, Nikaidou N, Miyashita K, Watanabe T. Comparison of enzymatic and antifungal properties between family 18 and 19 chitinases from S. coelicolor A3(2) Biosci Biotech Bioch. 2006;70:988–998. doi: 10.1271/bbb.70.988. [DOI] [PubMed] [Google Scholar]
- Karlsson M, Stenlid J. Evolution of family 18 glycoside hydrolases: Diversity, domain structures and phylogenetic relationships. J Mol Microbiol Biotechnol. 2009;16:208–223. doi: 10.1159/000151220. [DOI] [PubMed] [Google Scholar]
- Saito A, Ishizaka M, Francisco PB, Fujii T, Miyashita K. Transcriptional co-regulation of five chitinase genes scattered on the Streptomyces coelicolor A3(2) chromosome. Microbiol Sgm. 2000;146:2937–2946. doi: 10.1099/00221287-146-11-2937. [DOI] [PubMed] [Google Scholar]
- Heggset EB, Hoell IA, Kristoffersen M, Eijsink VGH, Vårum KM. Degradation of chitosans with chitinase G from Streptomyces coelicolor A3(2): Production of chito-oligosaccharides and insight into subsite specificities. Biomacromolecules. 2009;10:892–899. doi: 10.1021/bm801418p. [DOI] [PubMed] [Google Scholar]
- Ihrmark K, Asmail N, Ubhayasekera W, Melin P, Stenlid J, Karlsson M. Comparative molecular evolution of Trichoderma chitinases in response to mycoparasitic interactions. Evol Bioinform. 2010;6:1–26. doi: 10.4137/ebo.s4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamarabadi M, Jensen B, Lübeck M. Three endochitinase-encoding genes identified in the biocontrol fungus Clonostachys rosea are differentially expressed. Curr Genet. 2008;54:57–70. doi: 10.1007/s00294-008-0199-5. [DOI] [PubMed] [Google Scholar]
- Bishop JG, Dean AM, Mitchell-Olds T. Rapid evolution in plant chitinases: Molecular targets of selection in plant-pathogen coevolution. P Natl Acad Sci USA. 2000;97:5322–5327. doi: 10.1073/pnas.97.10.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffin P. Comparative evolutionary histories of chitinase genes in the genus Zea and family Poaceae. Genetics. 2004;167:1331–1340. doi: 10.1534/genetics.104.026856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2008;36:D25–D30. doi: 10.1093/nar/gkn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comp Appl Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. Clustal-W - Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr Sec A. 1991;47:110–119. doi: 10.1107/S0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA. In: International tables for crystallography, Vol F Crystallography of biological macromolecules. Rossmann MG, Arnold E, editor. Kluwer Academic, Dordrecht; 2001. Around O; pp. 353–356. 366–367. [Google Scholar]
- Kraulis PJ. Molscript - a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. doi: 10.1107/S0021889891004399. [DOI] [Google Scholar]
- Harris M, Jones TA. Molray - a web interface between O and the POV-Ray ray tracer. Acta Crystallogr Sec D. 2001;57:1201–1203. doi: 10.1107/s0907444901007697. [DOI] [PubMed] [Google Scholar]
- Lee T. Reverse conservation analysis reveals the specificity determining residues of cytochrome P450 family 2 (CYP 2) Evol Bioinform. 2008;4:7–16. doi: 10.4137/ebo.s291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I, Graur D, Ben-Tal N, Pupko T. Comparison of site-specific rate-inference methods for protein sequences: Empirical Bayesian methods are superior. Mol Biol Evol. 2004;21:1781–1791. doi: 10.1093/molbev/msh194. [DOI] [PubMed] [Google Scholar]
- Pupko T, Bell R, Mayrose I, Glaser F, Ben-Tal N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics. 2002;18:S71–S77. doi: 10.1093/bioinformatics/18.suppl_1.S71. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. A simple hierarchical approach to modeling distributions of substitution rates. Mol Biol Evol. 2005;22:223–234. doi: 10.1093/molbev/msi009. [DOI] [PubMed] [Google Scholar]
- Poon A, Lewis F, Kosakovsky Pond S, Frost S. An evolutionary-network model reveals stratified interactions in the V3 loop of the HIV-1 envelope. PLoS Comp Biol. 2007;3:e231. doi: 10.1371/journal.pcbi.0030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Fujii T, Miyashita K. Distribution and evolution of chitinase genes in Streptomyces species: Involvement of gene-duplication and domain-deletion. Antonie Van Leeuwenhoek. 2003;84:7–15. doi: 10.1023/A:1024463113606. [DOI] [PubMed] [Google Scholar]
- Bokma E, Rozeboom HJ, Sibbald M, Dijkstra BW, Beintema JJ. Expression and characterization of active site mutants of hevamine, a chitinase from the rubber tree Hevea brasiliensis. Eur J Biochem. 2002;269:893–901. doi: 10.1046/j.0014-2956.2001.02721.x. [DOI] [PubMed] [Google Scholar]
- Viterbo A, Haran S, Friesem D, Ramot O, Chet I. Antifungal activity of a novel endochitinase gene (chit36) from Trichoderma harzianum Rifai TM. FEMS Microbiol Lett. 2001;200:169–174. doi: 10.1111/j.1574-6968.2001.tb10710.x. [DOI] [PubMed] [Google Scholar]
- Mamarabadi M, Jensen B, Jensen DF, Lübeck M. Real-time RT-PCR expression analysis of chitinase and endoglucanase genes in the three-way interaction between the biocontrol strain Clonostachys rosea IK726, Botrytis cinerea and strawberry. FEMS Microbiol Lett. 2008;285:101–110. doi: 10.1111/j.1574-6968.2008.01228.x. [DOI] [PubMed] [Google Scholar]
- Viterbo A, Montero M, Ramot O, Friesem D, Monte E, Llobell A, Chet I. Expression regulation of the endochitinase chit36 from Trichoderma asperellum (T. harzianum T-203) Curr Genet. 2002;42:114–122. doi: 10.1007/s00294-002-0345-4. [DOI] [PubMed] [Google Scholar]
- Arai N, Shiomi K, Yamaguchi Y, Masuma R, Iwai Y, Turberg A, Kolbl H, Omura S. Argadin, a new chitinase inhibitor, produced by Clonostachys sp FO-7314. Chem Pharm Bull. 2000;48:1442–1446. doi: 10.1248/cpb.48.1442. [DOI] [PubMed] [Google Scholar]
- Hurtado-Guerrero R, van Aalten DMF. Structure of Saccharomyces cerevisiae chitinase 1 and screening-based discovery of potent inhibitors. Chem Biol. 2007;14:589–599. doi: 10.1016/j.chembiol.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Sakuda S, Isogai A, Matsumoto S, Suzuki A. Search for microbial insect growth-regulators .2. Allosamidin, a novel insect chitinase inhibitor. J Antibiot. 1987;40:296–300. doi: 10.7164/antibiotics.40.296. [DOI] [PubMed] [Google Scholar]
- Vaaje-Kolstad G, Horn SJ, van Aalten DMF, Synstad B, Eijsink VGH. The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J Biol Chem. 2005;280:28492–28497. doi: 10.1074/jbc.M504468200. [DOI] [PubMed] [Google Scholar]
- Saito A, Miyashita K, Biukovic G, Schrempf H. Characteristics of a Streptomyces coelicolor A3(2) extracellular protein targeting chitin and chitosan. Appl Environ Microb. 2001;67:1268–1273. doi: 10.1128/AEM.67.3.1268-1273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MF, Gaucher EA. Exploiting models of molecular evolution to efficiently direct protein engineering. J Mol Evol. 2011;72:193–203. doi: 10.1007/s00239-010-9415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Protein structures used for structural alignment.
Alignment of Streptomyces sp.ChiJ sequences.
Predicted catalytic and substrate binding residues in ChiJ.