

Abstract
Cytokines and transcription factors play key roles in dendritic cell (DC) development, yet information about regulatory interactions between these signals remains limited. Here we show that the cytokines GM-CSF and Flt3L induce the transcriptional mediators Id2 and E2-2 and control DC lineage diversification by STAT–dependent pathways. We found that STAT5 is required for tissue CD103+ DC generation and plasmacytoid DC (pDC) suppression in steady state or response to GM-CSF. STAT5 stimulates GM-CSF–dependent expression of Id2, which controls CD103+ DC production and pDC inhibition. By contrast, pDCs, but not CD103+ DCs, are dependent on STAT3. Consistently, STAT3 stimulates Flt3L-responsive expression of the pDC regulator Tcf4 (E2-2). These data suggest that STATs contribute to DC development by controlling transcription factors involved in lineage differentiation.
Introduction
Dendritic cells (DCs) are crucial immune sensors residing in lymphoid organs and peripheral tissues that regulate adaptive immune responses. Although numerous DC subsets have been identified, the mechanisms that control their diversity remain unclear. DCs arise from hematopoietic stem cells via multipotent progenitor subsets, including common DC progenitors (CDPs) localized in bone marrow (BM).1–4 CDPs differentiate into plasmacytoid DCs (pDCs) or mature to pre-DCs, which disperse to lymphoid organs or peripheral tissues and develop into conventional DC (cDC) populations, including the lymphoid organ-resident CD8α+, CD4+, and CD4− CD8α− DC subsets, and migratory or tissue-resident CD103+ DCs.4,5 Although primarily found in nonlymphoid tissues, CD103+ DCs are also detectable within lymph nodes (LNs), which reflects the fact that they migrate from tissues to LNs after activation. The pDCs, found in bone marrow, blood, and lymphoid organs secrete abundant quantities of type I IFNs after activation via Toll-like receptors (TLRs) and regulate antiviral immunity and immune tolerance.6 By contrast, lymphoid organ- and tissue-resident cDCs are adept at phagocytosis, antigen presentation, and stimulating adaptive immune responses.4 In particular, CD103+ DCs and CD8α+ DCs have garnered attention because of their ability to cross-present exogenous antigens and activate cytotoxic CD8+ T cells, an important facet of antiviral and antitumor immunity.5,7,8
The cytokines GM-CSF and Flt3 ligand (Flt3L) mediate homeostatic and demand-driven DC development.4,9 In DC progenitors, GM-CSF activates the signal transducer STAT5 and Flt3L stimulates STAT3.10,11 GM-CSF and STAT5 inhibit Flt3L–dependent pDC maturation by repressing Irf8,10 a transcription factor necessary for pDC differentiation.12 GM-CSF also drives cDC generation, including specific regulation of CD103+ DCs in steady state.13–16 By contrast, STAT3 controls homeostatic pDC production10,11; however, the molecular mechanisms involved in these responses remain to be determined. GM-CSF and Flt3L affect the expression of transcription factors with important roles in DC development, including Sfpi1 (PU.1), Tcf4 (E2-2), Irf8, Irf4, and Spib,10,11 in DC progenitors. Furthermore, STAT3 overexpression in multipotent BM progenitors induces Sfpi1 and enhances pDC and cDC production.17 These results suggest that STATs influence DC development by mediating expression of transcription factors that are crucial for DC lineage specification and/or differentiation.
Members of the inhibitor of DNA binding protein (Id) family also regulate transcription factors that direct hematopoietic lineage specification and commitment.18 Id proteins antagonize the function of basic helix-loop-helix transcriptional regulators, or E proteins, via their ability to bind E proteins and abrogate association with DNA.18 Id2 is required for generation of splenic CD8α+ DCs, tissue-resident CD103+ DCs, and epidermal Langerhans cells.19,20 By contrast, Id2 has been implicated as a negative regulator of pDCs.20–22 The distinct roles for Id2 suggest that manipulation of its expression level may influence DC progenitor cell fate; however, mechanisms that control Id2 expression during DC development have not been defined.
The E protein E2-2 (Tcf4) is highly expressed in pDCs compared with other DCs; consistently, E2-2 drives pDC development but does not regulate cDCs.21,23 Both copies of Tcf4 are required to generate functional pDCs.23 Enforced expression of E2-2 in human CD34+ CD1a− thymic progenitors promotes pDC development, suggesting E2-2 can direct pDC lineage specification.21 E2-2 controls a subset of genes with important roles in pDCs, including Tlr7, Tlr9, and Irf7.23,24 Moreover, E2-2 is critical for maintaining pDC cell identity, as conditional Tcf4 ablation in pDCs leads to spontaneous cDC differentiation.24 These data argue the importance of E2-2 in initiating and sustaining commitment and development of pDCs.
Here we address the mechanisms by which cytokines generate DC subset diversity. Our results show that STAT5 and STAT3 regulate CD103+ DC and pDC development, respectively; this occurs in conjunction with cytokine-responsive and STAT-dependent Id2 or Tcf4 induction. These data suggest that cytokine–activated STATs influence expression of transcription factors that mediate DC differentiation.
Methods
Mice and HGT
C57BL/6 mice were purchased from NCI or The Jackson Laboratory. Hematopoietic Stat5- or Stat3-deficient mice [ie, Tg(Tek-cre)12Flv Stat5f/Δ or Tg(Tek-cre)12Flv Stat3f/Δ] were bred in a specific pathogen-free facility via a strategy described previously.11,25 Age-matched Stat-sufficient (Stat5+/+ or Stat3+/+) littermates were used as controls. Experiments were performed with animals at 5-8 weeks of age; Stat5-deficient mice showed no obvious signs of disease, whereas this timeframe preceded severe inflammation in Stat3-deficient mice. Id2−/− and Id2+/+ chimeric mice were generated by reconstituting irradiated CD45.2+ recipients with CD45.1+ Id2−/− or Id2+/+ fetal liver.26 CD45.1+ Id2−/− or Id2+/+ BM cells were isolated by FACS. Hydrodynamic gene transfer (HGT) was performed as described previously.11 Animal experiments were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center. Animal numbers used in each experiment (n) are indicated in the legends.
Purification of DCs and DC progenitors, quantitative PCR, and immunoblotting assays
FACS was used to isolate CDPs and pDCs from BM; CD8α+, CD4+, and CD4− CD8α− DCs from spleen; and CD103+ DCs from liver, as described in supplemental Figure 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article). CDPs were cultured ex vivo with Flt3L (100 ng/mL) or GM-CSF (50 ng/mL) as indicated.10 RNA isolation and quantitative PCR were performed as described,11 using primers listed in supplemental Table 1. The relative amount of target gene expression was calculated with the equation 1.8(Rpl13a-GENE) × (100 000) where Rpl13a = average threshold cycle value for Rpl13a mRNA and GENE = average threshold cycle value for the gene of interest, per experiment. Immunoblotting of whole cell lysates was performed with phospho-STAT, total STAT, or C/EBPβ antibodies (Cell Signaling or Santa Cruz Biotechnology).
Retroviral infection, D2SC/mFlt3 cells, and reporter assays
D2SC/1 cells expressing murine Flt3 (D2SC/mFlt3) were generated by retroviral infection with a bicistronic vector (pCLXSN) encoding red fluorescent protein (RFP) and murine Flt3, followed by purification of RFP+ Flt3+ cells by FACS. The Id2 and Tcf4 proximal promoter regions were identified from the Ensembl database. Transcription factor binding sites were predicted by analysis with AliBaba and TF Search software programs. Reporter plasmids contained the Id2 (−2248 to +84) or Tcf4 (−1026 to +27) proximal promoter sequences inserted 5′ of luciferase coding sequences in the PGL3 or PGL4.12 vectors, respectively. D2SC/1 or D2SC/mFlt3 cells were transiently transfected with pGL3/Id2 or pGL4.12/Tcf4 reporter plasmids, phRL-TK-Renilla, and plasmids encoding wild-type (WT) or mutant STAT5A, STAT3, or C/EBPβ as indicated.10,11 Cells were treated with GM-CSF or Flt3L for 2 hours, and luciferase activity was measured by the Dual-Luciferase Reporter Assay System (Promega).
EMSAs and ChIPs
Electrophoretic mobility shift assays (EMSAs) used nuclear extracts from D2SC/1 or D2SC/mFlt3 cells stimulated with or without GM-CSF or Flt3L, as described.10 STAT ChIPs were performed with a mixture of antibodies for total and phosphorylated STAT5 or STAT3 (Santa Cruz Biotechnology and Cell Signaling), using the ChIP Assay Kit (Upstate Biotechnology) and PCR primers spanning STATx consensus sites in the Id2 or Tcf4 proximal promoter regions. Histone H3 ChIPs used antibodies against total H3, H3K9ac, H3K4me3, or H3K27me3 (Cell Signaling and Abcam), followed by PCR amplification of proximal promoter sequences. Primer sequences are provided in supplemental Table 1. ChIP amplification products were normalized to 1% total chromatin input or total H3 ChIPs, as indicated in the legends.
Statistical analysis
The 2-tailed t test was performed with GraphPad Prism Version 4 software (www.graphpad.com) to determine statistical significance between groups. Data are presented as mean ± SEM.
Results
STAT5 and STAT3 regulate unique DC subsets
To evaluate mechanisms by which GM-CSF and Flt3L control DC production, we used mice with conditional deletion of genes encoding the major cytokine signal transducers STAT5 or STAT3 within the hematopoietic compartment, mediated by cre expression under control of the Tie2 promoter (referred to as hematopoietic-Stat5Δ/Δ and hematopoietic-Stat3Δ/Δ mice, respectively). Hematopoietic-Stat5Δ/Δ mice were viable, with relatively normal amounts of circulating blood cells. We confirmed STAT5 protein depletion in BM, indicating Tie2cre-mediated removal of Stat5a/b is effective and compatible with adult hematopoiesis (supplemental Figure 2). Similarly, hematopoietic-Stat3Δ/Δ mice showed efficient BM STAT3 depletion.25 To induce circulating cytokine amounts, we used cytokine-encoding plasmids delivered by HGT.11 DC subset amounts in mice that received empty HGT vector were indistinguishable from untreated animals (data not shown); therefore, mice that received vector only were used as controls.
In hematopoietic-Stat5Δ/Δ mice, we found comparable amounts of pDCs and CD11b+ DCs in the BM, relative to Stat5+/+ animals, after vector delivery (Figure 1 top panels). In addition, splenic CD8α+, CD4+, and CD4− CD8α− DC amounts were similar in vector–treated hematopoietic-Stat5Δ/Δ mice and Stat5+/+ animals, yet splenic pDC numbers were increased ∼ 2-fold in hematopoietic-Stat5Δ/Δ mice (Figure 1 middle panels). By contrast, CD103+ DCs were depleted in the liver as well as the peripheral and mesenteric LNs of hematopoietic-Stat5Δ/Δ mice in the absence of cytokine treatment (Figure 1 bottom panels; supplemental Figure 3). These results suggest STAT5 is required for CD103+ DC homeostasis yet dispensable for pDCs and other cDC lineages. Indeed, STAT5 appears to have a suppressive role in steady-state pDC accrual in spleen or liver, consistent with our previous report.10
Figure 1.

STAT5 mediates CD103+ DC development and pDC inhibition. Hematopoietic-Stat5Δ/Δ and Stat5+/+ animals were treated by HGT with GM-CSF–encoding plasmid (5 μg) or empty pORF vector, as indicated. The proportion and absolute number of specific DC subsets in BM, spleen, and liver were determined after 7 days by flow cytometry (A) and enumeration (B). DC populations were defined by the following criteria: PDCs, CD11c+ CD11b- B220+; CD8α+ DCs, CD11c+ B220- CD8α+; CD4+ DCs, CD11c+ B220- CD4+; CD4- CD8α- DCs, CD11c+ B220- CD4- CD8α-; CD103+ DCs, CD11c+ CD45+ B220- MHC II+ CD103+. (A) Results indicate the percentage of each DC subset within the total BM, spleen, or liver population and represent data from 3 independent experiments. (B) Results indicate absolute numbers of each DC subset within BM (2 femurs, 2 tibiae), spleen, or liver. n = 3-6 mice/group. Error bars represent SEM of average values. P values of indicated comparisons are shown.
After GM-CSF induction by HGT, pDC amounts were severely reduced in BM, spleen, and liver in Stat5+/+ animals, whereas this inhibition was partially blocked in hematopoietic-Stat5Δ/Δ mice, in agreement with the negative effect of STAT5 in pDCs (Figure 1).10 GM-CSF enhanced the amount of BM CD11b+ DCs, splenic CD8α+, and CD4− CD8α− DCs, and liver CD103+ DCs. GM-CSF-responsive production of liver CD103+ DCs was inhibited by Stat5 deficiency; splenic CD8α+ DC induction also depended in part on STAT5, yet accrual of other DCs appeared to be STAT5-independent (Figure 1). Although CD103 is controlled by GM-CSF,15 and we show this is a STAT5-dependent response, CD103 is detectable on Stat5Δ/Δ cells confirming its suitability as a DC marker in hematopoietic-Stat5Δ/Δ mice (supplemental Figure 4A). Moreover, to confirm CD103+ DC identity, we also tested expression of the surrogate marker CD2416 as well as CD11b, an integrin subunit that is low to undetectable on most CD103+ DC populations, yet is inducible by cytokines, such as GM-CSF and G-CSF.16,27 We found that liver CD103+ DCs purified from vector- or GM-CSF–treated mice showed similar amounts of CD24 cell surface expression, consistent with their identification (supplemental Figure 4B left panel).16 In addition, we found that liver CD103+ DCs lack significant CD11b expression in homeostatic conditions, as expected (supplemental Figure 4B right panel). CD11b expression was uniformly induced, however, in CD103+ DCs by GM-CSF HGT in vivo or during ex vivo culture in GM-CSF (supplemental Figure 4B-C). These data suggest that CD103+ DCs are able to acquire CD11b expression in certain conditions, including exposure to GM-CSF. Collectively, our results indicate that STAT5 is uniquely required for the generation of CD103+ DCs in homeostasis or during response to GM-CSF.
In hematopoietic-Stat3Δ/Δ mice, we found that vector–treated animals contained 20%-40% pDC amounts in BM, spleen, and liver compared with Stat3+/+ mice, in agreement with previous work.11 No apparent differences were found in other DC subsets, including BM CD11b+ DCs; splenic CD8α+, CD4+ or CD4− CD8α− DCs; or liver CD103+ DCs (Figure 2). As expected, Flt3L HGT up-regulated pDCs and cDCs in BM, spleen, and liver. The induction of pDCs and cDCs in BM and spleen was abrogated in hematopoietic-Stat3Δ/Δ mice; however, Flt3L-responsive accumulation of liver CD103+ DCs did not require STAT3 (Figure 2). These results indicate that STAT3 is critical for pDC homeostasis and for Flt3L-dependent pDC and cDC production, with the exception of tissue CD103+ DCs. Taken together, our data suggest that STAT5 and STAT3 have specialized roles in regulating CD103+ DC or pDC populations, respectively.
Figure 2.

STAT3 controls pDCs and Flt3L-driven cDC generation. Hematopoietic-Stat3Δ/Δ and Stat3+/+ animals were treated by HGT with Flt3L-encoding plasmid (10 μg) or empty pORF vector, as indicated. The proportion and absolute number of specific DC subsets in BM, spleen, and liver were determined after 7 days by flow cytometry (A) and enumeration (B). DC populations were defined as indicated in Figure 1. (A) Results indicate the percentage of each DC subset within the total BM, spleen, or liver population and represent data from 4 independent experiments. (B) Results indicate absolute numbers of each DC subset within BM (2 femurs, 2 tibiae), spleen, or liver. n = 3-6 mice/group. Error bars represent SEM of average values. P values of indicated comparisons are shown.
Analysis of Id2 and Tcf4 expression and proximal promoter regions
The distinct roles for STAT5 and STAT3 were reminiscent of the functions of the HLH regulators Id2 and E2-2, which have been shown previously to control CD103+ DC and pDC production, respectively.19–23 These data suggested the potential for STATs to participate in Id2 (Id2) and E2-2 (Tcf4) regulation. To examine this possibility, we inspected 2 kb of sequence 5′ to the Id2 and Tcf4 transcriptional start sites, which we refer to as the proximal promoter regions, for transcription factor consensus elements. We identified consensus STATx sites as well as putative binding sites for other DC-related transcription factors (Figure 3A; supplemental Figure 5). We confirmed differential Id2 and Tcf4 expression in CD103+ DCs and pDCs; as expected, Id2 was enriched in liver CD103+ DCs as well as splenic CD8α+ DCs, whereas Tcf4 mRNA was highly expressed in pDCs (Figure 3B).19,20,23
Figure 3.

Promoter analysis and expression of Id2 and Tcf4 in DCs. (A) Schematic diagrams showing the location of putative STATx (open box) and C/EBPβ (gray box) consensus sites in the murine and human Id2 (left) and Tcf4 (right) promoter regions. Arrows indicate the approximate locations of primers used for STAT ChIPs. Primer sequences are listed in supplemental Table 1. (B) FACS-purified pDC and cDC subsets were analyzed for Tcf4 and Id2 mRNA expression by quantitative PCR, as indicated. DC purification strategies are shown in supplemental Figure 1. Results for Tcf4 and Id2 were normalized to Rpl13a. Results are shown as mean ± SEM of 3 independent experiments. (C-D) Liver CD103+ DCs and BM pDCs were purified from mice 7 days after GM-CSF or Flt3L HGT by FACS. Histone H3 modifications at the Id2 and Tcf4 proximal promoters were analyzed by ChIPs using antibodies against H3K9ac, H3K4me3, or H3K27me3; results were normalized to data from ChIPs with total H3 antibody. Normalized data were plotted versus the corresponding predicted ChIP amplification products from the Id2 and Tcf4 2 kb proximal promoter regions, as indicated by the schematic diagrams in the bottom panel, which include the approximate location of putative transcription factor binding sites. Results from CD103+ DCs (○) or pDCs (●) are presented as mean ± SEM of 3 independent experiments for individual modifications, as indicated.
We first evaluated whether the sequences encompassing the putative STAT sites in the Id2 and Tcf4 proximal promoters were accessible in CD103+ DCs and pDCs by examining post-translational histone H3 modifications associated with gene expression (H3K9ac, H3K4me3) or silencing (H3K27me3) at promoter regions.28 Because these experiments required significant DC numbers that we were unable to collect without affecting sample integrity from unmanipulated animals, we used liver CD103+ DCs or BM pDCs purified from mice treated by GM-CSF– or Flt3L-HGT, respectively. Investigation of a representative promoter region in each gene suggested that cytokine treatment did not affect H3 modifications significantly in differentiated DCs (supplemental Figure 6A).
Our analysis of the Id2 promoter showed an overall greater abundance of H3K4me3 in CD103+ DCs versus pDCs, accompanied by modest accumulation at the transcriptional start site in both DC subsets (Figure 3C). H3K9ac marks were enhanced across the entire promoter region in CD103+ DCs relative to pDCs, whereas the repressive H3K27me3 mark showed a reduced abundance at the Id2 promoter in CD103+ DCs (Figure 3C). The general enhancement of activating marks and decrease in repressive marks on the Id2 promoter in CD103+ DCs agrees with Id2 expression patterns (Figure 3B); however, the relative similarity in H3K27me3 abundance at the transcriptional start site in both lineages prompted us to investigate whether Id2 expression was inducible in pDCs. Indeed, stimulation of pDCs with the TLR9 agonist CpGA enhanced Id2 expression (supplemental Figure 6B). These results suggest a degree of accessibility for the Id2 promoter region in pDCs.
The Tcf4 promoter contained an enrichment of H3K9ac and H3K4me3 in the vicinity of the transcriptional start site in both DC subsets, with a modest enhancement of these activating marks in pDCs versus CD103+ DCs (Figure 3D). By contrast, H3K27me3 modifications were elevated across the distal portion and transcriptional start site of the Tcf4 promoter region in CD103+ DCs relative to pDCs (Figure 3D). These data are consistent with the preferred expression of Tcf4 in pDCs compared with CD103+ DCs. Therefore, the Id2 and Tcf4 promoters display similar patterns of chromatin marks in pDCs and CD103+ DCs, yet the amplitude of activating and repressive modifications is lineage-specific, in line with gene expression.
Id2 gene regulation by GM-CSF and STAT5
We next assessed the potential for STAT involvement in Id2 gene regulation by measuring Id2 mRNA amounts in CDPs purified from hematopoietic-Stat5Δ/Δ or Stat5+/+ animals after GM-CSF induction by HGT or control vector treatment. These experiments showed an ∼ 1.5-fold increase in Id2 mRNA amounts in CDPs from Stat5+/+ mice in response to GM-CSF, relative to Id2 expression in CDPs from vector–treated mice (Figure 4A). Id2 induction was abrogated in CDPs of hematopoietic-Stat5Δ/Δ animals (Figure 4A). Moreover, analysis of Id2 mRNA in ex vivo cultures of DC progenitors suggested that STAT5 controlled the amplitude of Id2 induction on GM-CSF stimulation (Figure 4B). By contrast, STAT5 did not appear to regulate Id2 in CDPs in steady-state conditions, as judged by comparison of vector–treated CDPs (Figure 4A). Furthermore, STAT3 was dispensable for Id2 induction by GM-CSF (supplemental Figure 7A). These results suggested a role for STAT5 in GM-CSF–responsive Id2 expression in CDPs.
Figure 4.

Regulation of Id2 by GM-CSF-responsive STAT5 and C/EBPβ. (A) Hematopoietic-Stat5Δ/Δ mice and Stat5+/+ control animals were treated by GM-CSF HGT or HGT pORF vector, as indicated. CDPs were isolated from BM after 2 days and analyzed for Id2 expression by quantitative PCR, using primers listed in supplemental Table 1. Id2 mRNA was normalized to Rpl13a mRNA amounts. Results represent average values from 2 independent experiments. Error bars represent SEM of average values. P values of indicated comparisons are shown. (B) Purified CDPs from hematopoietic-Stat5Δ/Δ mice and Stat5+/+ controls were cultured ex vivo with GM-CSF for 24 hours, 72 hours, or left untreated, as indicated. Id2 expression was analyzed by quantitative PCR as described in panel A. Results are presented as the fold induction of GM-CSF–treated samples relative to untreated samples. Results represent average values from 3 independent experiments. (C) D2SC/1 cells were transfected with the PGL3/Id2 reporter construct or empty vector, together with phRL-TK-Renilla and plasmids expressing WT STAT5A or a STAT5A mutant containing deletion of the transactivation domain (TAD). Cells were treated with GM-CSF for 2 hours. Results are expressed as normalized values (normalized values = relative light units [RLU] from GM-CSF–treated cells/RLU from untreated cells). Data represent the mean ± SEM of 3 independent experiments. (D) EMSAs were performed with nuclear extracts from D2SC/1 cells treated with or without GM-CSF for 30 minutes, using a 32P-labeled oligonucleotide probe corresponding to the Id2 distal STATx site (region −574 to −566; Figure 3A) with or without competitor oligonucleotides (I indicates Id2 probe WT; S5, an oligonucleotide containing the STAT5 consensus binding site; m, mutant Id2 probe) or anti-STAT5 antibody (☆ represents STAT5/DNA complexes; and ★, antibody supershifted STAT5/DNA complexes), as indicated (left). Control EMSAs were performed with a consensus STAT5 probe as indicated (right). (E) ChIP assays were performed with D2SC/1 cells treated with or without GM-CSF for 1 hour, using anti-STAT5 or IgG control antibody. Results were normalized to total cell lysate (input). (D-E) Results represent data from 3 independent experiments. (F) Hematopoietic-Stat3Δ/Δ mice and Stat3+/+ control animals were treated by Flt3L HGT or HGT vector, as indicated. Id2 mRNA amounts were measured in CDPs after 2 days, as described in panel A. (G-H) Lin− Flt3+ cells were cultured with GM-CSF or Flt3L for 24 hours, 72 hours, or left untreated. (G) Cebpb mRNA was analyzed by quantitative PCR. (H) C/EBPβ amounts were determined by immunoblotting. (I-J) Reporter assays were performed in D2SC/1 (I) or D2SC/mFlt3 cells (J) with the pGL3/Id2 reporter construct, in the presence of plasmids expressing STAT5A (WT), STAT5 TAD mutant (TAD), C/EBPβ (WT), or C/EBPβ T188A (T188A) as indicated, after GM-CSF (I) or Flt3L (J) stimulation for 2 hours. The fold change in RLU in GM-CSF– or Flt3L–treated cells relative to untreated cells is presented as mean ± SEM of 3 independent experiments.
We then evaluated whether STAT5 was directly involved in GM-CSF–dependent Id2 transcriptional regulation. For these experiments, we used D2SC/1 cells, which show GM-CSF–stimulated STAT5 activation and GM-CSF–dependent up-regulation of Id2 (supplemental Figure 8). Using reporter assays, we found a role for STAT5 transcriptional function in GM-CSF–responsive Id2 gene induction, as judged by increased Id2 reporter activity on coexpression of WT STAT5A, but not a mutant lacking the C-terminal transactivation domain29 (Figure 4C). EMSAs revealed that GM-CSF–activated STAT5 bound to the distal STATx site in the Id2 promoter (Figure 4D). ChIPs demonstrated STAT5 accumulation at the endogenous Id2 promoter on GM-CSF stimulation (Figure 4E). These data suggest that STAT5 induces Id2 gene transcription in response to GM-CSF by direct promoter binding.
To evaluate whether STAT5 regulates Id2 promoter accessibility, we surveyed H3 modifications in the vicinity of the Id2 promoter STAT element. These experiments revealed a similar abundance of H3K9ac, a reduction in H3K4me3, and an increase in H3K27me3 amounts in cDCs from hematopoietic-Stat5Δ/Δ mice versus cDCs from Stat5+/+ animals (supplemental Figure 6C). However, the reduction in H3K4me3 appeared to reflect an overall decrease in H3K4me3 abundance in BM cells from hematopoietic-Stat5Δ/Δ animals, as judged by ChIPs at candidate gene promoter regions and immunoblot analysis of H3K4me3 amounts in whole cell lysates (supplemental Figure 6E-F). Thus, our data suggest STAT5 may contribute to global H3K4me3 regulation as well as the abundance of H3K4me3 and H3K27me3 marks on the Id2 promoter.
GM-CSF–responsive Id2 up-regulation in CDPs during ex vivo culture (24-72 hours) was only partially dependent on STAT5 (Figure 4B). In addition, we observed Flt3L-responsive induction of Id2 in CDPs, which was regulated independently of STAT3 or STAT5 (Figure 4F; supplemental Figure 7A), suggesting the potential for other pathways to contribute to Id2 induction. C/EBPβ is a cytokine-responsive factor with a known role in controlling Id2.30–32 We found that Flt3L and GM-CSF induced Cebpb mRNA and C/EBPβ protein amounts in lin− Flt3+ cells, a BM population containing DC progenitors (Figure 4G-H). Id2 reporter assays suggested that C/EBPβ contributes to GM-CSF– and Flt3L-driven expression of Id2; in the GM-CSF pathway, this appears to be independent of the role for STAT5 (Figure 4I-J). These results suggest that Id2 is regulated by C/EBPβ as well as STAT5.
Control of Tcf4 expression by STAT3
We used a similar approach of analyzing gene expression in CDPs to evaluate whether Tcf4, such as Id2, was induced by cytokine treatment through a STAT-dependent mechanism. We found that Tcf4 mRNA was increased ∼ 1.5-fold in CDPs from Stat3+/+ mice on Flt3L treatment, relative to Tcf4 expression in CDPs from HGT vector-treated animals, whereas Flt3L–mediated induction of Tcf4 was abrogated in CDPs of hematopoietic-Stat3Δ/Δ animals (Figure 5A). Furthermore, Flt3L stimulated Tcf4 mRNA expression ex vivo in purified Stat3+/+ CDPs, and this induction was completely abolished in Stat3Δ/Δ CDPs (Figure 5B). STAT3 appears to be dispensable for steady-state Tcf4 expression (Figure 5A). We did not observe GM-CSF– or STAT5-responsive induction of Tcf4 in CDPs; indeed, GM-CSF appeared to suppress Tcf4 mRNA amounts in some experiments (supplemental Figure 7B-C). Thus, Flt3L stimulates Tcf4 expression by a mechanism dependent on STAT3.
Figure 5.

Control of Tcf4 expression by Flt3L-responsive STAT3. (A) Hematopoietic-Stat3Δ/Δ mice and Stat3+/+ control animals were treated by Flt3L HGT or pORF HGT vector as indicated. CDPs were isolated from BM after 2 days and analyzed for Tcf4 expression by quantitative PCR, using primers listed in supplemental Table 1. Tcf4 mRNA was normalized to Rpl13a mRNA amounts. Results represent average values from 2 independent experiments. Error bars represent SEM of average values. P values of indicated comparisons are shown. (B) Purified CDPs from hematopoietic-Stat3Δ/Δ and Stat3+/+ controls were cultured ex vivo with Flt3L for 24 hours, 72 hours, or left untreated. Tcf4 expression was analyzed by quantitative PCR as described in panel A. Results are presented as the fold induction of Flt3L-treated samples relative to untreated samples. Results represent average values from 3 independent experiments. (C) D2SC/mFlt3 cells were transfected with the PGL4.12/Tcf4 reporter construct or empty vector, together with phRL-TK-Renilla and plasmids expressing WT STAT3, a STAT3 isoform defective in DNA binding (DN) or a STAT3 isoform lacking the C-terminal transactivation domain (TAD). Cells were treated with Flt3L for 2 hours or left unstimulated. Results are expressed as normalized values (normalized values = RLU from Flt3L-treated cells/RLU from untreated cells). Data represent the mean ± SEM of 3 independent experiments. (D) EMSAs were performed with nuclear extracts isolated from D2SC/mFlt3 cells after stimulation with or without Flt3L for 30 minutes, using 32P-labeled oligonucleotide probes corresponding to the STATx consensus site in Tcf4, with or without competitor oligonucleotides (T indicates Tcf4 probe; S3, STAT3 binding site in murine Socs3 promoter; and m, mutant Tcf4 probe), or anti-STAT3 antibody (☆ represents STAT3/DNA complexes; and ★, antibody supershifted STAT3/DNA complexes), as indicated (left). Control EMSAs were performed with a consensus STAT3 probe from the Socs3 promoter as indicated (right). (E) ChIPs were performed with D2SC/mFlt3 cells treated with or without Flt3L for 1 hour, using anti-STAT3 or IgG control antibody, as indicated. Purified DNA from immunoprecipitated samples was analyzed by quantitative PCR. Results were normalized to total cell lysate (input). (D-E) Results represent data from 3 independent experiments.
We examined whether STAT3 directly regulates Flt3L-responsive Tcf4 expression using D2SC/1 cells that stably express murine Flt3 (D2SC/mFlt3); these cells demonstrate an intact Flt3 signaling cascade culminating in Tcf4 induction (supplemental Figure 8). Flt3L stimulated Tcf4 reporter activity in D2SC/mFlt3 cells expressing WT STAT3, but not cells expressing STAT3 mutants defective in DNA binding or transactivation function (Figure 5C). In addition, Flt3L induced STAT3 binding to the Tcf4 promoter STATx consensus site in EMSAs, whereas ChIPs demonstrated that STAT3 accumulated at the endogenous Tcf4 promoter on Flt3L stimulation (Figure 5D-E). Flt3L treatment of D2SC/1 cells, which lack detectable Flt3 expression, failed to activate STAT3 phosphorylation or induce STAT3 binding to the Tcf4 promoter (supplemental Figure 8). Moreover, STAT3 did not have a major influence on Tcf4 promoter accessibility, as judged by analysis of H3 modifications in pDCs from hematopoietic-Stat3Δ/Δ or Stat3+/+ mice, although STAT3 depletion appeared to affect the total cellular abundance of H3K4me3, leading to enriched H3K4me3 at Tcf4 as well as other gene promoters (supplemental Figure 6). Collectively, our data suggest that STAT3 directly stimulates Tcf4 transcription in response to Flt3L-Flt3 signaling, whereas distinct molecular cues are responsible for deposition of H3 modifications at the Tcf4 proximal promoter.
Functional roles for Id2 in cytokine-driven DC generation
Previous studies have delineated the importance of Id2 and E2-2 in homeostatic DC development20,23; however, their roles in cytokine-driven DC production are less well understood. We found that Id2−/− BM generated an increased number of pDCs in response to Flt3L or GM-CSF (Figure 6A), whereas the number of CD103+ DCs was reduced relative to Id2+/+ controls (Figure 6B). As expected, Tcf4 haploinsufficiency impaired Flt3L-driven pDC development yet did not affect Flt3L- or GM-CSF–mediated production of CD11c+ CD11b+ DCs (supplemental Figure 9; data not shown). These results indicate that Id2 and E2-2 are integral factors in GM-CSF– and Flt3L-driven DC generation, with Id2 necessary for either cytokine to mediate pDC suppression or to stimulate CD103+ DC production, and E2-2 specifically required for Flt3L-dependent pDC development. Taken together, our results suggest that cytokine-driven DC development involves the induction of Id2 and Tcf4 via STAT-regulated pathways (Figure 7).
Figure 6.

Roles for Id2 in ex vivo generation of pDCs and CD103+ DCs. CD45.1+ BM cells from Id2−/− or Id2+/+ chimeric mice were cultured with GM-CSF or Flt3L for 6 days, as indicated. The proportion and absolute number of pDCs (A) and CD103+ DCs (B) were determined by flow cytometry and enumeration. Surface expression of SiglecH on pDCs derived from Flt3L cultures (A) and MHC II on CD103+ DCs derived from GM-CSF cultures (B) was analyzed to confirm cell identity; similar expression patterns were observed in GM-CSF (SiglecH on pDCs) or Flt3L (MHC II on CD103+ DCs) conditions (not shown). Results represent data from 3-6 independent experiments. DC numbers shown are mean ± SEM values of 3-6 independent experiments.
Figure 7.
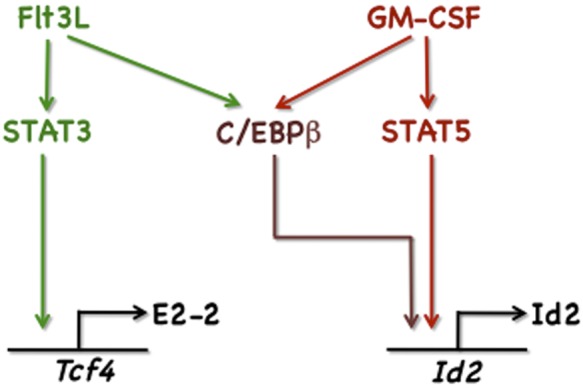
Schematic diagram of cytokine-STAT pathways regulating Id2 and Tcf4. Illustrated are GM-CSF– and Flt3L-responsive signaling cascades defined in this study that control Id2 and Tcf4 expression.
Discussion
Cytokines guide the production and diversification of DC subsets,4,9 yet the molecular mechanisms they use are poorly understood. Here we present evidence that GM-CSF and Flt3L stimulate expression of the DC transcriptional regulators Id2 and E2-2 within CDPs via STAT-dependent pathways. STAT5 controls GM-CSF-responsive Id2 induction, whereas STAT3 activates Flt3L-inducible Tcf4 expression. Both STATs operate by binding STATx elements in the Id2 and Tcf4 promoters, indicating a direct mechanism of gene regulation. Correspondingly, GM-CSF promotes STAT5- and Id2-dependent CD103+ DC development, and Flt3L stimulates STAT3- and Tcf4-dependent pDC generation. These results suggest that cytokine signaling via the STATs influences DC lineage differentiation by regulating Id2 and Tcf4 expression.
It is well established that precise expression of particular transcriptional regulators, including Id2 and E2-2, is important in lineage commitment and differentiation in the hematopoietic system.15,18,20,23,33–35 The role for cytokine-responsive STATs, however, appears complex and potentially lineage-specific. In B lymphocyte commitment, STAT5 controls progenitor survival but is not necessary for expression of the lineage-specifying factors E2A and EBF1, or the commitment factor Pax5.36 By contrast, STATs regulate expression of transcription factors that direct CD4+ T-cell differentiation, including RORγt and Foxp3.37,38 STAT3 also induces the myeloid-regulator PU.1 in multipotent hematopoietic progenitors and associates with regulatory sequences in the 5′ region of the PU.1 gene.17,39 Nonetheless, STATs are likely to be one of many signals that control transcription factor gene expression during immune cell development. For example, the Id2 and Tcf4 promoters contain consensus sites for factors that are involved in early stages of DC development, including GATA-2, IRF8, and PU.1,12,40,41 suggesting Id2 and Tcf4 as potential targets of factors that mediate the divergence of DC progenitors from other hematopoietic lineages. Cytokine-STAT signals may contribute by participating in transcriptional feed-forward or negative feedback loops that direct or sustain DC differentiation.
The Id family members are effective inhibitors of E proteins, with Id2 and Id3 specifically able to abrogate E2-2 binding to target DNA sequences.18,42,43 This agrees with the suppressive role for Id2 in pDCs,20 which rely on E2-2 for their development and function,23 and suggests that the ratio of Id2 to E2-2 in DC progenitors may influence pDC versus cDC lineage decisions. This concept is supported by work that showed E2-2 overexpression in human CD34+ progenitors enhanced pDC development.21 By contrast, increased expression of Id2 in CDPs by GM-CSF-STAT5 may antagonize the E2-2–dependent transcriptional program that leads to pDC differentiation. This idea is consistent with the rapid loss of pDCs from BM, the site of pDC development, during GM-CSF exposure, and the negative role for STAT5 and Id2 in pDC generation. Id2 is also induced by Flt3L and acts to suppress Flt3L-driven pDC production, although Id2 up-regulation in this case appears STAT–independent. Furthermore, because E2-2 is critical for maintaining pDC lineage identity,24 Id2 induction in differentiated pDCs may affect their phenotype and/or function. Although the reduction in lymphoid organ and tissue pDCs after GM-CSF treatment could be the result of suppressed pDC differentiation in BM coupled with loss of the pDC phenotype in peripheral populations because of Id2 up-regulation, our previous studies indicated that GM-CSF did not affect pDC marker expression,10 suggesting a major role for GM-CSF in blocking pDC development without effects on the mature pDC phenotype. However, TLR agonist-induced Id2 expression may promote pDC maturation, a process accompanied by acquisition of a phenotype resembling cDCs.6,44 The collective evidence therefore suggests that manipulation of Id2 expression by discrete stimuli during development may fine-tune DC progenitor commitment to the pDC lineage as well as pDC functional responses.
Among the cDC lineages, Id2 is critical for homeostatic CD103+ DC and CD8α+ DC development.15,19,20 Our data suggest the importance of a STAT5-Id2 signaling cascade in regulating CD103+ DCs during GM-CSF exposure. This pathway may also control CD8α+ DC production in response to GM-CSF, as Stat5 deficiency partially inhibited induction of this subset in spleen. By contrast, in steady state, CD103+ DCs, but not CD8α+ DCs, are dependent on GM-CSF.16 Our results demonstrate that CD103+ DCs are also uniquely dependent on STAT5 in homeostatic conditions. This suggests that local GM-CSF cues might regulate steady-state generation of CD103+ DCs by inducing STAT5-Id2–activating signals.45 Alternatively, CD103+ DCs may be more sensitive than other DC subsets to depletion of STAT5 and Id2. Nonetheless, in either homeostatic or GM-CSF conditions, it is not clear whether Id2 suppresses an E protein with antagonist activity toward CD103+ DC and CD8α+ DC lineage development, or whether Id2 has other, as yet unidentified, targets. E2-2 can be ruled out as its removal does not appear to affect cDC development in steady state or cytokine-driven conditions (data not shown).23 Thus, evaluating the expression and function of other E proteins as well as novel Id2-interacting proteins in cDCs should clarify the molecular role of Id2 in CD103+ and CD8α+ DCs.
Our observation that Flt3L and GM-CSF stimulate Id2 expression is consistent with their function in eliciting cDC production; however, the results were surprising as Flt3L and Id2 have opposing roles in pDCs.20,46,47 Both Flt3L and GM-CSF use C/EBPβ to induce Id2, whereas GM-CSF also uses STAT5. Parallel activities of C/EBPβ and STAT5 control expression of Id1 on IL-3 simulation.48 Moreover, STAT5 mediates cytokine-responsive Id2 expression in memory CD8+ T cells.49 Thus, STAT5 may enhance Id2 expression and potentially other Id family members in multiple cell types. By contrast, Flt3L uses STAT3 to induce Tcf4 along with simultaneous, yet STAT-independent, induction of Id2. These results suggest that the nature of the cytokine stimulus, and the specific intracellular pathways elicited (eg, GM-CSF-STAT5 or Flt3L-STAT3), affects the amplitude and relative ratio of transcription factors expressed in DC progenitors, with corresponding effects on lineage differentiation.
E2-2 has a unique role in pDCs but not other DC lineages.23 In homeostatic conditions, we found a similar function for STAT3 in pDCs, whereas generation of lymphoid organ or tissue CD103+ DCs appears to be STAT3-independent (results herein).11,50 We define E2-2/Tcf4 as a target of STAT3 in response to Flt3L stimulation. However, Tcf4 mRNA is found at near-normal levels in Stat3Δ/Δ CDPs or pDCs in steady-state conditions, and Stat3Δ/Δ pDCs retain the ability to produce type I IFNs on TLR ligation,10 a response that has been shown to require E2-2.23 Furthermore, unlike hematopoietic-Stat3Δ/Δ animals, mice lacking E2-2 exhibit a nearly complete absence of pDCs.23 These results suggest that E2-2 expression and development of a proportion of the pDC population are STAT3-independent in steady state. Similarly, Id2 mRNA was found at comparable amounts in Stat5Δ/Δ and Stat5+/+ CDPs. Thus, the principal role for STATs may be to control Tcf4 and Id2 in demand conditions driven by cytokine overexpression, or in microenvironments with enhancement of specific cytokines, with consequent effects on DC generation.
The physical interaction of STAT5 and STAT3 with the Id2 and Tcf4 promoters, respectively, suggests a direct mechanism of gene induction. Moreover, because CDPs were isolated on the basis of Flt3 expression and express GM-CSF receptor, it is unlikely that the failure to observe Id2 or Tcf4 induction in Stat5Δ/Δ or Stat3Δ/Δ CDPs is the result of selective loss of a cytokine-responsive population, supporting the idea that STATs directly regulate Id2 and Tcf4. The pattern of H3 modifications at the Id2 and Tcf4 promoter regions in CD103+ DCs and pDCs showed an overall similarity; however, the amplitude of activating and repressive marks appears to be lineage-specific. Furthermore, examination of the proximal promoter region of both genes in StatΔ/Δ DCs suggested that H3 marks were deposited primarily via a STAT-independent manner. Thus, our results suggest that the chromatin architecture at the Tcf4 and Id2 promoters is established during DC development independent of STAT function.
In conclusion, our study revealed functions for STAT5 and STAT3 in regulating Id2 and Tcf4 expression and DC lineage development. Further investigation of STAT-dependent and -independent pathways that influence DC development is of great interest for exploring mechanisms to redirect DC generation and function for therapeutic use in the treatment of cancer or immune disease.
Supplementary Material
Acknowledgments
The authors thank Drs Sharon Dent and Michelle Barton for helpful discussions; Drs Curt Horvath, Lothar Hennighausen, Yoshifumi Yokota, Paul J. Bertics, Demin Wang, and Jessica Schwartz for providing reagents; YeJi Jun and Ling Zhang for technical assistance; and Karen Ramirez and David He for assistance with cell sorting.
The flow core facility at the MD Anderson Cancer Center is supported by the National Cancer Institute (P30CA16672). This work was supported by the National Institutes of Health (AI072117, A.W.G.; AI073587 and AI098099, S.S.W.), a Pew Scholar Award (A.W.G.), the University of California, San Diego (CMG training grant; C.Y.Y.), and the R. E. “Bob” Smith Education Fund (H.S.L.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H.S.L. designed and performed experiments, analyzed data, and wrote the manuscript; C.Y.Y. and A.W.G. shared unpublished results and Id2−/− bone marrow; K.C.N. and H.Z. performed experiments; Y.-J. L. supervised the project; and S.S.W. designed experiments, supervised the project, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stephanie S. Watowich, Department of Immunology, MD Anderson Cancer Center, PO Box 301402, Houston, TX 77030; e-mail: swatowic@mdanderson.org; and Haiyan S. Li, Department of Immunology, MD Anderson Cancer Center, PO Box 301402, Houston, TX 77030; e-mail: haiyanli@mdanderson.org.
References
- 1.D'Amico A, Wu L. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J Exp Med. 2003;198(2):293–303. doi: 10.1084/jem.20030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3(+)M-CSFR(+) plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. 2007;8(11):1207–1216. doi: 10.1038/ni1518. [DOI] [PubMed] [Google Scholar]
- 3.Naik SH, Sathe P, Park HY, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. 2007;8(11):1217–1226. doi: 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 4.Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113(15):3418–3427. doi: 10.1182/blood-2008-12-180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234(1):45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 6.Liu YJ. Ipc: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 7.del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Forster R. Development and functional specialization of CD103+ dendritic cells. Immunol Rev. 2010;234(1):268–281. doi: 10.1111/j.0105-2896.2009.00874.x. [DOI] [PubMed] [Google Scholar]
- 8.Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev. 2010;234(1):18–31. doi: 10.1111/j.0105-2896.2009.00870.x. [DOI] [PubMed] [Google Scholar]
- 9.Watowich SS, Liu YJ. Mechanisms regulating dendritic cell specification and development. Immunol Rev. 2010;238(1):76–92. doi: 10.1111/j.1600-065X.2010.00949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. 2008;28(4):509–520. doi: 10.1016/j.immuni.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li HS, Gelbard A, Martinez GJ, et al. Cell intrinsic role for interferon-alpha-STAT1 signals in regulating murine Peyer patch plasmacytoid dendritic cells and conditioning an inflammatory response. Blood. 2011;118(14):3879–3889. doi: 10.1182/blood-2011-04-349761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiavoni G, Mattei F, Sestili P, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med. 2002;196(11):1415–1425. doi: 10.1084/jem.20021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Keeffe M, Hochrein H, Vremec D, et al. Effects of administration of progenipoietin 1, Flt-3 ligand, granulocyte colony-stimulating factor, and pegylated granulocyte-macrophage colony-stimulating factor on dendritic cell subsets in mice. Blood. 2002;99(6):2122–2130. doi: 10.1182/blood.v99.6.2122. [DOI] [PubMed] [Google Scholar]
- 15.Jackson JT, Hu Y, Liu R, et al. Id2 expression delineates differential checkpoints in the genetic program of CD8alpha+ and CD103+ dendritic cell lineages. EMBO J. 2011;30(13):2690–2704. doi: 10.1038/emboj.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greter M, Helft J, Chow A, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36(6):1031–1046. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onai N, Obata-Onai A, Tussiwand R, Lanzavecchia A, Manz MG. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J Exp Med. 2006;203(1):227–238. doi: 10.1084/jem.20051645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kee BL. E and Id proteins branch out. Nat Rev Immunol. 2009;9(3):175–184. doi: 10.1038/nri2507. [DOI] [PubMed] [Google Scholar]
- 19.Ginhoux F, Liu K, Helft J, et al. The origin and development of nonlymphoid tissue CD103+ dcs. J Exp Med. 2009;206(13):3115–3130. doi: 10.1084/jem.20091756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hacker C, Kirsch RD, Ju XS, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol. 2003;4(4):380–386. doi: 10.1038/ni903. [DOI] [PubMed] [Google Scholar]
- 21.Nagasawa M, Schmidlin H, Hazekamp MG, Schotte R, Blom B. Development of human plasmacytoid dendritic cells depends on the combined action of the basic helix-loop-helix factor E2-2 and the Ets factor Spi-B. Eur J Immunol. 2008;38(9):2389–2400. doi: 10.1002/eji.200838470. [DOI] [PubMed] [Google Scholar]
- 22.Spits H, Couwenberg F, Bakker AQ, Weijer K, Uittenbogaart CH. Id2 and Id3 inhibit development of CD34(+) stem cells into predendritic cell (pre-DC)2 but not into pre-DC1: evidence for a lymphoid origin of pre-DC2. J Exp Med. 2000;192(12):1775–1784. doi: 10.1084/jem.192.12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cisse B, Caton ML, Lehner M, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135(1):37–48. doi: 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous expression of the transcription factor E2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity. 2010;33(6):905–916. doi: 10.1016/j.immuni.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panopoulos AD, Zhang L, Snow JW, et al. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108(12):3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannarile MA, Lind NA, Rivera R, et al. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat Immunol. 2006;7(12):1317–1325. doi: 10.1038/ni1403. [DOI] [PubMed] [Google Scholar]
- 27.Devereux S, Bull H, Campos-Costa D, Saib R, Linch D. Granulocyte macrophage colony stimulating factor induced changes in cellular adhesion molecule expression and adhesion to endo-helium: in vitro and in vivo studies in man. Br J Haematol. 1989;71(3):323–330. doi: 10.1111/j.1365-2141.1989.tb04287.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 29.Wang D, Stravopodis D, Teglund S, Kitazawa J, Ihle JN. Naturally occurring dominant negative variants of STAT5. Mol Cell Biol. 1996;16(11):6141–6148. doi: 10.1128/mcb.16.11.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karaya K, Mori S, Kimoto H, et al. Regulation of Id2 expression by CCAAT/enhancer binding protein beta. Nucleic Acids Res. 2005;33(6):1924–1934. doi: 10.1093/nar/gki339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirai H, Zhang P, Dayaram T, et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7(7):732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116(14):2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenbauer F, Koschmieder S, Steidl U, Tenen DG. Effect of transcription-factor concentrations on leukemic stem cells. Blood. 2005;106(5):1519–1524. doi: 10.1182/blood-2005-02-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothenberg EV, Scripture-Adams DD. Competition and collaboration: GATA-3, PU.1, and Notch signaling in early T-cell fate determination. Semin Immunol. 2008;20(4):236–246. doi: 10.1016/j.smim.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol. 2012;12(2):101–113. doi: 10.1038/nri3149. [DOI] [PubMed] [Google Scholar]
- 36.Malin S, McManus S, Busslinger M. STAT5 in B cell development and leukemia. Curr Opin Immunol. 2010;22(2):168–176. doi: 10.1016/j.coi.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine–mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 38.Yao Z, Kanno Y, Kerenyi M, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109(10):4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hegde S, Ni S, He S, et al. Stat3 promotes the development of erythroleukemia by inducing PU.1 expression and inhibiting erythroid differentiation. Oncogene. 2009;28(38):3349–3359. doi: 10.1038/onc.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carotta S, Dakic A, D'Amico A, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose–dependent manner. Immunity. 2010;32(5):628–641. doi: 10.1016/j.immuni.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Dickinson RE, Griffin H, Bigley V, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–2658. doi: 10.1182/blood-2011-06-360313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langlands K, Yin X, Anand G, Prochownik EV. Differential interactions of Id proteins with basic-helix-loop-helix transcription factors. J Biol Chem. 1997;272(32):19785–19793. doi: 10.1074/jbc.272.32.19785. [DOI] [PubMed] [Google Scholar]
- 43.Liu YP, Burleigh D, Durning M, Hudson L, Chiu IM, Golos TG. Id2 is a primary partner for the E2-2 basic helix-loop-helix transcription factor in the human placenta. Mol Cell Endocrinol. 2004;222(1):83–91. doi: 10.1016/j.mce.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 44.Kadowaki N, Antonenko S, Lau JY, Liu YJ. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med. 2000;192(2):219–226. doi: 10.1084/jem.192.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kingston D, Schmid MA, Onai N, Obata-Onai A, Baumjohann D, Manz MG. The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood. 2009;114(4):835–843. doi: 10.1182/blood-2009-02-206318. [DOI] [PubMed] [Google Scholar]
- 46.Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med. 2003;198(2):305–313. doi: 10.1084/jem.20030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking Flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95(11):3489–3497. [PubMed] [Google Scholar]
- 48.Xu M, Nie L, Kim SH, Sun XH. STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. EMBO J. 2003;22(4):893–904. doi: 10.1093/emboj/cdg094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang CY, Best JA, Knell J, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12(12):1221–1229. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melillo JA, Song L, Bhagat G, et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol. 2010;184(5):2638–2645. doi: 10.4049/jimmunol.0902960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.