

Summary
Mutations in WNK1 and WNK4 kinase genes have been shown to cause a human hereditary hypertensive disease, pseudohypoaldosteronism type II (PHAII). We previously discovered that WNK kinases phosphorylate and activate OSR1/SPAK kinases that regulate renal SLC12A family transporters such as NKCC2 and NCC, and clarified that the constitutive activation of this cascade causes PHAII. WNK3, another member of the WNK kinase family, was reported to be a strong activator of NCC/NKCC2 when assayed in Xenopus oocytes, suggesting that WNK3 also plays a major role in regulating blood pressure and sodium reabsorption in the kidney. However, it remains to be determined whether WNK3 is in fact involved in the regulation of these transporters in vivo. To clarify this issue, we generated and analyzed WNK3 knockout mice. Surprisingly, phosphorylation and expression of OSR1, SPAK, NKCC2 and NCC did not decrease in knockout mouse kidney under normal and low-salt diets. Similarly, expression of epithelial Na channel and Na/H exchanger 3 were not affected in knockout mice. Na+ and K+ excretion in urine in WNK3 knockout mice was not affected under different salt diets. Blood pressure in WNK3 knockout mice was not lower under normal diet. However, lower blood pressure was observed in WNK3 knockout mice fed low-salt diet. WNK4 and WNK1 expression was slightly elevated in the knockout mice under low-salt diet, suggesting compensation for WNK3 knockout by these WNKs. Thus, WNK3 may have some role in the WNK-OSR1/SPAK-NCC/NKCC2 signal cascade in the kidney, but its contribution to total WNK kinase activity may be minimal.
Keywords: Na-K-Cl cotransporter, Na-Cl cotransporter, WNK3, mouse kidney, WNK
Introduction
Pseudohypoaldosteronism type II (PHAII) is an autosomal-dominant disease characterized by hypertension due to increased renal salt reabsorption, hyperkalemia and metabolic acidosis (Gordon, 1986; Schambelan et al., 1981; Achard et al., 2001). Mutations in with-no-lysine kinase 1 (WNK1) and with-no-lysine kinase 4 (WNK4) have been reported to cause PHAII (Wilson et al., 2001). We generated WNK4D561A/+ knock-in mice, an ideal mouse model of PHAII, and observed increased phosphorylation of oxidative stress-responsive kinase-1 (OSR1), STE20/SPS1-related proline/alanine-rich kinase (SPAK) and thiazide-sensitive Na-Cl co-transporter (NCC) (Yang, S. S. et al., 2007). We previously demonstrated in in vitro experiments that WNK1 and WNK4 phosphorylated and activated OSR1 and SPAK kinases, and that OSR1 and SPAK could phosphorylate NCC (Moriguchi et al., 2005; Vitari et al., 2005; Vitari et al., 2006; Richardson et al., 2008). Furthermore, Pacheco-Alvarez et al. reported that phosphorylation of NCC at Thr 53 and 58, and at Ser 71 was important for NCC function in Xenopus oocytes (Pacheco-Alvarez et al., 2006), and we showed that phosphorylated NCC is concentrated on the apical membranes of distal convoluted tubules in the WNK4D561A/+ knock-in mice, which suggests that phosphorylation may also be important for intracellular localization of NCC (Yang, S. S. et al., 2007). Based on the above evidence, we postulated that WNK, OSR1/SPAK and NCC constitute a signal cascade in the in vivo kidney, which is important for NaCl homeostasis and blood pressure regulation. Recently, we mated WNK4D561A/+ knock-in mice with SPAK and OSR1 kinase-dead knock-in mice, in which the T-loop Thr residues in SPAK (Thr 243) and OSR1 (Thr 185) were mutated to Ala to prevent activation by WNK kinases (Rafiqi et al., 2010). In these triple knock-in mice, PHAII phenotypes and increased phosphorylation of NCC were completely corrected (Chiga et al., 2011). Based on the definitive genetic data, we clearly established the presence of the WNK-OSR1/SPAK-NCC kinase cascade in the in vivo kidney.
Although the signal cascade was established, it remains unclear which WNK kinase is responsible in the kidney. It is also uncertain whether a single dominant WNK kinase is present in each different type of cell, or whether multiple WNKs are present in the same cells and function as a WNK kinase complex, as postulated by Yang, C. L. et al. (Yang, C. L. et al., 2007). In fact, in addition to WNK1 and WNK4, whose mutations cause PHAII, WNK3 mRNA expression was reported to be present in the kidney (Holden et al., 2004). Therefore, although WNK3 mutation has not been observed in PHAII, WNK3 could be an important component of WNK kinase-mediated signal cascade in kidney. Previous in vitro data found that WNK3 regulates SLC12A cotransporters. WNK3 was shown to be an activator of Na-K-Cl cotransporter (NKCC1 and 2) and NCC (Kahle et al., 2005; Rinehart et al., 2005; Yang, C. L. et al., 2007; San-Cristobal et al., 2008; Ponce-Coria et al., 2008; Glover et al., 2009; Cruz-Rangel et al., 2011), and a repressor of K-Cl cotransporters (KCC 1-4) (Kahle et al., 2005; de Los Heros et al., 2006), when co-expressed in Xenopus laevis oocytes. Similar to WNK1 and WNK4, WNK3 was found to phosphorylate SPAK in Xenopus laevis oocytes (Ponce-Coria et al., 2008).
Previously, WNK4 hypomorphic mice and WNK1 heterozygous mice reportedly showed low blood pressure (Ohta et al., 2009; Zambrowicz et al., 2003). Therefore, we aimed to determine the contribution of WNK3 to WNK-mediated kidney functions by generating WNK3 knockout mice. The data obtained suggest that WNK3 may not play a major role in the WNK kinase cascade in the kidney.
Results
Generation of WNK3 knockout mice
In order to generate WNK3 knockout mice, we planned to delete exon 2 (Fig. 1A), as exon 2 contains the catalytic domain of mouse WNK3 (Holden et al., 2004; Verissimo et al., 2006). We crossed chimeric mice from recombinant ES clones with C57BL/6 mice to produce WNK3 (flox/+) mice. The generation of WNK3 (flox/+) mice was verified by PCR (Fig. 1B). Next, to delete exon 2 from the Wnk3 gene, we crossed WNK3 (flox/+) female mice with Cre recombinase transgenic male mice. The Cre-mediated excision of exon 2 and Neo cassette was verified by PCR, as shown in Fig. 1C. The absence of WNK3 protein was confirmed by immunoblotting in brain and testis (Fig. 1D). However, due to the low level of WNK3 protein expression in the kidney, WNK3 was not detected by immunoblotting, even in wild-type mouse kidney. To verify that WNK3 is also disrupted in the kidney, we performed RT-PCR of WNK3 and confirmed the absence of WNK3 mRNA in the kidneys of WNK3 knockout mice (Fig. 1E).
Fig. 1. Generation of WNK3 knockout mice.

(A) Targeting strategy for Wnk3 gene interruption. The diagram shows the targeting construct, the wild-type WNK3 locus, and the targeted locus before and after Cre recombination. Three loxPs were inserted to flank exon 2 and the LacZ-Neo-selective marker. Exon 2 was deleted by mating the flox mice with CAG promoter Cre recombinase mice. (B) Verification of homologous recombination by PCR of genomic DNA derived from tails of mice. The primer set is indicated by dotted arrows in A. The 227-bp band and 339-bp band represent the wild-type allele and flox allele, respectively. (C) Genotyping PCR after Cre recombination, using a primer set indicated by solid arrows in A. A 516-bp PCR product was specific to the mutant allele. (D) Immunoblot of brain, kidney and testis homogenates probed with anti-WNK3 antibody. Absence of WNK3 protein in WNK3 knockout mouse was confirmed by immunoblotting in brain and testis. WNK3 was not detected by immunoblotting, even in wild-type mouse kidney, due to the low level of WNK3 protein expression in the kidney. (E) RT-PCR of brain, kidney and testis of wild-type and WNK3 knockout mice.
Segmental expression of WNK3 along mouse nephron
First, we aimed to determine where WNK3 is expressed along the mouse nephron, as we had to identify the transporters present in the segment where WNK3 is expressed. It was previously reported that, on immunofluorescence, WNK3 is present in all nephron segments (Rinehart et al., 2005). However, because the same antibody did not work both in immunofluorescence and immunoblotting in our hand, we performed laser capture microdissection (LCM) and RT-PCR in order to confirm segmental expression of WNK3 along nephron. As shown in Fig. 2, we confirmed that WNK3 is expressed in proximal tubules, thick ascending limb of Henle's loop (TAL), distal convoluted tubules (DCT) and collecting ducts, where Na/H exchanger 3 (NHE3), NKCC2, NCC and epithelial Na channel (ENaC) are expressed, respectively.
Fig. 2. Segmental expression of WNK3 along mouse nephron.
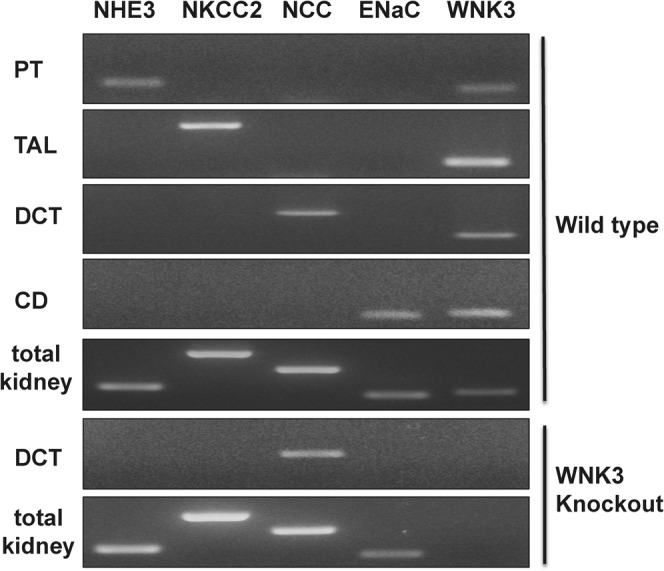
Primers for NHE3, NKCC2, NCC and ENaC were used as markers for each segment. WNK3 was positive in proximal tubules, thick ascending limb of Henle's loop, distal tubules and collecting ducts of wild type mouse. As a negative control, distal tubules and total kidney homogenate from WNK3 knockout mouse were used. PT, proximal tubule; TAL, thick ascending limb of Henle's loop; DCT, distal convoluted tubule; CD, collecting duct.
Blood and urine analysis
We observed no obvious differences between the WNK3 knockout mice and wild-type littermates in survival, gross physical appearance and organ morphology. There were no significant differences in the plasma K+ and 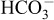 levels (Table 1). Urine volume and urinary excretion of Na+ and K+ under normal conditions were not significantly affected in WNK3 knockout mice. To more thoroughly characterize the phenotypes, we fed WNK3 knockout and wild-type mice low-salt (0.01% NaCl) and high-salt (4% NaCl) diets, and monitored urinary Na+ and K+ excretion, particularly focusing on the transition periods while changing diets. However, as shown in Fig. 3A–C, sodium excretion in the WNK3 knockout mice did not change during the experimental period.
levels (Table 1). Urine volume and urinary excretion of Na+ and K+ under normal conditions were not significantly affected in WNK3 knockout mice. To more thoroughly characterize the phenotypes, we fed WNK3 knockout and wild-type mice low-salt (0.01% NaCl) and high-salt (4% NaCl) diets, and monitored urinary Na+ and K+ excretion, particularly focusing on the transition periods while changing diets. However, as shown in Fig. 3A–C, sodium excretion in the WNK3 knockout mice did not change during the experimental period.
Table 1. Biochemical analysis of blood from wild-type and WNK3 knockout mice.
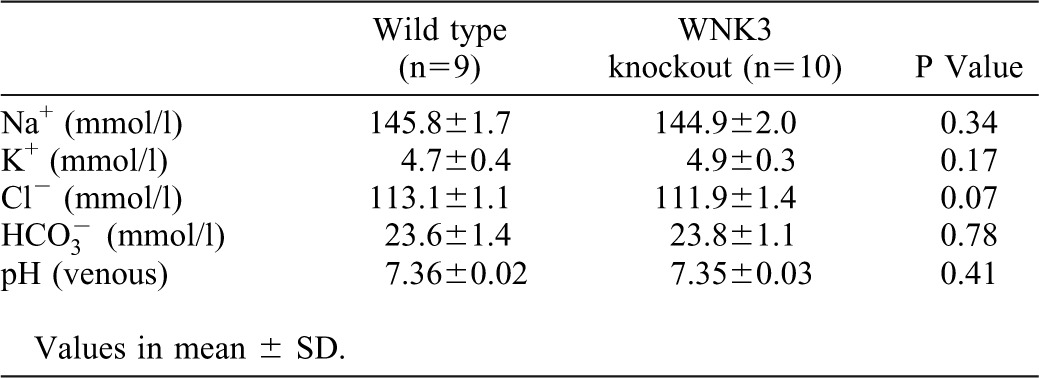
Fig. 3. Urinary excretion of Na+ and K+ in WNK3 knockout mice.

There were no significant differences in urine volume (A), urinary Na+ excretion (B) or K+ excretion (C) between WNK3 knockout mice (squares, n = 8) and their wild-type littermates (circles, n = 8). On day 0, mice were switched from low-salt diet (0.01% NaCl) to high-salt diet (4.0% NaCl). Before switching diet, mice were fed low-salt diet for 1 week. n.s. not significant.
Blood pressure
We used a tail-cuff system to measure blood pressure. As shown in Fig. 4A, blood pressure in WNK3 knockout mice did not show any significant differences when compared with wild-type mice under normal diet (109.7±1.6 vs. 111.2±1.6 mmHg, knockout: n = 11, wild-type: n = 14). However, when mice were fed with low-salt diet, blood pressure in WNK3 knockout mice was lower compared with wild-type mice (105.8±1.1 vs. 110.7±0.7 mmHg, knockout: n = 11, wild-type: n = 9, P<0.01) (Fig. 4B).
Fig. 4. Lower blood pressure in WNK3 knockout mice fed low-salt diet.

(A) Blood pressure in WNK3 knockout mice under normal diet. Blood pressure was measured using a tail-cuff system. WNK3 knockout mice (n = 9) did not show significantly decreased systolic blood pressure, as compared to their wild-type littermates (n = 14). (B) Blood pressure in WNK3 knockout mice (n = 11) under low-salt diet. WNK3 knockout mice showed lower blood pressure, as compared to their wild-type littermates (n = 9). *P<0.05. n.s. not significant.
Expression and phosphorylation of NCC and NKCC2 were not affected in WNK3 knockout mouse kidney
WNK3 reportedly activates NCC and NKCC2 function when co-expressed in Xenopus oocytes. Therefore, we investigated whether expression and phosphorylation of NCC and NKCC2 were decreased in the kidneys of WNK3 knockout mice. However, as shown in Fig. 5A–B, we could not see any significant difference in the protein abundance or the magnitude of phosphorylation of NKCC2 and NCC between WNK3 knockout and wild-type mice under a normal diet. Next, to investigate whether phosphorylation of OSR1 and SPAK was lower due to an absence of WNK3, we examined phosphorylation of OSR1 at 325S and SPAK at 380S, phosphorylation sites for WNK kinases, using phospho-specific antibodies. As shown in Fig. 5C–D, phosphorylation of OSR1 and SPAK at their WNK phosphorylation sites was not lower in kidneys from WNK3 knockout mice.
Fig. 5. Expression and phosphorylation of NKCC2 and NCC in kidneys from WNK3 knockout mice under normal diet.

(A) Representative immunoblots of total- and phosphorylated- NKCC2 and NCC in kidneys from wild-type and WNK3 knockout mice. (B) Densitometry analyses of expression and phosphorylation of NKCC2 and NCC in kidney from wild-type and WNK3 knockout mice. For densitometry analysis, values (n = 15) are expressed as ratios against the average of signals in the wild-type group. There were no significant decreases in the expression and phosphorylation of NKCC2 and NCC in kidneys from WNK3 knockout mice, as compared to wild-type littermates. (C) Representative immunoblots of p-OSR1 and p-SPAK in kidneys from wild-type and WNK3 knockout mice. (D) Densitometry analyses of p-OSR1 and p-SPAK in kidneys from wild-type and WNK3 knockout mice. For densitometry analysis, values (n = 15) are expressed as ratios against the average of signals in the wild-type group. There were no significant decreases in phosphorylation of OSR1 and SPAK in kidneys from WNK3 knockout mice, as compared to wild-type littermates. n.s. not significant.
Furthermore, we examined the phosphorylation status of NCC and NKCC2 in WNK3 knockout mice under low-salt diet, since blood pressure in WNK3 knockout mice was lower, when mice were fed with low-salt diet. However, even under low-salt diet, WNK3 knockout mice did not show decreased phosphorylation of either NCC or NKCC2, when compared with wild-type mice (Fig. 6A–B). These results indicated that WNK3 does not play a major role in regulation of NCC and NKCC2 in vivo mouse kidney, in contrast to several over-expression studies in Xenopus oocytes. Phosphorylation of OSR1 and SPAK at their WNK phosphorylation sites was not lower in kidneys from WNK3 knockout mice, even under low-salt diet (Fig. 6C–D).
Fig. 6. Expression and phosphorylation of NKCC2 and NCC in kidneys from WNK3 knockout mice fed low-salt diet.

(A) Representative immunoblots of total and phosphorylated NKCC2 and NCC in kidneys from wild-type and WNK3 knockout mice fed with low-salt diet. (B) Densitometry analyses of expression and phosphorylation of NKCC2 and NCC in kidneys from wild-type and WNK3 knockout mice fed low-salt diet. For densitometry analysis, values (n = 10) are expressed as ratios against the average of signals in the wild-type group. There were no significant decreases in the expression and phosphorylation of NKCC2 and NCC in kidneys from WNK3 knockout mice, as compared to wild-type littermates, even under low-salt diet. (C) Representative immunoblots of p-OSR1 and p-SPAK in kidneys from wild-type and WNK3 knockout mice fed low-salt diet. (D) Densitometry analyses of p-OSR1 and p-SPAK in kidneys from wild-type and WNK3 knockout mice fed low-salt diet. For densitometry analysis, values (n = 10) are expressed as ratios against the average of signals in the wild-type group. There were no significant decreases in phosphorylation of OSR1 and SPAK in kidneys from WNK3 knockout mice, as compared to wild-type littermates. n.s. not significant.
Expression of NHE3 and γ-ENaC in the kidneys of WNK3 knockout mice was not significantly different from that in wild-type mice (supplementary material Fig. S1).
Expression levels of WNK4 were elevated in kidneys of WNK3 knockout mice
The WNK kinase family phosphorylates and activates OSR1 and SPAK, and activated OSR1 and SPAK kinases phosphorylate NCC. We hypothesized that other WNKs could compensate for the absence of WNK3 in the kidney. To examine this hypothesis, we examine the expression levels of WNK1 and WNK4 in the kidneys of WNK3 knockout mice. As shown in Fig. 7A–B, we found that WNK4 expression was slightly but significantly elevated in kidneys from WNK3 knockout mice, as compared to those from wild-type mice. We also examined WNK1 and WNK4 expression in WNK3 knockout mice fed low-salt diet, as compensation would become clearer when the WNK-OSR1/SPAK-NCC phosphorylation cascade is activated (Chiga et al., 2008). As expected, under low-salt diet, both WNK1 and WNK4 expression increased significantly in kidneys from WNK3 knockout mice, as compared to wild-type mice (Fig. 7C–D). These results indicate that increased expression of WNK1 and WNK4 compensate for the absence of WNK3 in kidneys from WNK3 knockout mice.
Fig. 7. WNK1 and WNK4 were elevated in kidneys from WNK3 knockout mice fed low-salt diet.

(A) Representative immunoblots of WNK1 and WNK4 in kidneys from wild-type and WNK3 knockout mice fed normal diet. (B) Densitometry analyses of WNK1 and WNK4 in kidneys from wild-type and WNK3 knockout mice fed normal diet. For densitometry analysis, values (n = 15) are expressed as ratios against the average of signals in the wild-type group. Expression of WNK4 was elevated in kidneys from WNK3 knockout mice, as compared to wild-type littermates. (C) Representative immunoblots of WNK1 and WNK4 in kidneys from wild-type and WNK3 knockout mice fed low-salt diet. (D) Densitometry analyses of WNK1 and WNK4 in kidneys from wild-type and WNK3 knockout mice fed low-salt diet. For densitometry analysis, values (n = 10) are expressed as ratios against the average of signals in the wild-type group. Expression of WNK1 and WNK4 was elevated in kidneys from WNK3 knockout mice, as compared to wild-type littermates. *P<0.05. n.s. not significant.
Discussion
One of the major issues in hypertension research is regulation of renal sodium transporters that control sodium reabsorption in the kidney. NKCC2 and NCC are kidney-specific members of the SLC12A family of electroneutral cation-chloride co-transporters (Haas and Forbush, 1998; Gamba, 2005). They have been shown to play an important role in regulation of blood pressure and sodium reabsorption in the kidney.
Through analyses using genetically engineered mice, we have established the existence of the WNK-OSR1/SPAK-NCC kinase cascade in the in vivo kidney (Yang, S. S. et al., 2007; Chiga et al., 2008; Chiga et al., 2011; Ohta et al., 2009). To date, several physiological regulators of WNK-OSR1/SPAK-NCC phosphorylation cascade have been reported. We have reported that the WNK-OSR1/SPAK-NCC cascade is activated by low-salt diet and inhibited by high-salt diet, mainly via the action of aldosterone (Chiga et al., 2008). Angiotensin II has also been shown to be a regulator of their phosphorylation (San-Cristobal et al., 2009; Talati et al., 2010; van der Lubbe et al., 2011). Thus, WNK-OSR1/SPAK-NCC is a regulator of NaCl homeostasis in the kidney through the renin-angiotensin-aldosterone system. Another vasoactive hormone, vasopressin, was shown to regulate this cascade (Pedersen et al., 2010). We recently demonstrated that insulin is another potent regulator of the WNK-OSR1/SPAK-NCC phosphorylation cascade in the kidney (Sohara et al., 2011). This discovery is important when considering mechanisms of salt-sensitive hypertension in hyperinsulinemic patients, such as those with metabolic syndrome. Moreover, Vallon et al. and Naito et al. reported that potassium intake and extracellular potassium ions, respectively, regulate this cascade (Vallon et al., 2009; Naito et al., 2010). Taken together, these reports indicate that the WNK-OSR1/SPAK-NCC phosphorylation cascade is very important in kidney, not only for the pathogenesis of PHAII, but also for the homeostatic regulation of sodium, potassium, and blood pressure under various pathophysiological conditions.
Similar to NCC, WNKs, OSR1 and SPAK reportedly regulate NKCC2. OSR1 and SPAK kinases interact with an RFQV motif on NKCC2 and directly phosphorylate NKCC2 in in vitro kinase assays (Moriguchi et al., 2005; Richardson et al., 2008; Richardson et al., 2011). In addition to OSR1 and SPAK, reduced expression of WNK1 by siRNA inhibits endogenous NKCC1 activity in HeLa cells, measured by 86Rb influx assays (Anselmo et al., 2006). WNK3 is another WNK kinase that is reported to regulate NKCC2 as well as NCC. Rinehart et al. reported that WNK3 increases NKCC2 phosphorylation at Thr-184 and Thr-189 residues, which had been identified to be necessary for vasopressin-mediated plasma membrane translocation and activation of NKCC2 in Xenopus laevis oocytes (Rinehart et al., 2005). In addition, overexpression of WNK3 in Xenopus laevis oocytes leads to the activation of NKCC2, which is dependent upon the interaction of SPAK and OSR1 with one of the three RFx[V/I]-motifs that are present in WNK3 (Ponce-Coria et al., 2008; Richardson and Alessi, 2008). Several other groups have reported that co-expression of NCC with WNK3 in Xenopus laevis oocytes increased its transport activity (Rinehart et al., 2005; Yang, C. L. et al., 2007; San-Cristobal et al., 2008; Glover et al., 2009; Cruz-Rangel et al., 2011). Therefore, in the kidney, it was expected that WNK3 could regulate sodium reabsorption along the nephron by activating NCC and NKCC2 via phosphorylation of OSR1 and SPAK.
Recently, we re-evaluated the immunolocalization of WNK4 along the mouse nephron. In this study, we confirmed that WNK4 is present in DCT, collecting duct and other segments of the nephron, but not in TAL, where NKCC2 and SPAK are present (Ohno et al., 2011). The lack of WNK4 protein in TAL is consistent with our observation that increased phosphorylation of NKCC2 is not observed in WNK4 knock-in mice (manuscript in preparation), and the fact that PHAII is sensitive to thiazide, but not to furosemide. This indicates that activation of furosemide-sensitive NKCC2 is not observed in PHAII caused by WNK1 or WNK4 mutation, indicating that WNK1 and WNK4 may not play a major role in regulation of NKCC2 in the kidney, although WNK1 phosphorylates NKCC2 in in vitro kinase assays (Anselmo et al., 2006). Therefore, another WNK kinase could be present in TAL to regulate SPAK and NKCC2. This suggests that WNK3 is a regulator of NKCC2 in the kidney.
In this study, we successfully generated WNK3 knockout mice and analyzed their renal phenotype. However, in contrast to data obtained in the Xenopus oocytes system and our expectation that WNK3 is a major regulator of NKCC2, WNK3 knockout mice did not show any decrease in expression and phosphorylation of NKCC2 and NCC. Indeed, urinary excretion of Na+ and K+ was not significantly affected in WNK3 knockout mice, even though we focused on the transition periods when changing sodium diets. These results suggest that WNK3 is not a powerful regulator of NKCC2 and NCC in the kidney, unlike in Xenopus oocytes.
There are several possible explanations as to why expression and phosphorylation of NKCC2 and NCC were not lower in WNK3 knockout mouse kidney. It is possible that the expression level of WNK3 in individual tubular cells was too low to contribute to the overall WNK kinase activity. Our immunoblot data clearly showed that WNK3 protein abundance in kidney was below the detection limit. RT-PCR using dissected nephron segments did not show the presence of preferential expression sites for WNK3 along the nephron. These results suggest that WNK3 does not have a specific role in the kidney, in contrast to the role of WNK4 and kidney-specific WNK1 as regulators of NCC in DCT. Another possibility is that the other WNKs, WNK1 and WNK4, compensated for the absence of WNK3 in the NKCC2- and NCC-expressing nephrons. Considering that 1) phosphorylation of OSR1 and SPAK at their WNK phosphorylation sites was not lower in the kidneys of WNK3 knockout mice, and 2) WNK1 and WNK4 expression levels were slightly higher in the kidneys of WNK3 knockout mice, it is possible that WNK1 and WNK4 compensated for the absence of WNK3. Therefore, the lack of phenotype in the absence of WNK3 in the mouse kidney does not necessarily exclude a potential role for WNK3 in activation of NCC and NKCC2 in vivo. On the other hand, WNK4 hypomorphic mice showed decreased phosphorylation of NCC and lower blood pressure, and other WNKs could not compensate (Ohta et al., 2009). Therefore, as the absence of WNK3 is compensated for by other WNKs, the role of WNK3 in the regulation of NKCC2 and NCC in vivo may not be substantial. To clarify this issue further, WNK3 and WNK4 double-knockout mouse would be required; however, we have not obtained conditional WNK4 knockout mice to date.
WNK3 knockout mice showed lower blood pressure only when mice were fed low-salt diet, although urinary excretion of Na+ was not significantly affected in WNK3 knockout mice. Indeed, consistent with urinary data, WNK3 knockout mice did not show any decrease in expression and phosphorylation of NKCC2 and NCC in the kidney even under low-salt diet. These results clearly indicated that lower blood pressure in WNK3 knockout mice fed low-salt diet is not due to decreased sodium reabsorption in the kidney, but due to the other mechanism(s). Since WNK3 is abundantly expressed in brain, it is possible that the absence of WNK3 in brain might affect neuronal regulatory mechanisms of blood pressure, such as secretion of vasopressin, control of sympathetic nerve activity, etc., although the involvement of WNK3 in such processes has not been investigated. It is also possible that WNK3 knockout mice might have reduced vascular resistance since NKCC1, a substrate of SPAK/OSR1, reportedly regulated tonus of vascular smooth muscles, and decreased blood pressure was in fact in the NKCC1 knockout mouse (Akar et al., 2001; Meyer et al., 2002; Garg et al., 2007). Interestingly, similar to WNK3 knockout mice, NKCC1 knockout mice also showed lower blood pressure only when mice were fed with low-salt diet (Kim et al., 2008). Moreover, we recently found that SPAK knockout mice showed the reduced aortic contractility with decreased phosphorylation of NKCC1 (Yang et al., 2010). Accordingly, we tried to evaluate the phosphorylation status of NKCC1 in aorta of WNK3 knockout mice. Unfortunately, we have not yet detected an apparent decrease of NKCC1 phosphorylation in the aorta of WNK3 knockout mice (data not shown). Since the exact quantification of NKCC1 phosphorylation in tiny mouse aortic tissues by immunoblot is in fact very tricky, further investigation must be required to clarify this issue.
In this study, we generated and analyzed WNK3 knockout mice, focusing on their renal phenotypes. However, we did not observe any significant decreases in expression and phosphorylation of NKCC2 and NCC in the WNK3 knockout mouse kidney. Our results suggest that WNK3 only has a minor role in the regulation of NKCC2 and NCC in the in vivo mouse kidney.
Materials and Methods
Targeted disruption of the Wnk3 gene
For generation of WNK3 knockout mice, we prepared the targeting vector using PCR-amplified segments of the Wnk3 gene after verifying sequences. The targeting vector was transfected into J1 ES cells by electroporation, as described previously (Sohara et al., 2006). After selection with 150 µg/ml G418 and 2 µM ganciclovir, correctly targeted ES cell clones were selected by PCR and Southern blotting. Chimeric male mice were bred with C57BL/6 female mice to produce the heterozygous floxed mice, and the neo cassette was then deleted by crossing the mice with CAG-Cre recombinase-expressing transgenic mice (Sakai and Miyazaki, 1997). Wild-type controls and WNK3 knockout mice were bred and tail genomic DNA was applied for genotyping by PCR (forward primer; 5′-GATATGTAAGCACTACTACC-3′, reverse primer-A; 5′-TCTAATAGCTCAACTGAGTG-3′, reverse primer-B; 5′-GTTCTCAAGTTCTACATCTC-3′). The mice were raised in a 12-hour day and night cycle, fed with normal rodent diet and plain drinking water. The phenotype of male mice was evaluated at the age of 8–14 weeks. The experiment was approved by the Animal Care and Use Committee of Tokyo Medical and Dental University.
Laser capture microdissection (LCM) and RT-PCR
For LCM, mouse kidneys were cut along the long axis and embedded in Tissue-Tek OCT compound (Sakura Finetechnical Co., Ltd., Tokyo, Japan) and immediately frozen in dry ice. Tissue samples were then immersed in nitrogen oxide. Frozen tissue blocks were cut into 10-µm sections, and were mounted on uncoated, uncharged glass slides. Sections were stained for 20 s in Histogene Staining Solution (Arcturus, Mountain View, CA, USA), subjected to dehydration in a graded alcohol series, cleared for 5 min in fresh xylene, and air-dried for 5 min. LCM was performed using a Pixcell II laser capture system (Arcturus). Tubular cells in each nephron were visualized and captured using CapSure LCM macrocaps (Arcturus). Laser setting ranged between 70 to 90 mW in power and 0.6 to 1.0 ms in duration. Total RNA from LCM samples was extracted from captured cells using a PicoPure RNA Isolation kit (Arcturus) and total RNA from mouse kidneys was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Total RNA was reverse-transcribed using Omniscript reverse transcriptase (Qiagen, Hilden, Germany). We confirmed the presence of nephron cells using region-specific primers; NHE3 (sense; 5′-GCTGTCATTGGCACTATATGG-3′ (exon 2) and antisense; 5′-GAGGACTTCATTGACATGGAC-3′ (exon 3)), NKCC2 ((sense; 5′-AACTCAGTGCCCAGTAGTGC-3′ (exon 2) and antisense; 5′-AGGCATCCCATCTCCATTAG -3′ (exon 3)), NCC ((sense; 5′- GTCATCATGGTCTCCTTTGC -3′ (exon 7) and antisense; 5′-TAGCTGAGATGGCAAGGTAG-3′ (exon 9)), and ENaC α subunit (sense; 5′-TCAACATTCTGTCCAGACTGC-3′ (exon 3) and antisense; 5′-GTAGCATGGCCCATACATGG-3′ (exon 4)). Finally, we investigated the presence of WNK3 in these tissues; WNK3 (sense; 5′-GCTGTTGCAACTTCCCCTAGT-3′ (exon 1) and antisense; 5′-CCGTTGCTGCTCAGCTTTAG-3′ (exon 2)).
Blood and urine analysis and blood pressure measurement
Blood for electrolyte analyses was obtained from the submandibular venous plexus under light ether anesthesia. Electrolyte levels were determined with an i-STAT analyzer (Fuso, Osaka, Japan). Mice were kept in metabolic cages for urine collection. Low-salt (0.01% NaCl) and high-salt (4% NaCl) diets were obtained from Oriental Yeast Co., Ltd. (Tokyo, Japan). Urine samples were analyzed by DRI-CHEM (Fujifilm, Tokyo, Japan). Blood pressure in restrained conscious mice at steady state was measured with a programmable tail-cuff sphygmomanometer (MK-2000A; Muromachi, Tokyo, Japan).
Antibodies
We prepared an affinity-purified sheep antibody raised against a fragment of recombinant mouse WNK3 (residues 1145 – 1508). Primary antibodies used in this study were: rabbit anti-WNK1 (1:250) (Bethyl Laboratories, Montgomery, TX, USA); rabbit anti-WNK4 (1:250) (Ohno et al., 2011); rabbit anti-phosphorylated OSR1 (1:3000) (Ohta et al., 2009); rabbit anti-phosphorylated SPAK (1:500) (Yang et al., 2010); rabbit anti-NHE3 (1:200) (Alpha Diagnostic, San Antonio, TX, USA); rabbit anti-phosphorylated NCC (1:250) (Yang, S. S. et al., 2007); guinea pig anti-NCC (1:500) (Nomura et al., 2011), rabbit anti-phosphorylated NKCC2 (1:500) (Yang et al., 2010); rabbit anti-NKCC2 (1:1000) (kindly provided by K. Mutig, Charité-Universitätsmedizin Berlin, Campus Charité-Mitte, Germany); and rabbit anti-ENaC γ subunit (1:250) (kindly provided by M. Knepper, National Institutes of Health, USA). Alkaline-phosphatase-conjugated anti-IgG antibodies (Promega, Madison, WI, USA) were used as secondary antibodies for immunoblotting.
Immunoblotting
Semiquantitative immunoblotting was performed as described previously using whole kidney homogenates without the nuclear fraction (600 × g) or the crude membrane fraction (17000 × g) (Yang, S. S. et al., 2007). Band intensity was analyzed using Image J (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Statistical significance was evaluated using unpaired t-test. P-values <0.05 were considered to be significant.
Supplementary Material
Acknowledgments
We thank C. Iijima for help in the experiments. We thank K. Mutig and M. Knepper for provision of antibodies. This study was supported in part by Grants-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grants-in-Aid for Scientific Research on Creative Scientific Research from the Japan Society for the Promotion of Science, Grant-in-Aid for Scientific Research (A) from the Japan Society for the Promotion of Science, Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Japan-Taiwan Joint Research Program for Interchange Association Japan, the Salt Science Research Foundation (NO. 1026), Takeda Science Foundation, Kanae Foundation for the Promotion of Medical Science, and The Nakajima Foundation.
Footnotes
Competing interests: The authors declare no competing interests.
References
- Achard J. M., Disse-Nicodeme S., Fiquet-Kempf B., Jeunemaitre X. (2001). Phenotypic and genetic heterogeneity of familial hyperkalaemic hypertension (Gordon syndrome). Clin. Exp. Pharmacol. Physiol. 28, 1048–1052 10.1046/j.1440-1681.2001.03575.x [DOI] [PubMed] [Google Scholar]
- Akar F., Jiang G., Paul R. J., O'Neill W. C. (2001). Contractile regulation of the Na(+)-K(+)-2Cl(-) cotransporter in vascular smooth muscle. Am J. Physiol. Cell Physiol. 281, C579–C584 [DOI] [PubMed] [Google Scholar]
- Anselmo A. N., Earnest S., Chen W., Juang Y. C., Kim S. C., Zhao Y., Cobb M. H. (2006). WNK1 and OSR1 regulate the Na+, K+, 2Cl- cotransporter in HeLa cells. Proc. Natl. Acad. Sci. USA 103, 10,883–10,888 10.1073/pnas.0604607103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiga M., Rai T., Yang S. S., Ohta A., Takizawa T., Sasaki S., Uchida S. (2008). Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int. 74, 1403–1409 10.1038/ki.2008.451 [DOI] [PubMed] [Google Scholar]
- Chiga M., Rafiqi F. H., Alessi D. R., Sohara E., Ohta A., Rai T., Sasaki S., Uchida S. (2011). Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J. Cell Sci. 124, 1391–1395 10.1242/jcs.084111 [DOI] [PubMed] [Google Scholar]
- Cruz-Rangel S., Melo Z., Vazquez N., Meade P., Bobadilla N. A., Pasantes-Morales H., Gamba G., Mercado A. (2011). Similar effects of all WNK3 variants on SLC12 cotransporters. Am. J. Physiol. Cell Physiol. 301, C601–C608 10.1152/ajpcell.00070.2011 [DOI] [PubMed] [Google Scholar]
- de Los Heros P., Kahle K. T., Rinehart J., Bobadilla N. A., Vazquez N., San Cristobal P., Mount D. B., Lifton R. P., Hebert S. C., Gamba G. (2006). WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. USA 103, 1976–1981 10.1073/pnas.0510947103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba G. (2005). Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493 10.1152/physrev.00011.2004 [DOI] [PubMed] [Google Scholar]
- Garg P., Martin C. F., Elms S. C., Gordon F. J., Wall S. M., Garland C. J., Sutliff R. L., O'Neill W. C. (2007). Effect of the Na-K-2Cl cotransporter NKCC1 on systemic blood pressure and smooth muscle tone. Am J. Physiol. Heart Circ. Physiol. 292, H2100–H2105 10.1152/ajpheart.01402.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover M., Zuber A. M., O'Shaughnessy K. M. (2009). Renal and brain isoforms of WNK3 have opposite effects on NCCT expression. J. Am. Soc. Nephrol. 20, 1314–1322 10.1681/ASN.2008050542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R. D. (1986). Syndrome of hypertension and hyperkalemia with normal glomerular filtration rate. Hypertension 8, 93–102 [DOI] [PubMed] [Google Scholar]
- Haas M., Forbush B., 3rd (1998). The Na-K-Cl cotransporters. J. Bioenerg. Biomembr. 30, 161–172 10.1023/A:1020521308985 [DOI] [PubMed] [Google Scholar]
- Holden S., Cox J., Raymond F. L. (2004). Cloning, genomic organization, alternative splicing and expression analysis of the human gene WNK3 (PRKWNK3). Gene 335, 109–119 10.1016/j.gene.2004.03.009 [DOI] [PubMed] [Google Scholar]
- Kahle K. T., Rinehart J., de Los Heros P., Louvi A., Meade P., Vazquez N., Hebert S. C., Gamba G., Gimenez I., Lifton R. P. (2005). WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. USA 102, 16,783–16,788 10.1073/pnas.0508307102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. M., Eisner C., Faulhaber-Walter R., Mizel D., Wall S. M., Briggs J. P., Schnermann J. (2008). Salt sensitivity of blood pressure in NKCC1-deficient mice. Am. J. Physiol. Renal Physiol. 295, F1230–F1238 10.1152/ajprenal.90392.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J. W., Flagella M., Sutliff R. L., Lorenz J. N., Nieman M. L., Weber C. S., Paul R. J., Shull G. E. (2002). Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na(+)-K(+)-2Cl(−) cotransporter. Am. J. Physiol. Heart Circ. Physiol. 283, H1846–H1855 [DOI] [PubMed] [Google Scholar]
- Moriguchi T., Urushiyama S., Hisamoto N., Iemura S., Uchida S., Natsume T., Matsumoto K., Shibuya H. (2005). WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem. 280, 42,685–42,693 10.1074/jbc.M510042200 [DOI] [PubMed] [Google Scholar]
- Naito S., Ohta A., Sohara E., Ohta E., Rai T., Sasaki S., Uchida S. (2010). Regulation of WNK1 kinase by extracellular potassium. Clin. Exp. Nephrol. 15, 195–202 10.1007/s10157-010-0378-9 [DOI] [PubMed] [Google Scholar]
- Nomura N., Tajima M., Sugawara N., Morimoto T., Kondo Y., Ohno M., Uchida K., Mutig K., Bachmann S., Soleimani M. et al. (2011). Generation and analyses of R8L barttin knockin mouse. Am. J. Physiol. Renal Physiol. 301, F297–F307 10.1152/ajprenal.00604.2010 [DOI] [PubMed] [Google Scholar]
- Ohno M., Uchida K., Ohashi T., Nitta K., Ohta A., Chiga M., Sasaki S., Uchida S. (2011). Immunolocalization of WNK4 in mouse kidney. Histochem. Cell Biol. 136, 25–35 10.1007/s00418-011-0827-x [DOI] [PubMed] [Google Scholar]
- Ohta A., Rai T., Yui N., Chiga M., Yang S. S., Lin S. H., Sohara E., Sasaki S., Uchida S. (2009). Targeted disruption of the Wnk4 gene decreases phosphorylation of Na-Cl cotransporter, increases Na excretion and lowers blood pressure. Hum. Mol. Genet. 18, 3978–3986 10.1093/hmg/ddp344 [DOI] [PubMed] [Google Scholar]
- Pacheco-Alvarez D., Cristobal P. S., Meade P., Moreno E., Vazquez N., Munoz E., Diaz A., Juarez M. E., Gimenez I., Gamba G. (2006). The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 281, 28,755–28,763 10.1074/jbc.M603773200 [DOI] [PubMed] [Google Scholar]
- Pedersen N. B., Hofmeister M. V., Rosenbaek L. L., Nielsen J., Fenton R. A. (2010). Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 78, 160–169 10.1038/ki.2010.130 [DOI] [PubMed] [Google Scholar]
- Ponce-Coria J., San-Cristobal P., Kahle K. T., Vazquez N., Pacheco-Alvarez D., de Los Heros P., Juarez P., Munoz E., Michel G., Bobadilla N. A. et al. (2008). Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc. Natl. Acad. Sci. USA 105, 8458–8463 10.1073/pnas.0802966105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiqi F. H., Zuber A. M., Glover M., Richardson C., Fleming S., Jovanovic S., Jovanovic A., O'Shaughnessy K. M., Alessi D. R. (2010). Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2, 63–75 10.1002/emmm.200900058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C., Alessi D. R. (2008). The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J. Cell Sci. 121, 3293–3304 10.1242/jcs.029223 [DOI] [PubMed] [Google Scholar]
- Richardson C., Rafiqi F. H., Karlsson H. K., Moleleki N., Vandewalle A., Campbell D. G., Morrice N. A., Alessi D. R. (2008). Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 121, 675–684 10.1242/jcs.025312 [DOI] [PubMed] [Google Scholar]
- Richardson C., Sakamoto K., de los Heros P., Deak M., Campbell D. G., Prescott A. R., Alessi D. R. (2011). Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J. Cell Sci. 124, 789–800 10.1242/jcs.077230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J., Kahle K. T., de Los Heros P., Vazquez N., Meade P., Wilson F. H., Hebert S. C., Gimenez I., Gamba G., Lifton R. P. (2005). WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl- cotransporters required for normal blood pressure homeostasis. Proc. Natl. Acad. Sci. USA 102, 16,777–16,782 10.1073/pnas.0508303102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K., Miyazaki J. (1997). A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem. Biophys. Res. Commun. 237, 318–324 10.1006/bbrc.1997.7111 [DOI] [PubMed] [Google Scholar]
- San-Cristobal P., Ponce-Coria J., Vazquez N., Bobadilla N. A., Gamba G. (2008). WNK3 and WNK4 amino-terminal domain defines their effect on the renal Na+-Cl- cotransporter. Am. J. Physiol. Renal Physiol. 295, F1199–F1206 10.1152/ajprenal.90396.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San-Cristobal P., Pacheco-Alvarez D., Richardson C., Ring A. M., Vazquez N., Rafiqi F. H., Chari D., Kahle K. T., Leng Q., Bobadilla N. A. et al. (2009). Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc. Natl. Acad. Sci. USA 106, 4384–4389 10.1073/pnas.0813238106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schambelan M., Sebastian A., Rector F. C., Jr (1981). Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism): role of increased renal chloride reabsorption. Kidney Int. 19, 716–727 10.1038/ki.1981.72 [DOI] [PubMed] [Google Scholar]
- Sohara E., Rai T., Yang S. S., Uchida K., Nitta K., Horita S., Ohno M., Harada A., Sasaki S., Uchida S. (2006). Pathogenesis and treatment of autosomal-dominant nephrogenic diabetes insipidus caused by an aquaporin 2 mutation. Proc. Natl. Acad. Sci. USA 103, 14,217–14,222 10.1073/pnas.0602331103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohara E., Rai T., Yang S. S., Ohta A., Naito S., Chiga M., Nomura N., Lin S. H., Vandewalle A., Ohta E. et al. (2011). Acute insulin stimulation induces phosphorylation of the Na-Cl cotransporter in cultured distal mpkDCT cells and mouse kidney. PLoS ONE 6, e24277 10.1371/journal.pone.0024277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talati G., Ohta A., Rai T., Sohara E., Naito S., Vandewalle A., Sasaki S., Uchida S. (2010). Effect of angiotensin II on the WNK-OSR1/SPAK-NCC phosphorylation cascade in cultured mpkDCT cells and in vivo mouse kidney. Biochem. Biophys. Res. Commun. 393, 844–848 10.1016/j.bbrc.2010.02.096 [DOI] [PubMed] [Google Scholar]
- Vallon V., Schroth J., Lang F., Kuhl D., Uchida S. (2009). Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am. J. Physiol. Renal Physiol. 297, F704–F712 10.1152/ajprenal.00030.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lubbe N., Lim C. H., Fenton R. A., Meima M. E., Jan Danser A. H., Zietse R., Hoorn E. J. (2011). Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 79, 66–76 10.1038/ki.2010.290 [DOI] [PubMed] [Google Scholar]
- Verissimo F., Silva E., Morris J. D., Pepperkok R., Jordan P. (2006). Protein kinase WNK3 increases cell survival in a caspase-3-dependent pathway. Oncogene 25, 4172–4182 10.1038/sj.onc.1209449 [DOI] [PubMed] [Google Scholar]
- Vitari A. C., Deak M., Morrice N. A., Alessi D. R. (2005). The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 391, 17–24 10.1042/BJ20051180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari A. C., Thastrup J., Rafiqi F. H., Deak M., Morrice N. A., Karlsson H. K., Alessi D. R. (2006). Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem. J. 397, 223–231 10.1042/BJ20060220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson F. H., Disse-Nicodeme S., Choate K. A., Ishikawa K., Nelson-Williams C., Desitter I., Gunel M., Milford D. V., Lipkin G. W., Achard J. M. et al. (2001). Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112 10.1126/science.1062844 [DOI] [PubMed] [Google Scholar]
- Yang C. L., Zhu X., Ellison D. H. (2007). The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J. Clin. Invest. 117, 3403–3411 10.1172/JCI32033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. S., Morimoto T., Rai T., Chiga M., Sohara E., Ohno M., Uchida K., Lin S. H., Moriguchi T., Shibuya H. et al. (2007). Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 5, 331–344 10.1016/j.cmet.2007.03.009 [DOI] [PubMed] [Google Scholar]
- Yang S. S., Lo Y. F., Wu C. C., Lin S. W., Yeh C. J., Chu P., Sytwu H. K., Uchida S., Sasaki S., Lin S. H. (2010). SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 21, 1868–1877 10.1681/ASN.2009121295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrowicz B. P., Abuin A., Ramirez-Solis R., Richter L. J., Piggott J., BeltrandelRio H., Buxton E. C., Edwards J., Finch R. A., Friddle C. J. et al. (2003). Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc. Natl. Acad. Sci. USA 100, 14,109–14,114 10.1073/pnas.2336103100 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}