

Summary
Topoisomerase II creates a double-strand break intermediate with topoisomerase covalently coupled to the DNA via a 5′-phosphotyrosyl bond. These intermediate complexes can become cytotoxic protein-DNA adducts and DSB repair at these lesions requires removal of topoisomerase II. To analyse removal of topoisomerase II from genomic DNA we adapted the trapped in agarose DNA immunostaining assay. Recombinant MRE11 from 2 sources removed topoisomerase IIα from genomic DNA in vitro, as did MRE11 immunoprecipitates isolated from A-TLD or K562 cells. Basal topoisomerase II complex levels were very high in A-TLD cells lacking full-length wild type MRE11, suggesting that MRE11 facilitates the processing of topoisomerase complexes that arise as part of normal cellular metabolism. In K562 cells inhibition of MRE11, PARP or replication increased topoisomerase IIα and β complex levels formed in the absence of an anti-topoisomerase II drug.
Key words: Topoisomerase II, MRE11, DSB repair, Protein-DNA adducts, A-TLD
Introduction
Human topoisomerase II is a target for important anti-cancer drugs including etoposide, mAMSA, mitoxantrone, daunorubicin and idarubicin (Kaufmann, 1998; Cowell et al., 2011a). The normal catalytic cycle of topoisomerase II produces a transient enzyme-bridged DNA double-strand break containing a covalent protein-DNA reaction intermediate, where attachment of the topoisomerase II to the DNA is via a 5′ tyrosyl phosphodiester linkage. Drugs such as etoposide stabilise this intermediate resulting in dead-end complexes that can lead to cell death. Thus, drugs that stabilise the topoisomerase-DNA complexes are referred to as topoisomerase II poisons (Kaufmann, 1998) and constitute a clinically important class of anti-cancer drugs.
These topoisomerase-DNA dead-end complexes can be repaired in cells. Topoisomerase II poison-induced DSBs are repaired predominantly through homologous recombination in the yeast S. cerevisiae (Sabourin et al., 2003), whilst vertebrates predominantly employ non-homologous end joining (NHEJ). The role of NHEJ to repair topoisomerase II damage has been demonstrated in cells from several species, including Chinese Hamster, Chicken and Human (Jeggo et al., 1989; Caldecott et al., 1990; Adachi et al., 2003; Adachi et al., 2004; Willmore et al., 2004; Ayene et al., 2005). For example, we previously reported that inhibition of the catalytic subunit of DNA dependent protein kinase (DNA-PKcs) with the small molecule inhibitor NU7026 massively potentiates the cytotoxicity of anti-topoisomerase II agents such as etoposide, mitoxantrone and mAMSA (Willmore et al., 2004).
During NHEJ, DNA dependent protein kinase (DNA-PK) is activated by DNA breaks. However, topoisomerase-linked DSBs do not activate DNA-PK nor bind KU in vitro (Mårtensson et al., 2003) and various lines of evidence suggest cellular processing is required before topoisomerase-induced breaks elicit a DNA damage response (Mao et al., 2001; Zhang et al., 2006; Fan et al., 2008; Alchanati et al., 2009). Thus, removal of the 5′ topoisomerase protein adducts from the DNA is presumably necessary for repair of DNA breaks in vivo by NHEJ. The cellular mechanism(s) of human topoisomerase II-DNA complex removal are still being elucidated.
Humans possess two separately encoded type II topoisomerases, the -α and -β isoforms. We have previously shown that both topoisomerase IIα and -β form stabilised enzyme-DNA complexes in the presence of drugs such as etoposide (Willmore et al., 1998). It is possible though that the complexes formed with each isoform are differentially distributed in the nucleus, differently affected by pre-existing DNA damage (Bigioni et al., 1996; Kingma et al., 1997; Wilstermann and Osheroff, 2001) or other cellular processes such as transcription or replication (Mao et al., 2001; Niimi et al., 2001), and/or that their resulting adducts are removed by different mechanisms.
While 5′ phosphotyrosyl-linked topoisomerase must be removed prior to DSB repair, the mechanism to achieve this may differ depending on the context. For example, topoisomerase II protein-DNA covalent complexes can form and be resolved in G1, but are also present in S-phase and interfere with replication leading to replication fork stalling, DSB generation, intra-S phase checkpoint signalling and dispersal of replication proteins (Kaufmann, 1998; Rossi et al., 2006). Processes implicated in removal of 5′-topoisomerase complexes involve: (1) a specific 5′ tyrosyl DNA phoshodiesterase (TDP2) cleaving the phosphodiester bond between the 5′ phosphate and the tyrosine (Cortes Ledesma et al., 2009; Zeng et al., 2011); (2) cleavage of the DNA end bearing the topoisomerase II by a nuclease such as MRE11 (Neale et al., 2005; Hartsuiker et al., 2009a); (3) cleavage by an AP lyase activity such as Ku (Ayene et al., 2005; Roberts et al., 2010); (4) a proteolytic mechanism (Mao et al., 2001; Sunter et al., 2010), (5) or a sequential combination of several of these activities.
Genetic studies in S. cerevisiae have provided a useful starting point for human studies and support a role for MRE11 in the removal of stabilised 5′-topoisomerase II-DNA complexes (Neale et al., 2005; Hartsuiker et al., 2009a). Mre11 is part of the MR complex (Mre11/Rad50), which is involved in the essential process of repairing double strand breaks and is conserved through evolution. The MR complex associates with NBS1 in humans (MRN complex) and Xrs in yeast (MRX complex). Mre11 is a nuclease with both exonuclease and endonuclease activities, the nuclease motifs are located in the N-terminal domain and are evolutionarily highly conserved. The single-strand endonuclease acts on a number of substrates including 5′ overhangs, 3′ flaps, 3′ branches and closed hairpins. The 3′–5′ exonuclease acts on double stranded DNA (D'Amours and Jackson, 2002). In vitro, the activity is highest in the presence of manganese but magnesium also supports some activity (Hopkins and Paull, 2008). In mammals the nuclease activity of MRE11 is required for homology directed double strand break repair, but the nuclease function is not required for ATM activation after DNA damage or telomere deprotection (Buis et al., 2008).
During meiosis a topoisomerase II like protein, Sporulation 11 (Spo11) is required to introduce double strand breaks. Unlike topoisomerase II, which normally re-ligates the transiently cleaved double stranded DNA (dsDNA), Spo11 is endonucleolytically removed from the 5′-end of the cleaved DNA revealing free DNA strands allowing meiotic recombination to take place, and releasing Spo11 attached to an oligonucleotide with a free 3′-end (Neale et al., 2005). In budding yeast two different Spo11-oligonucleotide complexes are produced; this was an important observation suggesting the DSB ends were biochemically distinct with asymmetric cleavage at each DSB. Notably, this removal of Spo11 requires the nuclease activity of the MRX complex and its functional partner, Sae2/Ctp1 in budding and fission yeast (Neale et al., 2005; Hartsuiker et al., 2009b; Cole et al., 2010).
Topoisomerase II-oligonucleotide complexes were also isolated from vegetative S. cerevisiae, but these were not genetically dependent upon MRE11 and SAE2 and the significance of these complexes is not known. Functional redundancy between nucleases has been proposed and possible nucleases include Sae2/Ctp1, Exo1 and Dna2 (Nicolette et al., 2010). In S. pombe Mre11 nuclease-dead or Ctp1 null strains are both hypersensitive to the etoposide derivative Top53 and the levels of covalent topoisomerase II-DNA complexes induced in these strains are approximately two fold higher than in wild type cells, implicating Mre11 and Ctp1 in removal of topoisomerase II covalent complexes (Malik and Nitiss, 2004; Hartsuiker et al., 2009a). Extending this analysis to bacterial and bacteriophage systems, the E. coli MR complex, SbcCD is able to remove an avidin protein adduct in vitro (Connelly and Leach, 2004) and the T4 endonuclease VII was shown to remove topoisomerase II complexes at stalled replication forks in vitro and in vivo (Hong and Kreuzer, 2003).
A number of studies also implicate MRE11 in the removal of topoisomerase II complexes in human cells. For instance, during adenovirus infections in human cells, the 5′-end of the linear viral genome is protected by a terminal protein. Without this protein viral DNA is joined into concatemers too large to be packaged effectively. However, in the presence of MRE11, RAD50 and NBS1 (which localise in foci next to viral replication centres) this protein is removed preventing viral packaging. Part of the viral defence mechanism, is to both degrade and reorganise the expression of the MRN complex (Stracker et al., 2002). In addition, a role for the MRN complex in processing topoisomerase II lesions has been suggested by the fact that murine cells with mutated Rad50 show elevated sensitivity to etoposide (Morales et al., 2008).
The precise role of the MR complex in processing topoisomerase II covalent DNA complexes is not clear. The nuclease activity of MRE11 may directly remove DNA bearing the covalently attached topoisomerase II adduct or it may play a role as a DNA damage sensor and signal transducer to activate a component downstream of the MR complex and trigger removal of the DNA adduct (Connelly and Leach, 2004).
We previously developed the trapped in agarose DNA immunostaining (TARDIS) assay to detect topoisomerase complexes formed in individual live cells on genomic DNA (Willmore et al., 1998). Cells are embedded in agarose and the cellular membranes and non-covalently bound proteins are removed; the covalently bound topoisomerase II remains on the DNA and can be visualised by immunofluorescence and quantified (Willmore et al., 1998; Cowell et al., 2011a). We have used this methodology to generate genomic DNA bearing 5′ topoisomerase II drug stabilised cleavage complexes. Using this as a substrate, we show that MRE11 can remove topoisomerase II-DNA complexes from genomic DNA in vitro. We also show that MRE11 plays a role in the maintaining low basal levels of topoisomerase II complexes in cells.
Results
Adaption of the TARDIS assay
To determine if MRE11 has a role in removing human topoisomerase II from DNA, we have modified the TARDIS assay. The TARDIS assay has previously been used extensively to study the kinetics of formation and removal of topoisomerase II complexes within cells (Cowell et al., 2011a). Here we have adapted the method to use the TARDIS slides as a source of non-recombinant topoisomerase II protein covalently attached to genomic DNA. K562 leukaemia cells were treated with 100 µM etoposide to generate genomic DNA bearing covalent topoisomerase II protein-DNA complexes. The cells were then embedded in agarose and lysed to remove non-covalently bound cellular proteins including histones. We refer to this in vitro substrate of genomic DNA bearing topoisomerase II complexes on slides, as DNA-topoisomerase II adducts, to distinguish them from complexes modulated within cells and then analysed on slides. The slides bearing the topoisomerase II adducts were incubated with immunoprecipitates or purified protein, and then washed prior to immunofluorescent visualisation and quantification steps (Fig. 1A). Non-specific proteolysis that could remove the protein signal was prevented by adding a cocktail of protease inhibitors to the buffers. Adducts were detected using anti-topoisomerase II antisera and visualised with an FITC labelled secondary antibody. An outline of the assay is shown in Fig. 1A. Data were obtained for large numbers of individual cells and the data are either shown in scattergrams where each point represents a single nucleus, or as histograms where the mean and standard error of the mean are shown. Experiments were normalised to the positive control (100 µM etoposide, no post-treatment), which was set at 100%. This adaption of the trapped in agarose DNA immunostaining assay was validated by showing that incubation with proteinase K resulted in a significant drop in the topoisomerase II immunofluorescence signal. With proteinase K the reduction for topoisomerase IIα was 68%±0.5, whilst for topoisomerase IIβ the reduction was 87%±0.3. Proteinase K did not remove DNA from the slides as determined by Hoechst fluorescence (Fig. 1B,C). Incubation with 200 units of purified mung bean nuclease removed DNA but not topoisomerase II protein (Fig. 1B,C).
Fig. 1. Use of TARDIS to assay activities capable of removing topoisomerase II covalent DNA complexes from genomic DNA.

(A) Scheme of assay method. The TARDIS assay allows quantitative assessment of topoisomerase II-DNA covalent complexes that are formed in vivo on genomic DNA (Willmore et al., 1998; Cowell et al., 2011a). Cells treated with topoisomerase poison (in this case 100 µM etoposide) are embedded in agarose and spread onto a microscope slide. After salt/detergent extraction of soluble components, covalently bound topoisomerase II remaining in the embedded genomic DNA is detected by immunofluorescence. To assay for components capable of removing topoisomerase II covalent DNA complexes, slides are incubated with extracts or purified proteins, before antibody probing and immunofluorescence (see Materials and Methods). (B,C) Validation of assay. K562 cells were either untreated (first column) or treated with etoposide (100 µM, 2 hr, columns 2–6). After extraction slides were either untreated or treated as shown with mung bean nuclease (MB) or proteinase K (PK) in the relevant buffer or with buffer alone, prior to immunofluorescence. Mean values of integrated fluorescence per nucleus were determined and the mean of all the cells analysed is shown ± standard error of the mean. All treatments were normalised to the mean values obtained for cells treated with 100 µM etoposide alone. Left graph, Hoechst fluorescence for DNA content; right graph, FITC fluorescence for remaining topoisomerase II. *** = p value <0.0001.
MRE11 immunoprecipitates remove topoisomerase II adducts in vitro
Immunoprecipitation was used to obtain non-recombinant, native human MRE11. MRE11 was immunoprecipitated from whole cell extracts of K562 leukaemia cells. The presence of MRE11 in the immunoprecipitate was confirmed by Western blotting (Fig. 2A) and mass spectrometry (supplementary material Fig. S1). Mass spectrometry indicated the immunoprecipitate also contained RAD50, but NBS1, CTIP, EXO1, DNA2, TDP1 or TDP2 were not detectable by mass spectrometry. Slides containing genomic DNA bearing topoisomerase II adducts derived from K562 cells treated with 100 µM etoposide were incubated with MRE11 immunoprecipitates from K562 cells for 90 minutes at 37°C. The slides were then washed and topoisomerase II adducts visualised and quantified by immunofluorescence. Topoisomerase IIα adducts were partially removed, with 45% reduction in FITC signal, in contrast an increase in topoisomerase IIβ FITC signal was seen (Fig. 2). As a control, MRE11 was immunodepleted from the MRE11 immunoprecipitate (Costanzo et al., 2004); following MRE11 immunodepletion the topoisomerase IIα adduct removal activity was reduced to only 9% removal. Other controls included immunoprecipitation with no antisera or whole IgG. Neither of these control immunoprecipitates removed as much topoisomerase IIα as the MRE11 immunoprecipitate (Fig. 2). In ten replicate experiments topoisomerase IIα adducts were partially removed by MRE11 immunoprecipitates from K562 cells, but topoisomerase IIβ was not removed by the same MRE11 immunoprecipitates, indeed the signal increased. The MRE11 immunoprecipitate reduced the Hoechst signal (supplementary material Fig. S2) as did the IgG control immunoprecipitate. The adduct removal activity was dependent upon divalent metal ions as 20 mM EDTA abolished the topoisomerase II adduct reduction by the MRE11 immunoprecipitate (data not shown). The simplest explanation of these results is that the immunoprecipitated MRE11 is responsible for the TOP2A adduct removal; however, we cannot totally exclude the possibility that the MRE11 immunoprecipitate contains a co precipitating metal ion dependent activity responsible for removing topoisomerase IIα, therefore we looked at purified recombinant MRE11.
Fig. 2. Immunopurified MRE11 removes topoisomerase IIα covalent complexes from genomic DNA.
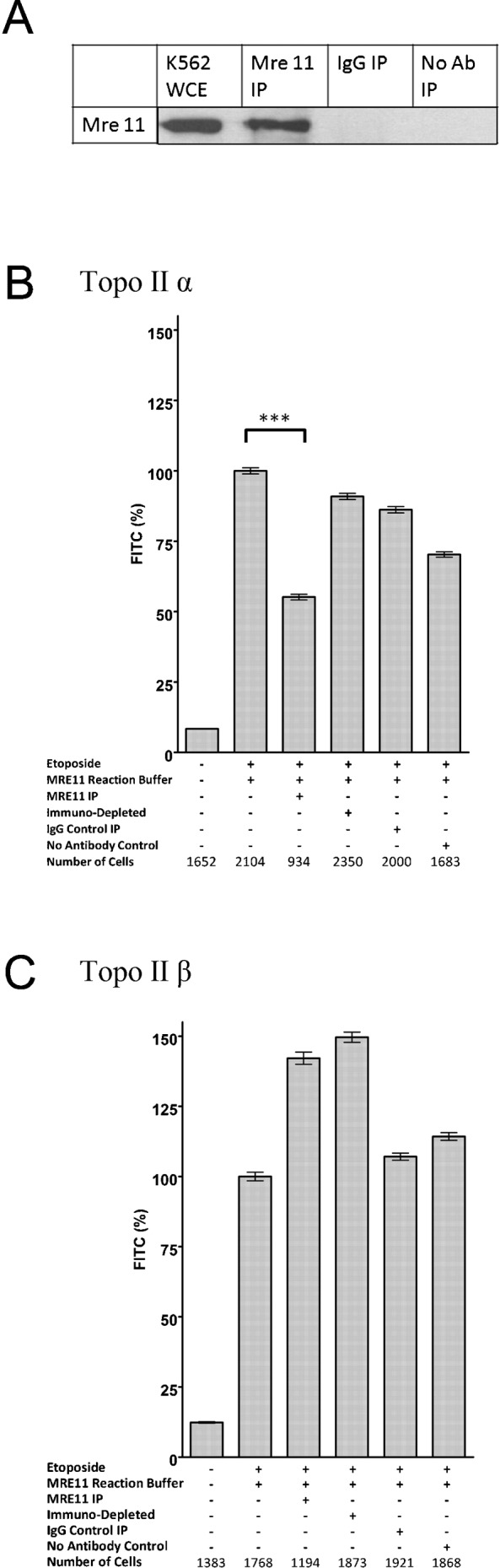
(A) Western blot probing K562 whole cell extract, K562 MRE11 immunoprecipitate and IgG and no antibody control immunoprecipitates for MRE11 (B,C) K562 cells were treated with 100 µM etoposide for two hours prior to embedding in agarose on microscope slides. The slides were incubated with various immunoprecipitates, the fluorescence levels for the topoisomerase II covalent complexes remaining after the incubations were measured, the fluorescence value for each nucleus was determined and normalised to the mean of the positive control incubated in buffer (100%). These are shown as percentage of FITC signal remaining on a slide after incubation with MRE11 IP = eluted components from MRE11 IP; Depl MRE11 IP = immunodepleted MRE 11 IP eluate, Ig cont IP = IgG control IP, No Ab cont IP = control IP without antibody. *** = p value <0.0001.
Recombinant MRE11 removes topoisomerase IIα adducts in vitro
To determine whether recombinant MRE11 could remove etoposide stabilised topoisomerase II adducts from genomic DNA in vitro we incubated slides bearing topoisomerase II adducts on genomic DNA with recombinant MRE11. Human recombinant MRE11 residues 1–206 encompassing the nuclease domain fused to GST was obtained from Abnova. In addition, TmMre11 from Thermotoga maritima was analysed. Wild type and three mutated forms of TmMre11 were used, one nuclease dead form TmMre11 H94S and two partially active forms TmMre11 H61S and TmMre11 H180S (Das et al., 2010).
A commassie stained gel, an in gel nuclease and Western of the truncated MRE11 from Abnova is shown in Fig. 3A. There was no evidence of any contaminating nucleases. Recombinant human truncated MRE11 removed 49% of non-recombinant human etoposide-stabilised topoisomerase IIα adducts from genomic DNA in vitro (Fig. 3B), but only 7% of the human topoisomerase IIβ adducts (Fig. 3C). Inclusion of mirin, an inhibitor of the nuclease activity of MRE11 (Dupré et al., 2008; Garner et al., 2009) significantly reduced the adduct removal (Fig. 3B,C). Addition of EDTA abolished adduct removal supporting the idea that the adduct removal is via a cation dependent nuclease (supplementary material Fig. S3). The recombinant truncated MRE11 did not remove DNA from the slides, as judged by Hoechst fluorescence (Fig. 3D,E). This truncated MRE11 was unable to remove 5′phosphotyrosine from an oligonucleotide, indicating that it is not acting as a tyrosyl phosphodiesterase (data not shown).
Fig. 3. Purified recombinant MRE11 removes topoisomerase IIα covalent complexes from genomic DNA.

(A). Coommassie, in-gel nuclease and Western analysis of recombinant MRE11. (B–E) K562 cells were treated with 100 µM etoposide for two hours. Slides bearing agarose-embedded cells (1–2×106 cells in 100 µl 1% agarose in PBS spread across the slide surface) were incubated with MRE11 buffer or 1 µg MRE11 in MRE11 buffer in the presence or absence of the MRE11 nuclease inhibitor mirin and quantitative immunofluorescence was carried out for topoisomerase IIα or -β. (B,C) The mean FITC fluorescence for each nucleus was normalised to the 100 µM etoposide positive control, and the mean±SEM are shown. *** = p value, 0.0001, * = p value <0.05. (D,E) The mean hoechst fluorescence for each nucleus was normalised to the positive control, and the mean±SEM are shown.
Wild type recombinant Mre11 from Thermotoga maritima, and three different mutated forms of TmMre11 protein were also studied (Fig. 4A) (Das et al., 2010). The three mutated proteins were TmMre11 H94S an endonuclease dead protein and TmMre11 H61S and H180S mutations that result in partial loss of function (Fig. 4B), consistent with analogous mutations in Pyrococcus furiosus and fission yeast Mre11 (Williams et al., 2008; Das et al., 2010). These proteins were applied to slides bearing etoposide stabilised covalent DNA-topoisomerase II adducts (Fig. 4). Like human MRE11, wild type TmMre11 removed a proportion of human topoisomerase IIα adducts from genomic DNA and the TmMre11 H180S protein also removed some topoisomerase IIα but less than wild type TmMre11. None of the four variants of TmMre11 removed human topoisomerase IIβ etoposide stabilised adducts under these conditions (Fig. 4D).
Fig. 4. Purified recombinant MRE11 from Thermotoga maritima removes topoisomerase IIα covalent complexes from genomic DNA.

Purified recombinant MRE11 from Thermotoga maritima (A) SDS-PAGE of TmMre11, TmMre11H94S, TmMre11H61S or TmMre11H180S proteins. (B) Endonuclease assays were performed with single-stranded bacteriophage φX174 DNA as a substrate with TmMre11, TmMre11H94S, TmMre11H61S or TmMre11H180S. K562 cells were treated with 100 µM etoposide for two hours prior to being embedded in agarose on microscope slides. Slides bearing agarose-embedded cells (1–2×106 cells in 100 µl 1% agarose in PBS spread across the slide surface) were incubated with MRE11 buffer (positive control) or 1 µg TmMre11, TmMre11H94S, TmMre11H61S or TmMre11H180S. Quantitative immunofluorescence was carried out for topoisomerase IIα (C) or -β (D). The mean fluorescence for each nucleus was normalised to the positive control, and the mean±SEM are shown. *** = p value, 0.0001.
A-TLD cells
As a human cell line model for the role of MRE11, we used A-TLD fibroblasts derived from a 20 year old male patient with Ataxia-Telangiectasia-Like Disorder 2 now referred to as A-TLD(S). These cells are homozygous for an MRE11 allele where codon 633 is altered to a stop codon, resulting in a truncated MRE11 protein with a molecular weight of 72 kDa rather than full-length, wild type MRE11 protein with a molecular weight of 81 kDa. The truncated protein retains a functional nuclease domain, but its cellular distribution is different to that of full-length MRE11, as it does not form a functional MRN complex (supplementary material Fig. S4) (Uziel et al., 2003; Stewart et al., 1999). In addition to immortalised A-TLD cells (A-TLD hTERT) (Uziel et al., 2003) we employed three isogenic variants A-TLD wtMRE11, A-TLD mre3 and A-TLD GFP which had been transduced with retroviruses expressing wild-type MRE11, MRE11-3 or GFP respectively (Fig. 5A; supplementary material Fig. S4). The mre3 protein is nuclease deficient due to amino acid changes in the phosphodiesterase domain III at positions 129 and 130 (Arthur et al., 2004). Western blotting confirmed that the control cells did not express full-length MRE11 and that the A-TLD cells lines transduced with the full-length MRE11 or mre3 contained a full-length MRE11 protein in addition to the truncated protein (Fig. 5B). The full-length nuclease-deficient protein has previously been shown to adopt the correct sub-cellular localisation pattern (Stewart et al., 1999; Uziel et al., 2003; Arthur et al., 2004). We confirmed by immunofluorescence that the truncated MRE11 in the A-TLD hTERT cells has a largely cytoplasmic distribution, in contrast to the nuclear distribution of the wild type MRE11 in the transduced cells (supplementary material Fig. S4). Although the nuclease activity is abrogated in the mre3 protein, a heterodimer with the nuclease-functional 72 kDa protein would produce a functional MRN complex with nuclease activity, albeit half that of a homodimer of full length wild type protein. All these A-TLD cells grew very slowly in culture.
Fig. 5. Functional MRE11 is required to maintain low topoisomerase II covalent complex levels.

(A) Schematic of the MRE11 variants expressed in the respective A-TLD cell lines. (B) Western blot showing the full length MRE11 in K562 whole cell extracts, full length and 72 kDa MRE11 in A-TLDmre-3 whole cell extract and only the truncated 72 KDa MRE11 in A-TLD whole cell extracts. (C) K562 cells were treated with 100 µM etoposide or vehicle alone. Cells were prepared for TARDIS analysis as described in Fig. 1 and slides were treated with IP eluates from cells expressing the MRE11 variants shown (columns 3–5 of each graph). Topoisomerase II immunofluorescence was measured and mean values for each nucleus were normalised to the mean signal obtained with 100 µM etoposide with no IP treatment, the mean of the cell means is shown ±SEM. (D,E) Topoisomerase IIα complex levels were determined by TARDIS assay in A-TLD (s) cells. The levels of complexes in the cells were determined in the absence and presence of etoposide. The mean level of complexes as determined by FITC fluorescence, with 100 µM etoposide was set at 100% for each cell line. (D). The untreated control levels are shown for each of the 3 A-TLD cell lines. For comparison the levels in untreated K562 cells are also shown as a percentage of the signal obtained in cells treated with 100 µM etoposide. (E) Change in topoisomerase IIα complex levels in cells, indicative of the rate of removal of the complexes are shown for K562, A-TLDwtMRE11, A-TLDmre-3 and A-TLD cells following removal from medium containing etoposide. *** = p value, 0.0001.
MRE11 immunoprecipitates were prepared from three A-TLD cell lines, A-TLD-GFP, A-TLD-wtMRE11 and A-TLD-mre3 (Fig. 5C). To analyse whether these immunoprecipitates were able to remove drug stabilised topoisomerase II adducts from DNA in vitro, we used the adapted TARDIS assay described previously. Incubation with MRE11 immunoprecipitates from any of the three A-TLD cell types resulted in significantly reduced topoisomerase IIα (P≤0.0001) but no reduction in topoisomerase IIβ adduct levels, similar to the results obtained with K562 immunoprecipitates (Fig. 2). As expected the MRE11 immunoprecipitate from the A-TLDwtMRE11 reduced the etoposide stabilised topoisomerase IIα adduct levels to a greater extent than those from the A-TLDmre3 or A-TLD cell lines. Nevertheless, substantial adduct removal was observed with immunoprecipitates from each A-TLD cell line, including A-TLD-GFP which expresses no full-length MRE11. However, the immunoprecipitations were carried out from whole cell lysates, and thus the cytoplasmic nuclease proficient truncated MRE11 present in all three A-TLD cell lines used will be captured by the immunoprecipitation protocol (supplementary material Fig. S4).
A-TLD cells have high background levels of topoisomerase II complexes
Topoisomerase IIα complex levels within A-TLD cells were analysed to determine whether dysfunctional cellular MRE11 affects the level of topoisomerase IIα complexes within cells. In preliminary experiments we noticed that the basal level of topoisomerase IIα adducts in A-TLD cells was very high. Subsequently, A-TLD cells were treated with etoposide or vehicle and the topoisomerase II complex levels within the A-TLD cells were visualised using the TARDIS assay. A number of replicate experiments were performed for A-TLD wt MRE11 (n = 6), A-TLD (n = 9) and A-TLD mre3 (n = 3) cells. In each case fluorescence values were normalised, setting the treatment with 100 µM etoposide for 2 hours as 100%. The level of topoisomerase IIα complexes in the absence of etoposide were very high in the A-TLD cells lacking wild type MRE11 (Fig. 5D), whilst the A-TLDwtMRE11 cells contained significantly lower levels of complexes.
We have previously noted that following etoposide treatment, the initial high level of topoisomerase IIα DNA covalent complexes that are detected by TARDIS falls rapidly when drug is removed (Errington et al., 2004). This is due either to processing of the complexes and repair or completion of the reaction cycle and resolution of the complexes following reversal of topoisomerase-etoposide interaction. We measured topoisomerase IIα complex levels in A-TLD cells at different time points following treatment with and removal of etoposide (Fig. 5E). In the A-TLDwtMRE11 cells, which contain functional MRE11 the topoisomerase II complex levels reduced as did those in K562 cells. The level of complexes in the A-TLDmre3 decreased but more slowly. It should be borne in mind that the A-TLDmre3 cells also express truncated nuclease proficient MRE11 with which it can heterodimerize (Fig. 5A), reconstituting a functional MRE11 activity. In contrast, the complexes in the A-TLD cells remained high, consistent with the very high levels of topoisomerase complexes in these cells prior to etoposide exposure. These data imply that cells lacking functional MRE11 accumulate topoisomerase-DNA covalent complexes in the absence of drug, either through an increased rate of production or decreased rate of resolution.
Inhibition of MRE11 nuclease increases topoisomerase II complex levels
To address whether it is a lack of nuclease activity that leads to increased topoisomerase complex levels in cells we treated K562 cells with mirin, a small molecule inhibitor of MRE11 nuclease activity. Inhibiting MRE11 nuclease activity for 24 hours significantly increased the background levels of topoisomerase IIα complexes and topoisomerase IIβ complexes (Fig. 6).
Fig. 6. The MRE11 nuclease inhibitor mirin increases topoisomerase II DNA covalent complexes.
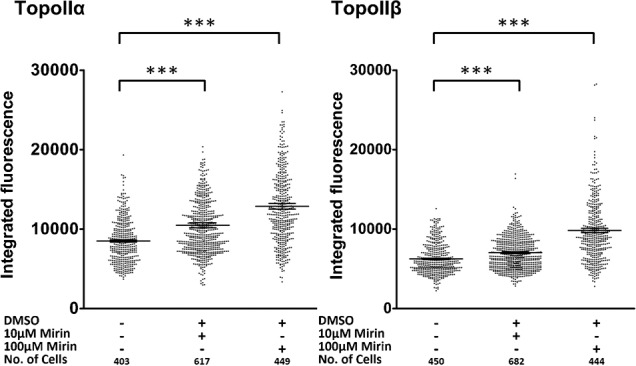
K562 cells were exposed to mirin for 24 hours at the concentrations shown and topoisomerase II complexes were quantified by TARDIS. Mann-Whitney significance values are shown. *** = p value, 0.0001.
Inhibition of replication increases topoisomerase II complex levels
MRE11 has been reported to be important for the restart of stalled replication forks in association with PARP (Robison et al., 2004; Bryant et al., 2009) and notably, inhibition of PARP increased the topoisomerase II complex levels in the absence of topoisomerase poison, as did inhibition of ATM in the TARDIS assay (data not shown). This led us to consider whether the elevated cellular topoisomerase II complex levels associated with MRE11 deficiency in cells is related to replication stress. To test if stalled replication forks cause the increase in topoisomerase II complexes, we treated cells with hydroxyurea (HU), which leads to replication stress through depletion of nucleotide pools. HU affects both initiation and elongation in replication and average rates of replication are reduced. HU generates stalled replication forks, which on prolonged exposure convert to DSBs, also extensive single-stranded regions accumulate in the presence of HU. Topoisomerase IIα and -β covalent complex levels increased following HU treatment (Fig. 7). Not all forms of replication stress increased topoisomerase II complex levels as the topoisomerase I poison camptothecin, which generates replication stress through collision of replisomes with CPT-stabilised topoisomerase I complexes, blocking replication fork progression (Furuta et al., 2003) did not lead to elevation of topoisomerase II complex levels.
Fig. 7. Hydroxyurea exposure leads to elevated topoisomerase II DNA covalent complex levels.
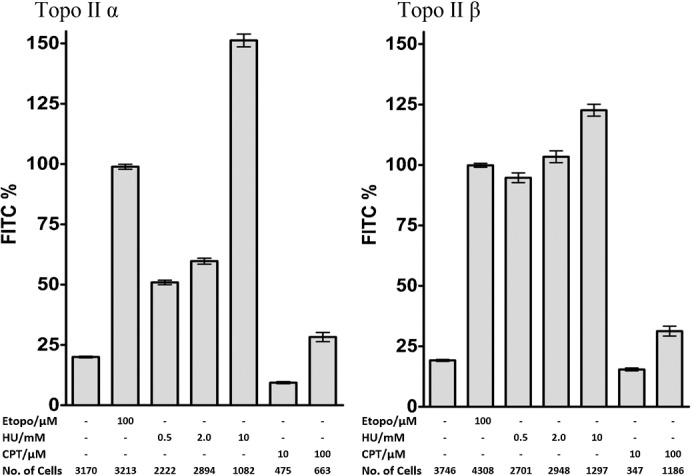
K562 cells were treated with etoposide for 2 hours (Etop), hydroxyurea for 18 hours (HU) or camptothecin for 18 hours (CPT). Topoisomerase II levels were measured by TARDIS. Mean fluorescence signals were normalised to the mean level obtained with 100 µM etoposide.
Discussion
MRE11 immunoprecipitates containing non-recombinant MRE11 removed etoposide-stabilised topoisomerase IIα protein adducts from genomic DNA in vitro, immunodepletion of immunoprecipitates reduced adduct removal. Thus, MRE11 or something that co-precipitates with it is able to remove etoposide stabilised topoisomerase IIα adducts from genomic DNA (Fig. 2). Addition of EDTA abolished this activity, indicating the activity was dependent upon divalent cations. The same immunoprecipitates and in vitro incubation conditions did not decrease the topoisomerase IIβ complexes, in fact the immunofluorescent signal increased. This may be due to increased accessibility to the anti-topoisomerase IIβ antisera following detergent and salt treatment of heterochromatin (Cowell et al., 2011b). Etoposide stabilised topoisomerase II adducts were also removed from genomic DNA in vitro by recombinant MRE11, 49% of the topoisomerase IIα adducts were removed (P>0.0001), but only 7% of the topoisomerase IIβ complexes (p = 0.0302). This activity was again abolished by EDTA. The MRE11 nuclease inhibitor mirin prevented MRE11-mediated in vitro topoisomerase IIα adduct removal, confirming the removal was via MRE11 nuclease activity (Fig. 3). In addition, TmMre11 removed etoposide stabilised topoisomerase IIα adducts (Fig. 4). These data strongly support a role for MRE11 nuclease activity in removing etoposide stabilised topoisomerase IIα adducts by cleaving the DNA to remove the protein adduct and a fragment of DNA, analogous to MRE11's role in removing SPO11 (Lange et al., 2011).
In contrast to the in vitro situation where recombinant MRE11 protein removed topoisomerase IIα etoposide stabilised complexes much more efficiently than topoisomerase IIβ (49% removal compared to 7% removal), lack of MRE11 activity within cells led to comparable increases in topoisomerase IIα and β complexes in the absence of etoposide. A-TLD cells, which have a truncated MRE11 that does not form an MRN complex, exhibited very high basal levels of topoisomerase II complexes. These high basal levels of complexes were substantially reduced when A-TLD cells were transduced with wild type MRE11 (Fig. 5D). Consistent with this, inhibition of MRE11 in K562 cells by inclusion of mirin in the culture medium substantially increased the normally low basal level of topoisomerase II complexes detected by TARDIS assay in this cell line, 100 µM mirin produced a 50% increase in the mean FITC signal for both isoforms compared to the untreated cells (Fig. 6). These data strongly suggest that MRE11 is in some way necessary for homeostasis of topoisomerase II-DNA complexes that arise as part of normal cellular metabolism, in the absence of drugs. The source of these complexes in the absence of etoposide is not yet clear. Abasic sites represent one candidate trigger for topoisomerase II complexes resulting from normal cellular metabolism (Wilstermann and Osheroff, 2001). Complexes arising during cellular metabolism may represent a substrate for MRE11-dependent processing, and thus lack of MRE11 activity would raise basal topoisomerase II complex levels in A-TLD cells (Fig. 5) and after mirin treatment of K562 cells (Fig. 6).
Alternatively replication stress and stalled replication forks may trigger generation of abortive topoisomerase II complexes. MRE11 nuclease activity is required for replication fork restart in conjunction with PARP (Robison et al., 2004; Bryant et al., 2009) and MRE11 co-localizes with proliferating cell nuclear antigen (PCNA) throughout S phase and appears to be loaded onto chromatin at the replication fork. Also MRE11 is localised to single stranded regions in hydroxyurea treated cells (Mirzoeva and Petrini, 2003), and can degrade stalled forks in an activity blocked by mirin (Schlacher et al., 2011). Topoisomerase II also co-localizes with PCNA (Niimi et al., 2001; Gilljam et al., 2009) and is associated with nascent DNA (Qiu et al., 1996). In support of the idea that stalled replication forks may trigger generation of abortive topoisomerase II complexes, we found that treatment of K562 cells with HU generated a topoisomerase IIα and β signal comparable to or greater than that obtained when cells were treated with etoposide (Fig. 7).
5′-phosphotyrosyl-linked topoisomerase protein arising from enzyme poisoning or through spontaneous abortive cleavage reactions is a potentially lethal obstacle to DNA repair and to cellular processes including transcription and replication. Evidence exists for more than one pathway to remove such complexes. Support for the involvement of MRE11 together with CtIP in processing topoisomerase II DNA complexes derives from genetic studies in yeast (Neale et al., 2005; Hartsuiker et al., 2009a) and in mammalian cells (Nakamura et al., 2010; Quennet et al., 2011). Here we show for the first time that MRE11 can remove topoisomerase IIα complexes from DNA in vitro, and that this is dependent on MRE11 nuclease activity (Figs 3, 4). Since genetic or inhibitor induced MRE11 nuclease deficiency leads to elevated basal topoisomerase II-DNA complexes in cultured cells (Figs 5, 6), we suggest that an MRE11 dependent pathway is involved in removing topoisomerase II complexes that arise through cellular metabolism.
Materials and Methods
Cell culture
The K562 cell line was derived from a patient with chronic myelogenous leukemia (CML) in terminal blast phase and its metabolism is therefore similar to that of AML blasts. Cells were maintained as a suspension culture in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum, penicillin (50 U/ml), and streptomycin (50 µg/ml) and were grown at concentrations between 1×105 and 1×106/mL and were free of mycoplasma contamination. The doubling time of the cells was approximately 24 hours (K562). A-TLD cells were kindly supplied by Yosef Shiloh and Yaniv Lerenthal, these cells were fibroblasts derived from a 20 year old male patient with Ataxia-Telangiectasia-Like Disorder 2 now referred to as A-TLD(S). They are homozygous for a truncated version of MRE11 72 kDa (residue 633 altered to a stop codon) rather than full-length 81 kDa MRE11. These cells are hypomorphic, express truncated protein and they are genetically unstable. The lifespan of primary A-TLD fibroblasts is short, so primary A-TLD(S) cells were immortalised by ectopic expression of the catalytic subunit of human telomerase (hTERT). These immortalised cells were then transduced with a retroviral vector expressing recombinant wild type MRE11. In the transduced cells the MRE11 co-localised with RAD50 and NBS1, suggesting reassembly of the nuclear MRN complex. A-TLD mre3 cells were transduced with nuclease deficient version of the MRE11 protein that still formed an MRN complex, but the nuclease activity was completely abrogated. Cell culture reagents were obtained from Invitrogen (Paisley, UK).
Drugs
Etoposide was dissolved in methanol as a 2 mM stock and was purchased from Sigma (Poole, Dorset, UK). Mirin was dissolved in DMSO, it was supplied by Alan Eastman or Biomol. Hydroxyurea was purchased from Sigma (Poole, Dorset, UK).
Recombinant MRE11
Human MRE11 amino acids 1–206 fused to GST was purchased from Abnova (H00004361-P01) (Taipei, Taiwan).
Wild type and three mutated forms of TmMre11 protein from eubacteria Thermotoga maritima was produced as detailed in (Das et al., 2010). In TmMre11 H94S is an endonuclease dead protein, whilst TmMre11 H61S and H180S are partially active (Das et al., 2010).
Antibodies
The anti-topoisomerase II polyclonal antibodies used in these studies were raised in rabbits. Antibody 18511 was raised to recombinant human topoisomerase IIα and antibody 18513 to a recombinant carboxyl-terminal fragment of human topoisomerase IIβ. Western blots demonstrated that 18511 detected the α isoform specifically and 18513 detected the β isoform specifically (Cowell et al., 1998). In the TARDIS assays, 18511(α) was used at a 1:50 dilution and 18513 (β) at 1:150. The anti-rabbit FITC-conjugated second antibody (F(ab′)2 fragment (Sigma, Poole, Dorset, UK) was used at 1:100 dilution. MRE11 antibody (ab397) (Abcam) was a rabbit polyclonal, raised to human MRE11 (full length) fusion protein.
Immunoprecipitations
Cell pellets containing 1.6×108 cells per ml were resuspended in RIPA buffer (phosphate-buffered saline (PBS) with 1% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) sodium dodecyl sulphate (SDS) and 10% DNase 1 in 0.15 M NaCl), at 4°C. The cell pellet was then centrifuged at 18,000 ×g for 10 minutes and the cell supernatant removed. 100 µl of protein A sepharose beads (GE Healthcare, Little Chalfont, Bucks, UK) was added to the supernatant and incubated for 1 hour. The solution was centrifuged at 18,000 ×g for 10 seconds and supernatant removed. 3 µl of MRE11 antibody ab397 was added to the supernatant and incubated for 2 hours. Next, 600 µl of protein A sepharose beads (GE Healthcare, Little Chalfont, Bucks, UK) were added and incubated for 2 hours. The solution was centrifuged at 18,000 ×g for 10 seconds and the supernatant removed. The beads with bound protein, were washed three times with PBS plus protease inhibitors (2 µg/ml pepstatin A, 2 µg/ml leupeptin, 1 mM phenyl methyl sulphonyl fluoride, 1 mM benzamidine) and 1 mM DTT. The washed beads were stored at −80°C. Proteins were extracted from the beads and dialysed. 0.1 M glycine (pH 3) was added to the samples for five minutes, to displace the protein from the protein A sepharose beads. The solution was centrifuged for 10 seconds and the supernatant removed. 6.9× elution buffer (5 M Tris, 414 mM KCl, 6.9 mM MnCl2 and 1.38% (v/v) Tween 20) plus 0.1% (w/v) DTT and protease inhibitors (2 µg/ml pepstatin A, 2 µg/ml leupeptin, 1 mM phenyl methyl sulphonyl fluoride, 1 mM benzamidine) were added to the supernatant. In-gel nuclease assays showed no detectable contaminating nuclease activity in the Mre11 immunoprecipitate. These immunoprecipitates were used in TARDIS assays as detailed in the results.
TARDIS assay
TARDIS assays were performed as described previously (Willmore et al., 1998; Cowell et al., 2011a). Briefly, images were taken on an Olympus IX81 microscope system with a Hamamatsu Orca-AG camera. Captured microscopy images were analysed by Volocity 64 software (Perkin Elmer). Statistical analysis was carried out using GraphPad Prism software (Cherwell Scientific, Oxford, UK). Statistical analysis (t-test and p-value) utilised the two tailed Mann-Whitney test. The mean fluorescence signal for cells incubated with 100 µM etoposide was used as a positive control in each experiment. The integrated fluorescence signal from individual cells was normalised to the positive control mean. The total number of cells analysed is shown in the figures.
For assays to quantify removal of topoisomerase adducts from genomic DNA, slides bearing agarose-embedded cells were incubated for 90 minutes at 37°C with the immunoprecipitate (extracted from 8.5×107 cells per slide) or purified proteins described in the text or with the following buffers: MB buffer (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM Magnesium acetate, 1 mM DTT, pH 7.9); PK buffer (30 mM Tris-HCl, pH 8.0); MRE11 buffer (25 mM MOPS pH 7.0, 60 mM KCl, 5 mM MnCl2, 2 mM DTT, 0.2% Tween 20).
Western blotting
Western blots were carried out by standard procedures using ECL detection (GE Healthcare).
In-gel nuclease assay
12.5% SDS-PAGE gels were prepared containing 200 µg/ml denatured salmon sperm DNA. MRE11 (1 µg) was run along with a positive control of 5 µg of DNaseI. After electrophoresis gels were left overnight at 4°C in 50 mM Tris-HCl pH 7.5 and 1 mM EDTA. Gels were washed washes in fresh 50 mM Tris-HCl pH 7.5 for 20 minutes at room temperature and then in the same buffer at 37°C. Gels were then incubated in MRE11 buffer containing ethidium bromide (25 mM MOPS pH 7.0, 60 mM KCl, 0.2% Tween 20, 5 mM MnCl2, 2 mM DTT, 0.5 µg/ml EtBr) for 4 hours at 37°C and then visualized using a UV transilluminator.
Endonuclease assays
Endonuclease assays were performed with single-stranded bacteriophage φX174 DNA as a substrate as detailed in (Das et al., 2010).
Immunofluorescence (in support of supplementary material)
Immunofluorescence was carried out essentially as described by Cowell et al., 2011b. MRE11 immunofluorescence was carried out using rabbit anti-AB397 (Abcam). Images were collected at 150 µm z-steps using an Olympus IX81 microscope fitted with an Orca AG greyscale camera and 60× NA0.95 oil immersion objective. Extended-focus images are displayed.
Supplementary Material
Acknowledgments
This work was funded by LRF grant 04037 and LLR grant 07038 (K.C.L., K.P., H.C., I.G.C., Z.S., G.H.J., N.J.M., S.J.C. and C.A.A.). The efforts of D.M. and J.A.T. on Mre11 mutant preparations were supported by National Cancer Institute grant CA117638 (J.A.T.).
Footnotes
Competing interests: The authors have no competing interests to declare.
References
- Adachi N., Suzuki H., Iiizumi S., Koyama H. (2003). Hypersensitivity of nonhomologous DNA end-joining mutants to VP-16 and ICRF-193: implications for the repair of topoisomerase II-mediated DNA damage. J. Biol. Chem. 278, 35897–35902 10.1074/jbc.M306500200 [DOI] [PubMed] [Google Scholar]
- Adachi N., Iiizumi S., So S., Koyama H. (2004). Genetic evidence for involvement of two distinct nonhomologous end-joining pathways in repair of topoisomerase II-mediated DNA damage. Biochem. Biophys. Res. Commun. 318, 856–861 10.1016/j.bbrc.2004.04.099 [DOI] [PubMed] [Google Scholar]
- Alchanati I., Teicher C., Cohen G., Shemesh V., Barr H. M., Nakache P., Ben-Avraham D., Idelevich A., Angel I., Livnah N. et al. (2009). The E3 ubiquitin-ligase Bmi1/Ring1A controls the proteasomal degradation of Top2α cleavage complex - a potentially new drug target. PLoS ONE 4, e8104 10.1371/journal.pone.0008104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur L. M., Gustausson K., Hopfner K. P., Carson C. T., Stracker T. H., Karcher A., Felton D., Weitzman M. D., Tainer J., Carney J. P. (2004). Structural and functional analysis of Mre11-3. Nucleic Acids Res. 32, 1886–1893 10.1093/nar/gkh343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayene I. S., Ford L. P., Koch C. J. (2005). Ku protein targeting by Ku70 small interfering RNA enhances human cancer cell response to topoisomerase II inhibitor and γ radiation. Mol. Cancer Ther. 4, 529–536 10.1158/1535-7163.MCT-04-0130 [DOI] [PubMed] [Google Scholar]
- Bigioni M., Zunino F., Tinelli S., Austin C. A., Willmore E., Capranico G. (1996). Position-specific effects of base mismatch on mammalian topoisomerase II DNA cleaving activity. Biochemistry 35, 153–159 10.1021/bi951736p [DOI] [PubMed] [Google Scholar]
- Bryant H. E., Petermann E., Schultz N., Jemth A.-S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., Helleday T. (2009). PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 10.1038/emboj.2009.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buis J., Wu Y., Deng Y., Leddon J., Westfield G., Eckersdorff M., Sekiguchi J. M., Chang S., Ferguson D. O. (2008). Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 135, 85–96 10.1016/j.cell.2008.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott K., Banks G., Jeggo P. (1990). DNA double-strand break repair pathways and cellular tolerance to inhibitors of topoisomerase II. Cancer Res. 50, 5778–5783. [PubMed] [Google Scholar]
- Cole F., Keeney S., Jasin M. (2010). Evolutionary conservation of meiotic DSB proteins: more than just Spo11. Genes Dev. 24, 1201–1207 10.1101/gad.1944710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly J. C., Leach D. R. (2004). Repair of DNA covalently linked to protein. Mol. Cell 13, 307–316 10.1016/S1097-2765(04)00056-5 [DOI] [PubMed] [Google Scholar]
- Cortes Ledesma F., El Khamisy S. F., Zuma M. C., Osborn K., Caldecott K. W. (2009). A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461, 674–678 10.1038/nature08444 [DOI] [PubMed] [Google Scholar]
- Costanzo V., Paull T., Gottesman M., Gautier J. (2004). Mre11 assembles linear DNA fragments into DNA damage signaling complexes. PLoS Biol. 2, e110 10.1371/journal.pbio.0020110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell I. G., Willmore E., Chalton D., Marsh K. L., Jazrawi E., Fisher L. M., Austin C. A. (1998). Nuclear distribution of human DNA topoisomerase IIβ: a nuclear targeting signal resides in the 116-residue C-terminal tail. Exp. Cell Res. 243, 232–240 10.1006/excr.1998.4150 [DOI] [PubMed] [Google Scholar]
- Cowell I. G., Tilby M. J., Austin C. A. (2011a). An overview of the visualisation and quantitation of low and high MW DNA adducts using the trapped in agarose DNA immunostaining (TARDIS) assay. Mutagenesis 26, 253–260 10.1093/mutage/geq094 [DOI] [PubMed] [Google Scholar]
- Cowell I. G., Papageorgiou N., Padget K., Watters G. P., Austin C. A. (2011b). Histone deacetylase inhibition redistributes topoisomerase IIβ from heterochromatin to euchromatin. Nucleus 2, 61–71 10.4161/nucl.2.1.14194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amours D., Jackson S. P. (2002). The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 3, 317–327 10.1038/nrm805 [DOI] [PubMed] [Google Scholar]
- Das D., Moiani D., Axelrod H. L., Miller M. D., McMullan D., Jin K. K., Abdubek P., Astakhova T., Burra P., Carlton D. et al. (2010). Crystal structure of the first eubacterial Mre11 nuclease reveals novel features that may discriminate substrates during DNA repair. J. Mol. Biol. 397, 647–663 10.1016/j.jmb.2010.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré A., Boyer-Chatenet L., Sattler R. M., Modi A. P., Lee J. H., Nicolette M. L., Kopelovich L., Jasin M., Baer R., Paull T. T. et al. (2008). A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 4, 119–125 10.1038/nchembio.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington F., Willmore E., Leontiou C., Tilby M. J., Austin C. A. (2004). Differences in the longevity of topo IIα and topo IIβ drug-stabilized cleavable complexes and the relationship to drug sensitivity. Cancer Chemother. Pharmacol. 53, 155–162 10.1007/s00280-003-0701-1 [DOI] [PubMed] [Google Scholar]
- Fan J. R., Peng A. L., Chen H. C., Lo S. C., Huang T. H., Li T. K. (2008). Cellular processing pathways contribute to the activation of etoposide-induced DNA damage responses. DNA Repair (Amst.) 7, 452–463 10.1016/j.dnarep.2007.12.002 [DOI] [PubMed] [Google Scholar]
- Furuta T., Takemura H., Liao Z. Y., Aune G. J., Redon C., Sedelnikova O. A., Pilch D. R., Rogakou E. P., Celeste A., Chen H. T. et al. (2003). Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 278, 20303–20312 10.1074/jbc.M300198200 [DOI] [PubMed] [Google Scholar]
- Garner K. M., Pletnev A. A., Eastman A. (2009). Corrected structure of mirin, a small-molecule inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 5, 129–130, author reply 130 10.1038/nchembio0309-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilljam K. M., Feyzi E., Aas P. A., Sousa M. M., Müller R., Vågbø C. B., Catterall T. C., Liabakk N. B., Slupphaug G., Drabløs F. et al. (2009). Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 186, 645–654 10.1083/jcb.200903138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsuiker E., Neale M. J., Carr A. M. (2009a). Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell 33, 117–123 10.1016/j.molcel.2008.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsuiker E., Mizuno K., Molnar M., Kohli J., Ohta K., Carr A. M. (2009b). Ctp1CtIP and Rad32Mre11 nuclease activity are required for Rec12Spo11 removal, but Rec12Spo11 removal is dispensable for other MRN-dependent meiotic functions. Mol. Cell. Biol. 29, 1671–1681 10.1128/MCB.01182-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong G., Kreuzer K. N. (2003). Endonuclease cleavage of blocked replication forks: An indirect pathway of DNA damage from antitumor drug-topoisomerase complexes. Proc. Natl. Acad. Sci. USA 100, 5046–5051 10.1073/pnas.0835166100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins B. B., Paull T. T. (2008). The P. furiosus mre11/rad50 complex promotes 5′ strand resection at a DNA double-strand break. Cell 135, 250–260 10.1016/j.cell.2008.09.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeggo P. A., Caldecott K., Pidsley S., Banks G. R. (1989). Sensitivity of Chinese hamster ovary mutants defective in DNA double strand break repair to topoisomerase II inhibitors. Cancer Res. 49, 7057–7063. [PubMed] [Google Scholar]
- Kaufmann S. H. (1998). Cell death induced by topoisomerase-targeted drugs: more questions than answers. Biochim. Biophys. Acta 1400, 195–211 10.1016/S0167-4781(98)00136-5 [DOI] [PubMed] [Google Scholar]
- Kingma P. S., Greider C. A., Osheroff N. (1997). Spontaneous DNA lesions poison human topoisomerase IIα and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry 36, 5934–5939 10.1021/bi970507v [DOI] [PubMed] [Google Scholar]
- Lange J., Pan J., Cole F., Thelen M. P., Jasin M., Keeney S. (2011). ATM controls meiotic double-strand-break formation. Nature 479, 237–240 10.1038/nature10508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M., Nitiss J. L. (2004). DNA repair functions that control sensitivity to topoisomerase-targeting drugs. Eukaryot. Cell 3, 82–90 10.1128/EC.3.1.82-90.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y., Desai S. D., Ting C. Y., Hwang J., Liu L. F. (2001). 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 276, 40652–40658 10.1074/jbc.M104009200 [DOI] [PubMed] [Google Scholar]
- Mårtensson S., Nygren J., Osheroff N., Hammarsten O. (2003). Activation of the DNA-dependent protein kinase by drug-induced and radiation-induced DNA strand breaks. Radiat. Res. 160, 291–301 10.1667/0033-7587(2003)160[0291:AOTDPK]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Mirzoeva O. K., Petrini J. H. (2003). DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 1, 207–218. [PubMed] [Google Scholar]
- Morales M., Liu Y., Laiakis E. C., Morgan W. F., Nimer S. D., Petrini J. H. (2008). DNA damage signaling in hematopoietic cells: a role for Mre11 complex repair of topoisomerase lesions. Cancer Res. 68, 2186–2193 10.1158/0008-5472.CAN-07-2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K., Kogame T., Oshiumi H., Shinohara A., Sumitomo Y., Agama K., Pommier Y., Tsutsui K. M., Tsutsui K., Hartsuiker E. et al. (2010). Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 6, e1000828 10.1371/journal.pgen.1000828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale M. J., Pan J., Keeney S. (2005). Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057 10.1038/nature03872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolette M. L., Lee K., Guo Z., Rani M., Chow J. M., Lee S. E., Paull T. T. (2010). Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat. Struct. Mol. Biol. 17, 1478–1485 10.1038/nsmb.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A., Suka N., Harata M., Kikuchi A., Mizuno S. (2001). Co-localization of chicken DNA topoisomerase IIα, but not β, with sites of DNA replication and possible involvement of a C-terminal region of α through its binding to PCNA. Chromosoma 110, 102–114 10.1007/s004120100140 [DOI] [PubMed] [Google Scholar]
- Qiu J., Catapano C. V., Fernandes D. J. (1996). Formation of topoisomerase IIα complexes with nascent DNA is related to VM-26-induced cytotoxicity. Biochemistry 35, 16354–16360 10.1021/bi9619637 [DOI] [PubMed] [Google Scholar]
- Quennet V., Beucher A., Barton O., Takeda S., Löbrich M. (2011). CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucl. Acids Res. 39, 2144–2152 10.1093/nar/gkq1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. A., Strande N., Burkhalter M. D., Strom C., Havener J. M., Hasty P., Ramsden D. A. (2010). Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 464, 1214–1217 10.1038/nature08926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison J. G., Elliott J., Dixon K., Oakley G. G. (2004). Replication protein A and the Mre11·Rad50·Nbs1 complex co-localize and interact at sites of stalled replication forks. J. Biol. Chem. 279, 34802–34810 10.1074/jbc.M404750200 [DOI] [PubMed] [Google Scholar]
- Rossi R., Lidonnici M. R., Soza S., Biamonti G., Montecucco A. (2006). The dispersal of replication proteins after Etoposide treatment requires the cooperation of Nbs1 with the ataxia telangiectasia Rad3-related/Chk1 pathway. Cancer Res. 66, 1675–1683 10.1158/0008-5472.CAN-05-2741 [DOI] [PubMed] [Google Scholar]
- Sabourin M., Nitiss J. L., Nitiss K. C., Tatebayashi K., Ikeda H., Osheroff N. (2003). Yeast recombination pathways triggered by topoisomerase II-mediated DNA breaks. Nucleic Acids Res. 31, 4373–4384 10.1093/nar/gkg497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 10.1016/j.cell.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G. S., Maser R. S., Stankovic T., Bressan D. A., Kaplan M. I., Jaspers N. G., Raams A., Byrd P. J., Petrini J. H., Taylor A. M. (1999). The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99, 577–587 10.1016/S0092-8674(00)81547-0 [DOI] [PubMed] [Google Scholar]
- Stracker T. H., Carson C. T., Weitzman M. D. (2002). Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418, 348–352 10.1038/nature00863 [DOI] [PubMed] [Google Scholar]
- Sunter N. J., Cowell I. G., Willmore E., Watters G. P., Austin C. A. (2010). Role of topoisomerase IIβ in DNA damage response following IR and etoposide. J. Nucleic Acids 2010, 710589 10.4061/2010/710589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh Y. (2003). Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22, 5612–5621 10.1093/emboj/cdg541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. S., Moncalian G., Williams J. S., Yamada Y., Limbo O., Shin D. S., Groocock L. M., Cahill D., Hitomi C., Guenther G. et al. (2008). Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135, 97–109 10.1016/j.cell.2008.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmore E., Frank A. J., Padget K., Tilby M. J., Austin C. A. (1998). Etoposide targets topoisomerase IIα and IIβ in leukemic cells: isoform-specific cleavable complexes visualized and quantified in situ by a novel immunofluorescence technique. Mol. Pharmacol. 54, 78–85. [DOI] [PubMed] [Google Scholar]
- Willmore E., de Caux S., Sunter N. J., Tilby M. J., Jackson G. H., Austin C. A., Durkacz B. W. (2004). A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 103, 4659–4665 10.1182/blood-2003-07-2527 [DOI] [PubMed] [Google Scholar]
- Wilstermann A. M., Osheroff N. (2001). Base excision repair intermediates as topoisomerase II poisons. J. Biol. Chem. 276, 46290–46296 10.1074/jbc.M105733200 [DOI] [PubMed] [Google Scholar]
- Zeng Z., Cortés-Ledesma F., El Khamisy S. F., Caldecott K. W. (2011). TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 286, 403–409 10.1074/jbc.M110.181016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A., Lyu Y. L., Lin C.-P., Zhou N., Azarova A. M., Wood L. M., Liu L. F. (2006). A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J. Biol. Chem. 281, 35997–36003 10.1074/jbc.M604149200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}