

Summary
The respiratory (tracheal) system of the Drosophila melanogaster larva is an intricate branched network of air-filled tubes. Its developmental logic is similar in some ways to that of the vertebrate vascular system. We previously described a unique embryonic tracheal tubulogenesis phenotype caused by loss of both of the Type III receptor tyrosine phosphatases (RPTPs), Ptp4E and Ptp10D. In Ptp4E Ptp10D double mutants, the linear tubes in unicellular and terminal tracheal branches are converted into bubble-like cysts that incorporate apical cell surface markers. This tube geometry phenotype is modulated by changes in the activity or expression of the epidermal growth factor receptor (Egfr) tyrosine kinase (TK). Ptp10D physically interacts with Egfr. Here we demonstrate that the Ptp4E Ptp10D phenotype is the consequence of the loss of negative regulation by the RPTPs of three growth factor receptor TKs: Egfr, Breathless and Pvr. Reducing the activity of any of the three kinases by tracheal expression of dominant-negative mutants suppresses cyst formation. By competing dominant-negative and constitutively active kinase mutants against each other, we show that the three RTKs have partially interchangeable activities, so that increasing the activity of one kinase can compensate for the effects of reducing the activity of another. This implies that SH2-domain downstream effectors that are required for the phenotype are likely to be able to interact with phosphotyrosine sites on all three receptor TKs. We also show that the phenotype involves increases in signaling through the MAP kinase and Rho GTPase pathways.
Keywords: DEP-1, PTPRJ, Epistasis, Tumor suppressor, Tubulogenesis
Introduction
Receptor tyrosine kinase (RTK) signaling controls cell proliferation and orchestrates the assembly of cells into organs during embryonic development. Most receptors for mitogenic growth factors are RTKs. The activities of these RTKs are tightly regulated in adult animals, so as to permit only those cell divisions that are necessary for organ maintenance. Cancer cells, which escape from normal growth control mechanisms, often display dysregulated growth factor receptor TK activity due to mutation or overexpression of RTK genes.
RTKs are usually dimerized by ligand binding, which brings the kinase domains into proximity and allows transphosphorylation of each cytoplasmic domain by the kinase of its partner. Transphosphorylation creates docking sites for phosphotyrosine (PTyr)-binding SH2-domain effectors, and can also increase kinase activity through phosphorylation-induced conformational changes. In some cases, binding to the RTK serves only to relocalize an effector to the plasma membrane, while in other cases the RTK phosphorylates the bound effector and alters its activity. RTKs interact with many different effectors and transmit signals through many different pathways.
RTK signaling is regulated by protein tyrosine phosphatases (PTPs), which dephosphorylate the RTKs themselves as well as their substrates (reviewed by Tonks, 2006). PTP activity is normally in excess over TK activity, and chemical inhibition of PTPs causes dramatic increases in cellular PTyr content, far beyond those that can be produced by overexpressing any TK (for an example, see Bugga et al., 2009). It is generally thought that the enzymes that maintain PTyr at low levels in the cell are cytoplasmic PTPs, while receptor PTPs (RPTPs), which are transmembrane proteins, regulate signaling by specific membrane-associated TKs.
There are 19 RPTPs encoded in the human genome, four of which are classified as Type III. Type III RPTPs are characterized by extracellular (XC) domains composed of chains of fibronectin type III repeats attached to single cytoplasmic PTP domains. Biochemical and genetic studies have suggested that Type III RPTPs are regulators of RTK activity, although they also have other targets (reviewed by Matozaki et al., 2010). ‘Substrate-trapping’ mutants (Flint et al., 1997) of Type III RPTPs, which bind to target PTyr sites but cannot complete the dephosphorylation reaction, form stable complexes with RTKs, suggesting that RTKs are direct targets for RPTP enzymatic activity.
One Type III RPTP, PTPRJ (DEP-1/CD148/R-PTP-eta) regulates a variety of growth factor receptors, including the epidermal growth factor receptor (EGFR), the platelet-derived growth factor receptor (PDGFR), the vascular-endothelial growth factor receptor (VEGFR), and the colony stimulating factor-1 receptor (CSF1R or MET), among others (Arora et al., 2011; Berset et al., 2005; Chabot et al., 2009; Lampugnani et al., 2003; Kappert et al., 2007; Palka et al., 2003; Tarcic et al., 2009). Negative regulation of these and other PTPRJ targets may be important for tumor suppression, because mouse PTPRJ corresponds to the Suppressor of colon cancer1 (Scc1) gene (Ruivenkamp et al., 2002). Two other Type III RPTPs, PTPRB (VE-PTP) and PTPRO, negatively regulate the angiopoietin receptor Tie-2 and the NT-3 receptor TrkC, respectively (Hower et al., 2009; Winderlich et al., 2009).
Drosophila has provided a valuable system in which to investigate RPTP function, because its genome encodes only six RPTPs, and three of these (Lar, Ptp69D, Ptp52F) have single-gene loss-of-function (LOF) phenotypes affecting axon guidance and synaptogenesis (reviewed by Johnson and Van Vactor, 2003). There are two Type III RPTPs in Drosophila, Ptp4E and Ptp10D, which are closely related, being the products of a recent gene duplication (Jeon et al., 2008). Ptp4E is widely expressed during embryogenesis (Jeon et al., 2008; Oon et al., 1993), while Ptp10D is restricted to central nervous system axons and the tracheal system (Jeon and Zinn, 2009; Tian et al., 1991; Yang et al., 1991). Ptp4E and Ptp10D single mutants are viable and fertile, and have no detectable embryonic defects (Jeon et al., 2008; Sun et al., 2000). Ptp4E Ptp10D double mutants, however, die at the end of embryogenesis due to respiratory failure. They display a unique tracheal phenotype in which unicellular and terminal branches develop bubble-like cysts in place of their normal tubular lumens (Jeon and Zinn, 2009). This phenotype may have never been found in genetic screens for mutations causing tracheal defects because it requires the loss of both RPTPs. There may also be no single component downstream of the RPTPs that could be mutated to generate this phenotype, since the RPTPs are likely to regulate multiple RTK signaling pathways.
In our previous paper, we characterized the cell biology of the Ptp4E Ptp10D phenotype in detail. A unicellular tracheal tube has a lumen that is surrounded by the apical surface of a single cell (for reviews of tracheal tubulogenesis, see Affolter and Caussinus, 2008; Ghabrial et al., 2011; Swanson and Beitel, 2006). Ptp4E and Ptp10D are apically localized in tracheae (Jeon and Zinn, 2009). In Ptp4E Ptp10D mutants, apical membrane markers that are normally localized to the lumen appear in the cysts. EM analysis demonstrated that the cysts in unicellular branches are extracellular compartments with adherens junctions, and are therefore distorted and enlarged versions of normal tubular lumens. We hypothesized that the phenotype arises because the apical actin cytoskeleton fails to interact correctly with the apical membrane during the cell remodeling processes that accompany tube formation in unicellular branches. These interactions would normally constrain the lumen into a cylindrical shape, and the interaction defects in the mutants result in the generation of spherical cysts in place of tubes. Interestingly, terminal branches, which contain ‘seamless’ tubes (lacking adherens junctions) within cells, also develop cysts (Jeon and Zinn, 2009). In terminal cells, apical membrane grows inward to form an intracellular lumen (Gervais and Casanova, 2010). This new apical membrane aligns along cytoskeleton elements, so the geometry of the seamless tubes might be altered by the same types of membrane-cytoskeleton interaction defects that affect tube formation in unicellular branches.
The Ptp4E Ptp10D phenotype involves a loss of negative regulation of the Egfr ortholog, and Ptp10D physically associates with Egfr. Further elevation of Egfr activity by tracheal expression of a constitutively activated (CA) Egfr mutant in the Ptp4E Ptp10D background causes cyst expansion, as does expression of a CA mutant of Drosophila Raf kinase, a MAP kinase pathway component which is downstream of Egfr (Brand and Perrimon, 1994). However, expression of CA mutants of Egfr or Raf in a wild-type background does not generate any cysts (Jeon and Zinn, 2009).
There are four clear growth factor receptor TK orthologs in Drosophila, three of which are expressed in tracheal cells during embryogenesis. These are: Egfr, Breathless (Btl; FGFR ortholog) and Pvr (PDGFR/VEGFR/CSF1R ortholog) (Cela and Llimargas, 2006; Cho et al., 2002; Glazer and Shilo, 1991) (for a review of Drosophila RTK gene sequences, see Morrison et al., 2000); also see http://bio.illinoisstate.edu/kaedwar/Flycellbio/PTKandPTPlists.shtml. In our earlier paper, we showed that cyst size in Ptp4E Ptp10D mutants is increased by expression of a CA mutant of Btl and decreased when one copy of wild-type Btl is removed, suggesting that hyperactivation of Btl might also contribute to the phenotype (Jeon and Zinn, 2009). In this paper, we analyze genetic interactions between all three growth factor RTKs and the Type III RPTPs by simultaneously expressing dominant-negative (DN) and CA RTK constructs in tracheal cells. We evaluate the effects of these constructs on Ptp4E Ptp10D cyst phenotypes on four different tracheal branches, and find that RTK perturbations affect each branch in a different manner. Our data show that all three RTKs are involved in determination of tube geometry, and that they have partially interchangeable activities. An increase in the activity of one RTK can compensate for a reduction in the activity of another.
We also examine genetic interactions between the RTKs and two downstream signaling pathways, the Ras/MAP kinase pathway and the Rho pathway. Both are involved in the tube formation processes that are affected in the Rptp double mutant.
Materials and Methods
Fly stocks and genetics
The following Drosophila stocks were obtained from the Bloomington Stock Center (Bloomington, IN, USA): UAS-EgfrElp, UAS-EgfrDN, UAS-Phl-CA, UAS-InR-R418P, UAS-Rho1V14, UASRho1N19, UAS-Rac1, UAS-Rac1V12, UAS-Rac1N17, UAS-Cdc42V12, UAS-Cdc42N17, UAS-mCD8GFP. UAS-λBtl, UAS-BtlDN, Btl-GAL4 were kindly provided by M. Krasnow (Stanford University, Stanford, CA, USA), and UAS-λPvr, UAS-PvrDN were kindly provided by P. Rorth (IMCB, Singapore). Ptp4E1Ptp10D1/FM7-GFP; Btl-GAL4 female flies were crossed to UAS-transgenic male flies and progeny were examined for genetic interactions. All crosses were carried out at 25°C.
To verify the presence of transgenes, PCR was used to amplify the regions spanning the deleted domains in dominant negative constructs UAS-EgfrDN (FlyBase, personal communication), UAS-BtlDN (Reichman-Fried and Shilo, 1995), UAS-PvrDN (Duchek et al., 2001). Primers were designed to amplify the λ dimerization domains in UAS-λBtl (Lee et al., 1996) and UAS-λPvr (Duchek et al., 2001). UAS-EgfrElp and UAS-Phl-CA were recognized using forward primers that annealed to the corresponding genes and reverse primers that recognized the pUAST and pUASP vectors, respectively. The following primer pairs were used: EgfrElp (forward 5′-GTTAGTGTGGACAATCCGG, reverse 5′-TTGTCCAATTATGTCACACCAC), λBtl (forward 5′-GCCCATGGTTACCTGGAGG, reverse 5′-CAGCCGACGCAGCATGAACG), λPvr (forward 5′-GCCCATGGTTACCTGGAGG, reverse-5′-TCCTCGAAGTTGGCCAATCC), EgfrDN (forward 5′-TACTGTGCAGCTAGTCCGC, reverse 5′-TTGTCCAATTATGTCACACCAC), BtlDN (forward 5′-GTCTACTCTGATCTGCATCC, reverse 5′-TTGTCCAATTATGTCACACCAC), PvrDN (forward 5′-TCTGCTGGGCAACATTTCCG, reverse 5′-TGGTGCTATGTTTATGGCGC), Phl-CA (forward 5′-CATTGAAGCGCTTGGCCG, reverse 5′-TGGTGCTATGTTTATGGCGC).
Immunohistochemistry and microscopy
A standard protocol as reported in (Jeon et al., 2008) was used for whole-mount antibody staining of stage 15 embryos. The following primary antibodies were used: monoclonal antibody (mAb) 8B2 against Ptp10D [1:3; Developmental Studies Hybridoma Bank (DSHB)], mAb 4G10 against pTyr (1:300; Millipore), and rabbit or chicken anti-GFP (1:1000; Invitrogen), mAb Cq4 against Crb [1:3; Developmental Studies Hybridoma Bank (DSHB)], mAb DCAD2 against DE-cadherin [1:3; Developmental Studies Hybridoma Bank (DSHB)]. Rabbit anti-Sas (1:1000) was kindly provided by D. Cavener (Vanderbilt University, TN, USA). Secondary antibodies used were goat anti-mouse, goat anti-rabbit, and goat-anti-chicken antibodies coupled to Alexa Fluor 488, 568, or 594 (1:1000; Invitrogen). 20–40 dissected embryos were mounted per slide in 10 µl of Vectashield (Vector Laboratories) using 22×22 mm coverslip. Images were acquired on an inverted laser scanning confocal miscroscope (DMIRE2; Leica) using 40× objective lens. Images were processed using Adobe Photoshop.
Quantification of tracheal phenotypes
Fixed and stained stage 15 embryos were dissected according to protocols described in http://www.its.caltech.edu/~zinnlab/motoraxons/protocols.html. Samples were imaged using confocal microscopy and z-stack projections were obtained. Measurements were made according to protocols described in (Jeon and Zinn, 2009). Tr4-7 segments had equivalent phenotypes, so we pooled data from these segments. We measured or counted cysts in Tr4-7 segments for at least five embryos for each genotype. Error bars in figures were obtained by calculating the standard error of the mean using the formula (s/√n; s = standard deviation, n = sample size).
Results
Three growth factor receptor tyrosine kinases participate in tracheal tube formation
To analyze genetic interactions between the Type III RPTPs and growth factor RTK orthologs, we used CA and DN transgenes for Egfr, Btl, and Pvr. The Egfr-CA transgene carries the Elp mutation, which changes a single amino acid in the cytoplasmic domain and generates a constitutively active receptor (Lesokhin et al., 1999). The CA Btl and Pvr constructs were engineered with a lambda receptor dimerization domain in the N-terminus that forces ligand-independent dimerization, thereby activating the receptors (Duchek et al., 2001; Lee et al., 1996).
The DN versions of all three receptors are cytoplasmic (kinase) domain deletions. DN RTK mutants were devised about 20 years ago (Amaya et al., 1991; Kashles et al., 1991), and have since been used in hundreds of papers, primarily in vertebrate systems, to selectively inhibit specific RTKs. When expressed at high levels, these DN mutants reduce or eliminate RTK activity by dimerizing with wild-type receptors in a ligand-dependent manner, producing inactive complexes that cannot undergo transphosphorylation because they have only one kinase domain. To ensure signaling specificity, RTK XC domains have evolved to dimerize only with themselves or with specific heterodimeric partners within the same family, so DN mutants are highly selective inhibitors of the RTK families from which they are derived. The three RTK-DN mutants used in this paper have been employed by many groups to perturb development of a variety of different tissue and organs in Drosophila. They have been repeatedly shown to produce phenotypes that match those of LOF mutations of the same RTK, thereby demonstrating that they have the appropriate specificities (Buff et al., 1998; Cela and Llimargas, 2006; Duchek et al., 2001; Kumar and Moses, 2001; Nagaraj et al., 1999; Reichman-Fried and Shilo, 1995; Scholz et al., 1997; Szüts et al., 1997).
The three DN mutants are capable of generating null or near-null phenotypes when expressed early in tissue development, so that they can block the activity of newly synthesized wild-type RTK in that tissue. However, when expressed after wild-type RTK has been made and executed some of its functions, they produce weaker phenotypes. Most relevant to the present paper, (Reichman-Fried and Shilo, 1995) showed that early expression of DN Btl from heat-shock-GAL4 blocks all branch migration, while later heat shock produces weaker phenotypes that affect only specific branches. In this paper, we are expressing DN Btl (and other RTK mutants) from the Btl-GAL4 driver, so it comes on later than the onset of wild-type Btl expression due to the time lag associated with the GAL4 regulatory circuit. The phenotypes we observe affect extension of specific branches, but no branches are missing (see Fig. 1). Similarly, we observe subtle tissue integrity phenotypes for DN Egfr that match those seen by (Cela and Llimargas, 2006), who also drove expression in tracheae using Btl-GAL4. By contrast, Egfr null mutant embryos, which lack all zygotically synthesized Egfr, have severe tracheal defects (Cela and Llimargas, 2006).
Fig. 1. Effects of dominant-negative and constitutively activated receptor tyrosine kinase mutants on tracheal tube formation.
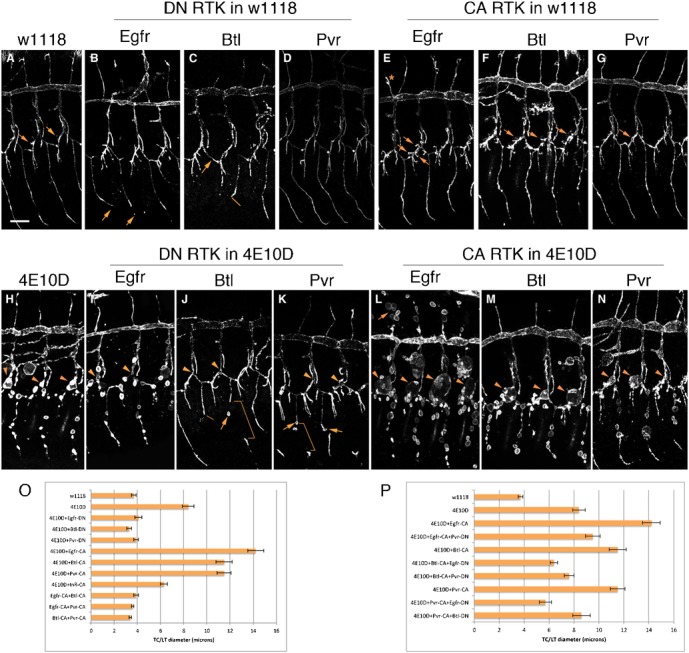
In all panels, the tracheal system was labeled in stage 15 embryos using an antibody against the apical cell-surface protein Sas. Three hemisegments are shown in each panel, with dorsal up and anterior to the left. In all panels, the DT is the large horizontal tube, with DBs extending upwards from it and TCs extending down. The GBs are the thin vertical branches at the bottom of each panel (B, arrows). The tracheal-specific driver Btl-GAL4 was used to express all mutant UAS-RTK constructs. When DN constructs were expressed in the wild-type (w1118) background (A), we observed shortening of GBs with Egfr-DN (B, arrows); missing GBs (C, arrow) and GBs that failed to extend (C, line) with Btl-DN; and relatively normal tracheae with Pvr-DN (D). With CA Egfr and Btl, ectopic branches were observed, including extra terminal branches along the LT (terminal branches indicated by arrows in A,E,F,G) and ectopic DBs (E, asterisk). Pvr-CA was indistinguishable from wild-type. In Ptp4E Ptp10D (4E10D) mutants, cysts were observed at all TC/LT junctions (H, arrowheads), and along the GBs (the three bottom branches in H). DN RTKs produced strong suppression of the TC/LT cyst phenotype (I–K, arrowheads). GB cysts were still present with Egfr-DN, while there were fewer with Btl-DN and Pvr-DN (J,K, arrows) (see Fig. 2F for quantitation). Brackets in (J,K) indicate GB regions with weak or absent staining for the apical marker. As in the wild-type background, there were shorter GBs with Egfr-DN and missing GBs with Btl-DN (J, line). Enhancement of the TC/LT junction phenotype (L–N, arrowheads) was observed with all three RTK CA mutants. The strongest enhancement was seen with Egfr-CA, which also produced numerous cysts on DBs (L, arrow) (see Fig. 3F for quantitation). (O) Quantitative analysis of cyst phenotypes produced by DN or CA RTK constructs. The diameters of TC/LT junctions in segments Tr4-7 were measured at the points indicated by the arrowheads. (P) Quantitative analysis of RTK competition experiments. Error bars in all figures indicate the standard error of the mean (see Materials and Methods for formula). Scale bar: 20 µm.
We first characterized the effects of the RTK transgenes when they were driven by the tracheal-specific Btl-GAL4 driver in a wild-type (w1118) background (Btl>RTK-DN or Btl>RTK-CA). In all figures in this paper, the tracheal system was labeled using an antibody against the apical cell surface protein Stranded at second (Sas) (Schonbaum et al., 1992). The strongest DN phenotype was produced by the Btl-DN transgene. This caused defects in ganglionic branch (GB) extension, so that many GBs failed to extend to the ventral nerve cord (VNC) in stage 15 embryos (Fig. 1C). Egfr-DN caused a weaker GB extension defect, in which all GBs grew but some were shorter than in wild-type (Fig. 1B). Pvr-DN did not generate any obvious phenotypes (Fig. 1D).
In stage 15 wild-type embryos, there are usually two terminal branches (Fig. 1A, arrow) extending away from each lateral trunk (LT) segment. In embryos expressing Egfr-CA or Btl-CA, there were many extra terminal branches along each LT (Lee et al., 1996) (Fig. 1E,F, arrows). Ectopic terminal branches were also observed along the visceral branch (VB), which was dissected away in the embryos shown in Fig. 1. There were also occasional ectopic unicellular branches attached to the dorsal trunk (DT) (Fig. 1E, asterisk) and the GB. Pvr-CA produced no visible tracheal phenotypes.
We then combined each DN transgene with the Ptp4E1 Ptp10D1 double null mutation, to generate Ptp4E Ptp10D, Btl>RTK-DN embryos. Ptp4E Ptp10D double mutant embryos have cysts instead of tubes at all transverse connective (TC)/LT junctions (Fig. 1H, arrowheads), resulting in a >2-fold increase in average TC/LT junction diameter relative to wild-type (Fig. 1O). As in our previous paper, we quantitatively analyzed the effects of the transgenes on the cyst phenotype by measuring the diameters of these junctions. Three different Egfr manipulations (Egfr-DN, removing one copy of wild-type Egfr, and a tracheally expressed Egfr dsRNA (RNAi) transgene) partially or completely suppress the increase in TC/LT junction diameter (Jeon and Zinn, 2009). Here we compared three different RTK-DN transgenes in the same set of experiments, and observed that all three transgenes completely suppressed the TC/LT diameter increase phenotype (Fig. 1O). Btl-DN and Pvr-DN eliminated all cysts in the TC/LT region (Fig. 1J,K), while small cysts were still present with Egfr-DN (Fig. 1I). These data show that a reduction in activity of any of the three RTKs suppresses the TC/LT phenotype produced by loss of negative regulation by the Type III RPTPs.
Expressing Egfr-CA from the Btl-GAL4 driver in the Ptp4E Ptp10D background produced a dramatic enhancement of the TC/LT phenotype, generating huge cysts (some are >20 µm in diameter) at TC/LT junctions (Fig. 1L). Here we compared the effects of expressing the three growth factor RTK-CA transgenes, as well as a CA transgene for the Drosophila insulin receptor (InR), in the Ptp4E Ptp10D background. Egfr-CA, Btl-CA, and Pvr-CA-expressing embryos all had expanded cysts at TC/LT junctions (Fig. 1L–N). InR-CA, however, did not enhance the Ptp4E Ptp10D phenotype, and in fact produced a slight decrease in TC/LT junction diameter (Fig. 1O).
These data show that all three growth factor RTKs are involved in the Ptp4E Ptp10D phenotype, and suggest that this phenotype might be caused by simultaneous increases in the activities of multiple RTKs. If so, perhaps one could generate a Ptp4E Ptp10D-like cystic phenotype in a wild-type background by expressing multiple RTK-CA transgenes in tracheal cells using Btl-GAL4. We tested this idea by expressing Egfr-CA+Btl-CA, Egfr-CA+Pvr-CA, and Btl-CA+Pvr-CA. However, none of these manipulations produced any cysts, and there was no increase in TC/LT junction diameter over wild-type (Fig. 1O). We also expressed all three transgenes together (Egfr-CA+Btl-CA+Pvr-CA). Because of the genetic manipulations necessary to make triple transgene embryos bearing the Btl-GAL4 driver, we could not obtain enough of these embryos to perform a quantitative analysis of TC/LT diameter, but it was clear that they did not have Ptp4E Ptp10D-like cystic phenotypes (data not shown).
Competing receptor tyrosine kinase mutants against each other
Having demonstrated that all three RTKs influence the Ptp4E Ptp10D phenotype, we now wished to ask if each RTK is uniquely required for the phenotype, or if they can substitute for each other. We evaluated this by simultaneously expressing DN and CA RTK transgenes in the Ptp4E Ptp10D background and determining the effects of these manipulations on TC/LT junction diameter. The results described above show that DN mutants of each RTK suppress the Ptp4E Ptp10D cyst phenotype. If the generation of the phenotype requires binding of SH2-domain effectors that can interact with only one phosphorylated RTK, then expressing a DN mutant of one RTK in the presence of a CA mutant of another RTK should still cause complete or almost-complete suppression, returning the embryos to a near-wild-type TC/LT phenotype. On the other hand, if the relevant effectors can bind to PTyr residues on two or more RTKs, the CA mutant should partially rescue the suppression caused by the DN mutant, returning the phenotype to the strength of unmodified Ptp4E Ptp10D.
We examined five of the six possible CA/DN combinations (we could not make Egfr-CA+Btl-DN because of the chromosomal positions of the transgene insertions). In contrast to their ability to suppress the TC/LT diameter phenotype in Ptp4E Ptp10D embryos to wild-type values (Fig. 1O), none of the DN mutants was able to suppress a Ptp4E Ptp10D, Btl>RTK-CA phenotype back to wild-type (Fig. 1P). For three of the five combinations, the phenotype was approximately the same as in unmodified Ptp4E Ptp10D embryos. The other two combinations (Btl-CA+Egfr-DN and Pvr-CA+Egfr-DN) exhibited somewhat stronger suppression, generating phenotypes that were intermediate between wild-type and Ptp4E Ptp10D (Fig. 1P). Based on these results, we conclude that none of the three RTKs is uniquely required to generate the Ptp4E Ptp10D cyst phenotype. This conclusion is strengthened by phenotypic analysis of the GB.
Effects of kinase activity manipulations on other tracheal branches
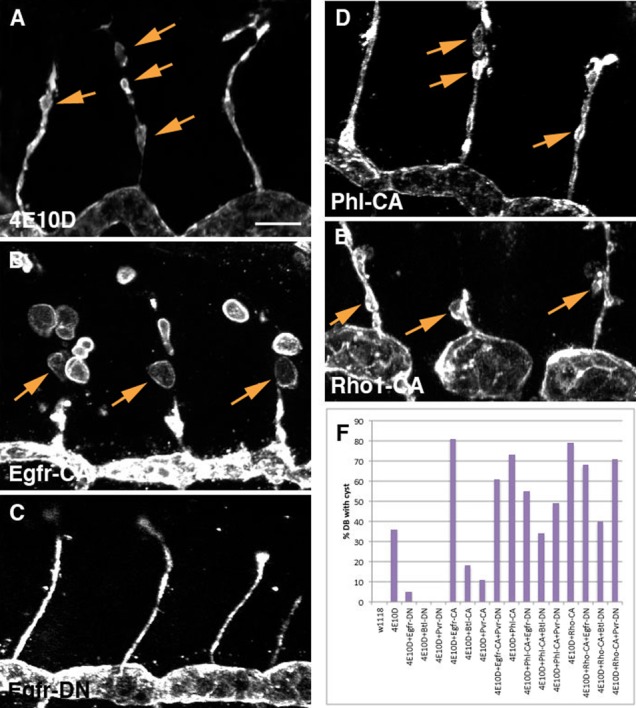
To quantitatively analyze the effects of kinase manipulations on the unicellular GB, we counted GB cysts. All GBs in Ptp4E Ptp10D embryos had cysts (Fig. 1H), but they were never observed in wild-type embryos, or in embryos where any of the three CA RTK mutants were expressed in a wild-type background. Egfr-CA and Pvr-CA both enhanced the Ptp4E Ptp10D GB cyst phenotype, increasing cyst number by ∼1.5-fold (Fig. 2F) and also increasing cyst diameters (not quantitated; compare Fig. 2A and Fig. 2E). Btl-CA did not significantly enhance the Ptp4E Ptp10D phenotype. When we examined the DN mutants, we found that they had somewhat different effects from those seen for the TC/LT phenotype (Fig. 1). Btl-DN almost completely suppressed the phenotype, decreasing cyst numbers to almost zero. However, Pvr-DN only produced a ∼1.5-fold decrease, and Egfr-DN did not affect cyst number (Fig. 2F).
Fig. 2. Effects of DN and CA RTK mutant constructs on the GB cyst phenotype.

Magnified images of single GBs stained with anti-Sas are shown in panels (A–E). Yellow lines indicate the beginning and the end of each GB, and orange arrowheads indicate GB cysts. Strong suppression of the Ptp4E Ptp10D phenotype was observed only for Btl-DN (C). Egfr-CA produced strong enhancement for both cyst number and diameter (E). (F) Quantitative analysis of the GB cyst number phenotype.
The DN mutant results might be interpreted as suggesting that there is a unique requirement for Btl activity for generation of GB cysts. However, when we examined embryos in which Btl-DN was expressed together with Pvr-CA, we found that the phenotype was suppressed only back to that of unmodified Ptp4E Ptp10D embryos (Fig. 2F). Thus, Pvr was able to substitute for Btl when Pvr activity levels were increased, even though the presence of wild-type Pvr could not prevent Btl-DN from completely suppressing the GB cyst phenotype of Ptp4E Ptp10D embryos.
The unicellular DB displays a different dependence on RPTP and RTK function than the GB. DB cysts are rare in Ptp4E Ptp10D mutants (only about 30% of DBs have any cysts), and are common only in Ptp4E Ptp10D, Btl>Egfr-CA embryos (Fig. 3B,F). The other two CA mutants did not enhance the Ptp4E Ptp10D DB phenotype.
Fig. 3. Effects of RTK, Raf (Phl), and Rho mutant constructs on the DB cyst phenotype.
Magnified images of DBs in three hemisegments stained with anti-Sas are shown in panels (A–E); cysts are indicated by arrows. Enhancement of the Ptp4E Ptp10D phenotype was observed for Egfr-CA (B), Phl-CA (D), and Rho1-CA (E). All DN RTKs produced strong suppression of the phenotype (shown for Egfr-DN in C). (F) Quantitative analysis of the DB cyst phenotype. Because DB cysts are rare in most genotypes, we scored the percentage of DBs that displayed any cysts rather than counting cysts. Scale bar: 10 µm.
All three DN mutants completely suppressed the Ptp4E Ptp10D DB cyst phenotype (Fig. 3C,F). However, Pvr-DN only produced a slight suppression of the enhanced Ptp4E Ptp10D, Btl>Egfr-CA phenotype, indicating that loss of Pvr activity cannot prevent excess Egfr activity from increasing cyst number on the DB (Fig. 3F).
To evaluate the effects of RTK manipulations on seamless (intracellular) tubes, we examined the LG terminal branch. Although seamless tubes are created by the inward growth of membrane into terminal cells (Gervais and Casanova, 2010), they also have apical membrane lining the lumen, suggesting that there may be parallels between the mechanisms of formation of seamless and unicellular tubes. In seamless tubes, a new apical compartment is created within the cell, while the existing apical side of a cell becomes the luminal face of a unicellular tube (for reviews, see Affolter and Caussinus, 2008; Swanson and Beitel, 2006).
We performed a qualitative assessment of the effects of the kinase manipulations on this branch, because the number of LG tubes is variable, making it difficult to count cysts. All of the CA RTK mutants enhanced the phenotype, creating larger cysts (compare Fig. 4A and Fig. 4D). As for the GB, Btl-DN produced complete suppression of the cyst phenotype (Fig. 4C), while the other two DN mutants had relatively little effect. However, Btl-DN could not completely suppress the cyst phenotype in Ptp4E Ptp10D, Btl>Pvr-CA embryos, indicating that, as for the GB and TC/LT phenotypes, increased activity of Pvr can compensate for the loss of Btl activity.
Fig. 4. Effects of RTK and Raf (phl) mutant constructs on the LG (terminal branch) cyst phenotype.
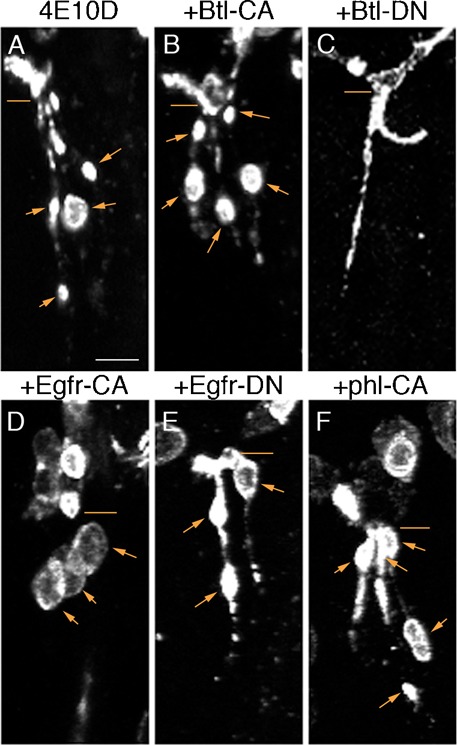
Each panel shows the LG branches in a single hemisegment, stained with anti-Sas. Lines indicate the points where the LGs branch from the LT, and arrows indicate LG cysts. In stage 15 Ptp4E Ptp10D embryos, most LGs had small cysts (A). All CA RTK mutants and Phl-CA produced larger cysts (B,D,F). Btl-DN completely suppressed the phenotype, eliminating all cysts (C), while Egfr-DN (E) and Pvr-DN (not shown) did not produce strong suppression. Scale bar: 5 µm.
Localized accumulation of phosphotyrosine in Ptp4E Pt10D mutant embryos
Since Ptp4E and Ptp10D appear to allow normal tube formation by negatively regulating RTK activity, we wondered if PTyr levels in the tracheal system might be increased in the absence of these RPTPs. To evaluate this idea, we double-stained stage 14 embryos with anti-PTyr and anti-Sas. Anti-PTyr outlines the entire tracheal system in wild-type embryos (Fig. 5B). The overall intensity of anti-PTyr staining was not changed in Ptp4E Ptp10D embryos (data not shown). This suggests that other PTPs, probably widely expressed cytoplasmic PTPs, are responsible for setting the overall levels of PTyr.
Fig. 5. Anti-phosphotyrosine staining of the tracheal system in wild-type and in Ptp4E Ptp10D mutants.
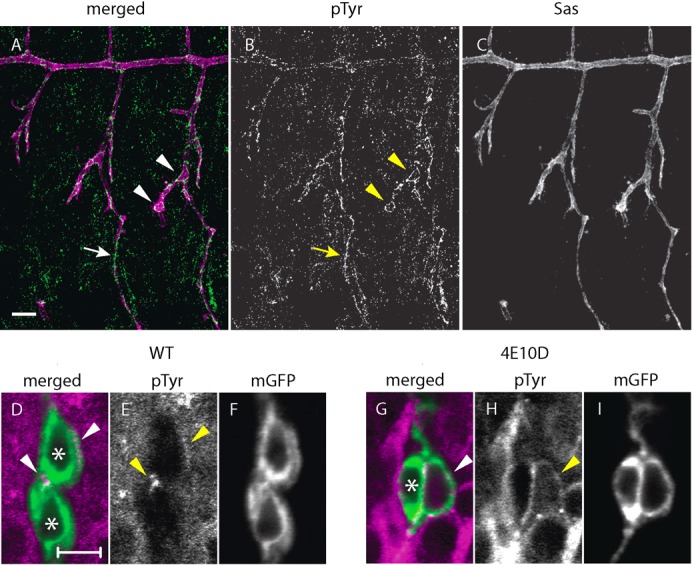
(A–C) A wild-type (w1118) embryo at stage 14 double-stained with monoclonal antibody 4G10 against PTyr (green/white) and anti-Sas (magenta/white). PTyr staining outlined the tracheal system at this stage; Ptp4E Ptp10D embryos showed no changes from this pattern (not shown). The LT, which has not fused across segment borders at this stage, is indicated by arrowheads; a GB is indicated by an arrow. (D–I) GB cells expressing mCD8GFP from Btl-GAL4 were visualized with anti-PTyr (magenta/white) and anti-GFP (green/white). Nuclei are indicated by asterisks. In wild-type, PTyr staining above baseline levels was detected at the autocellular junction (D,E, upper arrowhead) and at the junction between the two cells shown (D,E, lower arrowhead). (G–I) A single GB cell with a cyst is shown in a Ptp4E Ptp10D mutant. PTyr levels were usually higher around the entire cell than in wild-type, and cysts were outlined by high PTyr staining (arrowhead). The cyst membrane in this cell has three PTyr hot spots. Scale bars in (A–C): 10 µm; in (D–I): 5 µm.
We observed revealing local changes in PTyr in Ptp4E Ptp10D embryos. PTyr levels were low in wild-type GB cells, but there was a hotspot of labeling at junctions between the cells (Fig. 5E, arrowheads). Ptp4E Ptp10D GB cells that had cysts displayed bright anti-PTyr staining on the apical membrane that surrounds the lumen of the cyst (Fig. 5H, arrowhead). This staining might be due to elevated activity of one or more of the RTKs that are regulated by Ptp4E and Ptp10D.
The MAP kinase pathway is involved in generation of the Ptp4E Ptp10D cyst phenotype
To evaluate the role of Ras/MAP kinase signal transduction, which is downstream of RTKs, we expressed a constitutively activated mutant of Drosophila Raf (also known as Pole hole or Phl), called Phl-CA (Brand and Perrimon, 1994), in the tracheae using Btl-GAL4. Raf/Phl is a serine/threonine kinase that is an effector for Ras. It is also called MAP kinase kinase kinase (MAPKKK), and is the first enzyme in the MAP kinase cascade. Expression of Phl-CA in a wild-type background did not generate any cysts, but it was able to enhance the TC/LT cyst phenotype when expressed in the Ptp4E Ptp10D background (Fig. 6A,E). Like Egfr-CA, Phl-CA enhanced the DB cyst phenotype (Fig. 3D,F), but it did not produce a major increase in the number of GB cysts.
Fig. 6. Interactions between DN RTK mutants and elevated MAP kinase signaling.
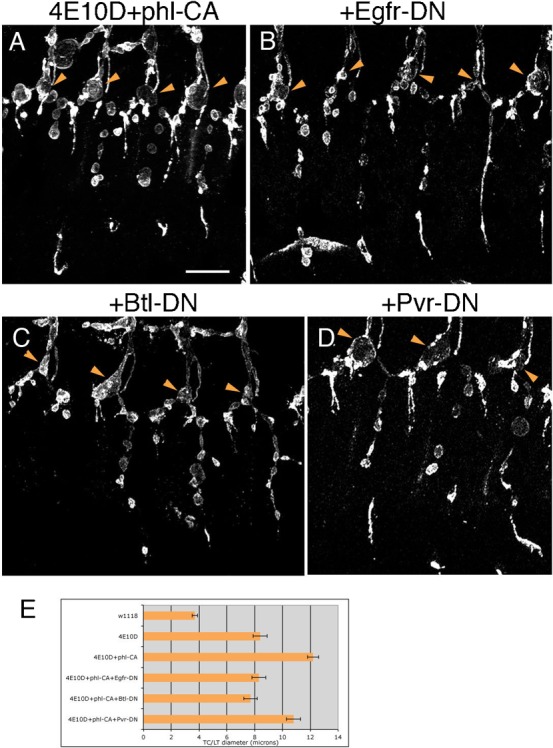
(A) An enhanced phenotype was produced by expressing Phl-CA in the Ptp4E Ptp10D background. This enhancement was suppressed back to the strength of unmodified Ptp4E Ptp10D by Egfr-DN (B) and Btl-DN (C). Pvr-DN produced weaker suppression (D). In all panels, arrowheads indicate the positions of TC/LT junctions. (E) Quantitative analysis of enhancement and suppression of TC/LT phenotypes by Phl-CA alone and combined with DN RTK mutant constructs. Scale bar: 20 µm.
If elevated RTK activity produces the cyst phenotype solely via increased signaling through the MAP kinase pathway, one might expect that expressing DN RTKs together with Phl-CA in the Ptp4E Ptp10D background would not suppress the Phl-CA-enhanced phenotype, since RTKs are upstream of Raf. On the other hand, if other signaling pathways downstream of RTKs must also be overactivated to generate cysts, then DN RTKs might be expected to suppress the Ptp4E Ptp10D, Btl>Phl-CA phenotype. The actual results observed were that Egfr-DN and Btl-DN affected the phenotype in the same manner as they altered Ptp4E Ptp10D, Btl>RTK-CA phenotypes: that is, they suppressed the phenotype back to approximately the same strength as that of unmodified Ptp4E Ptp10D (Fig. 6E). Suppression by Pvr-DN was very weak (at the margin of statistical significance).
The relative contributions of MAP kinase signaling vs. other pathways to the cyst phenotype, and the extent to which each of the RTKs drives signaling through each pathway, cannot easily be determined from these data, since we do not know how strongly MAP kinase signaling is activated by Phl-CA vs. the RTK-CA constructs. The major conclusion from the Egfr-DN and Btl-DN suppression of the Phl-GOF enhancement is that processes downstream of MAP kinase signaling are insufficient to explain the effects of these RTKs on the cyst phenotype.
Rho GTPase signaling is required for the Ptp4E Ptp10D cyst phenotype
Rho family GTPases are downstream of many RTKs in mammalian systems (reviewed by Schiller, 2006). They are activated by Rho family guanine nucleotide exchange factors (GEFs), some of which can bind to and/or become phosphorylated by activated RTKs. Rho family GTPases are logical candidates for signaling molecules that mediate the effects of Ptp4E and Ptp10D on formation of tubes in the tracheal system, because they are known to affect cytoskeletal and membrane compartments. The best hypothesis for the failure of tube formation in Ptp4E Ptp10D mutants is that elevation of RTK activity disrupts interactions between the apical membrane and the actin cytoskeleton.
The Rho family in Drosophila includes one Rho gene, three Rac genes, and one Cdc42 gene (reviewed by Johndrow et al., 2004). To examine the roles of these GTPases, we expressed DN mutants of Rho1, Rac1, and Cdc42, as well as Rho1 dsRNA (RNAi), in wild-type and Ptp4E Ptp10D mutant tracheal cells. In wild-type embryos, tracheal expression of Rho1-DN or Rac1-DN caused defects in branching morphogenesis. Embryos that expressed Rac1-DN had phenotypes in which many GBs failed to extend to their full lengths (Fig. 7C). In Rho1-DN-expressing embryos, GBs were full-length but some branches appeared to be discontinuous when visualized by staining for the apical marker Sas (Fig. 7B). Expression of Cdc42-DN caused defects in LT fusion (data not shown), but otherwise had little effect on tracheal branching.
Fig. 7. Rho family GTPase signaling is required for cyst formation in Ptp4E Ptp10D mutants.
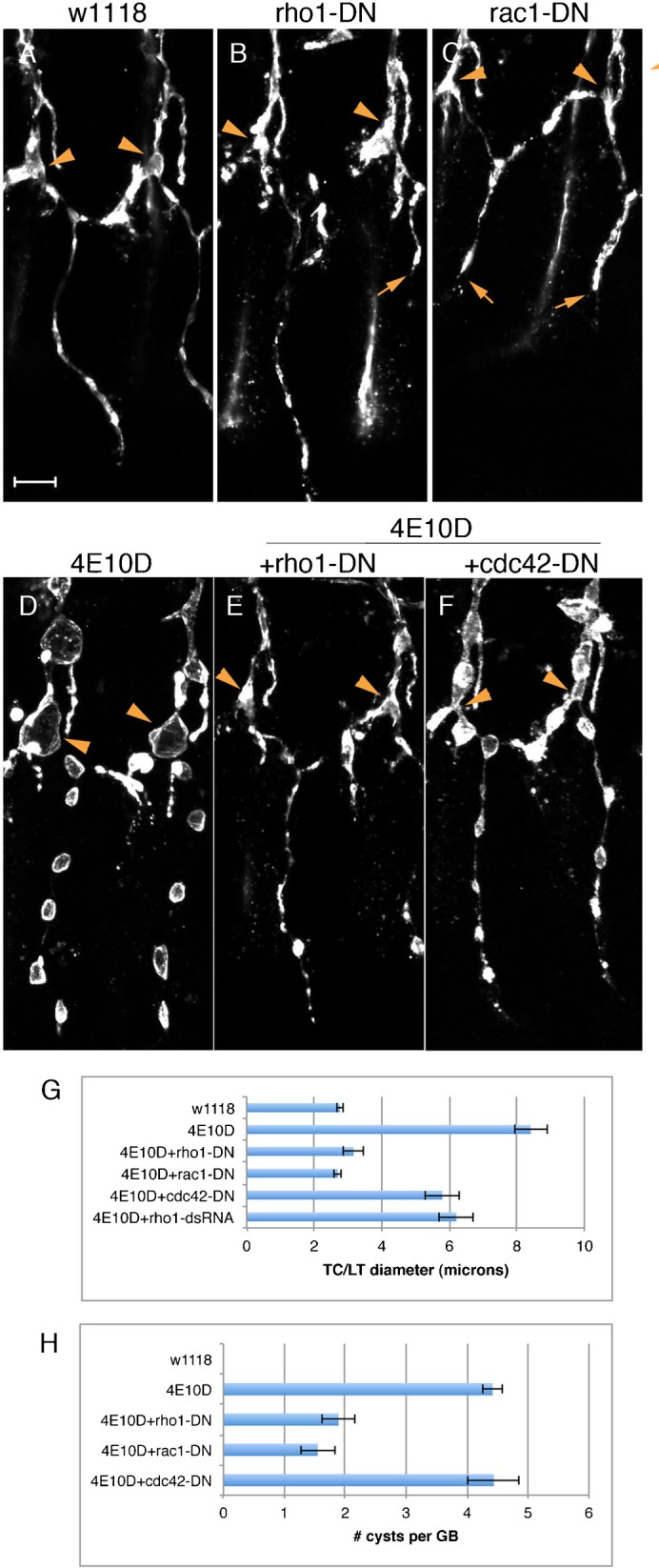
(A–C) Expression of DN Rho1 and Rac1 mutants in wild-type (w1118) embryos produced defects in tracheal branching, including failures of GB extension (B,C, arrows). In the Ptp4E Ptp10D background (D), expression of Rho1-DN (E) or Rac1-DN (not shown) produced complete suppression of the TC/LT diameter phenotype, and partial suppression of the GB cyst number phenotype. Cdc42-DN (F) and Rho1 dsRNA (not shown) produced partial suppression of the TC/LT phenotype, but did not affect GB cyst number. In all panels, arrowheads indicate the positions of TC/LT junctions. (G,H) Quantitative analysis of TC/LT (G) and GB cyst number (H) phenotypes. Scale bar: 10 µm.
In Ptp4E Ptp10D mutant embryos, we observed complete suppression of the phenotype, as assayed by the TC/LT junction diameter metric, when Rho1-DN or Rac1-DN was expressed. Rho1 RNAi and Cdc42-DN produced partial suppression (Fig. 7G). For GB cyst number, Rho1-DN (Fig. 7E,H) and Rac1-DN produced strong (>2-fold) suppression, but Cdc42-DN had no effect (Fig. 7F,H).
Next, we examined the effects of expressing CA mutants of Rho1, Rac1, or Cdc42. Tracheal expression of Rac1-CA in a wild-type background produced severe defects that prevented assessment of interactions with the Ptp4E Ptp10D phenotype. Wild-type embryos expressing Cdc42-CA had defects in DT and LT fusion and failures of GB extension in many hemisegments, and when Cdc42-CA was expressed in the Ptp4E Ptp10D background it caused a weak suppression of the phenotype (data not shown).
The most interesting effects were produced by Rho1-CA. When expressed in wild-type embryos, Rho1-CA caused failure of DT and LT fusion, and prevented GB branch extension (Fig. 8A). In the Ptp4E Ptp10D background, Rho1-CA produced DB cysts (Figs 3E, 8C). It also resulted in the conversion of the TC/LT region into networks of bubble-like structures (Fig. 8B). These bubbles are composed of apical membrane, and they also contain regions that express adherens junctions markers (Fig. 8D,E). We do not know whether these structures are cysts. In some cases, adherens junction markers were localized in circular structures surrounded by apical membrane, as in multicellular cysts (supplementary material Fig. S1), but other regions of apical membrane were not associated with junctional markers.
Fig. 8. Enhancement of the Ptp4E Ptp10D cyst phenotype by elevation of Rho activity.
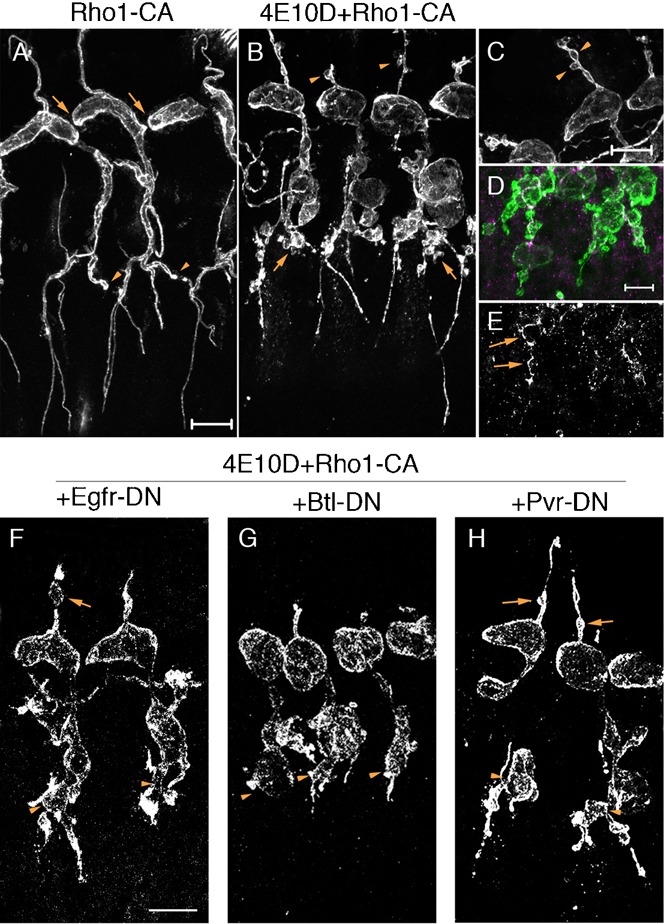
(A) Rho1-CA expression in a wild-type (w1118) background produced strong tracheal morphogenesis defects, including loss of intersegmental fusion of the DT (arrows) and LT (arrowheads). (B) Rho1-CA expression in the Ptp4E Ptp10D background converted the TC/LT region into bubble-like structures (arrows). Cysts were present on most DBs (B,C, arrowheads) (see also Fig. 3E). (D,E) Visualization of LT bubble networks at higher magnification, using double-staining with anti-Crumbs (apical marker; green) and anti-E-cadherin (junctional marker; magenta/white). (E) shows that adherens junction markers were present within some of the bubbles. (F–H) When DN RTKs were expressed together with Rho1-CA in Ptp4E Ptp10D embryos, the TC/LT regions still had abnormal morphologies like those in (B). Arrows in (F) and (H) indicate DB cysts in Egfr-DN and Pvr-DN-expressing embryos. Scale bars for (A,B,F–H): 20 µm; for (C–E): 10 µm).
To evaluate the potential relationships between Rho and the RTKs, we examined interactions between Rho1-CA and DN RTK mutants. We observed that Egfr-DN and Pvr-DN had little effect on the enhancement of the DB cyst phenotype by Rho1-CA (Fig. 3F). Btl-DN did suppress the effects of Rho1-CA, but it also shortened the DB branch, so we cannot be sure that it specifically blocked Rho1-CA-induced cyst formation. The odd bubble-like morphology of the TC/LT region seen in Ptp4E Ptp10D, Btl>Rho1-CA embryos was not obviously changed when any of the DN RTK mutants was expressed (Fig. 8F–H).
Discussion
The Drosophila tracheal system is an intricate branched network of air-filled tubes that delivers oxygen to tissues. Tube formation in the tracheal system involves complex morphogenetic events that differ between tube types. Multicellular tubes have lumens that are surrounded by the apical surfaces of several cells. Unicellular tubes are formed by rolling up of single cells to form junctions with themselves. Seamless tubes are intracellular structures within terminal cells. Many genes have been identified that affect the formation and morphology of tracheal tubes (for reviews, see Affolter and Caussinus, 2008; Ghabrial et al., 2011; Swanson and Beitel, 2006).
The absence of the two Type III RPTPs, Ptp4E and Ptp10D, changes the geometries of the tubes in unicellular and terminal branches, so that they form spherical cysts in place of continuous tubular lumen. The phenotype involves a loss of negative regulation of the Egfr RTK, and Ptp10D physically associates with Egfr (Jeon and Zinn, 2009).
One of the mammalian Ptp4E/Ptp10D orthologs, PTPRJ (DEP-1), is a direct regulator of multiple growth factor receptor TKs (Arora et al., 2011; Berset et al., 2005; Chabot et al., 2009; Lampugnani et al., 2003; Kappert et al., 2007; Palka et al., 2003; Tarcic et al., 2009). This led us to test the hypothesis that Btl and Pvr, the other two Drosophila growth factor receptor TK orthologs that are expressed in embryonic tracheae, are also required for the Ptp4E Ptp10D phenotype.
A quantitative analysis of cyst size at the TC/LT junction showed that tracheally expressed DN mutants of Egfr, Btl, and Pvr all suppress the Ptp4E Ptp10D phenotype almost to wild-type, suggesting that dysregulation of all three RTKs is required for the replacement of linear tubes by spherical cysts. Also, CA mutants of each RTK enhance the phenotype, producing enlarged cysts. These effects cannot be produced by all RTKs, since expression of a CA mutant of InR did not enhance the phenotype (Fig. 1O).
If the Ptp4E Ptp10D cyst phenotype is the consequence of simultaneous deregulation of all three RTKs, it might be possible to generate the phenotype in a wild-type background by expressing multiple RTK CA mutants. This did not work, even when we expressed all three CA mutants at once. Possible explanations include: 1) because PTP activity normally dominates over RTK activity, the effect on PTyr levels of removing negative regulation by the RPTPs is much greater than that produced by expressing CA RTK mutants in the presence of the RPTPs; 2) other TKs are important for the phenotype, and their activities must also be increased; 3) the RPTPs have PTP-independent activities as adhesion molecules, and generation of the phenotype requires both elevation of RTK activity and the absence of the PTP-independent functions of the RPTPs.
We investigated whether Ptp4E and Ptp10D both regulate all three RTKs. If they have specificity for particular RTKs, one might be able to generate the cyst phenotype by removing only one RPTP in the presence of a CA RTK mutant. We made such combinations for Egfr (Ptp4E, Btl>Egfr-CA and Ptp10D, Btl>Egfr-CA), but neither of them had cysts (M.J., unpublished). Since the PTP domains of Ptp4E and Ptp10D are 89% identical, it is likely that they have the same enzymatic targets. The idea that the RPTPs have redundant functions is also consistent with the observation that Ptp4E and Ptp10D single mutants have no detectable phenotypes, while the double mutant is lethal and has both tracheal (Jeon and Zinn, 2009) and nervous system defects (Jeon et al., 2008).
Models for RTK function in tube formation
When we expressed each of the DN RTK mutants together with a CA mutant of one of the other RTKs in the Ptp4E Ptp10D background, the DN mutant was now unable to suppress the phenotype back to wild-type. Instead the phenotype returned to the strength of unmodified Ptp4E Ptp10D mutants (Fig. 1P). This shows that if the activity of one RTK is sufficiently elevated, it can replace the requirement for another RTK. Thus, none of the three RTKs is uniquely required to generate the phenotype. Rather, the formation of tubes vs. cysts is controlled by the total amount of activity of certain RTKs in tracheal cells. This implies that the downstream pathways whose increased activity causes cyst formation use SH2-domain effectors that can bind to PTyr sites on any of the three RTKs.
It is interesting that the three RTKs can substitute for each other in regulating the tube vs. cyst decision when they are deregulated by loss of the RPTPs, since the RTKs do not seem to have interchangeable activities in wild-type tracheal cells (or in other tissues). Loss or gain of Btl function produces defects in primary, secondary, and terminal tracheal branching (Klämbt et al., 1992; Lee et al., 1996; Reichman-Fried and Shilo, 1995; Samakovlis et al., 1996). Loss of Egfr function produces a much more subtle tracheal phenotype affecting tissue integrity. Maintenance of tissue integrity requires signaling through the MAP kinase pathway downstream of Egfr, but is unaffected by reduction of MAP kinase signaling downstream of Btl (Cela and Llimargas, 2006).
These findings can be explained by the fact that growth factor receptor TKs are usually in an inactive state, due to insufficient levels of ligands and to negative regulation by PTPs. They become active only when they come into contact with elevated levels of their ligands at specific times and places. The activities of Type III RPTPs that dephosphorylate the RTKs might also be transiently reduced at some of these times and places, possibly through interactions of their XC domains with as yet unidentified ligands. As a consequence of the tight control of RTK activity, only those downstream signaling pathways that are most responsive to a particular RTK are likely to be activated by that RTK at any time in wild-type embryos, and the outcomes of signaling through these pathways may also be controlled by the subcellular distributions of ligands, RTKs, RPTPs, and downstream effectors. By contrast, in the absence of Ptp4E and Ptp10D, basal levels of RTK ligands may be able to drive all of the growth factor RTKs to a high level of activity, resulting in strong signaling through all of the downstream pathways they can control. Loss of negative regulation might also cause delocalization of signaling, so that effectors whose activity is normally restricted to particular parts of the cell become activated in a cell-wide manner. Under these conditions, a reduction in the activity of any one of the RTKs by a DN mutant will decrease signaling through multiple downstream pathways. Adding a CA mutant of another RTK can then turn signaling through all of these pathways back up, compensating for the effects of the DN mutant.
The ability of RTKs to substitute for each other in control of cyst formation is conceptually similar to cell transformation through elevation of RTK signaling. RTK activity in cultured cells is tightly controlled, and only a few endogenous RTKs are normally involved in cellular responses to the mitogenic growth factors in their culture medium. Many RTKs can signal through the Ras/MAP kinase pathway, however, and elevated Ras/MAP kinase transduction is sufficient to cause transformation of established cell lines. Thus, oncogenic (CA) mutants of a variety of RTKs are able to transform fibroblastic cell lines when expressed at high levels, regardless of whether the endogenous versions of the RTKs are normally used to regulate proliferation in those cell lines (or are even expressed there).
Our observations on the interchangeability of the RTKs suggests that the decision to form cysts rather than tubes in Ptp4E Ptp10D mutants is very sensitive to the levels of PTyr on the effector binding sites on autophosphorylated RTKs, and that cysts appear when total PTyr rises above a critical threshold. This conclusion is based on the complete suppression of the Ptp4E Ptp10D cyst phenotype that is produced by expression of any of the three DN mutants, even though each DN would eliminate only about 1/3 of total PTyr on the RTKs, if they have roughly equal activities. In wild-type embryos, negative regulation by the RPTPs keeps RTK signaling well below this threshold, so the system is insulated against random fluctuations in phosphorylation or downstream signaling.
The Ptp4E Ptp10D GB cyst number phenotype is completely suppressed by Btl-DN, but not suppressed at all by Egfr-DN and only slightly by Pvr-DN. This might be taken as evidence that elevation of Btl activity is uniquely required to replace GB tubes with cysts. However, when Btl-DN is competed against Pvr-CA, it is only able to suppress the Ptp4E Ptp10D, Btl>Pvr-CA phenotype back to that of unmodified Ptp4E Ptp10D, indicating that Btl can be replaced by Pvr if its activity is sufficiently elevated (Fig. 2F). These findings can be explained if Btl activity is much higher than Egfr or Pvr activity in GB cells, so that a DN mutant that knocks down endogenous Btl activity by a DN mutant has a greater effect on phosphorylation of effector binding sites than mutants that reduce Egfr or Pvr activities. The activity of CA RTK mutants is independent of the endogenous levels of RTKs, so the Pvr CA mutants could still reverse the effect of Btl-DN even if Pvr-DN has no effect on its own.
The MAP kinase and Rho GTPase pathways are involved in tube formation
To evaluate whether the MAP kinase signaling pathway is involved in the determination of tube geometry, we expressed a CA mutant of Phl, the Drosophila Raf kinase, in the Ptp4E Ptp10D background. Phl-CA enhances the TC/LT phenotype almost as strongly as Egfr-CA (Fig. 6A,E). When Phl-CA is combined with Egfr or Btl DN mutants, the phenotype is suppressed back to that of unmodified Ptp4E Ptp10D. Since these RTKs are upstream of Raf, the fact that suppression occurs suggests that pathways other than the MAP kinase pathway are required to generate the phenotype. However, these other pathways may be stimulated by elevation of MAP kinase signaling, since one would expect suppression back to a near-wild-type phenotype if they were completely independent of the MAP kinase pathway.
We tested the involvement of Rho GTPases by expressing DN mutants of Rho1, Rac1, and Cdc42 in the Ptp4E Ptp10D background. Rho1-DN and Rac1-DN completely suppress the TC/LT phenotype, and Cdc42-DN and a tracheally expressed Rho1 RNAi construct produces partial suppression (Fig. 7G). The DN mutant data do not necessarily show that Rho and Rac are both required for generation of the phenotype. DN mutants may occlude binding of wild-type Rho family GTPases to their GEFs, some of which can act on both Rho and Rac. Therefore high-level expression of Rac-DN might inhibit Rho activation, and vice versa. However, the suppression of the TC/LT phenotype by Rho1 RNAi, which is a specific inhibitor, implicates Rho1 itself in the phenotype. Rho1-CA enhances the Ptp4E Ptp10D phenotype, producing cysts on most DBs (Figs 3B, 8C).
The differences in the abilities of the three RTK DN mutants to suppress the effects of Phl-CA and Rho-CA may provide clues to the pathways that act downstream of these kinases. The MAP kinase and Rho pathways are not necessarily independent, since Ras/MAP kinase pathway activation can increase Rho-GTP in some cell lines (Sahai et al., 2001). Also, each of the three RTKs is likely to activate both pathways to some extent, as well as many other downstream pathways. Defining the specific outputs of the MAP kinase and Rho pathways that control tracheal tube geometry and identifying other RTK pathways that regulate tube formation is likely to require genome-wide screens for suppressors and enhancers of the Ptp4E Ptp10D phenotype.
Supplementary Material
Acknowledgments
We thank Lea Goentoro for helpful discussions, and our colleagues in the Drosophila community for stocks and reagents. This work was supported by NIH RO1 grant NS28182 to K.Z., and by support from the Howard Hughes Medical Institute to M.P.S.
Footnotes
Competing interests: The authors declare that there are no competing interests.
References
- Affolter M., Caussinus E. (2008). Tracheal branching morphogenesis in Drosophila: new insights into cell behaviour and organ architecture. Development 135, 2055–2064 10.1242/dev.014498 [DOI] [PubMed] [Google Scholar]
- Amaya E., Musci T. J., Kirschner M. W. (1991). Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell 66, 257–270 10.1016/0092-8674(91)90616-7 [DOI] [PubMed] [Google Scholar]
- Arora D., Stopp S., Böhmer S. A., Schons J., Godfrey R., Masson K., Razumovskaya E., Rönnstrand L., Tänzer S., Bauer R. et al. (2011). Protein-tyrosine phosphatase DEP-1 controls receptor tyrosine kinase FLT3 signaling. J. Biol. Chem. 286, 10918–10929 10.1074/jbc.M110.205021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berset T. A., Hoier E. F., Hajnal A. (2005). The C. elegans homolog of the mammalian tumor suppressor Dep-1/Scc1 inhibits EGFR signaling to regulate binary cell fate decisions. Genes Dev. 19, 1328–1340 10.1101/gad.333505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N. (1994). Raf acts downstream of the EGF receptor to determine dorsoventral polarity during Drosophila oogenesis. Genes Dev. 8, 629–639 10.1101/gad.8.5.629 [DOI] [PubMed] [Google Scholar]
- Buff E., Carmena A., Gisselbrecht S., Jiménez F., Michelson A. M. (1998). Signalling by the Drosophila epidermal growth factor receptor is required for the specification and diversification of embryonic muscle progenitors. Development 125, 2075–2086. [DOI] [PubMed] [Google Scholar]
- Bugga L., Ratnaparkhi A., Zinn K. (2009). The cell surface receptor Tartan is a potential in vivo substrate for the receptor tyrosine phosphatase Ptp52F. Mol. Cell. Biol. 29, 3390–3400 10.1128/MCB.01764-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cela C., Llimargas M. (2006). Egfr is essential for maintaining epithelial integrity during tracheal remodelling in Drosophila. Development 133, 3115–3125 10.1242/dev.02482 [DOI] [PubMed] [Google Scholar]
- Chabot C., Spring K., Gratton J. P., Elchebly M., Royal I. (2009). New role for the protein tyrosine phosphatase DEP-1 in Akt activation and endothelial cell survival. Mol. Cell. Biol. 29, 241–253 10.1128/MCB.01374-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho N. K., Keyes L., Johnson E., Heller J., Ryner L., Karim F., Krasnow M. A. (2002). Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 108, 865–876 10.1016/S0092-8674(02)00676-1 [DOI] [PubMed] [Google Scholar]
- Duchek P., Somogyi K., Jékely G., Beccari S., Rørth P. (2001). Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 107, 17–26 10.1016/S0092-8674(01)00502-5 [DOI] [PubMed] [Google Scholar]
- Flint A. J., Tiganis T., Barford D., Tonks N. K. (1997). Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 94, 1680–1685 10.1073/pnas.94.5.1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais L., Casanova J. (2010). In vivo coupling of cell elongation and lumen formation in a single cell. Curr. Biol. 20, 359–366 10.1016/j.cub.2009.12.043 [DOI] [PubMed] [Google Scholar]
- Ghabrial A. S., Levi B. P., Krasnow M. A. (2011). A systematic screen for tube morphogenesis and branching genes in the Drosophila tracheal system. PLoS Genet. 7, e1002087 10.1371/journal.pgen.1002087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer L., Shilo B. Z. (1991). The Drosophila FGF-R homolog is expressed in the embryonic tracheal system and appears to be required for directed tracheal cell extension. Genes Dev. 5, 697–705 10.1101/gad.5.4.697 [DOI] [PubMed] [Google Scholar]
- Hower A. E., Beltran P. J., Bixby J. L. (2009). Dimerization of tyrosine phosphatase PTPRO decreases its activity and ability to inactivate TrkC. J. Neurochem. 110, 1635–1647 10.1111/j.1471-4159.2009.06261.x [DOI] [PubMed] [Google Scholar]
- Jeon M., Zinn K. (2009). Receptor tyrosine phosphatases control tracheal tube geometries through negative regulation of Egfr signaling. Development 136, 3121–3129 10.1242/dev.033597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M., Nguyen H., Bahri S., Zinn K. (2008). Redundancy and compensation in axon guidance: genetic analysis of the Drosophila Ptp10D/Ptp4E receptor tyrosine phosphatase subfamily. Neural Dev. 3, 3 10.1186/1749-8104-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johndrow J. E., Magie C. R., Parkhurst S. M. (2004). Rho GTPase function in flies: insights from a developmental and organismal perspective. Biochem. Cell Biol. 82, 643–657 10.1139/o04-118 [DOI] [PubMed] [Google Scholar]
- Johnson K. G., Van Vactor D. (2003). Receptor protein tyrosine phosphatases in nervous system development. Physiol. Rev. 83, 1–24. [DOI] [PubMed] [Google Scholar]
- Kappert K., Paulsson J., Sparwel J., Leppänen O., Hellberg C., Ostman A., Micke P. (2007). Dynamic changes in the expression of DEP-1 and other PDGF receptor-antagonizing PTPs during onset and termination of neointima formation. FASEB J. 21, 523–534 10.1096/fj.06-6219com [DOI] [PubMed] [Google Scholar]
- Kashles O., Yarden Y., Fischer R., Ullrich A., Schlessinger J. (1991). A dominant negative mutation suppresses the function of normal epidermal growth factor receptors by heterodimerization. Mol. Cell. Biol. 11, 1454–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klämbt C., Glazer L., Shilo B. Z. (1992). breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes Dev. 6, 1668–1678 10.1101/gad.6.9.1668 [DOI] [PubMed] [Google Scholar]
- Kumar J. P., Moses K. (2001). The EGF receptor and notch signaling pathways control the initiation of the morphogenetic furrow during Drosophila eye development. Development 128, 2689–2697. [DOI] [PubMed] [Google Scholar]
- Lampugnani M. G., Zanetti A., Corada M., Takahashi T., Balconi G., Breviario F., Orsenigo F., Cattelino A., Kemler R., Daniel T. O. et al. (2003). Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161, 793–804 10.1083/jcb.200209019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T., Hacohen N., Krasnow M., Montell D. J. (1996). Regulated Breathless receptor tyrosine kinase activity required to pattern cell migration and branching in the Drosophila tracheal system. Genes Dev. 10, 2912–2921 10.1101/gad.10.22.2912 [DOI] [PubMed] [Google Scholar]
- Lesokhin A. M., Yu S. Y., Katz J., Baker N. E. (1999). Several levels of EGF receptor signaling during photoreceptor specification in wild-type, Ellipse, and null mutant Drosophila. Dev. Biol. 205, 129–144 10.1006/dbio.1998.9121 [DOI] [PubMed] [Google Scholar]
- Matozaki T., Murata Y., Mori M., Kotani T., Okazawa H., Ohnishi H. (2010). Expression, localization, and biological function of the R3 subtype of receptor-type protein tyrosine phosphatases in mammals. Cell. Signal. 22, 1811–1817 10.1016/j.cellsig.2010.07.001 [DOI] [PubMed] [Google Scholar]
- Morrison D. K., Murakami M. S., Cleghon V. (2000). Protein kinases and phosphatases in the Drosophila genome. J. Cell Biol. 150, F57–F62 10.1083/jcb.150.2.F57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj R., Pickup A. T., Howes R., Moses K., Freeman M., Banerjee U. (1999). Role of the EGF receptor pathway in growth and patterning of the Drosophila wing through the regulation of vestigial. Development 126, 975–985. [DOI] [PubMed] [Google Scholar]
- Oon S. H., Hong A., Yang X., Chia W. (1993). Alternative splicing in a novel tyrosine phosphatase gene (DPTP4E) of Drosophila melanogaster generates two large receptor-like proteins which differ in their carboxyl termini. J. Biol. Chem. 268, 23964–23971. [PubMed] [Google Scholar]
- Palka H. L., Park M., Tonks N. K. (2003). Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J. Biol. Chem. 278, 5728–5735 10.1074/jbc.M210656200 [DOI] [PubMed] [Google Scholar]
- Reichman-Fried M., Shilo B. Z. (1995). Breathless, a Drosophila FGF receptor homolog, is required for the onset of tracheal cell migration and tracheole formation. Mech. Dev. 52, 265–273 10.1016/0925-4773(95)00407-R [DOI] [PubMed] [Google Scholar]
- Ruivenkamp C. A., van Wezel T., Zanon C., Stassen A. P., Vlcek C., Csikós T., Klous A. M., Tripodis N., Perrakis A., Boerrigter L. et al. (2002). Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat. Genet. 31, 295–300 10.1038/ng903 [DOI] [PubMed] [Google Scholar]
- Sahai E., Olson M. F., Marshall C. J. (2001). Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 20, 755–766 10.1093/emboj/20.4.755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samakovlis C., Hacohen N., Manning G., Sutherland D. C., Guillemin K., Krasnow M. A. (1996). Development of the Drosophila tracheal system occurs by a series of morphologically distinct but genetically coupled branching events. Development 122, 1395–1407. [DOI] [PubMed] [Google Scholar]
- Schiller M. R. (2006). Coupling receptor tyrosine kinases to Rho GTPases-GEFs what’s the link. Cell. Signal. 18, 1834–1843 10.1016/j.cellsig.2006.01.022 [DOI] [PubMed] [Google Scholar]
- Scholz H., Sadlowski E., Klaes A., Klämbt C. (1997). Control of midline glia development in the embryonic Drosophila CNS. Mech. Dev. 64, 139–151 10.1016/S0925-4773(97)00078-6 [DOI] [PubMed] [Google Scholar]
- Schonbaum C. P., Organ E. L., Qu S., Cavener D. R. (1992). The Drosophila melanogaster stranded at second (sas) gene encodes a putative epidermal cell surface receptor required for larval development. Dev. Biol. 151, 431–445 10.1016/0012-1606(92)90183-H [DOI] [PubMed] [Google Scholar]
- Sun Q., Bahri S., Schmid A., Chia W., Zinn K. (2000). Receptor tyrosine phosphatases regulate axon guidance across the midline of the Drosophila embryo. Development 127, 801–812. [DOI] [PubMed] [Google Scholar]
- Swanson L. E., Beitel G. J. (2006). Tubulogenesis: an inside job. Curr. Biol. 16, R51–R53 10.1016/j.cub.2006.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szüts D., Freeman M., Bienz M. (1997). Antagonism between EGFR and Wingless signalling in the larval cuticle of Drosophila. Development 124, 3209–3219. [DOI] [PubMed] [Google Scholar]
- Tarcic G., Boguslavsky S. K., Wakim J., Kiuchi T., Liu A., Reinitz F., Nathanson D., Takahashi T., Mischel P. S., Ng T. et al. (2009). An unbiased screen identifies DEP-1 tumor suppressor as a phosphatase controlling EGFR endocytosis. Curr. Biol. 19, 1788–1798 10.1016/j.cub.2009.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S. S., Tsoulfas P., Zinn K. (1991). Three receptor-linked protein-tyrosine phosphatases are selectively expressed on central nervous system axons in the Drosophila embryo. Cell 67, 675–685 10.1016/0092-8674(91)90063-5 [DOI] [PubMed] [Google Scholar]
- Tonks N. K. (2006). Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 10.1038/nrm2039 [DOI] [PubMed] [Google Scholar]
- Winderlich M., Keller L., Cagna G., Broermann A., Kamenyeva O., Kiefer F., Deutsch U., Nottebaum A. F., Vestweber D. (2009). VE-PTP controls blood vessel development by balancing Tie-2 activity. J. Cell Biol. 185, 657–671 10.1083/jcb.200811159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. H., Seow K. T., Bahri S. M., Oon S. H., Chia W. (1991). Two Drosophila receptor-like tyrosine phosphatase genes are expressed in a subset of developing axons and pioneer neurons in the embryonic CNS. Cell 67, 661–673 10.1016/0092-8674(91)90062-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}