

Vps33, a member of the Sec1/Munc18 family of soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) chaperones, is a subunit of the homotypic fusion and protein sorting and class C core vacuole/endosome tethering complexes and essential for endolysosomal transport. In this study, Vps33 interactions with SNARE proteins are investigated using genetic and biochemical approaches.
Abstract
Soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) proteins catalyze membrane fusion events in the secretory and endolysosomal systems, and all SNARE-mediated fusion processes require cofactors of the Sec1/Munc18 (SM) family. Vps33 is an SM protein and subunit of the Vps-C complexes HOPS (homotypic fusion and protein sorting) and CORVET (class C core vacuole/endosome tethering), which are central regulators of endocytic traffic. Here we present biochemical studies of interactions between Saccharomyces cerevisiae vacuolar SNAREs and the HOPS holocomplex or Vps33 alone. HOPS binds the N-terminal Habc domain of the Qa-family SNARE Vam3, but Vps33 is not required for this interaction. Instead, Vps33 binds the SNARE domains of Vam3, Vam7, and Nyv1. Vps33 directly binds vacuolar quaternary SNARE complexes, and the affinity of Vps33 for SNARE complexes is greater than for individual SNAREs. Through targeted mutational analyses, we identify missense mutations of Vps33 that produce a novel set of defects, including cargo missorting and the loss of Vps33-HOPS association. Together these data suggest a working model for membrane docking: HOPS associates with N-terminal domains of Vam3 and Vam7 through Vps33-independent interactions, which are followed by binding of Vps33, the HOPS SM protein, to SNARE domains and finally to the quaternary SNARE complex. Our results also strengthen the hypothesis that SNARE complex binding is a core attribute of SM protein function.
INTRODUCTION
Fusion, the final step of membrane trafficking between cellular compartments, requires the coordinated action of multiple conserved protein families, including soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs), Rab small G proteins, tethering proteins, and SNARE cofactors, including the Sec1/Munc18 (SM) proteins. A typical docking and fusion sequence entails activation of Rab proteins and recruitment of tethering factors. After tethering, three Q-SNARE domains (designated Qa, Qb, Qc) and one R-SNARE assemble into a coiled-coil trans-complex spanning the docked membranes. trans-SNARE complex assembly is thought to drive membranes together, initiating a merger of the apposed bilayers and intermixing of lumenal contents. Following fusion, the universal factors Sec17/α-SNAP (α–soluble N-ethylmaleimide–sensitive factor attachment protein) and Sec18/NSF (N-ethylmaleimide–sensitive factor) disassemble cis-SNARE complexes (for a review, see Zhao et al., 2007).
Despite intense efforts, the roles of SM proteins in these processes are incompletely understood. Genetic ablation or biochemical interference with SM function typically results in cessation of traffic through one or more pathways (Sudhof and Rothman, 2009). In vitro, SM proteins stimulate fusion of liposomes reconstituted with SNARE proteins (Scott et al., 2004; Shen et al., 2007). At the biochemical level, SM proteins have divergent modes of association with SNAREs (Carr and Rizo, 2010). It remains unclear how these biochemical properties are linked to the nearly absolute requirement for SM function that prevails in vivo.
SM proteins differ in their ability to bind cognate Qa-SNAREs. Sly1 interacts with its cognate Qa-SNARE by binding an N-terminal motif called the N-peptide (Peng and Gallwitz, 2002, 2004; Yamaguchi et al., 2002). Vps45 and Munc18 also bind their cognate Qa-SNAREs through interactions involving the N-peptide. Primarily, this interaction is with the “closed” conformation of the Qa-SNARE, in which the trihelical N-terminal Habc domain folds back onto the SNARE domain (Dulubova et al., 2002). In these cases, the SM protein touches the N-peptide, the Habc domain, and the SNARE domain. Although it has been hypothesized that N-peptide engagement might allosterically activate SM proteins, recent work suggests that a major function of N-peptide binding is simple recruitment of the SM to the fusion site (Rathore et al., 2010; Shen et al., 2010). In contrast, other SM proteins, including Sec1 and, as shown in the present study, Vps33, do not engage their cognate Qa-SNAREs with high affinity. Furthermore, these Qa-SNAREs lack the N-peptide consensus sequence (Hu et al., 2007). Sec1 has primary affinity for the SNARE core complex and lesser affinities for individual SNAREs (Togneri et al., 2006; Morgera et al., 2011). In lieu of N-peptide binding, these SMs are probably recruited to SNAREs through alternative mechanisms. For example, Sec1 binds the Sec6 subunit of the exocyst tethering complex, suggesting that exocyst positions Sec1 at sites of exocytosis (Morgera et al., 2011). Another Sec1-binding protein, Mso1, also interacts with the Qa-SNARE Sso1 and contributes to correct Sec1 localization (Knop et al., 2005).
Relative to other SM proteins, the endolysosomal SM Vps33 is poorly understood. Direct biochemical characterization of Vps33 has proven challenging due to its critical roles within at least two highly conserved Vps-C tethering complexes, HOPS (homotypic fusion and protein sorting) and CORVET (class C core vacuole/endosome tethering; for review see Nickerson et al., 2009). In vivo and in vitro studies indicate that HOPS promotes tethering, docking, and fusion at the terminal lysosomal vacuole and further suggest that HOPS may proofread trans-SNARE complexes and shield trans-complexes from disassembly (Starai et al., 2008; Stroupe et al., 2009; Hickey and Wickner, 2010; Xu et al., 2010a). The specific contributions of the six HOPS subunits to each of these functions are not understood.
It has been unclear whether previously reported interactions between HOPS and SNAREs are mediated directly by Vps33, or by the other five HOPS subunits. Cell-free assays demonstrated that the Habc domain of the Qa-SNARE Vam3 promotes HOPS recruitment to the vacuole. However, mutational analyses suggested that Vps18, not Vps33, is necessary for HOPS binding to Vam3 (Laage and Ungermann, 2001). Moreover, overproduced Vps33 in yeast cell lysates binds the Vam3 SNARE domain but not the N-terminal domain (Dulubova et al., 2001). In apparent contrast to these studies, an interaction between Vps33 and the Vam3 Habc domain was postulated to occur (Pieren et al., 2010). Most recently, purified Vps33 was reported to bind preassembled 3Q- and 3Q:R-SNARE complexes but not individual SNAREs (Kramer and Ungermann, 2011). Purified HOPS was also reported to have affinity for the N-terminal PX-domain of Vam7 and weaker affinities for Vam3 and Nyv1 (Stroupe et al., 2006). In the present study, we reexamined the SNARE-binding properties of Vps33, both in isolation and in the context of the native HOPS complex. Our findings unify much of the previous literature on interactions among SNAREs, HOPS, and the SM protein Vps33 and suggest a working model for HOPS-SNARE interactions.
RESULTS
HOPS has multiple binding sites for the Qa-SNARE Vam3
To evaluate interactions between the Qa-SNARE Vam3 and native Saccharomyces cerevisiae HOPS (Figure 1A), we expressed and purified portions of Vam3 fused C-terminally to glutathione S-transferase (GST). Affinity-capture experiments were then performed using lysates from wild-type cells or lysates from deletion mutants lacking specific HOPS subunits. HOPS from wild-type cell lysates bound the full Vam3 cytoplasmic domain and the Habc domain (Figure 1B). HOPS derived from lysates lacking the Rab-binding subunits Vps39 or Vps41 still bound Vam3 Habc, indicating that Vps39 and Vps41 are not required for the Vam3-HOPS interaction (Supplemental Figure S1). Surprisingly, in the absence of Vps33 (vps33∆), HOPS still bound the Vam3 Habc domain, indicating that a HOPS subunit other than Vps33 mediates Habc binding. A partial loss of HOPS binding to the full Vam3 cytoplasmic domain in the absence of Vps33 led us to hypothesize that Vps33 cooperates with other HOPS subunits in binding Vam3 through its Habc and SNARE domains.
FIGURE 1:

HOPS has multiple interaction sites for Vam3. (A) Model of subunit interactions in HOPS of wild-type and vps16∆ cells. In vps16∆, Vps33 is separated from the remainder of HOPS. (B) Binding of native HOPS to GST-tagged Vam3 cytoplasmic domains. Equivalent loads (∼3.5 μM) of each C-terminally GST-tagged fusion protein (25–100 μg depending on the molecular weight) were incubated with glutathione sepharose (GSH) resin: Vam3 (residues 1–264), Habc (residues 1–145), linker (residues 116–186), and SNARE (residues 182–264). GST and Sso1 (residues 1–265) were negative controls. Coomassie Blue–stained gel of the of GST fusion proteins shown in (D). Yeast detergent lysate was incubated with GST fusion proteins for 2 h at 4°C, washed extensively, and eluted with 20 mM glutathione. Samples were analyzed by SDS–PAGE and Western blotting (WB) for Vps41, Vps11, and Vps33. (C) Size-exclusion chromatography shows that purified Vps33 migrates as expected for a monomer. Coomassie blue–stained SDS–PAGE of the pooled eluate. The faint band at 100 kDa is residual uncleaved GST-Vps33, which was removed prior to pull down by preincubating purified Vps33 with GSH resin for 1 h at 23°C. (D) 1 μg (∼25 nM) of purified Vps33 was incubated with Vam3 GST constructs, as in (B), and analyzed by SDS–PAGE and Western blotting for Vps33.
In vps16∆-null mutants, Vps33 cannot associate with the remainder of HOPS (Figure 1A; Rieder and Emr, 1997). In pull downs from vps16∆ cell lysates, Vps33 and HOPS had distinct binding profiles: Vps33 was retained on the Vam3 SNARE domain, while the remainder of HOPS was retained on the Vam3 Habc domain. Deletion of either Vps11 or Vps18 strongly reduced pull down of the remaining subunits with the Vam3 Habc from yeast detergent lysate (Figure S1). In addition to the interaction between HOPS and the Vam3 Habc domain, we detected weak interactions between HOPS and the Vam3 SNARE domain in the absence of Vps33 (Figure 1B). While this might be nonspecific binding, a recent report of interactions between the HOPS subunits Vps16 and Vps18 and vacuole SNAREs raises the possibility that HOPS subunits other than Vps33 contact SNARE domains (Kramer and Ungermann, 2011).
To test whether Vps33 directly binds the Vam3 SNARE domain, we expressed and purified S. cerevisiae Vps33 from insect cells (Figure 1C). In binding experiments, the purified Vps33 recapitulated results obtained with yeast cell lysates: Vps33 bound Vam3 directly through its SNARE domain (Figure 1D). When the binding conditions for the yeast lysate pull downs (2 h at 4°C) were used, purified Vps33 exhibited slight binding to the plasma membrane Qa-SNARE Sso1 (Figure 1D). With optimized binding conditions for purified Vps33 (2 h at 30°C), the Vam3-Vps33 interaction was more efficient, and there was no detectable binding to Sso1 or to two other noncognate SNAREs, Sed5 and Sec22 (Figures 2, B and D, S2, and S4). Taken together, the data indicate that HOPS has at least two Vam3-binding sites. Vps11 and Vps18 are necessary for binding the Vam3 Habc domain, while Vps33 binds the Vam3 SNARE domain. This working model is supported by the present results and most previous experiments addressing Vam3-HOPS interactions. We have so far been unable to detect binding of isolated Vps33, native or purified, to the Vam3 N-terminal Habc domain (Pieren et al., 2010).
FIGURE 2:

Purified Vps33 binds Qa-, Qc-, and R-SNAREs, and vacuole SNARE complexes. (A) Pull down of purified Vps33 by cytoplasmic domains of vacuole SNAREs or quaternary vacuole SNARE complex. Equivalent loads of GST fusion proteins (∼1 μM) were bound to GSH resin. Vacuole SNARE complexes were formed using 29 μg (∼1 μM) purified Vam3-GST cytoplasmic and purified cytoplasmic domains of His7-MBP-Vti1, His6-Vam7, and His6-Nyv1. Purified Vps33 (10.5 μg, ∼0.25 μM) was incubated for 2 h at 30°C with GST fusion proteins, washed extensively, and eluted with 20 mM glutathione. Samples were separated by SDS–PAGE and analyzed by Western blotting for Vps33. (B) Pull down of purified Vps33 by SNARE domains. Eighteen micrograms (∼1 μM) of the SNARE domains of Vam3 (residues 182–264), Vam7 (residues 190–316), and Nyv1 (residues 162–231) fused N-terminally to GST were used to pull down purified Vps33. GST and the SNARE domain of Sed5 (residues 170–319) were used as negative controls. Cleavage products of the Nyv1 SNARE domain are indicated (*). Pull downs with 10.5 μg (∼0.25 μM) of purified Vps33 were performed as described in (A). (C) Comparison of purified Vps33 pull down by Golgi or vacuole SNARE complexes. Vacuole SNARE complexes were formed as previously described. His6 fusions to cytoplasmic domains of Bos1, Bet1, and Sec22 were used to form complexes with 22 μg (1 μM) GST-tagged Sed5 SNARE domain (residues 170–319). Ten and one-half micrograms (∼0.25 μM) of purified Vps33 was used in the pull downs as in (A). (D) Pull downs of purified Vps33 with GST fusions to cytoplasmic domains of Nyv1 and Ykt6. Sec22 and GST were used as negative controls. SNARE complexes were formed as described for 3Q:R-Nyv1. For 3Q:R-Ykt6 complexes, a 2.5-fold greater amount of Vam3-GST was used to compensate for decreased SNARE complex formation with Ykt6. Pull downs with 10.5 μg (∼0.25 μM) of purified Vps33 were performed as described in (A). (E) Pull down of 70 μg (∼5 μM) Vps33 by GST, 12.8 μg (∼2 μM) Vam3 SNARE domain (residues 182–264), or ∼2 μM minimal SNARE complex lacking the N-terminal domains of Vam3 and Vam7. (F) Binding curve for Vps33 to the minimal SNARE complex. A one-site binding model fit to data from three replicates results in an apparent Kd of 2.8 ± 0.2 μM (mean ± SEM; r2 = 0.98).
Vps33 binds vacuolar Qa-, Qc-, and R-SNAREs and the quaternary SNARE complex
Interactions between SM proteins and individual non–Qa-SNAREs have been reported for Sly1, Vps45, Sec1, and Munc18 (Peng and Gallwitz, 2004; Xu et al., 2010b; Morgera et al., 2011). To test whether purified Vps33 directly binds vacuole SNAREs other than Vam3 (Qa), we prepared GST fusions to the cytoplasmic domains of Vti1 (Qb), Vam7 (Qc), and Nyv1 (R). We found (Figures 2A and S2) that purified Vps33 bound Vam3, Vam7, and Nyv1, but not Vti1 or GST alone. Because Vps33 binds Vam3 through its SNARE domain, we tested whether purified Vps33 could also bind SNARE domains of Vam7 and Nyv1, and observed direct binding to these domains (Figure 2B). We further tested whether Vps33 is responsible for the reported interaction between HOPS and the N-terminal PX domain of Vam7 (Stroupe et al., 2006). Although purified Vps33 bound full-length Vam7 and Vam7 SNARE domain, Vps33 did not bind to the Vam7 PX domain (Figure S4). It was previously reported that Vps33 could bind the late endosomal Qa-SNARE Pep12 (Subramanian et al., 2004). While little of the HOPS in a wild-type cell lysate was retained on Pep12 when compared with Vam3, purified Vps33 was retained equally on Pep12-GST and Vam3-GST resins (Figure S3, A and B).
Direct SM binding to SNARE complexes has been reported for Sec1, Vps45, and metazoan Munc18 (Carpp et al., 2006; Togneri et al., 2006; Xu et al., 2010b). To ascertain whether Vps33 binds SNARE complexes, quaternary complexes containing the cytoplasmic domains of Vti1, Vam7, and Nyv1 were assembled on solid supports bearing the Vam3-GST. Importantly, Vps33 bound quaternary 3Q:R-SNARE complexes, and was retained on SNARE complexes more efficiently than on any individual SNARE (Figure 2, A, D, and E; see Figures S2 and S4 for additional examples). To evaluate the selectivity of Vps33 binding to the vacuole SNARE complex, we assembled Golgi quaternary SNARE complexes containing the GST-Sed5 SNARE domain and cytoplasmic domains of Bos1, Bet1, and Sec22. Vps33 bound selectively to vacuole versus Golgi SNARE complex (Figure 2C).
The vacuole has two resident R-SNAREs. Nyv1 is thought to play a primary role in homotypic vacuole fusion, while Ykt6 functions in heterotypic fusion (Ungermann et al., 1999; Dilcher et al., 2001). To examine the ability of Vps33 to directly bind to Nyv1 or Ykt6, we prepared GST fusions to the full cytoplasmic domains of these R-SNAREs. Sec22, a Golgi R-SNARE, was used as negative control. Vps33 was retained on Ykt6 to a greater degree than the negative controls, though less efficiently than on Nyv1 (Figure 2D). Purified Vps33 bound vacuolar SNARE complexes containing Nyv1 or Ykt6 with comparable efficiency.
To compare the relative affinities of Vps33 for an individual SNARE domain or the quaternary SNARE complex, we examined Vps33 binding to the SNARE domain of Vam3 or a minimal SNARE complex lacking the N-terminal domains of Vam3 and Vam7. Using a higher concentration of Vps33 (5.0 μM in Figure 2E compared with 0.25 μM in Figure 2, A–C and F), we were readily able to detect binding of Vps33 to the minimal SNARE complex using Coomassie Blue–stained polyacrylamide gels (Figure 2E). Less Vps33 was retained by the Vam3 SNARE domain compared with the minimal SNARE complex, consistent with the interaction between Vps33 and the Vam3 SNARE domain being of lower affinity (Figure 2E). By assaying binding over a range of Vps33 concentrations, we estimated the dissociation constant for Vps33 and the minimal SNARE complex as ∼2.8 ± 0.2 μM (Figure 2F). This low micromolar apparent Kd falls between affinities reported for SNARE complex binding by Sec1 (∼0.3 μM) and Munc18 (∼6 μM; Togneri et al., 2006; Xu et al., 2010b). We were unable to measure an apparent dissociation constant for Vps33 and individual SNAREs due to the lower affinity of these interactions and instability of Vps33 at higher concentrations. Despite their relatively lower affinity, the interactions between Vps33 and monomeric SNARE domains are selective (Figure 2, A, B, and D; see Figures S2 and S4 for additional examples), and it is plausible that interactions between Vps33 and individual SNAREs contribute to the SNARE assembly cycle in vivo (see Discussion).
Vps33 binding sites within Qa- and R-SNARE domains
SNARE domains share a conserved architecture, with repeating hydrophobic packing layers and a central “zero” layer comprising polar (Q) and ionic (R) residues (Fasshauer et al., 1998). Negative numbering denotes packing layers N-terminal to the zero layer, while positive numbers are ascribed to the C-terminal (membrane-proximal) layers. Alignments of SNARE domains show substantial conservation of packing layers −8 to +8, but for clarity we have used the layer-based numbering system for our truncations outside this region. To map the interaction of Vps33 with Vam3 (Qa), we assayed binding of purified Vps33 to truncated Vam3 SNARE domains. When truncating from the N-terminal side of the Vam3 SNARE domain, Vps33 binding was lost upon truncation from layer −7 to layer −5 (Figure 3A). In the C-terminal truncation series (starting from the equivalent of layer +9), Vps33 binding decreased as the SNARE domain was truncated from layer +5 to +3, and Vps33 binding was lost when Vam3 was truncated to layer −5. The determinants for Vps33 binding reside within Vam3 SNARE layers −7 to +5. While it is possible that truncations of the Vam3 SNARE domain alter the conformation or presentation of the SNARE motif, like other Qa-SNARE domains, the Vam3 SNARE domain is largely unstructured in solution (Dulubova et al., 2001). Thus we define the outside boundaries of Vps33 binding site in Vam3 (i.e., the region bounded by layers −7 to +5) using only those truncation constructs that showed no loss of Vps33 binding.
FIGURE 3:

Mapping of Vps33-binding determinants on Vam3 and Nyv1 SNAREs. (A). Vam3 SNARE domain (residues 182–264) was fused to GST at the Vam3 C-terminus for N-terminal truncations and the N-terminus for the C-terminal truncations. Eighteen micrograms (∼1 μM) of GST fusion protein was bound to GSH resin, incubated with 10.5 μg (∼0.25 μM) of purified Vps33 for 2 h at 30°C, washed three times with SNARE Assay buffer, and eluted with SA buffer supplemented with 20 mM glutathione. Eluates were separated by SDS–PAGE and analyzed by Western blotting for Vps33. (B) Nyv1 (residues 1–231) cytoplasmic domain and the C-terminal truncation series were fused to GST at its N-terminus. Thirty micrograms (∼1 μM) of purified GST fusion protein was incubated with GSH resin. Ten and one-half micrograms (∼0.25 μM) of purified Vps33 was incubated with the GSH resin for 2 h at 30°C, washed three times with binding buffer, and eluted. Bound fractions and 2% of the load relative to the bound fractions were separated by SDS–PAGE and analyzed by Western blotting for Vps33.
Munc18 interacts with the SNARE domain of the R-SNARE synaptobrevin at the C-terminal juxtamembrane region, making contacts within layers +8 to +9 (Xu et al., 2010b). We asked whether truncation of the equivalent region of the vacuole R-SNARE Nyv1 would reduce Vps33 binding. Vps33 binding decreased when Nyv1 was truncated at its C-terminus to layer +9, and decreased further upon truncation from layer +7 to layer +5 (Figure 3B). We infer that residues required for Vps33 binding lie within the membrane-proximal region of the Nyv1 SNARE domain.
Phenotypic analyses of Vps33 point mutations
To further investigate the role of Vps33 in vacuole maintenance and fusion, we constructed VPS33 alleles encoding missense mutations that alter conserved amino acids and that have been identified as causing phenotypic defects in Vps33 or other SM proteins (Dascher et al., 1991; Sevrioukov et al., 1999; Suzuki et al., 2003; Gissen et al., 2004; Li et al., 2007; Hashizume et al., 2009). Evolutionary conservation of the targeted amino acid positions was verified using published and in-house sequence alignments (Gissen et al., 2004; Pieren et al., 2010; Table 1). Only an isoleucine residue mutated in a human patient with Hermansky-Pudlak syndrome falls at a position not identical to yeast Vps33. Instead of isoleucine, yeast Vps33 has phenylalanine at the equivalent position (Suzuki et al., 2003). In addition to the yeast Vps33 analogue of the mouse buff (bf) mutation, D300E, we also tested a glycine substitution at the same position (D300G).
TABLE 1:
Summary of Vps33 mutants used in this study.
| Vps33 mutant | Species/SM family member | Homologue/orthologue mutation | Name | Phenotype | Reference |
|---|---|---|---|---|---|
| L75P | Human/VPS33B | L30P | ARC syndrome | Multisystem disorder affecting the kidneys, liver, nervous system, and platelet function | Gissen et al., 2004 |
| D88K | Yeast/VPS33 | D88K | Fusion defect | Defect in content mixing and fusion | Pieren et al., 2010 |
| I278N | Yeast/SEC1 | I249N | Sec1-36 | Defects in growth and SNARE complex binding at restrictive temperature | Hashizume et al., 2009 |
| R281A | Yeast/SEC1 | R252A | Sec1-78 | No growth defect | Hashizume et al., 2009 |
| G297V | Fly/VPS33A | G249V | carnation | Defects in endocytic trafficking and pigment granules | Sevrioukov et al., 1999 |
| D300E/G | Mouse/VPS33A | D251E | buff | Defects in melanosome morphology, hypopigmentation, and a mild platelet-storage pool deficiency; as the mice age, worsening motor defects, loss of Purkinje cells, and reduction in size of the cerebellum | Suzuki et al., 2003; Chintala et al., 2009 |
| F305L | Human/VPS33A | I256L | HPS syndrome | Defects in the formation and maintenance of melanosomes, platelet-dense granules, and lysosomes result in albinism, bleeding disorder, and pulmonary fibrosis | Suzuki et al., 2003 |
| T553I | Yeast/SLY1 | T531I | Suppressor of Lethality of Ypt1 | Suppressor of Lethality of Ypt1 | Li et al., 2007 |
| E653A | Yeast/SEC1 | E604A | Sec1-50 | Defects in growth and SNARE complex binding at restrictive temperature | Hashizume et al., 2009 |
Yeast vacuole morphology is classified as A (wild-type), B (partially fragmented), C (severely fragmented), D (enlarged), and E (accumulation of a novel compartment adjacent to the vacuole; Raymond et al., 1992). Loss of Vps33 causes vacuolar cargo missorting, class C vacuole morphology, and a failure to grow at nonpermissive temperature (37°C; Banta et al., 1990). Based on these parameters, the phenotypic defects of the 10 Vps33 point mutations examined here ranged from no detected deficiency to the equivalent of a vps33∆-null mutant. The results from growth, trafficking, vacuole morphology, and Vps33-GFP localization assays are summarized in Table 2 and described below (Figures 4 and 5). Based on the results from these assays, the 10 mutants fall into four categories, referred to here as classes A, A´, B, and C.
TABLE 2:
Summary of phenotypic effects of Vps33 mutations on growth, vacuole morphology, and Vps33-GFP localization.
| Vps33 mutant | Growth at 30°C | Growth at 37°C | Growth at 37°C + 5 mM ZnCl2 | Cargo missorting at 30°C | Vacuole morphology at 30°C |
|---|---|---|---|---|---|
| vps33∆ | √ | No | No | Severe | C |
| WT | √ | √ | √ | None | A |
| L75P | √ | √ | No | None | A |
| D88K | √ | √ | √ | Detectable | A´ |
| I278N | √ | √ | Slow | Detectable | A´ |
| R281A | √ | No | No | Severe | C |
| G297V | √ | √ | No | Moderate | B |
| D300E | √ | √ | √ | None | A |
| D300G | √ | √ | No | Detectable | A´ |
| F305L | √ | √ | √ | None | A |
| T553I | √ | √ | Slow | Detectable | A´ |
| E653A | √ | No | No | Severe | C |
Four of the 10 mutants had a novel collection of phenotypic defects intermediate to class A and class B here referred to as class A´. While D88K, I278N, D300G, and T553I resembled the wild-type strain in growth at 30 and 37°C, these alleles showed slow or no growth at 37°C on media supplemented with 5 mM ZnCl2 (Figure 4A). Yeast with a wild-type copy of Vps33 showed no defect when grown under the same conditions. Furthermore, class A´ mutants had identifiable defects in trafficking of both CPY and ALP (Figure 4B). Although the vacuole morphology of class A´ mutants appeared similar to the wild-type, Vps33-GFP localization was cytoplasmic, rather than punctate, in these mutants, as described below (Figure 5).
FIGURE 4:
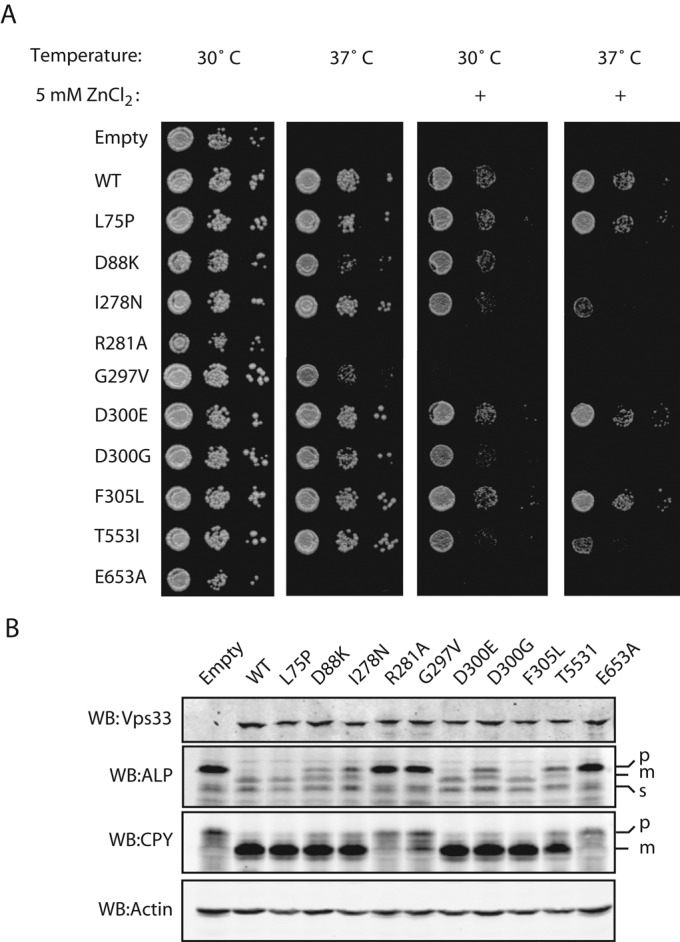
Characterization of Vps33 point mutants. (A) Cultures grown overnight at 24°C were plated onto SC-URA, YPD, or YPD + 5 mM ZnCl2 as 20-fold dilutions from 1.0 OD600 × ml. Plates were incubated for 48 h and imaged. (B) Lysates were prepared from SEY6210 cells expressing VPS33WT-ttx-GFP and each of the mutant alleles. Cell lysates were separated by SDS–PAGE and analyzed by Western blotting.
We saw no observable defects with three alleles associated with disease phenotypes in Vps33 orthologues: L75P, D300E, and F305L. Two of these encoded analogues of disease mutations in human VPS33B and VPS33A that cause ARC (arthrogryposis, renal dysfunction, and cholestasis) and HPS (Hermansky-Pudlak syndrome), respectively. For F305L, the yeast Vps33 analogue of the HPS mutation I256L in human VPS33a, the lack of conservation suggests a rationale for the absence of defects in the yeast F305L mutant. It is less clear why L75P has no effect in yeast despite the L30P mutation in human VPS33B causing multisystem disorders, although the L30P/L75P mutation falls within a region of Vps33 that is less well conserved. The amino acid mutated in the bf mouse is conserved in S. cerevisiae Vps33, but we observed no phenotypic defects from the analogous D300E mutation. We did observe phenotypic defects with a D300G mutation, indicating that the importance of this residue, if not its precise sensitivity to mutation, is conserved between mouse VPS33A and yeast Vps33.
Defects in growth and cargo sorting.
GFP-tagged Vps33 mutants were expressed in a vps33∆ background from single-copy plasmids under the native VPS33 promoter. As some vps33 mutants are temperature-sensitive for growth (Banta et al., 1990), we asked whether any of the 10 mutants produced similar sensitivity (Figure 4A). The vps33∆ mutant and two class C mutations, R281A and E653A, failed to grow at 37°C. Defects in vacuole biogenesis cause sensitivity to extracellular Zn2+, likely due to a failure to sequester metal ions within the vacuole (Subramanian et al., 2004). To test for more subtle defects in vacuole function, we stressed the cells by growing them in the presence of Zn2+ (Figure 4A). While the majority of Vps33 mutants had no growth defects at 37°C, several grew slowly or lost viability when grown at 37°C in the presence of 5 mM ZnCl2: the class A´ mutants D88K, I278N, D300G, and T553I (Figure 4A). This was not a universal effect of growth at 37°C in the presence of Zn2+, as strains containing wild-type Vps33 or the class A Vps33 mutants L75P, D300E, or F305L grew normally. The class C mutants R281A and E653A lost viability when grown 30°C with Zn2+. The class B mutant and carnation analogue G297V had strong growth defects at 37°C, and at 30° or 37°C in the presence of Zn2+ (Figure 4A).
Carboxypeptidase Y (CPY) traffics from the Golgi to the vacuole via endosomes, while alkaline phosphatase (ALP) traffics directly from the Golgi to the vacuole. As CPY and ALP take independent routes to the vacuole, it is possible to selectively impair one pathway, while leaving the other unaltered (Bowers and Stevens, 2005). Defects in trafficking to the vacuole result in accumulation of proenzymes (p) relative to active mature (m) forms. For further characterization of the vps33 alleles, detergent lysates from cells grown at 30°C were analyzed for Vps33 protein levels and defects in cargo transport (Figure 4B). While Vps33 levels were similar in each of the 10 strains, ALP and CPY maturation defects varied greatly among the mutants (Figure 4B). Consistent with the growth data, strains with a wild-type copy of VPS33 or class A mutations had normal levels of mature ALP and CPY. Partial accumulation of proALP and proCPY were noted in class A´ strains. Vps33-G297V, the sole class B mutant, had an intermediate cargo maturation phenotype: most of its ALP accumulated in the pro-form, but there was partial maturation of CPY. Strains lacking a copy of Vps33 or carrying class C Vps33 mutant alleles R281A or E653A failed to mature both CPY and ALP.
Defects in vacuole morphology and Vps33-GFP localization.
We next examined vacuole morphology and Vps33 localization in each of the mutants. The vacuole was visualized by pulse-chase labeling with the endocytic tracer FM4-64. In a strain carrying wild-type VPS33, vacuoles had normal (class A) morphology and Vps33-GFP accumulated in punctae adjacent to the vacuole, as expected (Figure 5; Wang et al., 2002; Jun et al., 2006). In accord with data from growth and trafficking assays, vacuole morphology and Vps33-GFP localization in L75P, D300E, and F305L mutants were indistinguishable from wild-type Vps33. In strains lacking Vps33, extreme fragmentation of the vacuole (class C morphology) was observed (Banta et al., 1990). Strains carrying the R281A or E653A mutations, which phenocopied the vps33∆ null in growth and cargo maturation assays, had class C morphology and lacked clearly identifiable Vps33-GFP punctae. The G297V mutation had partially fragmented, or class B, vacuole morphology without clear Vps33-GFP punctae. The class A´ mutants (D88K, I278N, D300G, and T553I) had normal, class A, vacuole morphology. Surprisingly, in these four class A´ strains, the fluorescent Vps33-GFP punctae observed in wild-type cells were absent. Instead, Vps33-GFP had a diffuse cytosolic localization.
FIGURE 5:

Vacuole morphology and Vps33-GFP localization. Strains were grown in minimal media to mid–log phase, and vacuoles were imaged by fluorescence microscopy after labeling with FM4-64.
Class A´ Vps33 mutants show loss of in vivo binding to the HOPS complex.
Despite their normal vacuole morphology, the four class A´ mutants (D88K, I278N, D300G, and T553I) had growth defects under stressful conditions (37°C and 5 mM ZnCl2), and impaired cargo trafficking under standard conditions (30°C). For further characterization of these mutants, Vps33-GFP alleles from different phenotypic classes (D88K, R281A, G297V, and D300G) were chromosomally integrated under the native VPS33 promoter in a protease-deficient pep4∆ background. Examination of the chromosomal integrants revealed defects identical to phenotypes from plasmid-borne vps33 alleles (Figure S5).
To test whether loss of Vps33-GFP punctae in the class A´ mutants was due to altered Vps33 association with membranes, we performed subcellular fractionation. Lysates of spheroplasted cells were centrifuged at 13,000 × g to obtain pellets (P13) highly enriched for vacuole membranes. The 13,000 × g supernatants (S13) were then centrifuged at 100,000 × g to yield a P100 fraction (enriched in endosomes, vesicles, and Golgi) and S100 cytosol. In wild-type cells, HOPS sedimented predominantly with the vacuole-enriched P13 fraction, with a smaller amount remaining in the S100 cytosol (Figure 6, A and B). In strains carrying the class A´ mutations D88K or D300G, however, most Vps33 shifted from the P13 to the S100 cytosol. Importantly, the other HOPS subunits remained in the vacuolar P13 (Figure 6, A and B). Together with microscopy, these subcellular fractionation experiments confirm that in the D88K and D300G class A´ mutants, Vps33—but not the remainder of HOPS—shifts from mainly vacuolar to mainly cytosolic.
FIGURE 6:

Vps33 mutations cause loss of steady-state association with HOPS. (A) Subcellular fraction was performed using differential centrifugation with ∼150 OD600 × ml. Spheroplasts were lysed and centrifuged to enrich for vacuoles (P13; 13,000 × g pellet); vesicles, endosomes, and Golgi (P100; 100,000 × g pellet); and cytoplasm (S100; 100,000 × g supernatant). Fractions were separated by SDS–PAGE and analyzed by Western blotting for Vps33 (WB). (B) Subcellular fraction as in (A), with Western blotting for HOPS subunits Vps41 and Vps11 and fractionation markers Ypt7 (found in the P13) and an actin subunit (found primarily in the S100). (C) Coimmunoprecipitation of HOPS complex with Vps33-GFP. Yeast detergent lysates from strains carrying a wild-type copy of Vps33-GFP, or the indicated Vps33 mutant, were incubated with protein A resins covalently linked to an anti-GFP antibody. Input of the cell lysate is shown as 10% relative to eluate of the immunoprecipitation.
In previous studies, Vps33 stably associated with HOPS in coimmunoprecipitations (Rieder and Emr, 1997; Seals et al., 2000). On the basis of the above data, we predicted that the Vps33 mutations D88K or D300G would abrogate the Vps33-HOPS interaction. Coimmunoprecipitation from detergent lysates verified the stable association of wild-type Vps33 with HOPS (Figure 6C). In contrast, coimmunoprecipitation from lysates from D88K or D300G mutant cells revealed a loss of association between Vps33 and HOPS. These results suggest that the altered Vps33 localization in class A´ mutants results from reduced steady-state association of Vps33 with HOPS. Together with the growth and cargo-trafficking defects, the class A´ mutants underscore the importance of proper steady-state recruitment of Vps33 to the vacuole and vacuole fusion machinery (see Discussion).
DISCUSSION
A central challenge in developing general models for SM protein function is to reconcile the apparently divergent modes of SNARE binding reported for the four SM subfamilies. In this study, we have shown that Vps33 interacts with SNAREs and SNARE complexes in a manner closely analogous to Sec1 (Togneri et al., 2006; Morgera et al., 2011). Specifically, Vps33 interacts with individual SNARE domains and, with significantly greater affinity, quaternary SNARE complexes. Furthermore, we found that HOPS binds the Vam3 N-terminal domain through an SM-independent mechanism that requires Vps11 and Vps18 but not Vps33. This is in contrast to the SM proteins Sly1, Vps45, and Munc18, which have high-affinity interaction sites for their cognate Qa-SNAREs. We also identified Vps33 mutants that exhibit a collection of phenotypic defects referred to here as class A´, including the loss of vacuole-associated Vps33 and reduced steady-state Vps33-HOPS interactions.
Although isolated Vps33 can bind SNAREs and SNARE complexes in vitro, recruitment of HOPS to membranes still occurs when Vps33 is absent. HOPS recruitment to docking junctions is enhanced by affinities of the HOPS complex for the Rab Ypt7 (Price et al., 2000; Brett et al., 2008; Plemel et al., 2011; Brocker et al., 2012), for specific lipids on the vacuole (Boeddinghaus et al., 2002; Fratti and Wickner, 2007), and for the N-terminal domains of the Qa- and Qc-SNAREs, Vam3 and Vam7 (Sato et al., 2000; Stroupe et al., 2006). Taken together, the available data suggest a model in which HOPS coordinates SNARE binding through its interactions with the N-terminal domains of the Qa- and Qc-SNAREs. This positions Vps33 to interact with the fusion-active SNARE domains of Qa-, Qc-, and R-SNAREs, and finally with the SNARE core complex (Figure 7).
FIGURE 7:

Summary of interactions between Ypt7 Rab, HOPS, and SNAREs. Together with the published literature, our data suggest that several subunits of the HOPS complex coordinate binding of multiple SNARE domains. Vps33 directly binds the Qa-, Qc-, and R-SNARE domains, while the N-terminal Habc and PX domains of the Qa- and Qc-SNAREs are bound by other subunits within HOPS. Interactions of HOPS subunits Vps39 and Vps41 with the Rab Ypt7 have been demonstrated previously (Brett et al., 2008; Plemel et al., 2011).
Consistent with this emerging picture, we identified four class A´ mutations in Vps33 that result in a change in the localization of Vps33, but not the remainder of HOPS, from the vacuole to the cytoplasm. Strains carrying class A´ alleles have reduced viability and cargo-trafficking defects. It is tempting to speculate, given the loss of association between HOPS and Vps33 D88K or D300G, that the phenotypic defects of class A´ mutants are due to a loss of HOPS-promoted recruitment of Vps33 to the vacuole. However, further characterization of these Vps33 alleles will be required to rule out the possibility that class A´ alleles have secondary defects that contribute to the observed phenotypes.
As with the interactions between Vps33 and individual SNAREs reported here, each of the other SM subfamilies has been reported to interact with non–Qa-SNAREs and SNARE domains. Sly1 binds SNARE domains of Qb- (Bos1 and Gos1) and Qc-SNAREs (Bet1 and Sft1; Peng and Gallwitz, 2004). The yeast endosomal SM Vps45 binds the SNARE domain of the R-SNARE Snc2 (Carpp et al., 2006). Munc18-1 binds the SNARE domains of the R-SNARE synaptobrevin and the Qa-SNARE syntaxin-1a (Xu et al., 2010b; Shi et al., 2011). Sec1 binds the Qb-c–SNARE Sec9 and the Qa-SNARE Sso1 (Morgera et al., 2011). As interactions between SM proteins and individual SNARE domains are typically weak (6–20 μM), the functional in vivo significance of these interactions is still unclear (Xu et al., 2010b; Morgera et al., 2011). One model consistent with this pattern of interactions is that the SM protein makes similar contacts with a subset of SNARE domains when the SNAREs are unpaired or in the assembling SNARE complex bundle. The low affinities for individual SNAREs would be synergistic in the context of the SNARE complex, and these SNARE domain contacts could in turn provide a basis for the hypothesized activity of SM proteins toward the assembling trans-complex (Sudhof and Rothman, 2009; Carr and Rizo, 2010).
While nearly every SM protein has been reported to interact with individual SNARE domains, no clear pattern emerges as to which specific SNARE domains SM proteins contact. As some SM proteins must contact SNARE complexes with multiple Qa-, Qb-, Qc-, and R-SNARE compositions, it seems possible that a strict arrangement of SM-to-SNARE domain contacts would not be conserved, while the general feature of interaction with at least two SNARE domains per SM would be maintained. A closely related question is whether SM proteins contribute to the specificity of fusion. We report here that Vps33 binds similarly to quaternary complexes with either of the physiologic R-SNAREs (Nyv1 and Ykt6), and binds the SNARE domains of both endosomal and vacuole Qa-SNAREs (Vam3 and Pep12). In contrast, Vps33 shows high selectivity in binding to vacuole versus Golgi SNARE complexes and in binding cognate versus noncognate individual SNAREs. This suggests, at least in principle, that SM proteins can contribute to the selectivity of membrane docking and fusion.
A recent study reported binding of Vps33 to 3Q and 3Q:R quaternary SNARE complexes, but not individual SNAREs (Kramer and Ungermann, 2011). As we observed stronger binding of Vps33 to SNARE complexes than to individual SNAREs, it is possible that differences in observed interactions reflect differences in experimental conditions. As 3Q complexes did not remain stably associated under our assay conditions, we did not examine their binding to Vps33. We emphasize that interactions between Vps33 and quaternary 3Q:R complexes were detected in both studies.
Our data suggest that compared with other SM proteins, Vps33 behaves in a manner most similar to Sec1. Both proteins can interact with isolated SNAREs but have greatest affinity for the SNARE complex. Moreover, both Vps33 and Sec1 interact with larger protein complexes (HOPS and exocyst), which in turn have additional affinities for SNAREs. In both cases, multisubunit tethering complexes play an important role in recruitment of the cognate SM protein. We suggest that SM proteins fall into two classes based on a primary affinity for the Qa-SNARE or SNARE complex. Class I SM proteins (including Munc18, Vps45, and Sly1) have a high-affinity interaction site for the cognate Qa-SNARE and that cognate Qa-SNARE contains the N-peptide consensus sequence. Class I SM proteins can be further divided into those that bind the closed conformation of the Qa-SNARE (Munc18, Vps45) or do not (Sly1). Class II SM proteins (including Sec1 and Vps33) do not directly bind the N-peptide and, in fact, their cognate Qa-SNAREs seem to lack the consensus N-peptide sequence (Hu et al., 2007). Instead, Sec1 and Vps33 have primary affinity for the SNARE complex. Recently it was suggested that the N-peptide of syntaxin-1 serves predominantly as a physical recruitment site for Munc18-1 (Rathore et al., 2010). In this model, class I SM proteins (Munc18, Vps45, and Sly1) are recruited through direct interactions with the N-peptide. Recruitment of class II SM proteins (including Sec1 and Vps33) to unpaired SNAREs may be enhanced by tethering complexes, such as HOPS and exocyst. Despite the divergence in SM protein affinity for cognate Qa-SNARE, all SM proteins studied to date have the ability to bind unpaired SNARE domains. Furthermore, Munc18 and Vps45 have been reported to interact with the SNARE complex bundle, suggesting that the primary SNARE-binding mode of Vps33 and Sec1—interaction with the SNARE complex bundle—is a shared feature of all SM proteins. This would suggest a unifying model in which the executive functions of SM proteins in docking and fusion are broadly conserved.
MATERIALS AND METHODS
Strain and plasmid construction
Yeast strains and plasmids are summarized in Supplemental Table S1. Briefly, VPS33 was deleted from SEY6210 BHY10 and BY4742 pep4∆. Vectors for expressing Vps33 under its native promoter were PCR-amplified from VPS33-ttx-GFPA207K (ttx: TEV [tobacco etch virus] protease, thrombin, and factor X recognition sites) and incorporated by homologous recombination into pRS416 vector (Wang et al., 2002). Point mutations in VPS33 were introduced through homologous recombination, rescued, verified by sequencing, and transformed into SEY6210 BHY10 vps33∆. For integration into BY4742 pep4∆vps33∆, wild-type and point mutants were PCR-amplified from the pRS416 vectors and integrated into the chromosomal locus of VPS33. Bacterial expression plasmids are summarized in Table S1. In brief, the GST-SNARE fusions were cloned in-frame with either an N- or C-terminal GST sequence separated by a short linker and TEV recognition site. GST-Vti1 was cloned as previously described (Stroupe et al., 2006). Full-length Vam7 was cloned as previously described (Schwartz and Merz, 2009). Bos1 was cloned as previously described (Stone et al., 1997). N-terminally His6-tagged SNARE fusions were cloned into pHIS-parallel1 vector. Vam7 SNARE domain was cloned into a pRSF-1b backbone in-frame with an N-terminal His6-GFP separated with a short linker and TEV recognition site. Vti1 was cloned into the pRSF-1b vector with a N-terminal His7-MBP (maltose-binding protein) tag separated with a short linker and TEV recognition site. Vps33 was cloned for expression in the insect cell baculovirus system as previously described (Brett et al., 2008).
Protein expression
Vps33.
Insect cells expressing GST-Vps33 were lysed by sonication or high-pressure disruption in buffer A (50 mM HEPES, 400 mM NaCl, 10% [wt/vol] glycerol, 5 mM 2-mercaptoethanol, 5 mM EDTA, 0.5% Triton X-100, pH 7.4) with protease inhibitors, and clarified lysate was bound to glutathione sepharose 4B (GSH) resin for 2 h at 4°C. The resin was washed extensively, and the buffer was changed stepwise into IEX binding buffer (20 mM Tris, 150 mM NaCl, 10% [wt/vol] glycerol, 5 mM 2-mercaptoethanol, pH 8.0). GST-Vps33 was eluted from GSH resin with 20 mM glutathione in IEX binding buffer and incubated with TEV protease at 4°C for 16–22 h. Cleaved Vps33 was further purified over UnoQ and Superdex200 columns, exchanged into Storage buffer (20 mM HEPES, 200 mM NaCl, 10% [wt/vol] glycerol, 5 mM 2-mercaptoethanol), and snap-frozen in liquid nitrogen.
SNAREs.
SNARE proteins were expressed in Escherichia coli that harbored a pRIL codon-bias correction plasmid. Cells were inoculated at 0.05 OD600, grown to 1.0–1.2 OD600 in Terrific Broth (TB; Tartof and Hobbs, 1987), and expression was induced with 100 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) for overnight expression at 21°C (His6-Nyv1 and His7-MBP-Vti1, GST-Vam3SNARE domain and truncations, GST-Nyv1SNARE domain and truncations, His6-Bos1, His6-Bet1, His6-Sec22), 500 μM IPTG for 4–5 h at 30°C (His6-GFP-Vam7 SNARE), or 1 mM IPTG for 3 h at 37°C (Vam3-GST). Full-length Vam7 was expressed and purified as previously described (Schwartz and Merz, 2009). Cells expressing His-tagged SNAREs were lysed by sonication in Buffer A without EDTA and supplemented with 20 mM imidazole and protease inhibitors. Clarified lysates were prepared by centrifugation at 18,500 × g for 25 min at 4°C and incubated with Ni-NTA HP resin (GE Healthcare, Piscataway, NJ) for 10 min at 4°C. The bound material was washed extensively, and His-tagged SNAREs were eluted with Storage buffer (20 mM HEPES, 200 mM NaCl, 10% [wt/vol] glycerol, 5 mM 2-mercaptoethanol, pH 7.4) supplemented with 400 mM imidazole (pH 7.4). His-tagged SNAREs were buffer-exchanged into Storage buffer without imidazole using PD-10 desalting columns (GE Healthcare) and snap-frozen in liquid nitrogen. Cells expressing GST-tagged SNAREs were lysed by sonication in Storage buffer supplemented with 5 mM EDTA and protease inhibitors, and clarified lysate was snap-frozen in liquid nitrogen.
Pull downs
GST-SNARE proteins were bound to GSH 4B (GE Healthcare) by incubating clarified lysate with the resin for 2 h at 4°C with rocking and were then washed extensively. For pull downs on SNARE complexes, a fivefold molar excess of purified Vti1, Vam7, and Nyv1 was incubated with Vam3 prebound to GSH 4B for 16–22 h at 4°C with rocking. Golgi SNARE complexes were prepared in a similar manner, with GST-Sed5 SNARE domain prebound to GSH 4B resin, and the purified cytoplasmic domains of the other three SNAREs incubated at fivefold molar excess for 16–22 h at 4°C with rocking. In all cases, resins were washed extensively before pull down. Prior to pull down, purified Vps33 was incubated with GSH 4B resin for 1 h at 23°C to remove residual uncleaved material. Pull downs with recombinant Vps33 were performed in SNARE Assay buffer (20 mM HEPES, 200 mM NaCl, 5 mM 2-mercaptoethanol, 0.5% Triton, pH 7.4). Pull downs with yeast lysate were performed as previously described (Angers and Merz, 2009). Briefly, lysis was performed in Yeast Lysis buffer (20 mM HEPES, 50 mM KOAc, 200 mM sorbitol) supplemented with protease inhibitors. Detergent lysates from ∼450 OD600 × ml of cells were incubated with resins prebound to GST fusion proteins for 2 h at 4°C, washed three times with Yeast Lysis buffer, and eluted by incubating the resins with Yeast Lysis buffer supplemented with 20 mM reduced glutathione (pH 7.4 final) for 10 min at 4°C. Analysis was performed by SDS–PAGE and Western blotting. Antibodies were prepared as previously described (Angers and Merz, 2009). All pull downs were repeated a minimum of three times; representative results are shown.
Quantification of saturation binding
Minimal SNARE complexes were formed on GSH resin using the SNARE domain of Vam3-GST, the SNARE domain of Vam7 (His6-GFP-Vam7), and the cytoplasmic domains of Vti1 (His7-MBP-Vti1) and Nyv1 (His6-Nyv1). Increasing concentrations of Vps33 (0.1–10 μM) were incubated with minimal SNARE complex for 2 h at 30°C in SNARE Assay buffer, washed three times, and eluted by incubating the resins with SNARE Assay buffer supplemented with 20 mM reduced glutathione (pH 7.4 final) for 10 min at room temperature. Samples were separated by SDS–PAGE, detected with SYPRO-Ruby (Invitrogen, Carlsbad, CA), and imaged using a Gel Doc XR+ (Bio-Rad, Hercules, CA). The data from three experiments were plotted as fractional saturation of Vps33 binding to the minimal SNARE complexes relative to the total concentration of Vps33 in the reaction. Kd was estimated by nonlinear fitting of a single-site binding model to the data (Prism 5; GraphPad, La Jolla, CA).
Growth assays
Strains were grown 16–22 h at 24°C, and three dilutions were prepared (1.0, 0.05, and 0.0025 OD600). Strains were plated on synthetic complete media lacking uracil or yeast–peptone–dextrose (YPD) plates, with or without 5 mM ZnCl2, and grown for 48 h at the indicated temperature.
Protein sorting and vacuole morphology
Yeast strains were grown 16–22 h at 24°C, and back-diluted to 0.2 OD600 × ml. Yeast were then grown at 30°C to 0.8–1.0 OD600, and cell pellets were washed in 10 mM NaN3. Cell pellets were resuspended in SDS–PAGE sample buffer. Samples were boiled for 15 min and then vortex-mixed with glass beads for 3 min. Samples were subjected to SDS–PAGE, and Western blotting were performed as previously described (Plemel et al., 2011). Vacuoles were imaged by FM4-64 (Invitrogen) pulse-chase as previously described (Plemel et al., 2011).
Subcellular fractionation
Yeast strains were grown at 30°C to ∼1.0 OD600. Cells were centrifuged and washed for 10 min at room temperature with 100 mM Tris-Cl (pH 9.4) and 10 mM 2-mercaptoethanol. Cells were centrifuged and resuspended in Spheroplasting buffer (50 mM Tris-Cl, pH 7.9, 8% YPD, 1M sorbitol) with Zymolyase 20T (Seikagaku, Tokyo, Japan); the Zymolyase was further purified by cation-exchange chromatography prior to use. Cells were spheroplasted for 25 min at 30°C, harvested by sedimentation, and resuspended in SF/IP buffer (20 mM HEPES, 200 mM sorbitol, 100 mM NaCl, pH 7.4) supplemented with protease inhibitors. Spheroplasts were lysed by Dounce homogenization and clarified by centrifugation at 1000 × g. Lysates were subsequently centrifuged at 13,000 × g for 15 min to obtain pellet (P13) and supernatant (S13) fractions. The S13 fraction was centrifuged for 45 min at 100,000 × g to obtain pellet (P100) and cytosol (S100) fractions. Samples were incubated with SDS-loading buffer for 7 min at 95°C and analyzed by SDS–PAGE and Western blotting. Subcellular fractionations were repeated a minimum of three times, and representative Western blots are reported.
Immunoprecipitation
Cells were grown and spheroplasted as described for subcellular fractionation. The S1 fraction was incubated with 0.5% Triton X-100, on ice, for 10 min and clarified by centrifugation at 20,000 × g for 15 min. Next, ∼450 OD600 × ml of lysate was incubated for 2 h at 4°C with protein A-Sepharose resin (Pierce, Rockford, IL) covalently coupled to affinity-purified rabbit polyclonal GFP antiserum. Resins were washed five times in SF/IP buffer and eluted by incubating the resin with SDS-loading buffer for 7 min at 95°C. Immunoprecipitations were repeated a minimum of three times, and representative Western blots are shown.
Supplementary Material
Acknowledgments
We thank W. Wicner for sharing antisera, S. Ferro-Novick for the Bet1 expression plasmid, and members of the Merz group for insightful discussions and comments on the manuscript. Our work on the biochemistry of membrane fusion is supported by NIH-NIGMS GM077349. B.T.L. was supported by NIH-NIGMS T32 GM07270, and A.J.M. is a Research Scholar of the American Cancer Society.
Abbreviations used:
- ARC
arthrogryposis, renal dysfunction, and cholestasis
- bf mutation
mouse buff mutation
- CORVET
class C core vacuole/endosome tethering
- GFP
green fluorescent protein
- GSH
glutathione sepharose resin
- GST
glutathione S-transferase
- HOPS
homotypic fusion and protein sorting
- HPS
Hermansky-Pudlak syndrome
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- MBP
maltose-binding protein
- NSF
N-ethylmaleimide–sensitive factor
- Q
glutamine
- R
arginine
- SM
Sec1/Munc18
- SNAP
soluble N-ethylmaleimide–sensitive factor attachment protein
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor
- TEV
tobacco etch virus
- ttx
TEV protease, thrombin, and factor X
- YPD
yeast–peptone–dextrose
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0343) on October 10, 2012.
REFERENCES
- Angers CG, Merz AJ. HOPS interacts with Apl5 at the vacuole membrane and is required for consumption of AP-3 transport vesicles. Mol Biol Cell. 2009;20:4563–4574. doi: 10.1091/mbc.E09-04-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta LM, Vida TA, Herman PK, Emr SD. Characterization of yeast Vps33p, a protein required for vacuolar protein sorting and vacuole biogenesis. Mol Cell Biol. 1990;10:4638–4649. doi: 10.1128/mcb.10.9.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeddinghaus C, Merz AJ, Laage R, Ungermann C. A cycle of Vam7p release from and PtdIns 3-P-dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J Cell Biol. 2002;157:79–89. doi: 10.1083/jcb.200112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K, Stevens TH. Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2005;1744:438–454. doi: 10.1016/j.bbamcr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Brett CL, Plemel RL, Lobinger BT, Vignali M, Fields S, Merz AJ. Efficient termination of vacuolar Rab GTPase signaling requires coordinated action by a GAP and a protein kinase. J Cell Biol. 2008;182:1141–1151. doi: 10.1083/jcb.200801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker C, Kuhlee A, Gatsogiannis C, Balderhaar HJ, Honscher C, Engelbrecht-Vandre S, Ungermann C, Raunser S. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc Natl Acad Sci USA. 2012;109:1991–1996. doi: 10.1073/pnas.1117797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpp LN, Ciufo LF, Shanks SG, Boyd A, Bryant NJ. The Sec1p/Munc18 protein Vps45p binds its cognate SNARE proteins via two distinct modes. J Cell Biol. 2006;173:927–936. doi: 10.1083/jcb.200512024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CM, Rizo J. At the junction of SNARE and SM protein function. Curr Opin Cell Biol. 2010;22:488–495. doi: 10.1016/j.ceb.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintala S, et al. The Vps33a gene regulates behavior and cerebellar Purkinje cell number. Brain Res. 2009;1266:18–28. doi: 10.1016/j.brainres.2009.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascher C, Ossig R, Gallwitz D, Schmitt HD. Identification and structure of four yeast genes (SLY) that are able to suppress the functional loss of YPT1, a member of the RAS superfamily. Mol Cell Biol. 1991;11:872–885. doi: 10.1128/mcb.11.2.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilcher M, Kohler B, von Mollard GF. Genetic interactions with the yeast Q-SNARE VTI1 reveal novel functions for the R-SNARE YKT6. J Biol Chem. 2001;276:34537–34544. doi: 10.1074/jbc.M101551200. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Yamaguchi T, Gao Y, Min SW, Huryeva I, Sudhof TC, Rizo J. How Tlg2p/syntaxin 16 “snares” Vps45. EMBO J. 2002;21:3620–3631. doi: 10.1093/emboj/cdf381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Yamaguchi T, Wang Y, Sudhof TC, Rizo J. Vam3p structure reveals conserved and divergent properties of syntaxins. Nat Struct Biol. 2001;8:258–264. doi: 10.1038/85012. [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci USA. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratti RA, Wickner W. Distinct targeting and fusion functions of the PX and SNARE domains of yeast vacuolar Vam7p. J Biol Chem. 2007;282:13133–13138. doi: 10.1074/jbc.M700584200. [DOI] [PubMed] [Google Scholar]
- Gissen P, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- Hashizume K, Cheng YS, Hutton JL, Chiu CH, Carr CM. Yeast Sec1p functions before and after vesicle docking. Mol Biol Cell. 2009;20:4673–4685. doi: 10.1091/mbc.E09-02-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey CM, Wickner W. HOPS initiates vacuole docking by tethering membranes before trans-SNARE complex assembly. Mol Biol Cell. 2010;21:2297–2305. doi: 10.1091/mbc.E10-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SH, Latham CF, Gee CL, James DE, Martin JL. Structure of the Munc18c/Syntaxin4 N-peptide complex defines universal features of the N-peptide binding mode of Sec1/Munc18 proteins. Proc Natl Acad Sci USA. 2007;104:8773–8778. doi: 10.1073/pnas.0701124104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun Y, Thorngren N, Starai VJ, Fratti RA, Collins K, Wickner W. Reversible, cooperative reactions of yeast vacuole docking. EMBO J. 2006;25:5260–5269. doi: 10.1038/sj.emboj.7601413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Miller KJ, Mazza M, Feng D, Weber M, Keranen S, Jantti J. Molecular interactions position Mso1p, a novel PTB domain homologue, in the interface of the exocyst complex and the exocytic SNARE machinery in yeast. Mol Biol Cell. 2005;16:4543–4556. doi: 10.1091/mbc.E05-03-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer L, Ungermann C. HOPS drives vacuole fusion by binding the vacuolar SNARE complex and the Vam7 PX domain via two distinct sites. Mol Biol Cell. 2011;22:2601–2611. doi: 10.1091/mbc.E11-02-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laage R, Ungermann C. The N-terminal domain of the t-SNARE Vam3p coordinates priming and docking in yeast vacuole fusion. Mol Biol Cell. 2001;12:3375–3385. doi: 10.1091/mbc.12.11.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Schmitt HD, Gallwitz D, Peng RW. Mutations of the SM protein Sly1 resulting in bypass of GTPase requirement in vesicular transport are confined to a short helical region. FEBS Lett. 2007;581:5698–5702. doi: 10.1016/j.febslet.2007.11.033. [DOI] [PubMed] [Google Scholar]
- Morgera F, Sallah MR, Dubuke ML, Gandhi P, Brewer DN, Carr CM, Munson M. Regulation of exocytosis by the exocyst subunit Sec6 and the SM protein Sec1. Mol Biol Cell. 2011;23:337–346. doi: 10.1091/mbc.E11-08-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson DP, Brett CL, Merz AJ. Vps-C complexes: gatekeepers of endolysosomal traffic. Curr Opin Cell Biol. 2009;21:543–551. doi: 10.1016/j.ceb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng R, Gallwitz D. Sly1 protein bound to Golgi syntaxin Sed5p allows assembly and contributes to specificity of SNARE fusion complexes. J Cell Biol. 2002;157:645–655. doi: 10.1083/jcb.200202006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng R, Gallwitz D. Multiple SNARE interactions of an SM protein: Sed5p/Sly1p binding is dispensable for transport. EMBO J. 2004;23:3939–3949. doi: 10.1038/sj.emboj.7600410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieren M, Schmidt A, Mayer A. The SM protein Vps33 and the t-SNARE H(abc) domain promote fusion pore opening. Nat Struct Mol Biol. 2010;17:710–717. doi: 10.1038/nsmb.1809. [DOI] [PubMed] [Google Scholar]
- Plemel RL, Lobingier BT, Brett CL, Angers CG, Nickerson DP, Paulsel A, Sprague D, Merz AJ. Subunit organization and Rab interactions of Vps-C protein complexes that control endolysosomal membrane traffic. Mol Biol Cell. 2011;22:1353–1363. doi: 10.1091/mbc.E10-03-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A, Seals D, Wickner W, Ungermann C. The docking stage of yeast vacuole fusion requires the transfer of proteins from a cis-SNARE complex to a Rab/Ypt protein. J Cell Biol. 2000;148:1231–1238. doi: 10.1083/jcb.148.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, Shen J. Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc Natl Acad Sci USA. 2010;107:22399–22406. doi: 10.1073/pnas.1012997108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CK, Howald-Stevenson I, Vater CA, Stevens TH. Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vps mutants. Mol Biol Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder SE, Emr SD. A novel RING finger protein complex essential for a late step in protein transport to the yeast vacuole. Mol Biol Cell. 1997;8:2307–2327. doi: 10.1091/mbc.8.11.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TK, Rehling P, Peterson MR, Emr SD. Class C Vps protein complex regulates vacuolar SNARE pairing and is required for vesicle docking/fusion. Mol Cell. 2000;6:661–671. doi: 10.1016/s1097-2765(00)00064-2. [DOI] [PubMed] [Google Scholar]
- Schwartz ML, Merz AJ. Capture and release of partially zipped trans-SNARE complexes on intact organelles. J Cell Biol. 2009;185:535–549. doi: 10.1083/jcb.200811082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott BL, Van Komen JS, Irshad H, Liu S, Wilson KA, McNew JA. Sec1p directly stimulates SNARE-mediated membrane fusion in vitro. J Cell Biol. 2004;167:75–85. doi: 10.1083/jcb.200405018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci USA. 2000;97:9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukov EA, He JP, Moghrabi N, Sunio A, Kramer H. A role for the deep orange and carnation eye color genes in lysosomal delivery in Drosophila. Mol Cell. 1999;4:479–486. doi: 10.1016/s1097-2765(00)80199-9. [DOI] [PubMed] [Google Scholar]
- Shen J, Rathore SS, Khandan L, Rothman JE. SNARE bundle and syntaxin N-peptide constitute a minimal complement for Munc18-1 activation of membrane fusion. J Cell Biol. 2010;190:55–63. doi: 10.1083/jcb.201003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Shi L, Kummel D, Coleman J, Melia TJ, Giraudo CG. Dual roles of Munc18-1 rely on distinct binding modes of the central cavity with Stx1A and SNARE complex. Mol Biol Cell. 2011;22:4150–4160. doi: 10.1091/mbc.E11-02-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Hickey CM, Wickner W. HOPS proofreads the trans-SNARE complex for yeast vacuole fusion. Mol Biol Cell. 2008;19:2500–2508. doi: 10.1091/mbc.E08-01-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Sacher M, Mao Y, Carr C, Lyons P, Quinn AM, Ferro-Novick S. Bet1p activates the v-SNARE Bos1p. Mol Biol Cell. 1997;8:1175–1181. doi: 10.1091/mbc.8.7.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroupe C, Collins KM, Fratti RA, Wickner W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 2006;25:1579–1589. doi: 10.1038/sj.emboj.7601051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroupe C, Hickey CM, Mima J, Burfeind AS, Wickner W. Minimal membrane docking requirements revealed by reconstitution of Rab GTPase-dependent membrane fusion from purified components. Proc Natl Acad Sci USA. 2009;106:17626–17633. doi: 10.1073/pnas.0903801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Woolford CA, Jones EW. The Sec1/Munc18 protein, Vps33p, functions at the endosome and the vacuole of Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:2593–2605. doi: 10.1091/mbc.E03-10-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Oiso N, Gautam R, Novak EK, Panthier JJ, Suprabha PG, Vida T, Swank RT, Spritz RA. The mouse organellar biogenesis mutant buff results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation. Proc Natl Acad Sci USA. 2003;100:1146–1150. doi: 10.1073/pnas.0237292100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartof KD, Hobbs CA. Improved media for growing plasmid and cosmid clones. Focus. 1987;9:12. [Google Scholar]
- Togneri J, Cheng YS, Munson M, Hughson FM, Carr CM. Specific SNARE complex binding mode of the Sec1/Munc-18 protein, Sec1p. Proc Natl Acad Sci USA. 2006;103:17730–17735. doi: 10.1073/pnas.0605448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, von Mollard GF, Jensen ON, Margolis N, Stevens TH, Wickner W. Three v-SNAREs and two t-SNAREs, present in a pentameric cis-SNARE complex on isolated vacuoles, are essential for homotypic fusion. J Cell Biol. 1999;145:1435–1442. doi: 10.1083/jcb.145.7.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Seeley ES, Wickner W, Merz AJ. Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell. 2002;108:357–369. doi: 10.1016/s0092-8674(02)00632-3. [DOI] [PubMed] [Google Scholar]
- Xu H, Jun Y, Thompson J, Yates J, Wickner W. HOPS prevents the disassembly of trans-SNARE complexes by Sec17p/Sec18p during membrane fusion. EMBO J. 2010a;29:1948–1960. doi: 10.1038/emboj.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J. Binding of Munc18-1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry. 2010b;49:1568–1576. doi: 10.1021/bi9021878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Dulubova I, Min SW, Chen X, Rizo J, Sudhof TC. Sly1 binds to Golgi and ER syntaxins via a conserved N-terminal peptide motif. Dev Cell. 2002;2:295–305. doi: 10.1016/s1534-5807(02)00125-9. [DOI] [PubMed] [Google Scholar]
- Zhao C, Slevin JT, Whiteheart SW. Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett. 2007;581:2140–2149. doi: 10.1016/j.febslet.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.