

Abstract
Yeast prions, based on self-seeded highly ordered fibrous aggregates (amyloids), serve as a model for human amyloid diseases. Propagation of yeast prions depends on the balance between chaperones of the Hsp100 and Hsp70 families. The yeast prion [PSI+] can be eliminated by an excess of the chaperone Hsp104. This effect is reversed by an excess of the chaperone Hsp70-Ssa. Here we show that the actions of Hsp104 and Ssa on [PSI+] are modulated by the small glutamine-rich tetratricopeptide cochaperone Sgt2. Sgt2 is conserved from yeast to humans, has previously been implicated in the guided entry of tail-anchored proteins (GET) trafficking pathway, and is known to interact with Hsps, cytosolic Get proteins, and tail-anchored proteins. We demonstrate that Sgt2 increases the ability of excess Ssa to counteract [PSI+] curing by excess Hsp104. Deletion of SGT2 also restores trafficking of a tail-anchored protein in cells with a disrupted GET pathway. One region of Sgt2 interacts both with the prion domain of Sup35 and with tail-anchored proteins. Sgt2 levels are increased in response to the presence of a prion when major Hsps are not induced. Our data implicate Sgt2 as an amyloid “sensor” and a regulator of chaperone targeting to different types of aggregation-prone proteins.
INTRODUCTION
Ordered cross-β fibrous protein aggregates (amyloids) are associated with a variety of human diseases, including fatal and incurable Alzheimer's and Huntington's diseases (reviewed in references 19 and 34). Amyloids spread by nucleated polymerization via immobilizing a soluble protein of the same sequence and converting it into an amyloid form. Prion diseases (such as Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy [BSE], or “mad cow disease,” in cattle) are caused by infectious amyloids, termed prions (15, 47). A number of endogenous prion proteins have been identified in the yeast Saccharomyces cerevisiae (reviewed in references 35 and 39). Most yeast prions are intracellular amyloids inherited in a non-Mendelian fashion (via cytoplasm) and containing QN-rich prion domains that resemble mammalian proteins associated with polyglutamine disorders such as Huntington's disease. Some yeast prions are pathogenic to the yeast “host” (41, 68), thus establishing yeast as a model for studying the pathogenic amyloids. Other yeast prions are widespread in nature, pointing to potentially positive biological effects (23). By transmitting phenotypic changes not associated with DNA variations, prions provide an additional mechanism of inheritance, which may play an important role in both pathology and evolution. Understanding the cellular control of prion propagation and clearance in yeast sheds light on mechanisms that control the spread of mammalian and human amyloids and may lead to the development of antiamyloid therapies.
[PSI+], one of the most extensively studied yeast prions, is a self-perpetuating polymer of the translation termination factor Sup35, or eRF3 (reviewed in reference 35). [PSI+]-carrying cells are defective in translation termination, thus providing a convenient phenotypic assay for [PSI+] based on the readthrough of stop codons (nonsense suppression). Much of our knowledge about the propagation of yeast prions, including the establishment of a crucial role of the chaperone machinery in prion propagation, initially came from experiments on the [PSI+] model that were later extended to other prions. [PSI+] propagation requires an intermediate level of the chaperone Hsp104 (12), as inactivation or overproduction of Hsp104 cures yeast cells of [PSI+] (see Fig. 1A). Most other known yeast amyloid-based prions also require Hsp104 but are not cured by Hsp104 overproduction (reviewed in references 35, 51, and 53). Hsp104 is known to promote disaggregation of heat-damaged proteins after stress (22), and existing evidence (35) supports a model (32) postulating that the requirement of Hsp104 for prion propagation is due to its ability to fragment prion polymers, thus generating “seeds” for new rounds of polymerization. It was also proposed that [PSI+] curing by Hsp104 overproduction occurs due to monomerization of polymers in the presence of high levels of Hsp104 (32). However, direct evidence for this mechanism is lacking, and other mechanisms, such as impairment of prion segregation in cell divisions at high Hsp104 levels, have also been discussed (35, 44).
Fig 1.

get deficiencies impair [PSI+] curing by excess Hsp104. (A) The known role of the chaperone Hsp104 in propagation of the [PSI+] prion. At normal levels, Hsp104 promotes fragmentation of Sup35 polymers (composed of a prion isoform [squares]) into oligomers, initiating new rounds of prion propagation (right part of the panel). A decrease (↓) in Hsp104 levels (left part, upper portion) impairs oligomer production, leading to eventual loss (“curing”) of prion polymers in future cell generations and to accumulation of newly synthesized Sup35 in a nonprion form (circles). An increase (↑) in Hsp104 levels (left part, bottom portion) directs Sup35 polymers to the “nonproductive” pathway, also resulting in prion curing and accumulation of a nonprion form of Sup35. (B) Screening for EMS-induced mutants defective in [PSI+] curing by excess Hsp104. One of the curing-defective mutants (mutant 156) was complemented by a plasmid with the GET2 gene. The initial mutation was identified as a G-to-A substitution at position 473 of the GET2 gene (get2-473), resulting in a premature stop codon at amino acid position 157. This mutation prevents synthesis of the C-terminal part of the Get2 protein (dotted lines), thus eliminating 2.5 of 3 transmembrane domains (gray boxes). (C) Deletion of GET1, -2, -3, -4, or -5 decreased [PSI+] curing by excess Hsp104, similar to the get2-473 mutation. [PSI+] curing in the wild-type (WT) and getΔ cultures was detected by a decrease or elimination of growth on −Ade medium selective for PGAL-HSP104 following 36 h of induction on galactose. (D) The [PSI+] curing defect was stronger in get1Δ and get2Δ strains than in the other getΔ strains. The same assay as that for panel C was performed after a longer (72 h) induction of PGAL-HSP104. For panels C and D, plates were photographed after 4 days of incubation. (E) The get2Δ deletion did not change the level of Hsp104 produced from the plasmid-borne PGAL-HSP104 construct after 24 h. Cells were grown in −Ura medium with galactose and raffinose. Amounts of total protein were normalized by use of Ade2, as shown. Per the densitometry results, Hsp104 levels in the get2Δ strain were identical (with a standard error of ±0.1) to those in the wild-type (WT) strain. (F) The get2Δ deletion increased background levels of endogenous Hsp104 in mid-exponential-phase cultures grown in YPD medium. Amounts of total protein were normalized by use of Ade2. Per the densitometry results, Hsp104 levels in the get2Δ cells were 12- ± 0.9-fold higher than those in wild-type (WT) cells. Note that different exposures were used for panels E and F; therefore, Hsp104 levels should not be compared between the panels. (G) GET2 alterations do not impair Hsp104's function in thermotolerance. Exponential-phase cultures of the strains with the get2 deletion (get2Δ) or mutation (get2-473) exhibited increased basal tolerance (top right image) and similar induced tolerance (after pretreatment at 37°C) (bottom right image) to a 50°C heat shock compared to the wild-type (WT) strain. Nontreated cultures are shown on the left, as a control. Decimal serial dilutions were plated onto YPD plates and incubated for 4 days at 25°C in each case. (H) Levels of Hsp104 induced by incubation at 37°C for 30 min were the same (within a standard error of ±0.1, as confirmed by densitometry) in the WT, get2Δ, and get2-473 strains. The ribosomal protein Rpl3 was used as a loading control.
Conserved chaperones of the Hsp70 and Hsp40 families cooperate with Hsp104 in both solubilization of stress-damaged proteins and prion propagation (reviewed in references 35, 51, and 54). Notably, members of the different yeast Hsp70 subfamilies exhibit different effects on [PSI+] curing by excess Hsp104. Hsp70-Ssb (represented by two proteins, Ssb1 and -2) promotes [PSI+] curing by excess Hsp104 (3, 13), while excess Hsp70-Ssa (represented by Ssa1, -2, -3, and -4) counteracts [PSI+] curing by excess Hsp104 (3, 45). Some other genetic alterations, such as elimination of the ubiquitin-conjugating enzyme Ubc4 (2) or one of the Hsp70/90 cochaperones, Sti1 or Cpr7 (43, 52), as well as the chemical inhibition of Hsp90 (52), also antagonize [PSI+] curing by excess Hsp104; however, the mechanisms of these effects remain unknown.
In addition to prions, a variety of other protein aggregates are detected in the yeast cytoplasm. One group of aggregation-prone proteins is represented by tail-anchored (TA) proteins (reviewed in references 8 and 49). These proteins are inserted into membranes (e.g., the endoplasmic reticulum [ER] membrane) via hydrophobic tails, which must be protected from aggregation until proteins reach their target destination. The guided entry of tail-anchored proteins (GET) pathway is responsible for trafficking of TA proteins (57, 65). This pathway includes cytoplasmic Get3, -4, and -5 proteins and transmembrane Get1 and -2 proteins. Failure of the GET pathway, due to genetic alteration of one of its components or under certain physiological conditions, results in the aggregation of TA proteins and remaining cytoplasmic Get proteins (26, 57, 60). Relationships between Get-TA aggregates and other aggregated proteins are poorly understood. Heat shock proteins (Hsps) are known to interact with TA proteins, and these interactions are mediated by the tetratricopeptide cochaperone Sgt2, which also binds Get5 (36, 65).
In this study, we attempted to identify the genetic factors that control elimination of the [PSI+] prion by excess Hsp104. We found that alterations of the GET pathway antagonize [PSI+] curing caused by excess Hsp104. This effect is associated with the simultaneous induction of the chaperone Hsp70-Ssa and the cochaperone Sgt2 by get alterations. Notably, Sgt2 also helps excess Ssa to efficiently reverse the effect of excess Hsp104 on [PSI+] in Get+ strains. Our data show that Sgt2 physically interacts with Sup35 and that Sgt2 levels are increased in response to the presence of a prion. These results implicate Sgt2 as a protein that coordinates chaperone interactions with various types of aggregates.
MATERIALS AND METHODS
Yeast strains.
Saccharomyces cerevisiae strains constructed in this work (see Table S1 in the supplemental material) originated from strain 74-D694, with the genotype MATa ade1-14 (UGA) his3 leu2 trp1 ura3 (12), or from the isogenic haploid strains of the GT81 series, with the genotype ade1-14 (UGA) his3 leu2 lys2 trp1 ura3 (11). The original strains typically carried both [PSI+] and [PIN+] prions. Usually, a “strong” variant of the [PSI+] prion, characterized by more translational readthrough and high mitotic stability (35), was used. A weak variant of [PSI+] was used for [PSI+] destabilization by mild heat shock (44). The [psi− PIN+] and [psi− pin−] derivatives were obtained after transient overproduction of Hsp104 and after growth on medium with 5 mM guanidine hydrochloride, respectively (14). Construction of the MATa GT81-derived strains GT146 (ssb1Δ ssb2Δ) and GT349 (ubc4Δ) was described previously (2, 13). Complete gene deletions were constructed by a one-step replacement with cassettes bearing the HIS5 gene of Schizosaccharomyces pombe (an ortholog of S. cerevisiae HIS3), a Kanr gene that determines resistance to G418 in yeast (38), or an hph gene that confers resistance to hygromycin B (25). The same procedure was used for tagging chromosomal copies of genes with hemagglutinin (HA) or green fluorescent protein (GFP) (38). Combinations of two or several deletions, as well as combinations of deletions with the tagged constructs, were combined together either via subsequent transplacement steps or through genetic crosses between the otherwise isogenic haploid strains of the GT81 series, followed by sporulation and dissection. Strain pJ69-4A (MATa trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ GAL2-ADE2 LYS2::PGAL1-HIS3 met2::GAL7-lacZ) (24) and its hsp104Δ derivative were used for two-hybrid assays.
Plasmids.
Plasmids pYS-GAL104 (11) and pGAL-SSA1 (56), bearing the HSP104 and SSA1 genes, respectively, under the control of the galactose-inducible GAL1,10 promoter (PGAL), and the multicopy plasmid pLH101 (45), bearing SSA1 under the control of its own promoter, were described previously. Multicopy plasmid pFL44-SSB1, bearing SSB1 under the control of its own promoter, was created by cloning the BamHI/SacI fragment of YCp-SSB1 (46) into pFL44 (7) cut with the same enzymes. Low-copy-number (centromeric) plasmids based on pRS315 (62) and bearing the SIS1 (33) or YDJ1 (20) gene under the control of the gene's own promoter were generously provided by D. Cyr. The centromeric plasmid pLH105 (13) contains the HSP104 gene under the control of a constitutively expressed GPD promoter. Plasmid pRS315-GAL104 was constructed by cloning the PGAL-HSP104 sequence from pYS-GAL104 into the BamHI site of pRS315. Plasmids carrying genes that code for proteins with fluorescent tags were generously provided by M. Schuldiner in the case of Get3 (57) and R. Parker in the case of Dcp2 (16), Edc3, and Pub1 (9). Plasmid pRS316-DCP2 was constructed by ligating the DCP-GFP-encoding XbaI fragment from pRP1175 (16) into pRS316 (62) cut by the same enzyme. The 103QP-GFP- and 103QP-RFP-encoding plasmids were described earlier (42). The GET3 and SGT2 genes were PCR amplified from genomic DNA of the S288C strain and then cloned into pRS316GAL (37), with the GFP coding sequence from plasmid pmCUP-sGFP (59) added to the 3′ ends of both genes, followed by recloning of the PGAL-GET3-GFP- or PGAL-SGT2-GFP-containing fragment into pRS315. Plasmid pRS315-PGAL-Sup35NM-dsRed is plasmid pRS315 containing the PGAL-carrying XhoI/BamHI fragment from plasmid pRS316-GAL (37) followed by the BamHI/SacII fragment from the plasmid pmCUPNMsGFP (59), coding for the NM domains of Sup35, and by the dsRed2-encoding SacII/HpaI fragment from plasmid pDsRed2-N1 (Clontech). Gene fusions for the two-hybrid analysis of protein interactions involving Sgt2 or its fragments were created by inserting the respective PCR-amplified DNA fragments into the plasmid pACT2 (a gift of S. Elledge [17]), in frame with the N-terminally attached activation domain of Gal4, or pG4BD-0, in frame with the C-terminally attached DNA-binding domain of Gal4 (a gift of R. Brazas). A list of oligonucleotide primers used in this study can be found in Table S2 in the supplemental material. Plasmid pACT2-SUP35N, which codes for the prion domain of Sup35 fused to the C-terminally attached activation domain of Gal4, was described earlier (6). The fusions of Sup35N to the DNA-binding domain of Gal4 were difficult to use due to a residual transcription activation capability. A yeast genomic library based on plasmid p366 was a gift from P. Hieter (21).
Growth conditions and phenotypic assays.
Standard yeast media and protocols were employed (61). Rich organic medium was yeast extract-peptone-dextrose (YPD) medium. Synthetic dropout media are designated by the supplements that were missing (e.g., “−Ade medium” indicates synthetic medium lacking adenine). The PGAL promoter was induced on solid medium containing 2% galactose instead of glucose or in liquid synthetic medium containing 2% galactose and 2% raffinose instead of glucose. The addition of raffinose to liquid medium was necessary because the strains used in this study grow poorly in liquid medium with galactose but no raffinose. The stringencies for chaperone effects measured in the liquid medium were different from those observed in the plate assay, possibly due to (at least in part) different parameters of PGAL induction in the presence and absence of raffinose. However, differences between the compared strain-plasmid combinations were always in the same direction both in liquid medium and on plates. Yeast cultures were normally incubated at 30°C, unless stated otherwise. The presence of [PSI+] was detected by white or light pink growth on YPD and growth on −Ade medium due to readthrough of the ade1-14 (UGA) reporter; in contrast, the [psi−] strains were red on YPD and did not grow on −Ade medium. Temperature sensitivity was tested on rich YPD medium at 37°C. For heat shock experiments, cultures were grown to mid-exponential phase at 25°C. A mild heat shock for [PSI+] curing was performed at 39°C, while a severe heat shock for viability assays was performed at 50°C. Induced thermotolerance was measured after incubating cultures, originally grown at 25°C, at 37°C for 30 min before shifting them to 50°C. Antibiotic sensitivities were detected on YPD medium supplemented with paromomycin (600 μg/ml) or hygromycin (40 μg/ml). Yeast cultures bearing the plasmid pYS-GAL104 were mutagenized in a phosphate-buffered saline (PBS) (pH 7.0) solution containing 3% (GT81 series) or 4% (74-D694) ethyl ester of methanesulfonic acid (EMS) for 1 to 2 h at a concentration of 107 cells/ml, followed by the addition of sodium thiosulfate to a final concentration of 5% (for inactivation of EMS) and spreading of cells on synthetic −Ura medium with galactose in order to maintain the plasmid and to induce PGAL-HSP104 expression. Grown colonies were replica plated onto YPD and −Ade/glucose media for [PSI+] detection. To assess [PSI+] curing by excess Hsp104 in the plate assay, cultures were replica plated either from synthetic plasmid-selective medium onto −Ade and YPD media (in the case of the plasmid carrying HSP104 under the control of the constitutive promoter) or from galactose medium onto −Ade/glucose medium selective for the plasmid and onto YPD medium (in the case of the plasmid carrying HSP104 under the control of the PGAL promoter). For quantitation of [PSI+] curing, yeast cultures bearing galactose-inducible constructs were grown in liquid synthetic medium with galactose and raffinose. After specified periods, aliquots were plated onto synthetic glucose-containing medium selective for the respective plasmid(s). The presence of [PSI+] in the resulting colonies was detected after replica plating onto YPD and −Ade media.
Protein analysis.
For protein isolation, cultures were grown in 50 ml of appropriate medium to an optical density at 600 nm (OD600) of between 0.5 and 0.7, precipitated by centrifugation, transferred to 1.5-ml centrifuge tubes, washed twice with water, and resuspended in 200 μl of lysis buffer (50 mM Tris-Cl, pH 7.5, 200 mM NaCl, 1% Triton X-100, 0.1% SDS, 1× Complete Mini protease inhibitor cocktail [Roche], 3 mM phenylmethylsulfonyl fluoride, 1 mM N-ethylmaleimide). Cells were disrupted by agitation with an equal volume of glass beads for a total of 5 min, with periodic cooling of the mixture on ice. Cell debris was spun down at 4,000 × g for 2 min, and the supernatant was used for electrophoresis and Western analysis as described below.
For fractionation of protein aggregates by semidenaturing detergent agarose gel electrophoresis (SDD-AGE) (31), yeast extracts were mixed with a 1/4 volume of 4× loading buffer (240 mM Tris-Cl, pH 6.8, 8% SDS, 40% glycerol, 12% 2-mercaptoethanol, and 0.002% bromophenol blue), loaded onto a 1.8% Tris-acetate-EDTA (TAE)-based agarose gel, and run in 1× TAE buffer with 0.1% SDS. Proteins were then transferred to a nitrocellulose Protran membrane (Whatman) via capillary blotting. The membrane was blocked in 5% dry milk and incubated with the appropriate antibody.
For denaturing polyacrylamide gel electrophoresis (SDS-PAGE) and Western analysis, protein samples in loading buffer (the same as that described above) were boiled in a water bath for 10 min, separated in a 10% SDS-polyacrylamide gel with a 4% stacking gel in Tris-glycine-SDS running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3), electrotransferred to a Hybond-ECL nitrocellulose membrane (Amersham), blocked in 5% dry milk, and incubated with the appropriate antibody.
For coimmunoprecipitation (co-IP), protein lysates were incubated overnight with anti-HA antibody, followed by a 2-h incubation with protein A agarose beads (Calbiochem). Beads were washed three times with PBS buffer (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.0) to remove unbound proteins. Proteins bound to beads were eluted by adding 1× loading buffer with SDS (60 mM Tris-Cl, pH 6.8, 2% SDS, 10% glycerol, 3% 2-mercaptoethanol, and 0.0005% bromophenol blue).
Antibodies.
Antibodies to the C-terminal part of Sup35, Hsp104, Hsp70s (Ssa and Ssb), Hsp40s (Ydj1 and Sis1), and Rpl3 (64) were kindly provided by D. Bedwell, S. Lindquist, E. Craig, D. Cyr, and J. Warner, respectively. The antibody to Ade2 was described previously (3). A monoclonal antibody to HA was purchased from Covance.
Fluorescence microscopy.
Fluorescence microscopy was performed using an Olympus BX41 microscope with an Olympus DP-71 camera. Green and red fluorescence was detected by Endow GFP/EGFP band-pass and Cy3/TRITC (rhodamine)/DiI filter sets, respectively, from Chroma Technology Corporation. All images were taken using a ×100 objective. Cells for fluorescence microscopy were precipitated from exponential-phase cultures by centrifugation, washed twice with water, resuspended in water to a final concentration of approximately 2 × 108 cells/ml, and placed onto a glass slide (10 μl per sample) under a coverslip for microscopy examination.
RESULTS
get deficiencies impair [PSI+] curing by excess Hsp104.
In order to identify mutations that antagonize [PSI+] curing by excess Hsp104, we performed EMS mutagenesis followed by a genetic screen (Fig. 1B). A total of 5,700 colonies, originating from the mutagenized cells of two “strong” [PSI+] strains of the 74-D694 and GT81 series, were tested. A total of 13 recessive mutants were uncovered after excluding mutations in the HSP104 plasmid, nonsense suppressors that mimicked effects of [PSI+], and other false-positive mutations. Two of these mutations, not allelic to each other and generated in the 74-D694 background, also caused a defect in growth at high temperature (37°C), referred to here as temperature sensitivity (ts). Plasmids with two overlapping genomic DNA inserts, able to compensate for both the ts phenotype and the [PSI+] curing defect in one of these mutants, were isolated from a yeast genomic library. (Identification of another mutant will be described elsewhere.) A deletion analysis of the overlapping portion of the inserts identified GET2, encoding one of the major membrane-associated components of the GET trafficking pathway, as a gene responsible for compensation of both phenotypes. Sequencing of the get2 allele from the mutant strain has shown that it contains a G-to-A transition at nucleotide position 473, converting a codon for tryptophan (TGG) at amino acid position 157 into a stop codon (TAG). This generates a shortened version of the Get2 protein which lacks most of the C-proximal region bearing the transmembrane domains (Fig. 1B). A deletion of the GET2 gene, as well as a deletion of any other GET gene, also impaired [PSI+] curing by excess Hsp104 (Fig. 1C). Notably, the elimination of either of the crucial membrane-associated components of the pathway (Get1 or Get2) inhibited [PSI+] curing more severely than the elimination of any cytosolic component (Get3, -4, or -5), as seen after prolonged Hsp104 induction (Fig. 1D). This agrees with the appearance of some other phenotypes, such as the previously described (and confirmed by us [see Fig. S1A in the supplemental material]) sensitivity to high temperature and the translational antibiotic hygromycin B, as well as a newly detected sensitivity to paromomycin (see Fig. S1A), in get1Δ and get2Δ strains but not get4Δ and get5Δ strains. However, the get3Δ strain resembled get1Δ and get2Δ strains rather than get4Δ and get5Δ strains in these phenotypes. Pairwise combinations of get deletions revealed epistatic interactions, with stronger effects on [PSI+] curing prevailing over weaker effects (data not shown).
Effects of get2 deletion or mutation on [PSI+] are not due to a decrease in Hsp104 levels or activity.
One potential explanation for the effect of get alterations on [PSI+] curing is a decrease in the Hsp104 level or activity in get-deficient strains. However, levels of overexpressed Hsp104 remained unchanged in the get2Δ strain compared to the wild type (Fig. 1E). Moreover, background Hsp104 levels in the absence of an overproducer plasmid were increased (about 10-fold or higher) rather than decreased in the get2Δ strain (Fig. 1F). Other get deletions also induced Hsp104 (see Fig. S1H in the supplemental material). Induced thermotolerance of an exponential-phase yeast culture to severe (50°C) heat shock after pretreatment at 37°C, a phenotype that is strictly dependent on Hsp104 (55), was not changed in the strains containing the get2 deletion or mutation compared to the isogenic wild-type strain (Fig. 1G). This is in good agreement with the observation that Hsp104 was induced to about the same level after a 37°C pretreatment in both wild-type and get2-defective strains (Fig. 1H). Notably, basal thermotolerance to 50°C (without pretreatment) was actually increased in the strains with get alterations, consistent with the increased basal Hsp104 levels in these strains. Thus, decreased [PSI+] curing by excess Hsp104 in strains with get deficiencies is not due to a general impairment of Hsp104 function.
Genetic interactions between get deficiencies and other alterations influencing [PSI+] curing.
The chaperone proteins Hsp70-Ssa, Hsp70-Ssb, Hsp40-Sis1, and Hsp40-Ydj1, as well as Hsp90 with its cochaperones, Sti1 and Cpr7, and the ubiquitin-conjugating enzyme Ubc4, are known to influence effects of Hsp104 on [PSI+] (reviewed in references 35 and 51). We checked for functional interactions between the respective genes and the get2Δ deletion in regard to [PSI+] curing. Overproduction of Ssa1 (Fig. 2A) or Ydj1 (see Fig. S1B and C in the supplemental material) ameliorated [PSI+] curing in the wild-type strain but did not have an effect on curing in the get2Δ strain, while excess Sis1 slightly increased [PSI+] curing in both the get2Δ and wild-type strains (see Fig. S1D and E). An overproduction of Ssb1, known to increase [PSI+] curing in the wild-type strain, also increased curing in the get2Δ strain (not shown), while the deletion of both Ssb-encoding genes, ssb1/2Δ, decreased curing only in the wild-type strain, not the get2Δ strain (Fig. 2B). This suggests that the Get2 and Hsp70 proteins influence [PSI+] curing by excess Hsp104 through the same pathway, with Ssa and Ssb acting in opposite directions and Hsp40s possibly participating in the pathway as well.
Fig 2.
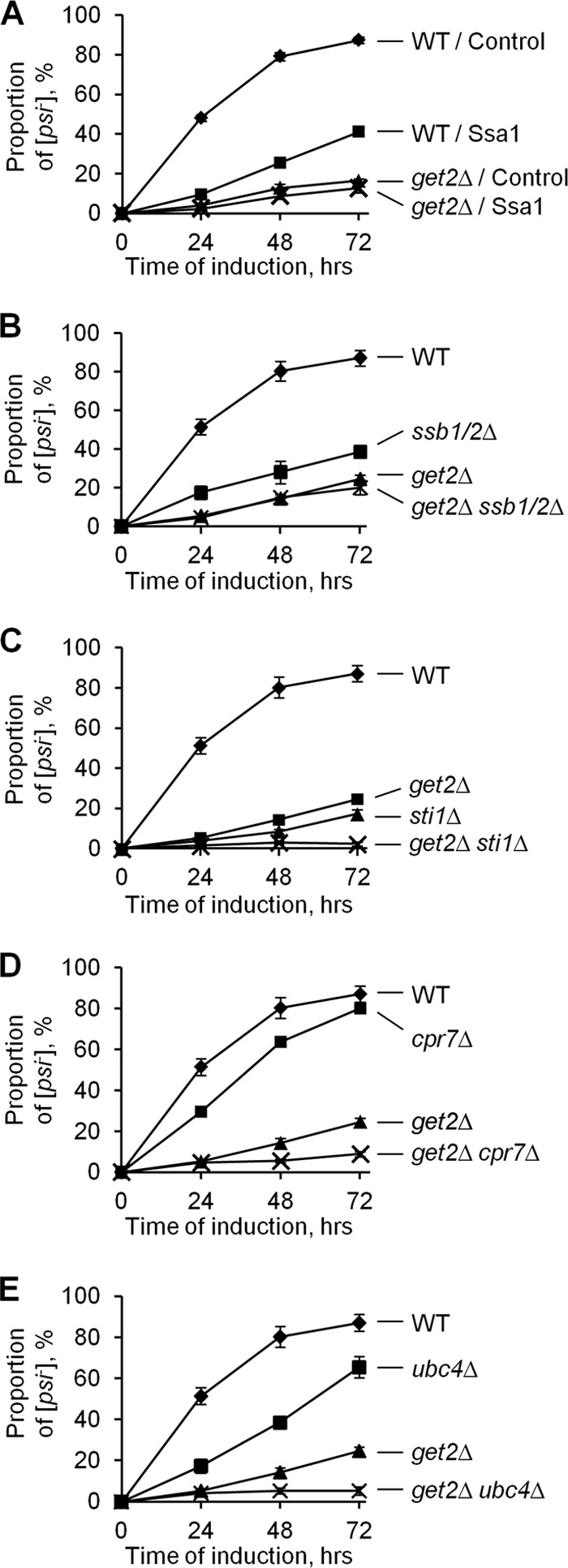
Genetic interactions of the get2Δ deletion with other genes known to influence [PSI+] curing. (A and B) Overproduction of Ssa1 (A) or deletion of both SSB genes (ssb1/2Δ) (B) antagonized [PSI+] curing by excess Hsp104 in the Get+ but not the get2Δ strain, thus revealing an epistatic interaction with the get2Δ deletion. (C to E) Deletion of STI1 (C), CPR7 (D), or UBC4 (E) antagonized [PSI+] curing by excess Hsp104 in both the wild-type and get2Δ strains, revealing an additive effect with the get2Δ deletion. In all experiments, cultures were grown in plasmid-selective medium with galactose and raffinose, and aliquots were taken and plated onto plasmid-selective medium with glucose after the specified periods. The presence or absence of [PSI+] was detected after replica plating to −Ade medium. At least three experiments were performed for each culture. Standard deviations are shown by error bars. In cases where error bars are not seen, the standard deviation was smaller than the symbol. WT, wild type; control, empty vector.
Effects of sti1 (Fig. 2C) or cpr7 (Fig. 2D) deletion on [PSI+] curing were additive to the effect of the get2Δ deletion. This shows that the action of get deficiencies on [PSI+] is not mediated by Hsp90's cochaperones. Likewise, a combination of the get2Δ deletion with the ubc4Δ deletion (Fig. 2E) exhibited a stronger defect of [PSI+] curing than that with either single deletion, indicating that the get2Δ deletion and defects of the ubiquitin-proteasome system are likely to affect [PSI+] curing via different pathways.
Because get deficiencies lead to aggregation of cytosolic Get and TA proteins (57), one could suggest that Hsp104 sequestration by aggregates impairs the effect of excess Hsp104 on [PSI+] curing. To test this possibility, we employed a construct containing the N-terminal region of the human huntingtin protein with an expanded (to 103 residues) polyglutamine stretch followed by a proline-rich region (103QP) and fusion to GFP. In yeast, aggregated 103QP is assembled into a perivacuolar (and typically perinuclear) deposit (67) resembling other quality control deposits of aggregated proteins, such as the mammalian aggresome (29) and yeast IPOD (27). Expression of 103QP-GFP did not inhibit [PSI+] curing by excess Hsp104 (see Fig. S1F in the supplemental material), indicating that the effect of get deficiencies on [PSI+] curing is not due to nonspecific sequestration of Hsp104 by protein aggregates.
Deletion of SGT2 ameliorates the [PSI+] curing defect in getΔ strains.
In contrast to get deletions, a deletion of the gene coding for the cochaperone Sgt2, which is also associated with the GET pathway (see above), did not antagonize [PSI+] curing by excess Hsp104 (Fig. 3A). Moreover, the sgt2Δ deletion ameliorated the [PSI+] curing defect of each get deletion (Fig. 3A and B). The average size of the Sup35 prion aggregates, as determined by SDD-AGE, was somewhat increased in the get2Δ strain; this increase was eliminated by the sgt2Δ deletion (see Fig. S1G in the supplemental material). Weak [PSI+] variants are known to be destabilized by a short-term (30 min) mild heat shock, possibly due to a more rapid accumulation of Hsp104 than of other Hsps (44). In agreement with these data, the get2Δ deletion reduced destabilization of the weak [PSI+] variant by mild heat shock, while the sgt2Δ deletion reversed this effect (Fig. 3C).
Fig 3.

Sgt2 aids Ssa in protecting [PSI+] from curing by excess Hsp104. (A and B) Deletion of SGT2 partly restored [PSI+] curing by excess Hsp104 in the getΔ strains, as seen from both a plate assay of all get deletion strains (A) and a quantitative assay of get2Δ strains (B). Experiments were performed as described in the legend to Fig. 1B (for panel A) or Fig. 2 (for panel B). (C) [PSI+] destabilization during short-term mild heat shock was significantly decreased by the get2Δ deletion, while the sgt2Δ deletion slightly decreased [PSI+] destabilization in the Get+ strain but almost restored it in the get2Δ strain. [PSI+] destabilization was scored by color on YPD; complete [psi−] and mosaic [PSI+]/[psi−] colonies were added together. Differences were concluded to be statistically significant at PHo values of <0.05 (*) or <0.01 (**), according to the t test (40). No significant [PSI+] loss was detected in the control cultures left at 25°C (not shown). (D) The stress response induced by get2Δ deletion was not ameliorated by sgt2Δ deletion. Protein levels, determined by densitometry of Western blots, are shown in relation to the wild-type strain level. Cultures were grown in YPD to the log phase. Ade2 was used as a loading control. (E) Deletion of SGT2 decreased protection of [PSI+] from excess Hsp104 by excess Ssa1 in the Get+ strain (plate assay). After induction of Ssa1 and Hsp104 from the PGAL promoter on galactose medium, plates were replica plated onto −Ade medium selective for both plasmids. (F) Deletion of SGT2 decreased protection of [PSI+] from excess Hsp104 by excess Ssa1 in the Get+ strain (quantitative assay). (G) Overproduction of Sgt2 increased [PSI+] protection from excess Hsp104 by excess Ssa1. All constructs were induced from the PGAL promoter. The experiment was performed as described for panel E, except for the longer incubation period on galactose, as indicated. WT, wild type; control, respective empty vector. Plate images were taken after 3 (A and G) or 4 (E) days of incubation.
Induction of stress response by get deletions does not depend on Sgt2.
In addition to Hsp104, get deletions induced expression of other Hsps, specifically Hsp70-Ssa, Hsp40-Sis1, and Hsp40-Ydj1 (Fig. 3D; see Fig. S1H in the supplemental material). This is consistent with the previously reported induction of the general stress response in strains deficient in the GET pathway (5). As expected, levels of Hsp70-Ssb, which is not stress inducible, were not changed. Notably, Sgt2 levels were increased in the strains with get deletions. However, the sgt2Δ deletion did not affect Hsp induction in the get-deficient strains (Fig. 3D; see Fig. S1H), indicating that induction of the stress response by get alterations is independent of Sgt2.
Sgt2 is involved in protection of [PSI+] from excess Hsp104 by excess Ssa.
Increased levels of Ssa in get deletion strains could provide the simplest explanation for the effect of get deficiencies on [PSI+] curing, as Ssa is known to reverse the effect of excess Hsp104 on [PSI+] (45). However, restoration of [PSI+] curing by the sgt2Δ deletion without affecting Ssa levels indicates that Sgt2 is needed for efficient [PSI+] protection by excess Ssa. To test this further, we compared the effects of Ssa1 and Hsp104 overproduction in Get+ [PSI+] strains that differed from each other by the presence, absence, or overproduction of Sgt2. Indeed, the sgt2Δ deletion interfered with the ability of excess Ssa1 to protect [PSI+] from excess Hsp104. This effect was clearly seen in the plate assay (Fig. 3E). Differences were also statistically significant in the quantitative assay in liquid medium, although the sgt2Δ deletion did not completely abolish [PSI+] protection by excess Ssa (Fig. 3F). Levels of the Hsp104 and Ssa proteins, induced from the PGAL promoter, were not affected by the sgt2Δ deletion, confirming that the decrease in the protective effect of Ssa on curing was not due to a change in chaperone accumulation (data not shown). Sgt2 overproduction did not have any effect on [PSI+] mitotic stability (not shown) or on [PSI+] curing by excess Hsp104 on its own; however, it increased [PSI+] protection from excess Hsp104 in the presence of excess Ssa (Fig. 3G).
Taken together, our data indicate that the defect in [PSI+] curing by excess Hsp104 in the strains with get alterations was due to simultaneously increased levels of both Ssa and Sgt2. The residual ability of excess Ssa to protect [PSI+] from excess Hsp104 in the absence of Sgt2 (Fig. 3F) explains why the sgt2Δ deletion did not completely abolish [PSI+] protection in the get2Δ strain (Fig. 3B).
Role of Sgt2 in the other phenotypes caused by get deletions.
The sgt2Δ deletion partly ameliorated the antibiotic sensitivities of get1, get2, and get3 deletion strains but did not have any effect on temperature sensitivity (an example with the get2Δ strain is shown in Fig. S1I in the supplemental material). When the GET pathway is disrupted, the cytosolic Get proteins and TA proteins form cytologically detectable aggregates in the cytoplasm (57). We confirmed the coaggregation of the Sgt2, Get3 (Fig. 4A), Get5 (see Fig. S2A in the supplemental material), and Get4 (data not shown) proteins within the same clumps in the get2Δ cells. We have also shown that cytoplasmic Get aggregates (visualized by tagged Get3) colocalize with neither Dcp2 (Fig. 4B) and Edc3 (see Fig. S2B), which are markers of P-bodies (9, 16), nor Pub1 (Fig. 4C), a marker of stress granules (9). Thus, Get-TA aggregates are distinct from other known cytoplasmic assemblies. Aggregation of Get3 (Fig. 4D and E), Get4 (see Fig. S2C), and Get5 (see Fig. S2D) in the get2Δ cells strictly depended on the presence of Sgt2, in agreement with a model suggesting that Sgt2 participates in linking the cytosolic Get proteins to TA proteins, and possibly to chaperones (65). Formation of Get aggregates was abolished (see Fig. S2E) in the presence of benomyl, an inhibitor of microtubule assembly in yeast (28). This suggests a direct or indirect dependence of Get-TA aggregate formation on the microtubular cytoskeleton, as demonstrated previously (67) and confirmed here (see Fig. S2F) for the aggresome-like deposits of 103QP in yeast. The TA protein Sbh1, a component of the Sec61 translocon complex normally localized to the ER membrane (63), was mislocalized and coaggregated with Get3 in the same get2Δ strain; notably, the sgt2Δ deletion restored the membrane localization of Sbh1 (Fig. 4F). Thus, some (though not all) phenotypes of get deletions depend on the presence of Sgt2.
Fig 4.
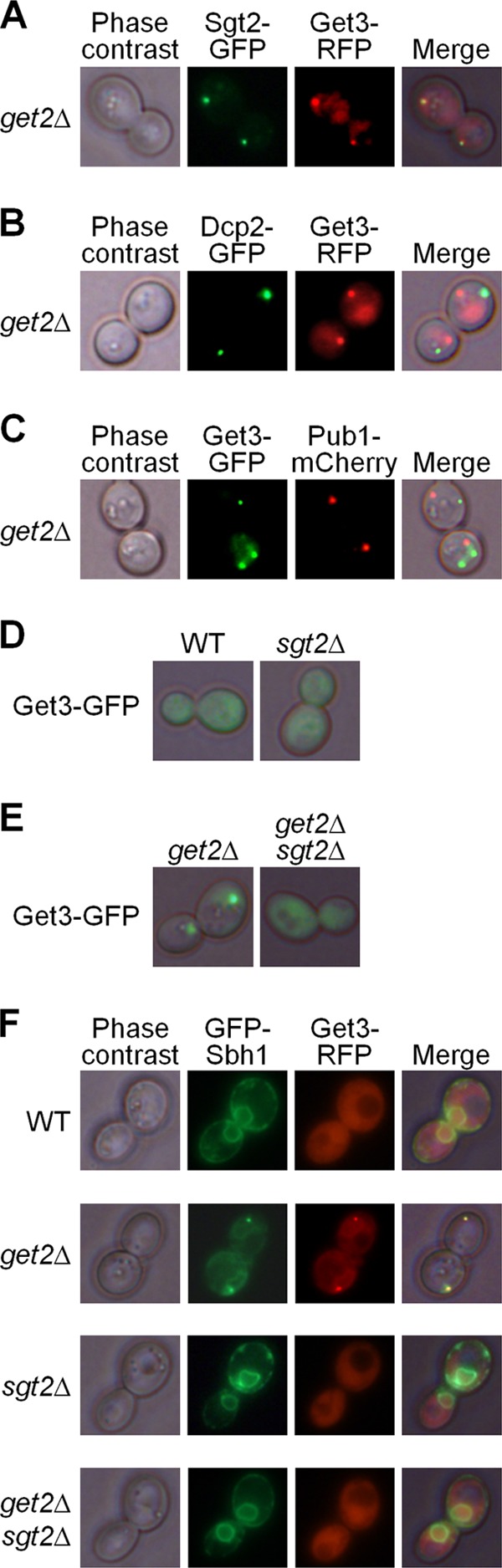
Aggregation patterns in strains with the get2Δ and/or sgt2Δ deletion. (A) In the get2Δ strain, Get3 (tagged by red fluorescent protein [RFP]) and Sgt2 (tagged by GFP) formed aggregates that were colocalized. (B and C) Get-TA aggregates, marked by fluorescently tagged Get3, did not colocalize with the P-body marker Dcp2 (B) or with the stress granule marker Pub1 (C). (D) The sgt2Δ deletion did not cause the formation of Get-TA aggregates (marked by Get3-GFP) in the Get+ strain. (E) The sgt2Δ deletion abolished aggregation of the cytosolic Get protein Get3-GFP in the get2Δ strains. (F) The tail-anchored protein Sbh1 (tagged by GFP) was localized to the endoplasmic reticulum membrane in the wild-type (WT) strain but was colocalized with the Get3 aggregates in the get2Δ strain. Membrane localization of Sbh1 was not altered by the sgt2Δ deletion and was restored in the get2Δ sgt2Δ strain.
Effects of prions on Sgt2 levels.
The [PSI+] prion was previously reported to induce Hsps (18, 58); however, these data were obtained with strains also containing the tRNA suppressor gene SUP16 (SUQ5), which further increases translational readthrough. In our strains, the presence of neither [PSI+] nor [PIN+] prions increased the level of Hsp104, Ssa, or Sis1 (Fig. 5A). Remarkably, the presence of both [PSI+] and [PIN+] simultaneously or [PIN+] alone led to a 4-fold or 2-fold increase, respectively, in the levels of Sgt2 in the same strains compared to the [psi− pin−] control (Fig. 5B). These data show that the Sgt2 level serves as a more sensitive detector of amyloid presence than Hsp levels, with different prions exhibiting additive effects on the accumulation of Sgt2.
Fig 5.
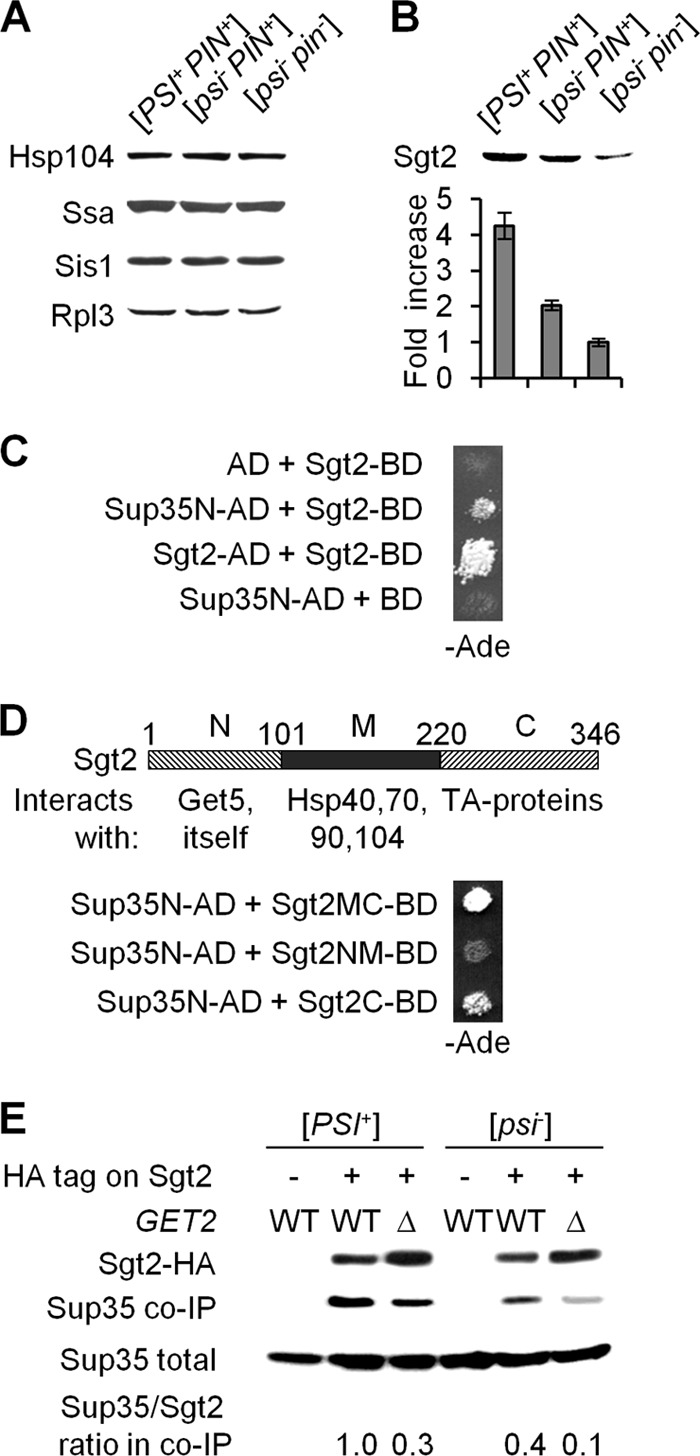
Physical interactions of Sgt2 with amyloidogenic proteins. (A and B) The presence of [PSI+] and/or [PIN+] prions did not increase levels of major Hsps in our strains (A); however, it did increase levels of Sgt2, in an additive fashion (B). Rpl3 served as a loading control. Densitometry data in panel B are shown in relation to the [psi− pin−] strain. Average results for three independent experiments are shown. For each pairwise combination, the difference is statistically significant. (C) Sgt2 interacts with the prion domain of Sup35 (Sup35N) and with itself in the two-hybrid assay. (D) Known information about the protein interaction capabilities of the Sgt2 domains (shown on the scheme) is compared to the abilities of these domains to interact with Sup35N in the two-hybrid assay. Numbers over the Sgt2 map indicate amino acid positions. In both panels C and D, two-hybrid interaction was detected by activation of the PGAL2-ADE2 reporter, resulting in growth on −Ade medium. Plate images were taken after 5 days of incubation. (E) Sup35 is coimmunoprecipitated with HA-tagged Sgt2. The absence or presence of the HA tag in the chromosomal copy of the SGT2 gene is indicated with “−” or “+,” respectively. Anti-HA antibodies were used to precipitate Sgt2-HA. Coimmunoprecipitated Sup35 was detected in the samples prepared from SGT2-HA strains but not in the samples prepared from strains with nontagged SGT2. Levels of Sgt2-associated Sup35 were determined by densitometry, using Sup35 in total lysate as a loading control, and are shown in relation to the strain with the highest level, whose level was considered 1.0.
Physical interactions between Sgt2 and prion/amyloid proteins.
A large-scale two-hybrid assay previously detected an interaction between Sgt2 and Rnq1 (70). We checked to see if Sgt2 interacts with Sup35. Indeed, a two-hybrid assay demonstrated that Sgt2 and the N-terminal (prion) domain of Sup35, Sup35N, interact with each other (Fig. 5C). Sgt2 also interacted with itself, confirming previous reports that it is a homodimer (10). The C-terminal domain of Sgt2 (amino acids 221 to 346), known to interact with the hydrophobic tails of TA proteins (65), was required for the two-hybrid interaction with Sup35N, while the N-terminal domain of Sgt2 (amino acids 2 to 101), known to interact with Get5 (10, 65), was dispensable for the interaction with Sup35N (Fig. 5D). The middle (M) domain of Sgt2 (amino acids 102 to 220), previously implicated in interactions with Hsps (10, 65), was not sufficient for a two-hybrid interaction with Sup35N; however, the presence of this domain increased the interaction.
Co-IP experiments also confirmed an interaction between HA-tagged Sgt2 (used as “bait”) and Sup35 (Fig. 5E). Co-IP was detected in both [PSI+] and [psi−] cells; however, it was more efficient in [PSI+] cells if normalized by the amount of precipitated Sgt2-HA. This indicates a higher affinity of Sgt2 for the prion form of Sup35 or could have resulted from higher Sgt2 levels in the [PSI+] strains. The latter possibility seems likely, as a 2-fold difference in Sgt2 levels between the [PSI+ PIN+] and [psi− PIN+] strains correlates well with an approximately 2.5-fold difference in the amounts of Sup35 coprecipitated with Sgt2 from these strains. Likewise, the ratio of coimmunoprecipitated Sup35 to Sgt2 was decreased in the get2Δ background, where Sgt2 levels were increased. Apparently, in cells lacking Get2, much of the excess Sgt2 was targeted to cytosolic Get-TA aggregates. Notably, Sup35 prion aggregates (marked by Sup35-dsRed) were colocalized with the Get-TA aggregates (marked by Get3-GFP or Sgt2-GFP) in 12 to 15% of the [PSI+] cells that exhibited Get-TA aggregate formation (see Fig. S2G and H in the supplemental material). This confirms at least an occasional association between these two types of aggregated structures, possibly mediated via Sgt2 and maybe reflecting an Sgt2-Sup35 interaction occurring in the get2Δ cells.
Previous works (66, 67) indicated that Sgt2 is associated with polyglutamines expressed in yeast cells containing an endogenous prion. We showed that about 8% of wild-type cells with no genetic disruption of the GET pathway but expressing 103QP-GFP and containing aggresome-like 103QP deposits (67) also contained Get3 clumps. Moreover, the Get3 clumps were always located next to (or partly overlapping) the 103QP-GFP deposit, indicating that polyglutamines nucleate aggregation of the Get3 protein in Get+ cells (see Fig. S2I in the supplemental material). In about 12% of the get2Δ cells bearing both 103QP-RFP and Sgt2-GFP aggregates, these aggregates were colocalized or juxtaposed with each other (see Fig. S2J). This points to an interaction between the quality control compartments containing amyloid-like aggregates and cytosolic Get (and possibly TA) proteins, presumably occurring via Sgt2.
DISCUSSION
Our data demonstrate that disruption of the GET pathway protects the prion [PSI+] from excess chaperone Hsp104 via inducing the chaperone Hsp70-Ssa and the cochaperone Sgt2. Moreover, Sgt2 also modulates the actions of Ssa and Hsp104 on [PSI+] in strains with a normally functioning GET pathway. Notably, Sgt2 does not have a noticeable effect on [PSI+] on its own, and its abundance becomes important for a prion only when Ssa is present in excess.
A postulated function of Sgt2 in the GET pathway is targeting of Hsps and cytoplasmic Get proteins to the newly synthesized TA proteins (10, 65). This ensures protection of the hydrophobic tails of TA proteins from intermolecular collapse. Interestingly, our data show that disruption of TA trafficking in cells with a defective GET pathway depends on Sgt2. Cytosolic Get proteins, TA proteins, and Sgt2 form aggregates in the cytoplasm of get2Δ cells (57) (Fig. 4). However, in a strain lacking both Get2 and Sgt2, the aggregation of Get proteins (Fig. 4E; see Fig. S2C and D in the supplemental material) and the TA protein Sbh1 (Fig. 4F) is abolished, and localization of Sbh1 to the ER membrane is restored. This shows that in GET-deficient cells lacking Sgt2, at least some TA proteins are not sequestered into aggregates, allowing trafficking via an alternative pathway, whose existence was proposed previously (48, 57). It is possible that such an alternative pathway utilizes Hsp70 and Hsp40 chaperones, which have been shown to facilitate in vitro integration of TA proteins into membranes (1, 50). This is consistent with the high levels of Hsp induction observed in the getΔ and getΔ sgt2Δ strains (Fig. 3D; see Fig. S1H in the supplemental material) and with the role of Sgt2 as a chaperone targeting factor.
Our data indicate that Sgt2 also plays a role in targeting Hsps (specifically Ssa) to prion aggregates. Sgt2 is known to interact with Hsps (10, 65) and with the prion protein Rnq1 (70), and our data uncovered an interaction of Sgt2 with the prion protein Sup35 (Fig. 5). Sgt2 was also detected among the proteins associated with polyglutamine aggregates in yeast cells (66, 67). Remarkably, one (C-terminal) domain of Sgt2 is involved in binding both hydrophobic tails of TA proteins and the prion domain of Sup35 (Fig. 5D). This suggests that the cellular function of Sgt2 is not confined to the GET pathway. Rather, Sgt2 targets chaperones to different types of aggregation-prone proteins. Indeed, Sgt2 manifests itself as a sensitive indicator of the presence of amyloid-based prions in the yeast cell, as its levels are increased in the presence of a prion(s) under conditions where conventional Hsps are not induced (Fig. 5A and B). A decrease in the proportion of Sgt2-associated Sup35 in get2Δ cells, in which Get-TA aggregates are formed (Fig. 5E), indicates that different types of aggregation-prone proteins compete for Sgt2 binding. However, occasional colocalization of Sup35 prion aggregates and cytosolic Get-TA aggregates (see Fig. S2G and H in the supplemental material), as well as occasional nucleation of the Get-TA aggregates by the polyglutamine-derived quality control deposits (see Fig. S2I), points to the possibility of Sgt2 linking different aggregated proteins to each other, thus forming more complex assemblies. Whether this plays a role in detoxification and/or elimination of aggregated proteins remains to be seen. However, taking into account that Get-TA aggregate formation depends on the microtubular cytoskeleton (see Fig. S2E) and that Sgt2 is involved in stress resistance (4), the possibility of Sgt2 triggering the specific assembly pathway that counteracts potential cytotoxicity of aggregation-prone proteins appears to be quite likely.
It is not yet entirely clear how excess Ssa reverses the effect of excess Hsp104 on [PSI+]; therefore, a molecular basis for the role of Sgt2 in this process remains hypothetical (Fig. 6). It has been proposed (35) that the proper balance between Ssa and Hsp104, rather than an intermediate level of Hsp104 per se, is required for the efficient fragmentation (and therefore propagation) of Sup35 prion aggregates. Indeed, recent experiments in various models indicated that aggregate fragmentation is initiated by Hsp70 (bacterial DnaK) and/or its Hsp40 cochaperone (bacterial DnaJ or yeast Sis1), while Hsp104 acts on further stages (69). In the case where Hsp104 is present in excess, it might interact with the aggregates without Ssa. Such a “nonproductive” interaction would not result in an efficient fragmentation of prion aggregates to oligomers. Analysis of [PSI+] loss after short-term heat shock (that is, also under conditions of Hsp104-Ssa imbalance) suggests that prion interaction with excess Hsp104 may lead to asymmetric accumulation of prion aggregates in the mother cell, and eventually to segregational loss of a prion (44). When both Ssa and Hsp104 are present in excess, the proper chaperone balance, and therefore proper fragmentation, is restored. However, this occurs only if Ssa has unrestricted access to Sup35 aggregates. In such a scenario, the role of Sgt2 would be to target excess Ssa to the prion. This could be achieved via promoting Ssa binding to an amyloid substrate and/or altering the structural parameters of amyloid aggregates, thus making them more accessible to Ssa. Modulation of the Hsp104 interaction with either Ssa or aggregates (or both) by Sgt2 also cannot be excluded. Further experiments aimed at determining which of these interactions provide a major input(s) into the observed outcomes are under way.
Fig 6.
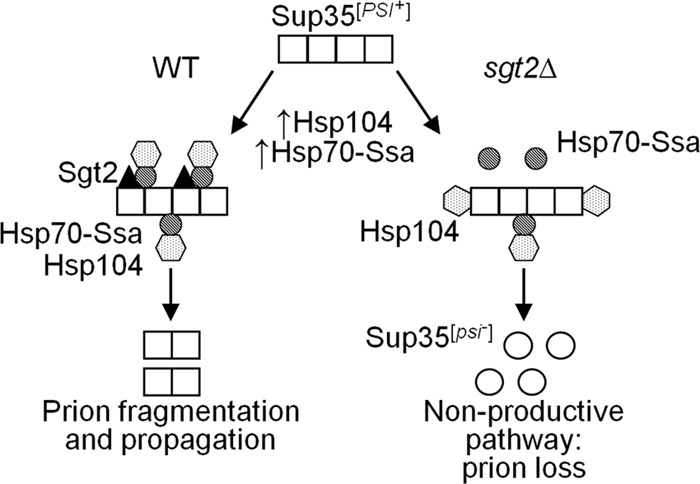
Model for the effects of Sgt2 and Hsp70-Ssa on [PSI+] curing by excess Hsp104. Hsp104 (dotted hexagons) promotes fragmentation of Sup35 prion polymers (squares) by binding them via the chaperone Hsp70-Ssa (striped circles). However, excess Hsp104 binds prion polymers on its own (without Hsp70-Ssa) at different sites and impairs prion propagation. In the sgt2Δ strain (right panel), overproduction of Hsp70-Ssa does not significantly increase the amount of polymer-bound Hsp70-Ssa, as the number of accessible sites is limited. However, when Sgt2 is present (left panel), it increases access of Hsp70-Ssa to prion polymers. This results in targeting of excess Hsp104 to polymer-bound Hsp70-Ssa and therefore in restoration of normal prion fragmentation and propagation. Some Hsp40 chaperones (not shown in the figure) serve as cochaperones of Hsp70-Ssa, and Hsp70-Ssb presumably antagonizes Hsp70-Ssa. Effects of disruptions of the GET pathway are due to induction of Hsps and Sgt2.
Taking into account that get disruption affects [PSI+] via Sgt2 and Ssa actions, one may draw additional interesting conclusions from the genetic interactions between get deletions and alterations of some other chaperone/cochaperone proteins in regard to their effects on [PSI+]. For example, the epistatic interaction between the double ssb1/2 deletion and the get2Δ deletion (Fig. 2B) suggests that Ssb might act on [PSI+] via antagonizing, directly or indirectly, the interaction of Ssa with prion aggregates. On the other hand, additive genetic interactions between the get2Δ deletion and deletions of the genes coding for the Hsp90/70 cochaperones Sti1 and Cpr7 (Fig. 2C and D) indicate that these cochaperones (and possibly their interacting partner, Hsp90) assist Hsp104 in [PSI+] curing via a mechanism that does not include direct competition with Ssa.
Since an Sgt2 ortholog is conserved across eukaryotes, including humans (30), the question of whether human Sgt2 has any effect on amyloids associated with human aggregation disorders arises. The intimate relationship between Sgt2 and the chaperone machinery that controls amyloid propagation in yeast makes Sgt2 involvement in human amyloidoses an attractive topic for further investigations.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Brazas, E. Craig, D. Cyr, S. Elledge, P. Hieter, S. Lindquist, R. Parker, M. Schuldiner, and J. Warner for plasmids and antibodies, Y. Nishida and A. Romanyuk for help with some experiments, K. Bruce and G. Newnam for proofreading the manuscript, Y. Pavlov for guidance on the EMS mutagenesis experiments, and J. Weissman for helpful discussions.
This work was supported by grants R01GM058763 and R01GM093294 from the NIH. J.C.P. was supported by an undergraduate research scholarship from the Institute for Bioengineering and Bioscience.
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 8 October 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Abell BM, Rabu C, Leznicki P, Young JC, High S. 2007. Post-translational integration of tail-anchored proteins is facilitated by defined molecular chaperones. J. Cell Sci. 15:1743–1751 [DOI] [PubMed] [Google Scholar]
- 2. Allen KD, Chernova TA, Tennant EP, Wilkinson KD, Chernoff YO. 2007. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J. Biol. Chem. 282:3004–3013 [DOI] [PubMed] [Google Scholar]
- 3. Allen KD, et al. 2005. Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 169:1227–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Angeletti PC, Walker D, Panganiban AT. 2002. Small glutamine-rich protein/viral protein U-binding protein is a novel cochaperone that affects heat shock protein 70 activity. Cell Stress Chaperones 7:258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Auld KL, et al. 2006. The conserved ATPase Get3/Arr4 modulates the activity of membrane-associated proteins in Saccharomyces cerevisiae. Genetics 174:215–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bailleul PA, Newnam GP, Steenbergen JN, Chernoff YO. 1999. Genetic study of interactions between the cytoskeletal assembly protein sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae. Genetics 153:81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonneaud N, et al. 1991. A family of low and high copy replicative, integrative and single-stranded S. cerevisiae/E. coli shuttle vectors. Yeast 7:609–615 [DOI] [PubMed] [Google Scholar]
- 8. Borgese N, Brambillasca S, Colombo S. 2007. How tails guide tail-anchored proteins to their destinations. Curr. Opin. Cell Biol. 19:368–375 [DOI] [PubMed] [Google Scholar]
- 9. Buchan JR, Muhlrad D, Parker R. 2008. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 183:441–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chartron JW, Gonzalez GM, Clemons WM., Jr 2011. A structural model of the Sgt2 protein and its interactions with chaperones and the Get4/Get5 complex. J. Biol. Chem. 286:34325–34334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chernoff YO, et al. 2000. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol. Microbiol. 35:865–876 [DOI] [PubMed] [Google Scholar]
- 12. Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268:880–884 [DOI] [PubMed] [Google Scholar]
- 13. Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. 1999. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI] prion. Mol. Cell. Biol. 19:8103–8112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chernoff YO, Uptain SM, Lindquist SL. 2002. Analysis of prion factors in yeast. Methods Enzymol. 351:499–538 [DOI] [PubMed] [Google Scholar]
- 15. Colby DW, Prusiner SB. 2011. Prions. Cold Spring Harb. Perspect. Biol. 3:a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coller J, Parker R. 2005. General translational repression by activators of mRNA decapping. Cell 122:875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durfee T, et al. 1993. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev. 7:555–569 [DOI] [PubMed] [Google Scholar]
- 18. Eaglestone SS, Cox BS, Tuite MF. 1999. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J. 18:1974–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eisenberg D, Jucker M. 2012. The amyloid state of proteins in human diseases. Cell 148:1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan CY, Lee S, Ren HY, Cyr DM. 2004. Exchangeable chaperone modules contribute to specification of type I and type II Hsp40 cellular function. Mol. Biol. Cell 15:761–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gerring SL, Spencer F, Hieter P. 1990. The CHL1 (CTF1) gene product of Saccharomyces cerevisiae is important for chromosome transmission and normal cell cycle progression in G2/M. EMBO J. 9:4347–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Glover JR, Lindquist S. 1998. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94:73–82 [DOI] [PubMed] [Google Scholar]
- 23. Halfmann R, et al. 2012. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 482:363–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. James P, Halladay J, Craig EA. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Janke C, et al. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21:947–962 [DOI] [PubMed] [Google Scholar]
- 26. Jonikas MC, et al. 2009. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323:1693–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaganovich D, Kopito R, Frydman J. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454:1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kilmartin JV. 1981. Purification of yeast tubulin by self-assembly in vitro. Biochemistry 20:3629–3633 [DOI] [PubMed] [Google Scholar]
- 29. Kopito RR. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10:524–530 [DOI] [PubMed] [Google Scholar]
- 30. Kordes E, et al. 1998. Isolation and characterization of human SGT and identification of homologues in Saccharomyces cerevisiae and Caenorhabditis elegans. Genomics 52:90–94 [DOI] [PubMed] [Google Scholar]
- 31. Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. 2003. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278:49636–49643 [DOI] [PubMed] [Google Scholar]
- 32. Kushnirov VV, Ter-Avanesyan MD. 1998. Structure and replication of yeast prions. Cell 94:13–16 [DOI] [PubMed] [Google Scholar]
- 33. Lee S, Fan CY, Younger JM, Ren H, Cyr DM. 2002. Identification of essential residues in the type II Hsp40 Sis1 that function in polypeptide binding. J. Biol. Chem. 277:21675–21682 [DOI] [PubMed] [Google Scholar]
- 34. Lee SJ, Lim HS, Masliah E, Lee HJ. 2011. Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci. Res. 70:339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liebman SW, Chernoff YO. 2012. Prions in yeast. Genetics 191:1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liou ST, Cheng MY, Wang C. 2007. SGT2 and MDY2 interact with molecular chaperone YDJ1 in Saccharomyces cerevisiae. Cell Stress Chaperones 12:59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu H, Krizek J, Bretscher A. 1992. Construction of a GAL1-regulated yeast cDNA expression library and its application to the identification of genes whose overexpression causes lethality in yeast. Genetics 132:665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Longtine MS, et al. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961 [DOI] [PubMed] [Google Scholar]
- 39. Maclea KS, Ross ED. 2011. Strategies for identifying new prions in yeast. Prion 5:263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McDonald JH. 2009. Handbook of biological statistics, 2nd ed, p 118–122 Sparky House Publishing, Baltimore, MD [Google Scholar]
- 41. McGlinchey RP, Kryndushkin D, Wickner RB. 2011. Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. U. S. A. 108:5337–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meriin AB, et al. 2007. Endocytosis machinery is involved in aggregation of proteins with expanded polyglutamine domains. FASEB J. 21:1915–1925 [DOI] [PubMed] [Google Scholar]
- 43. Moosavi B, Wongwigkarn J, Tuite MF. 2010. Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI+] prion but not for prion propagation. Yeast 27:167–179 [DOI] [PubMed] [Google Scholar]
- 44. Newnam GP, Birchmore JL, Chernoff YO. 2011. Destabilization and recovery of a yeast prion after mild heat shock. J. Mol. Biol. 408:432–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Newnam GP, Wegrzyn RD, Lindquist SL, Chernoff YO. 1999. Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol. Cell. Biol. 19:1325–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ohba M. 1997. Modulation of intracellular protein degradation by SSB1-SIS1 chaperon system in yeast S. cerevisiae. FEBS Lett. 409:307–311 [DOI] [PubMed] [Google Scholar]
- 47. Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144 [DOI] [PubMed] [Google Scholar]
- 48. Rabu C, High S. 2007. Membrane protein chaperones: a new twist in the tail? Curr. Biol. 17:R472–R474 [DOI] [PubMed] [Google Scholar]
- 49. Rabu C, Schmid V, Schwappach B, High S. 2009. Biogenesis of tail-anchored proteins: the beginning for the end? J. Cell Sci. 122:3605–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rabu C, Wipf P, Brodsky JL, High S. 2008. A precursor-specific role for Hsp40/Hsc70 during tail-anchored protein integration at the endoplasmic reticulum. J. Biol. Chem. 283:27504–27513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reidy M, Masison DC. 2011. Modulation and elimination of yeast prions by protein chaperones and co-chaperones. Prion 5:245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reidy M, Masison DC. 2010. Sti1 regulation of Hsp70 and Hsp90 is critical for curing of Saccharomyces cerevisiae [PSI+] prions by Hsp104. Mol. Cell. Biol. 30:3542–3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rikhvanov EG, Romanova NV, Chernoff YO. 2007. Chaperone effects on prion and nonprion aggregates. Prion 1:217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Romanova NV, Chernoff YO. 2009. Hsp104 and prion propagation. Protein Pept. Lett. 16:598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sanchez Y, Lindquist SL. 1990. HSP104 required for induced thermotolerance. Science 248:1112–1115 [DOI] [PubMed] [Google Scholar]
- 56. Sanchez Y, et al. 1993. Genetic evidence for a functional relationship between Hsp104 and Hsp70. J. Bacteriol. 175:6484–6491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schuldiner M, et al. 2008. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 134:634–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schwimmer C, Masison DC. 2002. Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol. Cell. Biol. 22:3590–3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Serio TR, Cashikar AG, Moslehi JJ, Kowal AS, Lindquist SL. 1999. Yeast prion [psi+] and its determinant, Sup35p. Methods Enzymol. 309:649–673 [DOI] [PubMed] [Google Scholar]
- 60. Shen J, Hsu CM, Kang BK, Rosen BP, Bhattacharjee H. 2003. The Saccharomyces cerevisiae Arr4p is involved in metal and heat tolerance. Biometals 16:369–378 [DOI] [PubMed] [Google Scholar]
- 61. Sherman F. 2002. Getting started with yeast. Methods Enzymol. 350:3–41 [DOI] [PubMed] [Google Scholar]
- 62. Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Toikkanen J, et al. 1996. Yeast protein translocation complex: isolation of two genes SEB1 and SEB2 encoding proteins homologous to the Sec61β subunit. Yeast 12:425–438 [DOI] [PubMed] [Google Scholar]
- 64. Vilardell J, Warner JR. 1997. Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol. Cell. Biol. 17:1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang F, Brown EC, Mak G, Zhuang J, Denic V. 2010. A chaperone cascade sorts proteins for posttranslational membrane insertion into the endoplasmic reticulum. Mol. Cell 40:159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang Y, Meriin AB, Costello CE, Sherman MY. 2007. Characterization of proteins associated with polyglutamine aggregates: a novel approach towards isolation of aggregates from protein conformation disorders. Prion 1:128–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Y, et al. 2009. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 23:451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wickner RB, Edskes HK, Bateman D, Kelly AC, Gorkovskiy A. 2011. The yeast prions [PSI+] and [URE3] are molecular degenerative diseases. Prion 5:258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Winkler J, Tyedmers J, Bukau B, Mogk A. 2012. Chaperone networks in protein disaggregation and prion propagation. J. Struct. Biol. 179:152–160 [DOI] [PubMed] [Google Scholar]
- 70. Yu H, et al. 2008. High-quality binary protein interaction map of the yeast interactome network. Science 322:104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.