

Abstract

A general protocol for the coupling of haloarenes with a variety of allylic acetates is presented. Strengths of the method are a tolerance for electrophilic (ketone, aldehyde) and acidic (sulfonamide, trifluoroacetamide) substrates and the ability to couple with a variety of substituted allylic acetates. Secondary alkyl bromides can also be allylated under slightly modified conditions, demonstrating the generality of the approach. Finally, the coupling of a reactive vinyl halide could be achieved by the use of a very hindered ligand and more reactive, branched allylic acetates.
1. Introduction
Allylarene derivatives are versatile synthetic intermediates to which a variety of approaches have been developed: Friedel–Crafts allylation, allylation of aryl nucleophiles by metal-mediated allylic substitution chemistry,1 metal-mediated cross-coupling of aryl halides with allyl nucleophiles,2 and Heck-reactions of olefins with aryl halides.3 We present here an alternative disconnection, the cross-coupling of two electrophiles, allylic acetates and aryl halides (Figure 1). While the coupling of simple allyl acetate with aryl halides has been reported several times, few examples with substituted allylic acetates have been reported. In our own studies, we have found that the ligands previously used for such reactions, pyridine and bipyridine ligands, were poorly selective. We report here a new ligand that promotes selective coupling of aryl halides with allylic acetates. These conditions, with a few modifications, can also be used to couple allylic acetates with secondary alkyl bromides and a vinyl bromide.
Figure 1.

Approaches to the synthesis of allylated arenes.
2. Background
The reductive approach to allylated arenes (Figure 1) is complementary to the other approaches, addressing some of the limitations present in each. Friedel-Crafts is direct, but precise control of the products is difficult and electron-rich arenes are required. The Heck reaction is also direct, and functional-group compatibility can be excellent, but selectivity for allylarene over vinyl arene is usually poor.3,4 In cases where selectivity is achieved, it is for the styrene, not the allylated arene.5 Methods that rely upon pre-formed organometallic reagents are limited by the low commercial availability of organometallic reagents and the reactivity of the organometallic reagent itself.
Allylic acetate derivatives and aryl halides are more convenient and affordable than organometallic reagents, but few reports on the reductive coupling of substituted allylic acetates with functionalized arenes exist. The reductive coupling of aryl bromides and chlorides with an excess (2.0–2.7 equiv) of allylic acetates has been reported to be catalyzed by 13–40 mol % cobalt under both electrochemical6 and chemical conditions (zinc or manganese reductant).7 The principle side reactions observed were reduction of the aryl halide and dimerization of the aryl halide to form biaryl. Although aryl bromides and electron-poor aryl chlorides worked well, no examples with aryl iodides were reported, and it was noted that even more biaryl formation occurred with these substrates. Selectivity for cross product was the major limitation.
Durandetti, Nédélec, and Périchon had noted in 1996 that bipyridine-ligated nickel catalysts could couple allylic acetates with aryl halides, but slow addition of the allylic acetate was required for high selectivity.8a Starting from a bipyridine catalyst we had developed for the coupling of aryl halides with alkyl halides,9 Gong recently reported that several additives significantly improved yields for the coupling of 3 different aryl bromides, but only with simple allyl acetate (CH2=CH2CH2OAc).8b
Only four successful reactions with substituted allylic acetates have been reported to date (Scheme 1). For the cobalt-catalyzed reactions, yields and selectivities were modest and relatively high catalyst loading was required. Similarly, two nickel catalyzed examples with crotyl acetate provided good yield and regioselectivity, but required the slow addition of the allylic acetate to achieve high cross-selectivity. No examples of substituted allylic acetates utilizing chemical reductants have been reported.
Scheme 1.

All previous examples of reductive allylations with substituted allylic acetates.
As evident in these prior studies, the major challenge associated with the reductive coupling of two electrophiles is the selective formation of cross-coupled product over competitive dimerization reactions (Figure 2). The nickel-catalyzed methods required slow addition of the allylic acetate over the course of the electrolysis8a or two equivalents of allyl acetate for high yield.8b These conditions were proposed to favor formation of arylnickel intermediates over allylnickel intermediates.8a The cobalt-catalyzed methods,6,7 on the other hand, were reported to suffer from large amounts of biaryl formation or hydrodehalogenation, even with excess allyl acetate.
Figure 2.

Selectivity challenge for reductive allylation.
More selective, general conditions for the allylation of substituted allylic acetates may require the development of new catalysts and strategies for selective oxidative addition of either the allylic acetate or the aryl halide. Few studies on the relative rates at which nickel complexes react with different electrophiles have been reported.8a In the course of studying dimerization reactions, we had found that terpyridine-ligated nickel complexes dimerized alkyl bromides and allylic acetates, but only slowly reacted with aryl halides.10 Because allylnickel(II) complexes are well-known to react with aryl halides to form allylarenes,11 we hypothesized that a terpyridine-ligated nickel complex could achieve better selectivity than bipyridine catalysts. We report here our results with this same catalyst for the coupling of substituted allylic acetates with a range of organic halides.
During the course of these studies, Gosmini reported a Co-catalyzed method for the allylation of alkyl halides,12 Gong reported the use of our terpyridine-nickel system10 for the allylation of alkyl halides,13 and Gong reported the coupling of simple allyl acetate with three aryl bromides.8b None of these manuscripts explored the coupling of substituted allylic acetates with aryl halides or vinyl halides.
3. Results
3.1 Reaction Optimization
Building on the observation that 4,4′,4″-tri-tert-butyl-2,2′:6′,2″-terpyridine (L1) and NiCl2(dme) formed a catalyst that was slow to dimerize haloarenes, but was reactive with allylic acetates, we examined it’s ability to mediate the coupling of cinnamyl acetate (2a) with iodobenzene (Table 1). We found that it is highly selective for cross-coupling over competing dimerization reactions (B and C in Table 1, entry 1), although some hydrodehalogenation competes (D). Our own studies on the coupling of aryl halides with unactivated alkyl halides had previously found bipyridine L3 to form a particularly selective catalyst,9 but biaryl formation was a problem in reactions with allylic acetates (entry 2). Our observed low selectivity mirrors previous reports with pyridine6, 7 as a co-solvent and ligand (entry 3) or a bipyridine8 (L3) as a ligand (entry 4). Finally, reactions conducted without ligand (entry 5), without nickel, or without zinc produced little or no cross-product, respectively (Table S6 in Supporting Information).
Table 1.
Ligand effect on cross-selectivitya
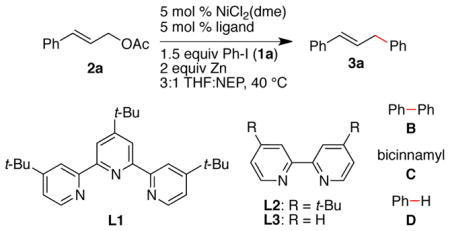
| |||
|---|---|---|---|
| Entry | ligand | GC yieldb | ratioc 3a:B:C:D |
| 1 | L1 | 90 | 100:1:2:15 |
| 2 | L2 | 62 | 100:37:5:5 |
| 3 | pyridine (10 equiv) | 45d | 100:11:0:20 |
| 4 | L3 | 73 | 100:17:1:4 |
| 5 | none | 4e | 100:0:0:86 |
Reaction conditions: 1:1.5:2 cinnamyl acetate:Ph-I:Zn, 5 mol % NiCl2(dme) in 3:1 THF:NEP (NEP = N-ethylpyrrolidinone). Reaction time was 15–24 h.
Corrected GC yields, see Supporting Information for an example calculation.
Ratios based upon GC Area % data.
Reaction time was 96 h.
Reaction was incomplete at 96 h, 31% 1a and 51% 2a remained.
Initial studies had found that dimethylacetamide (DMA) and N-ethylpyrrolidinone (NEP) provided the best yields out of various amide and urea solvents. In order to simplify product isolation, we examined THF solvent mixtures and found that NEP/THF provided the highest yields. In very cost-sensitive situations, DMA/THF can be substituted for NEP/THF with only a small decrease in yield (73% vs 90% in entry 1).
3.2 Aryl Halide Scope
In order to examine the compatibility of these conditions with various functional-groups, the optimized conditions were applied to a series of aryl iodides and aryl bromides (Table 2). Both electron-rich and electron-poor aryl iodides formed product in high yield (entries 3–11). Broad functional-group tolerance is notable, including an N-aryl trifluoroacetamide group (entry 6), which bears a strongly acidic proton (similar to acetic acid, see also entry 5) and is easily cleaved by nucleophiles. Although nickel is known to catalyze both pinacol coupling14 and allylation15 of ketones and aldehydes, products 3b and 3c were obtained in high yield (entries 3 and 4). Because bromobenzene was unreactive under these reaction conditions, we were able to couple 1-bromo-4-iodobenzene with high chemoselectivity (entry 14). Finally, meta- and ortho-substituted iodoarenes also coupled in good yield (entries 16–18).
Table 2.
Allylation of a variety of aryl halidesa
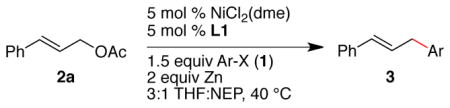
| |||
|---|---|---|---|
| Entry | X | product | yieldb |
| 1 | I |
1 mmol scale, set up in glovebox |
88 |
| 2 | I | 10 mmol scale, set up on benchtopc | 81 |
| 3 | I |
 R = C(O)Me (3b) |
71 |
| 4 | I | R = CHO (3c) | 70 |
| 5 | I | R = NHTs (3d) | 73 |
| 6 | I | R = NHC(O)CF3 (3e) | 64 |
| 7 | I | R = CH2OTBS (3f) | 80d |
| 8 | I | R = NMe2 (3g) | 55 |
| 9 | I | R = Me (3h) | 86 |
| 10 | I | R = OMe (3i) | 83 |
| 11 | I | R = Br (3j) | 64e |
| 12 | Br | R = CO2Me (3k) | 65 |
| 13 | Br | R = C(O)Me (3b) | 48 |
| 14 | Br | R = CF3 (3l) | 51f |
| 15 | Br | R = CN (3m) | 77 |
| 16 | I |
 (3n) |
78 |
| 17 | I | R = CN (3o) | 86 |
| 18 | I | R = OMe (3p) | 80 |
As in Table 1, but on 1 mmol scale in 2 mL of THF/NEP. Reaction times were 15–24 h.
Yield after purification. For complete selectivity data, see Tables S1 and S2 in the Supporting Information.
A 1 mmol-scale reaction run on the benchtop (Procedure B) gave an 82% GC yield.
Reaction was run on a 0.5 mmol scale.
NMR yield of 3j. Isolated product is contaminated with 9% of 3a from hydrodeiodination.
Yield is an average of two runs, one at 0.5 mmol scale and one at 1 mmol scale.
In contrast to electron-neutral bromoarenes, bromoarenes containing an electron-withdrawing group couple in reasonable yield (Table 2, entries 12–15).
3.3 Allylic Acetate Scope
A variety of different allylic acetates were also examined (Table 3), with promising results. Not only aryl-substituted cinnamyl acetate, but also primary alkyl (entry 1), secondary alkyl (entry 3), and vinyl substituted (entry 6) allylic acetates coupled in good yield, with selectivity for the linear, E-product. The high regioselectivity and stereoselectivity obtained for the linear isomer, regardless of starting material, provides flexibility in synthesis and suggests a common allylnickel intermediate (Table 2, entry 1 and Table 3, entries 2 and 3). Finally, cyclic and acyclic α-substituted allylic acetates provide good yields of product (entries 8 and 9). In the case of non-symmetrical substrate 2j, regioselectivity for α-substitution is high (entry 9). The coupling of a γ,γ-disubstituted olefin, geranyl acetate (entry 7), provided a high yield of allylated products (80% total yield), but the selectivity was poor (2:1 linear:branched, 1:1 E:Z for linear).
Table 3.
Reaction of iodoarenes with various allylic acetatesa

| |||
|---|---|---|---|
| Entry | allylic acetate | Product | yieldb |
| 1 |
(2b) |
(4a) |
81c |
| 2 |
 (2c) |
(3a) |
52 |
| 3 |
(2d) |
(3a) |
75 |
| 4 |
(2e) |
(4b) |
97d |
| 5 |
 (2f) |
 (4c) |
55 |
| 6 |
(2g) |
(4d) |
65e |
| 7 |
(2h) |
(4e) |
52f |
| 8 |
 (2i) |
 (4f) |
80 |
| 9 |
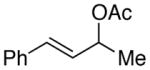 (2j) |
 (4g) |
73g |
Reaction conditions as in Table 3. Reaction times were 15–18 h. See Table S3 in the Supporting Information for full selectivity data.
Yield after purification.
Product has a 86:14 [linear]:[branched] ratio.
Product has a 93:7 [linear]:[branched] ratio.
Product is a mixture of olefin isomers: 6.9:1:1 [2E,4E]:[2E,4Z]:[unidentified olefin isomer].
NMR yield of the E/Z mixture of linear products. Product has a 1.9:1 [linear]:[branched] ratio. The linear product was a 1:1 mixture of E:Z isomers.
Product contains ~3% of an unidentified side product.
3.4 Secondary Alkyl Bromides
We also investigated the coupling of unactivated alkyl bromides with allylic acetates, which builds upon our previous studies with (L1)Ni catalysts.10 We found that cross-selectivity was poor for reactions with primary alkyl bromides due to rapid dimerization of the alkyl bromide. Secondary alkyl bromides could be cross-coupled with cinnamyl acetate derivatives with high selectivity and yield (Table 4). Conditions are nearly the same as those used for aryl halides except that THF/DMA mixtures performed best and manganese provided the highest yields. During the course of these experiments, the groups of Gosmini and Gong reported Co-12 and Ni-catalyzed13 approaches to the same products. Gong used very similar conditions, but found that the addition of large amounts of MgCl2 made the reaction more general. In light of their results, we did not pursue our additive-free conditions further.
Table 4.
Coupling of allylic acetates with alkyl bromidesa

| ||
|---|---|---|
| entry | product | yieldb |
| 1 |
|
79c |
| 2 |
|
88c |
| 3 | Set up in the benchtopd | 90 |
| 4 |
|
68c |
| 5 |
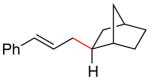 (5d)
(5d)
|
78c |
| 6 |
|
79c |
| 7 | Ar = p-F3 C-C6H4 (5f) | 66c |
Reaction conditions: 1.5:1:2 allyl acetate:R-Br:Mn. Reaction times were 15–18 h. See Table S4 in the Supporting Information for full selectivity data.
Yield after purification.
Yield is an average of two runs, one at 0.5 mmol scale and one at 1 mmol scale.
Reagents were weighed out on the bench top in a vial that was then sparged with argon gas through the septa cap.
3.5 Potential for RZnX, RMnX, or Radical Intermediates
We first considered the potential intermediacy of aryl zinc reagents, ArZnI, cecause we observed significant amounts of hydrodehalogenation of the aryl halide in some cases, (Up to 29% GC area %, Table 1, D and Tables S1–S5 in the Supporting Information).16 Although direct insertion of unactivated zinc into aryl iodides is reported to be slow,17 nickel can catalyze the process.17d Several observations lead us to believe that ArZnI intermediates are unlikely: (1) reactions without nickel only converted trace amounts of iodobenzene to benzene (2% in 48 h, Table 2 in Supporting Information); (2) a reaction quenched at intermediate conversion with D2O did not form increased amounts of C6H5D when compared to a reaction quenched with H2O (GC/MS); (3) functional groups reported to be reactive with ArZnI, such as aldehydes and acidic protons, are well tolerated (Table 3, entries 4–6).18
We also considered aryl radical intermediates to explain the observed hydrodehalogenation. An aryl radical could abstract a hydrogen atom from the solvent to form Ar-H. If this was the case, then a reaction conducted in a fully deuterated solvent mixture could be expected to form Ar-D instead of Ar-H.19 Because deuterated NEP and DMA are not commercially available, we conducted the experiment in a THF-d8/DMF-d7 mixture.20 We did not observe an increase in the amount of C6H5D produced when compared to a reaction in protic solvents (GC/MS). This argues against hydrogen atom abstraction from solvent, but does not completely rule out the presence of radical intermediates.21
3.6 Extension to Vinyl Halide
We could find no examples of the catalytic coupling of vinyl halides with allylic acetates in the literature, but the skipped diene products of such reactions are valuable in synthesis.22 Preliminary investigations on the coupling of cinnamyl acetate (2a) with 2-bromocyclohexenone provided only small amounts of product 7a. Monitoring reactions at low conversion revealed that consumption of the vinyl bromide was much faster than consumption of cinnamyl acetate. If our hypothesis that selective formation of an allylnickel intermediate was required for selective cross-coupling were correct, a more reactive allylic alcohol donor would be required.
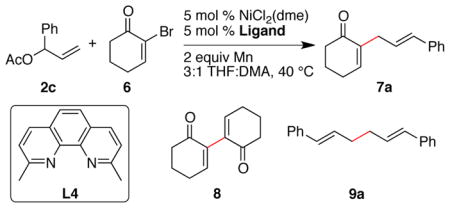
Based upon this hypothesis, we examined reactions with branched allylic acetate 2c because the less hindered olefin should facilitate coordination and ionization (Table 5). Reactions run with terpyridine L1 did not form the skipped diene 7a and failed to consume starting materials (entry 1). Bipyridine L2 provided better results, but significant allyl dimer (9a) and vinyl dimer (8) were formed. Kishi has reported that neocuproine (L4) nickel complexes are slow to dimerize aryl halides23 and we have also found that neocuproine was a selective ligand for the reductive conjugate addition of aryl and vinyl halides with enones, which we propose involves allylnickel intermediates.24 Indeed, L4 formed a highly selective catalyst for the cross-coupling of 2-bromocyclohexenone with the branched allylic acetate 2c (entry 3). The results appear related to sterics because the unsubstituted 1,10-phenanthroline provided poor results (entry 4).
Table 5.
Ligand effect on cross-selectivity for reactions with 2-bromo-cyclohex-2-enone a
| ||||||
|---|---|---|---|---|---|---|
| entry | ligand | GC Area % (Yield)b | ||||
| 2c | 6 | 7a | 8 | 9a | ||
| 1 | L1 | 66 | 34 | 0 | 0 | 0 |
| 2 | L2 | 0 | 0 | 78 | 6 | 16 |
| 3 | L4 | 0 | 0 | 94 (78) | 0 | 5 |
| 4 | phen | 66 | 34 | 0 | 0 | 0 |
Reactions run with a 1:1 ratio of reactants for 21 h. phen = 1,10-phenanthroline.
Results in table are GC Area % data. The number in parentheses is yield after purification.
The less reactive alkyl-substituted allylic substrates 2k and 2l provided low yields when the cross-coupling with the vinyl bromide was attempted (Table 6, entries 1 and 2). Again, low reactivity of the allyl derivative compared to vinyl bromide 6 appeared to be the problem. In order to further boost reactivity of the allylic donor, we examined the methyl carbonates 2m and 2n. These more reactive substrates coupled with higher selectivities and resulted in better yields (entries 3 and 4).
Table 6.
Effect of leaving group on selectivity for reactions with 2-bromo-cyclohex-2-enonea
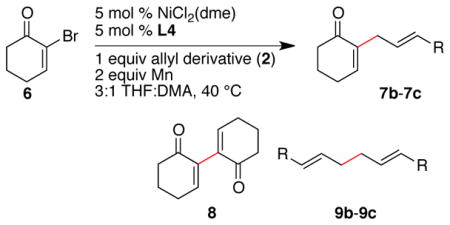
| ||||||
|---|---|---|---|---|---|---|
| entry | allyl derivative | GC Area % (Yield)b | ||||
| 2 | 6 | 7 | 8 | 9 | ||
| 1 |
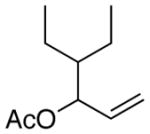 (2k)
(2k)
|
0 | 0 |
7b 21 |
26 | 51 |
| 2 |
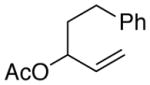 (2l)
(2l)
|
26 | 0 |
7c 36 |
17 | 19 |
| 3 |
 (2m)
(2m)
|
0 | 0 |
7b 68 (51) |
11 | 19 |
| 4 |
 (2n)
(2n)
|
0 | 0 |
7c 90 (67) |
3 | 6 |
Reactions run with a 1:1 ratio of reactants for 19 h.
Results in table are GC area % data. Numbers in parenthesis are yields after purification.
Although preliminary, this approach compares favorably with prior approaches to α-allylated enones (7). Those methods relied upon tin and selenium chemistry,25 organocuprates,26, 27 or a multistep sequence starting with the allylation of cyclohexane dione.26, 28 Product 7a has been used as a versatile precursor to the fused 6,3,5-tricyclic skeleton of Mycorrhizin A.28
4. Discussion
4.1 Mechanism
Both arylmetal6b, 8a and allylmetal8b intermediates have been proposed for cobalt- and nickel-catalyzed coupling of aryl halides with allylic acetates, but these studies were conducted with monodentate or bidentate ligands. Our previous studies with terpyridine nickel catalysts10 suggested an allylnickel intermediate (Figure 3) would be formed preferentially in reactions catalyzed by terpyridine nickel complexes.29
Figure 3.

Proposed mechanism.
Based upon studies by many groups, including Corey, Semmelhack, Hegedus, and Kochi, pre-formed allylnickel(II) reagents are known to react with aryl, alkyl, and vinyl halides, consistent with the observed generality of our nickel catalyzed process.30 The conversion of the allylnickel intermediate into product and a nickel(II) salt is proposed to involve a radical-chain-like process and has been studied extensively.30b–d Separately, Gong has proposed the intermediate reduction of allylnickel(II) intermediates into allylnickel(I) intermediates,31 but detailed mechanistic studies have not been conducted for this alternative mechanism. At this time, we cannot differentiate between the single-electron reduction mechanism and the radical-chain mechanism.
4.2 Scope of Reaction
Generally, reductive cross-electrophile coupling reactions have displayed broad functional-group tolerance.6–13 However, previous studies on reductive allylation had not demonstrated compatibility with enones, unprotected aldehydes, or highly acidic protons (Table 2).
Allylation of aryl halides with substituted allylic acetates had been limited to only four examples (Scheme 1), but we have found that reactions with (L1)Ni have broad scope, including primary and secondary alkyl, cyclic, vinyl, and 1,3-disubstituted allylic acetates allylic acetates (Table 3).
A wide variety of organic halides couple under similar conditions using (L1)Ni as the catalyst. This includes, for the first time, a vinyl bromide. Improvement of conditions for the allylation of vinyl halides will be the subject of future studies.
4.3 Selectivity
Although the true order of reactivity for the allylic acetate and haloarene with nickel has not yet been unambiguously determined, our studies show that the inherent reactivity of the substrate, ligand sterics, and ligand coordination number can all be manipulated in a rational manner to improve selectivity. For example, 1-phenyl-2-propenyl acetate (2c) is a poor match for ligand L2 and vinyl bromide 6, resulting in significantly more allyl dimerization. With a more hindered ligand (L4) only a small amount of allyl dimer is formed (Table 5).
Finally, looking over the comprehensive selectivity data in the Supporting Information, it is clear that hydrodehalogenation is the major side reaction for most examples and that this side reaction appears to happen more with bromoarenes. Development of catalysts and conditions that will suppress hydrodehalogenation are needed to further improve the scope and yields for arene allylation.
5. Conclusions
In conclusion, a new nickel-catalyzed coupling of aryl iodides, electron-poor aryl bromides, and secondary alkyl bromides with allylic acetates has been developed. This chemistry addresses the regioselectivity and substrate availability limitations observed for more well developed approaches. The functional-group compatibility, the scope of allylic acetate substitution, and the potential for the allylation of vinyl halides are promising. Finally, our results show that cross-selectivity can be controlled by the choice of ligand, which provides a clear path for future improvements.
6. Experimental Section
General
1H and 13C NMR spectra were acquired on 500 MHz or 400 MHz (proton) NMR instruments. NMR chemical shifts are reported in ppm and referenced to the residual solvent peak as an internal standard (for CDCl3 δ = 7.260 ppm, 1H; δ = 77.160 ppm, 13C). Analysis of reaction mixtures was accomplished by quenching an aliquot (10 μL) with 1 M NaHSO4(aq) (0.1 mL) and analysis of the filtered (silica or celite) ether extract (1 mL) by GC or GC/MS. Dodecane was used as the internal standard. GC analyses (FID detector) were performed on a DB-5 column (20 m × 0.18 mm × 0.18 μm) with hydrogen as the carrier gas. Std method: 1.8 mL/min flow at 20.3 psi, 300 °C inj., 325 °C detector, oven program: 50 °C (0.46 min), ramp at 65 °C/min to 300 °C, hold for 0.69 min. GC/MS analyses (EI+, quadrupole mass analyzer) were performed on an instrument equipped with an RTX-XLB column (30 m × 0.25 mm × 0.28 μm) with helium carrier gas. Std method: 1 mL/min flow at 7.8 psi, 225 ºC inj., 250 ºC interface/ion source, oven program: 50 ºC (3 min), ramp at 40 ºC/min to 280 ºC, hold for 3 min. Only the parent ion and the base peak are reported. High resolution mass spectra (HRMS) were acquired on an instrument with electron impact (EI) ionization and a magnetic sector mass analyzer. Compounds were either purified on an automated flash purification system on Redisep Rf Gold normal-phase silica columns or by standard chromatography on silica gel (EMD, silica gel 60, particle size 0.040–0.063 mm) using standard flash techniques. Products were visualized by UV light, KMnO4 stain, or by GC.
Unless otherwise noted, all reagents and solvents were purchased from commercial suppliers and were used as received. NiCl2(1,2-dimethoxyethane) (NiCl2(dme)) was purchased from Strem or synthesized.32 As we have reported elsewhere, the stoichiometry of the NiCl2(dme) can be variable.10 The amount of NiCl2(dme) used was corrected for the actual amount of dimethoxyethane present as determined by elemental analysis. 4,4′,4″-tri-tert-butyl-2,2′:6′,2″-terpyridine33 (L1), 4,4′-di-tert-butyl-2,2′-bipyridine (L2), and bipyridine (L3) were purchased or synthesized according to the literature procedure. Zinc flakes (−325 mesh, purity ≥99.9%) and manganese powder (−325 mesh, ≥99%) were stored under nitrogen. N-Ethyl-2- pyrrolidone (NEP) was purified by stirring over CaH2 (48 h) followed by distillation from CaH2. The purified NEP was stored under nitrogen over 4 Å molecular sieves. Tetrahydrofuran (THF), N,N-dimethylacetamide (DMA), dichloromethane (DCM), and pyridine (py) were purified by passage through alumina and molecular sieves and stored over 4 Å molecular sieves. 4-iodotoluene, 4-bromobenzotrifluoride, and 3-iodotoluene were filtered through a short plug of basic alumina (1 cm) in a glass pipette before use. 4-Iodobenzaldehyde,34 N-(4-iodophenyl)-4-methylbenzenesulfonamide,35 2,2,2-trifluoro-N-(4-iodophenyl)acetamide,36 tert-butyl(4-iodophenoxy)dimethylsilane,37 4-iodo-N,N-dimethylaniline,38 cinnamyl acetate39 (2a), (E)-hex-2-en-1-yl acetate37 (2b), (Z)-3-phenylallyl acetate40 (2d), (E)-3-cyclohexylallyl acetate41 (2e), 2-methylallyl acetate42 (2f), (2E,4E)-hexa-2,4-dien-1-yl acetate13a (2g), cyclohex-2-en-1-yl acetate43 (2i), (E)-4-phenylbut-3-en-2-yl acetate44 (2j), and (E)-3-(4-methoxyphenyl)allyl acetate39, 45 (2k), (E)-3-(4-trifluoromethyl)phenyl)allyl acetate46 (2l), and 2-Bromocyclohex-2-en-1-one47 (6) were synthesized according to literature procedures.
Allylic Acetates and Carbonates
1-Phenyl-2-propenyl acetate (2c)48
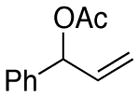
Benzaldehyde (2.31 mL, 22.73 mmol, Fluka) was filtered though basic alumina and added to a flame dried 100-mL 1-neck round bottomed flask. The flask was also charged with THF (25 mL) and a Teflon coated magnetic stirbar, then cooled to 0 °C. A 1.09 M solution of vinyl magnesium bromide in THF (25 mL, 25 mmol, 1.1 equiv) was then added slowly via an addition funnel. After 4 h no benzaldhyde remained and acetic anhydride (3.22 mL, 34.09 mmol, 1.5 equiv,) was added at 0 °C. After 2 hours an additional 1.0 equiv (2.15 mL) of acetic anhydride was added and the reaction was allowed to warm to room temperature overnight. The reaction mixture was poured into a separatory funnel, diluted with Et2O (50 mL), and the organic layer was washed with 1 M HCl until the pH of the aqueous layer was 1. The organic layer was then washed with saturated NaHCO3 until the pH of the aqueous layer was 7. The combined aqueous layers were then back extracted with Et2O (2 × 50 mL). The combined organic layers were then dried over MgSO4 and the supernatant was concentrated in vacuo. The product was then isolated by flash chromatography (5% EtOAc in hexanes, Rf = 0.24). Mixed fractions were rechromatographed (2.5% EtOAc in hexanes). The combined pure 1-phenyl-2-propenyl acetate (1.99 g, 88%) was isolated as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 7.37 (d, J = 4.5 Hz, 4H), 7.32 (t, J = 4.3 Hz, 1H), 6.28 (dt, J = 5.9, 1.3 Hz, 1H), 6.02 (ddd, J = 17.1, 10.5, 5.9 Hz, 1H), 5.33-5.24 (m, 2H), 2.12 (s, 3H). 13C NMR (101 MHz; CDCl3): δ 170.0, 139.0, 136.4, 128.6, 128.3, 127.2, 117.0, 76.3, 21.4.
4-Ethylhex-1-en-3-yl methyl carbonate (2m)
An oven-dried 250 mL, three-neck round bottom flask was equipped with a magnetic stirbar, a nitrogen inlet, an addition funnel, and a rubber septum. 2-Ethylbutyraldehyde (2.46 mL, 20 mmol, distilled over Na2SO4) and dry Et2O (140 mL) were added to this vessel, and the mixture was cooled to 0 °C. Vinylmagnesium bromide in THF (1.09 M, 22.02 mL, 24 mmol, 1.2 equiv) was added drop-wise via the addition funnel. The reaction was allowed to warm to rt and then stirred at rt for 1 hour. The white suspension was then poured onto ice cold water (50 mL) and the organic layer separated, dried over Na2SO4, and supernatant concentrated in vacuo in a 100 mL round bottom flask to give the allyl alcohol. The crude alcohol was then used without further purification in the next step.
In the 100 mL vessel used above, a solution of the allyl alcohol (assume 20 mmol from previous step), DCM (50 mL) and pyridine (4.85 mL, 3 equiv) was cooled to 0 °C, under nitrogen with stirring. Methyl chloroformate (3.09 mL, 2.0 equiv) was then added dropwise by syringe and the reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was treated with water (25 mL), and the aqueous layer was separated and extracted with additional Et2O (2 × 25 mL). The combined organic layers were washed with saturated NaHCO3 (25 mL), brine (25 mL), dried over Na2SO4, and the supernatant was concentrated in vacuo. Purification by flash chromatography (2% EtOAc in hexanes) yielded pure 4-ethylhex-1-en-3-yl methyl carbonate (1.50 g, 50%) as a clear oil. 1H NMR (400 MHz; CDCl3): δ 5.78 (ddd, J = 17.3, 10.5, 6.8 Hz, 1H), 5.30-5.21 (m, 2H), 5.10-5.07 (m, 1H), 3.76 (s, 3H), 1.50-1.36 (m, 4H), 1.25 (m, 1H), 0.89 (td, J = 7.4, 3.4 Hz, 6H). 13C NMR (101 MHz; CDCl3): δ 155.5, 134.6, 118.1, 81.0, 54.7, 44.9, 21.7 (d, J = 9.3 Hz), 11.5. Anal. Calcd for C10H18O3: 64.49 % C, 9.74% H, 0.00% N; found 64.69% C, 9.95% H, −0.065% N.
Methyl 5-phenylpent-1-en-3-yl carbonate (2n) [376647-56-2]49
An oven-dried 250 mL, three-neck round bottom flask was equipped with a magnetic stirbar, a nitrogen inlet, an addition funnel, and a rubber septum. Hydrocinnamaldehyde (2.63 mL, 20 mmol, distilled over Na2SO4) and dry Et2O (140 mL) were added to this vessel, and the mixture was cooled to 0 °C. Vinylmagnesium bromide in THF (1.09 M, 22.02 mL, 24 mmol, 1.2 equiv) was added drop-wise via the addition funnel. The reaction was then stirred at room temperature for 1 hour. The white suspension was then poured onto ice cold water (50 mL) and the organic layer separated, dried over Na2SO4, and supernatant concentrated in vacuo in a 100 mL round bottom flask to give the allyl alcohol. The crude alcohol was then used without further purification in the next step.
In the 100 mL vessel used above, a solution of the allyl alcohol (assume 20 mmol from previous step), DCM (50 mL) and pyridine (4.85 mL, 3 equiv) was cooled to 0 °C under nitrogen with stirring. Methyl chloroformate (3.09 mL, 2.0 equiv) was then added dropwise by syringe and the reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was treated with water (25 mL), and the aqueous layer was separated and extracted with additional Et2O (2 × 25 mL). The combined organic layers were washed with saturated NaHCO3 (25 mL), brine (25 mL), dried over Na2SO4, and the supernatant was concentrated in vacuo. Purification of the residue by flash chromatography (2% EtOAc in hexanes) yielded methyl 5-phenylpent-1-en-3-yl carbonate (2.60 g, 59%) as a clear oil. 1H-NMR (400 MHz; CDCl3): δ 7.31-7.18 (m, 5H), 5.85 (ddd, J = 17.2, 10.5, 6.7 Hz, 1H), 5.36-5.24 (m, 2H), 5.10 (q, J = 6.6 Hz, 1H), 3.79 (s, 3H), 2.76-2.64 (m, 2H), 2.11-1.90 (m, 2H). 13C NMR (101 MHz; CDCl3): δ 155.3, 141.2, 135.8, 128.6, 128.5, 126.1, 117.9, 78.6, 54.8, 36.0, 31.4.
Procedure A: Allylic acetates with aryl halides in a nitrogen glove box
A 1-dram vial containing a teflon-coated magnetic stir was sequentially charged with NiCl2(dme) (10.6 mg, 0.0483 mmol), L1 (20.0 mg, (0.0498 mmol), NEP (500 μL), THF (1500 μL), aryl halide (1.50 mmol), allylic acetate (1.00 mmol), and the zinc flakes (130 mg, 2.00 mmol). The vial was then capped with a screw cap fitted with a PTFE-faced silicone septum. After removal from the glove box, the vial was shaken for 15 seconds and then stirred (1300 rpm) at 40°C until judged complete (less than 1 % allylic acetate remaining) by GC analysis (12–15 h). In order to remove NEP, the reaction mixture was filtered through a short plug of silica gel (1 inch wide, ~ 3 inches high) in an 18 mL disposable polyethylene fritted filter funnel, eluting with either hexanes or diethyl ether, depending on the polarity of the expected product (200 mL). The filtrate was concentrated in vacuo and the residue purified by flash chromatography on a Teledyne Isco combiflash Rf-200.
Procedure B: Allylic acetates with aryl halides on the bench top under argon (1 mmol scale)
Reactions set up as in procedure A except that after the vial was sealed, the headspace was flushed with argon gas for 1 min. The needles were then removed and the vial was placed in a heating block at 40 °C in the usual manner.
Procedure C: Allylic acetates with aryl halides on the bench top under argon (10 mmol scale)
On the bench top, an oven-dried 50-mL Schlenk flask containing a teflon-coated magnetic stir bar was charged with NiCl2(dme) (106 mg, 0.483 mmol) and 4,4′,4″-tri-tert-butyl-2,2′:6′,2″-terpyridine (200 mg, (0.498 mmol). The flask was then sealed with a rubber septa. NEP (500 μL), THF (1500 μL), aryl halide (15.0 mmol), and allylic acetate (10.0 mmol) were then added sequentially under a low flow of argon with an outlet needle (bubbler). The flow of argon was then increased, the rubber septa removed, and the zinc flake reductant was quickly tipped into the flask (1.30 g, 20 mmol). The septa was then replaced onto the flask, and the flask was gently shaken for 15 seconds. The side-arm valve was closed, the argon line was removed, and the reaction mixture was then stirred at 40 °C until judged complete by GC analysis. The work-up and isolation is performed as in Procedure A except 400 mL of hexanes was used to elute the crude product from the silica gel plug, and one 120 g normal-phase “gold” silica column (Teledyne-Isco) was used for chromatography.
(E)-prop-1-ene-1,3-diyldibenzene (3a) (Table 2, Entry 1)50
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and iodobenzene (167 μL, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 171 mg (88% yield) of the title compound as a colorless oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.42-7.25 (m, 10H), 6.51 (d, J = 15.8 Hz, 1H), 6.42 (dd, J = 14.6, 7.8 Hz, 1H), 3.61 (d, J = 6.7 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 140.2, 137.5, 131.1, 129.3, 128.7, 128.5, 127.1, 126.2, 126.1, 39.4. GC-MS (EI) m/z (% rel. int., ion): 194.05 (100.00, M+). HRMS (EI) [M+] calc. for C15H14: 194.110; found: 194.110.
(E)-prop-1-ene-1,3-diyldibenzene (3a) (Table 2, Entry 2)50
Procedure C was followed for a large-scale reaction run outside the glove box with cinnamyl acetate (2a) (1.68 mL, 10 mmol) and iodobenzene (1.67 mL, 1.50 mmol). Purification by flash chromatography using (100% hexanes) afforded 157 mg (81% yield) of the title compound as a colorless oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.41-7.22 (m, 10 H), 6.50 (d, J = 15.8 Hz, 1H), 6.40 (dt, J = 15.1, 7.3 Hz, 1H), 3.59 (d, J = 6.6 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 140.6, 137.9, 131.5, 129.6, 129.1, 128.9, 127.5, 126.6, 126.5, 39.8. GC-MS (EI) m/z (% rel. int., ion): 194.05 (100.00, M+). HRMS (EI) [M+] calc. for C15H14: 194.110; found: 194.110.
(E)-1-[p-(3-phenyl-allyl)phenyl]ethanone (3b) (Table 2, Entry 3)51
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 4-iodoacetophenone (369 mg, 1.50 mmol). Purification by flash chromatography (8% ethyl acetate in hexanes) afforded 168 mg (71% yield) of the title compound as a pale yellow oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.97 (d, J = 8.2 Hz, 2H), 7.41-7.31(m, 9H), 6.53 (d, J = 15.8 Hz, 1H), 6.38 (t, J = 11.3 Hz, 1H), 3.66 (d, J = 6.8 Hz, 2H), 2.65 (s, 3H). 13C NMR (101 MHz; CDCl3) δ 198.2, 146.3, 137.6, 135.8, 132.3, 129.3, 129.1, 129.0, 128.3, 127.8, 126.6, 39.7, 27.0. GC-MS (EI) m/z (% rel. int., ion): 236.15 (100.00, M+). HRMS (EI) [M+] calc. for C17H16O: 236.120; found: 236.120.
(E)-p-(3-phenyl-allyl)benzaldehyde (3c) (Table 2, Entry 4)51
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.0 mmol) and 4-iodobenzaldehyde (348 mg, 1.50 mmol). Purification by flash chromatography (9% ethyl acetate in hexanes) afforded 155 mg (70% yield) of the title compound as a pale yellow oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 10.05 (s, 1H), 7.89 (d, J = 8.1 Hz, 2H), 7.48-7.27 (m, 9H), 6.54 (d, J = 15.7 Hz, 1H), 6.39 (dt, J = 15.7, 6.9 Hz, 1H), 3.69 (d, J = 6.7 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 192.0, 147.6, 137.1, 134.8, 132.1, 130.1, 129.4, 128.6, 127.6, 127.5, 126.2, 39.5. GC-MS (EI) m/z (% rel. int., ion): 222.05 (77.96, M+), 115 (100.00, M+ -C7H5O). HRMS (EI) [M+] calc. for C16H14O: 222.104; found: 222.105.
(E)-N-(4-cinnamylphenyl)-4-methylbenzenesulfonamide (3d) (Table 2, Entry 5)
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and N- (4-iodophenyl)-4-methylbenzenesulfonamide (560 mg, 1.50 mmol). Purification by flash chromatography (40% ethyl acetate in hexanes) afforded 265 mg (73% yield) of the title compound as a white solid. 1H NMR (400 MHz; CDCl3) δ 7.65-7.63 (m, 2H), 7.34-7.27 (m, 4H), 7.23-7.21 (m, 3H), 7.11 (d, J = 8.4 Hz, 2H), 7.00-6.98 (m, 2H), 6.42-6.38 (m, 2H), 6.29 (dd, J = 14.6, 7.9 Hz, 1H), 3.47 (d, J = 6.5 Hz, 2H), 2.38 (s, 3H). 13C NMR (101 MHz; CDCl3) δ 143.9, 137.7, 137.4, 136.3, 134.6, 131.4, 129.8, 129.7, 128.9, 128.7, 127.42, 127.37, 126.2, 122.4, 38.8, 21.7. Mp: 137–138 ° C. IR (cm−1): 3240 (N-H, strong), 3024, 2912, 2870 (C-H, weak), 1153 (S=O, strong), 1508, 1469 (C=C, medium). Product contains 0.21 equiv H2O by 1H NMR analysis (see Supporting Information for copy of spectrum). Anal. Calcd for C22H21NO2S + 0.21 H2O: 71.95 % C, 5.88% H, 3.81% N; found 72.07% C, 5.88% H, 3.79% N.
(E)-N-(4-cinnamylphenyl)-2,2,2-trifluoroacetamide (3e) (Table 2, Entry 6)
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 2,2,2-trifluoro-N-(4-iodophenyl)acetamide (472 mg, 1.50 mmol). Purification by flash chromatography (4% ethyl acetate in hexanes) afforded 195 mg (64% yield) of the title compound as a white solid. 1H NMR (400 MHz; CDCl3) δ 7.84 (s, 1H), 7.54-7.23 (m, 9H), 6.48 (d, J = 15.8 Hz, 1H), 6.35 (dt, J = 15.7, 6.8 Hz, 1H), 3.58 (d, J = 6.8 Hz, 2H). 13 C NMR (101 MHz; CDCl3) δ 154.8, 154.5, 138.5, 137.3, 133.2, 131.5, 129.6, 128.6, 127.3, 126.1, 120.7, 116.9, 38.7. 19F NMR (376 MHz; CDCl3) δ 76.2. Mp: 143–145 °C. IR (cm−1): 3302 (N-H, medium), 3213, 3151, 2955 (C-H, weak), 1701 (C=O, strong), 1554, 1512 (C=C, strong), 1145 (C-F, strong). Anal. Calcd for C17H14F3NO: 66.88% C, 4.62% H, 4.59% N; found 66.610% C, 4.705% H, 4.517% N.
(E)-tert-butyl(4-cinnamylphenoxy)dimethylsilane (3f) (Table 2, Entry 7)
Procedure A was followed with cinnamyl acetate (2a) (83 μL, 1.00 mmol) and tert-butyl(4-iodophenoxy)dimethylsilane (261 mg, 1.50 mmol). Purification by flash chromatography (1% ethyl acetate in hexanes) afforded 135 mg (80% yield) of the title compound as a colorless oil. 1H NMR (400 MHz; CDCl3) δ 7.40-7.23 (m, 9H), 6.46 (s, 1H), 6.40 (s, 1H), 4.76 (s, 2H), 3.57 (d, J = 6.6 Hz, 2H), 0.98 (s, 9H), 0.13 (s, 6H). 13C NMR (101 MHz; CDCl3) δ 139.7, 139.1, 137.9, 131.3, 129.8, 128.92, 128.88, 127.5, 126.7, 126.5, 65.2, 39.4, 26.4, 18.8, −4.8. GC-MS (EI) m/z (% rel. int., ion) 338.30 (0.83, M+), 117.10 (100.00, M+ -C13H21OSi). HRMS (EI) [M+] calc. for C22H30OSi: 338.207; found: 338.207.
(E)-3-(4-N,N-dimethylaminophenyl)-1-phenylpropene (3g) (Table 2, Entry 8)51
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 4-iodo-N,N-dimethylaniline (371 mg, 1.50 mmol). Purification by flash chromatography (3% ethyl acetate in hexanes) afforded 130 mg (55% yield) of the title compound as a yellow oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.40-7.14 (m, 7H), 6.75 (d, J = 8.7 Hz, 2H), 6.45 (s, 1H), 6.41 (d, J = 6.3 Hz, 1H), 3.49 (d, J = 6.4 Hz, 2H), 2.95 (s, 6H). 13C NMR (101 MHz; CDCl3) δ 149.3, 137.7, 130.3, 130.2, 129.3, 128.5, 128.2, 126.9, 126.1, 113.1, 40.9, 38.4. GC-MS (EI) m/z (% rel. int., ion): 238.15 (100.00, M+). HRMS (EI) [M+] calc. for C17H19N: 237.152; found: 237.152.
(E)-1-methyl-4-(3-phenyl-2-propen-1-yl)benzene (3h) (Table 2, Entry 9)51
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 4-iodotoluene (377 mg, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 179 mg (86% yield) of the title compound as a colorless oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.42-7.17 (m, 9H), 6.51 (d, J = 15.7 Hz, 1H), 6.44-6.38 (m, 1H), 3.57 (d, J = 6.6 Hz, 2H), 2.39 (s, 3H). 13 C NMR (101 MHz; CDCl3) δ 137.6, 137.1, 135.7, 130.8, 129.5, 129.2, 128.6, 128.5, 127.1, 126.1, 39.0, 21.1. GC-MS (EI) m/z (% rel. int., ion): 208.05 (100.00, M+). HRMS (EI) [M+] calc. for C16H16: 208.125; found: 208.125.
(E)-1-methoxy-4-(3-phenyl-2-propen-1-yl)benzene (3i) (Table 2, Entry 10)50
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1 mmol) and 4-iodoanisole (351 mg, 1.5 mmol). Purification by flash chromatography (4% hexanes) afforded 186 mg (83% yield) of the title compound as a yellow oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.39-7.17 (m, 7H), 6.90-6.86 (m, 2H), 6.48-6.33 (m, 2H), 3.81 (s, 3H), 3.51 (d, J = 6.5 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 158.2, 137.7, 132.3, 130.9, 129.8, 129.7, 128.6, 127.2, 126.2, 114.0, 55.4, 38.6. GC-MS (EI) m/z (% rel. int., ion): 224.10 (100.00, M+). HRMS (EI) [M+] calc. for C16H16O: 224.120; found: 224.121.
(E)-1-bromo-4-cinnamylbenzene (3j) (Table 2, Entry 11)1g
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 1-bromo-4-iodobenzene (424 mg, 1.50 mmol). Purification by flash chromatography (3% ethyl acetate in hexanes) afforded 191 mg (64% yield) of the title compound as a pale yellow oil; analytical data matched those reported in the literature. This product was isolated as an inseparable mixture with 9% 3a due to competing hydrodeiodination. Yield reported is the yield of 3j, calculated by NMR analysis and comparison with isolated 3a. 1H NMR (400 MHz; CDCl3) δ 7.47-7.15 (m, 9H), 6.48 (d, J = 15.8 Hz, 1H), 6.34 (dt, J = 15.7, 6.8 Hz, 1H), 3.59 (d, J = 6.7 Hz,), 3.53 (d, J = 6.7 Hz, 2H) 13C NMR (101 MHz; CDCl3) δ 139.1, 137.2, 131.5, 130.4, 129.2, 128.6, 128.4, 127.3, 126.1, 120.0, 38.7. GC-MS (EI) m/z (% rel. int., ion): 273.00 (6.88, M+), 115.05 (100.00, M+ -C6H4Br). HRMS (EI) [M+] calc. for C15H13Br: 272.020; found: 272.020.
(E)-p-(3-phenyl-allyl) methyl benzoate (3k) (Table 2, Entry 12)50
Procedure A was followed with cinnamyl acetate (2a) (166.7 μL, 1.00 mmol) and methyl 4-bromobenzoate (322.6 mg, 1.50 mmol). Purification by flash chromatography (2% ethyl acetate in hexanes) afforded 163.3 mg (65% yield) of the title compound as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 8.02-7.99 (m, 2H), 7.42-7.29 (m, 6H), 7.25-7.21 (m, 1H), 6.48 (d, J = 15.7 Hz, 1H), 6.34 (dt, J = 15.8, 6.8 Hz, 1H), 3.92 (s, 3H), 3.61 (d, J = 6.8 Hz, 2H). 13C NMR (101 MHz; CDCl3): δ 167.2, 145.7, 137.3, 131.9, 130.0, 128.8, 128.7, 128.3, 128.2, 127.4, 126.3, 52.1, 39.4. GC-MS (EI) m/z (% rel. int., ion): 252.10 (56.47, M+), 193.05 (100.00, M+-C2H3O2). HRMS (EI) [M+] calc. for C17H16O2: 252.115; found: 252.116.
(E)-1-[p-(3-phenyl-allyl)phenyl]ethanone (3b) (Table 2, Entry 13)51
Procedure A was followed with cinnamyl acetate (2a) (166.7 μL, 1.00 mmol) and 4′-bromoacetophenone (298.6 mg, 1.50 mmol). Purification by flash chromatography (5% ethyl acetate in hexanes) afforded 114.3 mg (48% yield) of the title compound as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 7.93 (d, J = 8.3 Hz, 2H), 7.39-7.21 (m, 7H), 6.48 (d, J = 15.8 Hz, 1H), 6.34 (dt, J = 15.8, 6.8 Hz, 1H), 3.61 (d, J = 6.7 Hz, 2H), 2.60 (s, 3H). 13C NMR (101 MHz; CDCl3): δ 197.9, 146.0, 137.2, 135.5, 132.0, 129.0, 128.8, 128.7, 128.0, 127.5, 126.3, 39.4, 26.7. GC-MS (EI) m/z (% rel. int., ion): 236.10 (100.00, M+). HRMS (EI) [M+] calc. for C17H16O: 236.120; found: 236.120.
(E)-1-phenyl-3-[4-(trifluoromethyl)phenyl]propene (3l) (Table 2, Entry 14)51
Run 1 (1.00 mmol): Procedure A was followed with cinnamyl acetate (2a) (166.7 μL, 1.00 mmol) and 4-bromobenzotrifluoride (210.0 μL, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 130.8 mg (50% yield) of the title compound as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 7.54 (d, J = 8.0 Hz, 2H), 7.35-7.18 (m, 7H), 6.45 (d, J = 15.8 Hz, 1H), 6.34-6.26 (m, 1H), 3.57 (d, J = 6.8 Hz, 2H). 13C NMR (101 MHz; CDCl3): δ 144.4, 137.3, 132.1, 129.1, 128.7, [literature 128.49 (q, J=10.8 Hz) was unresolved], 128.0, 127.5, 126.3, 125.8, 125.5 (q, J=3.7 Hz), 39.2. 19F NMR (376 MHz; CDCl3): δ −62.7. Run 2 (0.50 mmol): Procedure A (but on half scale) was followed with cinnamyl acetate (83.4 μL, 0.50 mmol) and 4-bromobenzotrifluoride (105.0 μL, 0.75 mmol). Purification by flash chromatography (100% hexanes) afforded 68.3 mg (52% yield) of the title compound as a clear oil; analytical data matched those above. GC-MS (EI) m/z (% rel. int., ion): 262.10 (100.00, M+). HRMS (EI) [M+] calc. for C16H13F3: 262.097; found: 262.097.
(E)-3-(4-cyanophenyl)-1-phenylpropene (3m) (Table 2, Entry 15)51
Procedure A was followed with cinnamyl acetate (2a) (166.7 μL, 1.00 mmol) and 4-bromobenzonitrile (273.0 mg, 1.50 mmol). Purification by flash chromatography (2% ethyl acetate in hexanes) afforded 168.3 mg (77% yield) of the title compound as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 7.58-7.55 (m, 2H), 7.34-7.18 (m, 7H), 6.44 (d, J = 15.8 Hz, 1H), 6.26 (dt, J = 15.7, 6.9 Hz, 1H), 3.57 (d, J = 6.8 Hz, 2H). 13C NMR (101 MHz; CDCl3): δ 145.9, 137.0, 132.5, 132.4, 129.5, 128.7, 127.6, 127.2, 126.3, 119.1, 110.2, 39.4. GC-MS (EI) m/z (% rel. int., ion): 219.05 (100.00, M+). HRMS (EI) [M+] calc. for C16H13N: 219.105; found: 219.106.
(E)-3-(3-methylphenyl)-1-phenylpropene (3n) (Table 2, Entry 16)51
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 3-iodotoluene (377 mg, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 162 mg (78% yield) of the title compound as a colorless oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.39-7.17 (m, 7H), 6.90-6.86 (m, 2H), 6.48-6.33 (m, 2H), 3.81 (s, 3H), 3.51 (d, J = 6.5 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 140.1, 138.1, 137.5, 131.0, 129.44, 129.38, 128.5, 128.4, 127.1, 126.9, 126.1, 125.7, 39.3, 21.4. GC-MS (EI) m/z (% rel. int., ion): 208.10 (100.00, M+). HRMS (EI) [M+] calc. for C16H16: 208.125; found: 208.125.
2-[(2E)-3-phenylprop-2-en-1-yl]benzonitrile (3o) (Table 2, Entry 17)52
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 2-iodobenzonitrile (343 mg, 1.50 mmol). Purification by flash chromatography (3% hexanes in ethyl acetate) afforded 188 mg (86% yield) of the title compound as a colorless oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.70 (dt, J = 7.7, 0.6 Hz, 1H), 7.59 (td, J = 7.7, 1.2 Hz, 1H), 7.46-7.26 (m, 7H), 6.58 (d, J = 15.8 Hz, 1H), 6.37 (dt, J = 15.7, 7.0 Hz, 1H), 3.83 (d, J = 6.9 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 144.4, 137.3, 133.3, 133.0, 130.1, 129.0, 127.9, 127.3, 126.9, 126.7, 126.0, 118.4, 112.9, 38.1. GC-MS (EI) m/z (% rel. int., ion): 218.10 (100.00, M+). HRMS (EI) [M+] calc. for C16H12N: 218.097; found: 218.097.
(E)-1-methoxy-2-(3-phenyl-2-propen-1-yl)benzene (3p) (Table 2, Entry 18)50
Procedure A was followed with cinnamyl acetate (2a) (166 μL, 1.00 mmol) and 2-iodoanisole (351 mg, 1.50 mmol). Purification by flash chromatography (4% hexanes in ethyl acetate) afforded 179 mg (80% yield) of the title compound as a yellow oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.41-7.39 (m, 2H), 7.34-7.29 (m, 2H), 7.28-7.21 (m, 3H), 6.97-6.91 (m, 2H), 6.50-6.41 (m, 2H), 3.89 (s, 3H), 3.59 (d, J = 6.0 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 157.3, 137.8, 130.7, 129.9, 128.9, 128.7, 128.4, 127.4, 126.9, 126.1, 120.5, 110.4, 55.4, 33.4. GC-MS (EI) m/z (% rel. int., ion): 224.10 (100.00, M+). HRMS (EI) [M+] calc. for C16H16O: 224.120; found: 224.120.
(E)-1-Phenylhex-2-ene (4a) (Table 3 Entry 1)53
Procedure A was followed with (E)-hex-2-en-1-yl acetate (2b) (142 mg, 1 mmol) and iodobenzene (167 μL, 1.5 mmol). Purification by flash chromatography (100% hexanes) afforded 129 mg (81% yield) of the title compound as a colorless oil. The isolated product had a 86:14 [linear]:[branched] ratio, as determined by analysis of the GC/MS fragmentation patterns and comparison with literature NMR data. 1H NMR (400 MHz; CDCl3) linear isomer28,29: δ 7.33-7.21 (m, 5H), 5.64-5.50 (m, 2H), 3.37 (d, J = 6.1 Hz, 2H), 2.08-2.01 (m, 2H), 1.43 (tq, J = 7.4 Hz, 2H), 0.95-0.90 (t, 3H); branched isomer: δ 7.33-7.21 (m, 5H), δ 6.02-5.90 (m, 1H), δ 5.07-5.03 (m, 2H), δ 3.32-3.26 (m, 1H), δ 1.75-1.68 (m, 2H), δ 1.42-1.30 (m, 2H), 0.95-0.90 (t, 3H). 13C NMR (101 MHz; CDCl3) linear isomer: δ 141.3, 132.1, 129.0, 128.6, 128.53, 128.48, 126.0, 39.2, 34.8, 22.8, 13.9; Partial 13C NMR for branched isomer, δ 127.7, 37.8, 20.8. GC-MS (EI) m/z (% rel. int., ion): 160.10 (54.39, M+), 117.05 (100.00, M+ C3H7). HRMS (EI) [M+] calc. for C12H16: 160.125; found: 160.125.
(E)-prop-1-ene-1, 3-diyldibenzene (3a) (Table 3, Entry 2)50
Procedure A was followed with 1-phenylallyl acetate (2c) (176 mg, 1.00 mmol) and iodobenzene (167 μL, 1.50 mmol). Purification by flash chromatography (100 % hexanes) afforded 101 mg (52% yield) of the title compound as a colorless oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.41-7.24 (m, 10H), 6.50 (d, J = 15.8 Hz, 1H), 6.40 (dt, J = 15.1, 7.3 Hz, 1H), 3.59 (d, J = 6.5 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 140.6, 137.9, 131.5, 129.6, 129.1, 128.9, 127.5, 126.6, 126.5, 39.8. GC-MS (EI) m/z (% rel. int., ion): 194.10 (100.00, M+). HRMS (EI) [M+] calc. for C15H14: 194.120; found: 194.120.
(E)-prop-1-ene-1,3-diyldibenzene (3a) (Table 3, Entry 3)50
Procedure A was followed with (Z)-3-phenylallyl acetate (2d) (176 mg, 1 mmol) and iodobenzene (167 μL, 1.5 mmol). Purification by flash chromatography (100% hexanes) afforded 146 mg (75% yield) of the title compound as a colorless oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3) δ 7.41-7.22 (m, 10H), 6.50 (d, J = 15.8 Hz, 1H), 6.40 (dt, J = 15.7, 6.7 Hz, 1H), 3.60 (d, J = 6.6 Hz, 2H). 13C NMR (101 MHz; CDCl3) δ 140.6, 137.9, 131.5, 129.6, 129.1, 128.9, 127.5, 126.6, 126.5, 39.8. GC-MS (EI) m/z (% rel. int., ion): 194.05 (100.00, M+). HRMS (EI) [M+] calc. for C15H14: 194.110; found: 194.110.
(E)-1-cyclohexyl-3-phenylpropene (4b) (Table 3, Entry 4)50
Procedure A was followed with 3-cyclohexylallyl acetate (2e) (182 mg, 1.00 mmol) and iodobenzene (167 μL, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 194 mg (97% yield) of the title compound as a colorless oil. The isolated product has a 93:7 [linear]:[branched] ratio, as determined by GC/MS fragmentation patterns and by comparison with literature NMR data. 1H NMR (400 MHz; CDCl3) Linear isomer: δ 7.35-7.18 (m, 5H), 5.60-5.49 (m, 2H), 3.36 (d, J = 5.8 Hz, 2H), 2.01-1.96 (m, 1H), 1.78-1.66 (m, 5H), 1.34-1.09 (m, 5H). Branched isomer: δ 7.35-7.18 (m, 5H), δ 6.06-5.98 (m, 1H), δ 5.06 (d, 1H), δ 5.03 (d, 1H), 2.97 (t, 1H), 1.97-2.01 (m, 1H), 1.65-1.59 (m, 3H), 1.48-1.44 (m, 1H), 1.34-1.09 (m, 5H). 13C NMR (101 MHz; CDCl3) linear isomer: δ 141.2, 138.1, 128.5, 128.3, 126.1, 125.8, 40.7, 39.1, 33.1, 26.2, 26.1; branched isomer: d 141.2, 138.1, 128.5, 128.3, 126.1, 125.8, 40.7, 39.1, 33.1, 26.2, 26.1 (one peak was not visible). GC-MS (EI) m/z (% rel. int., ion): 200.10 (14.90, M+), 109.10 (100.00, M+ -C7H7). HRMS (EI) [M+] calc. for C15H20: 200.157; found: 200.157.
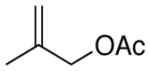
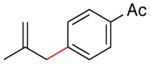
1-(4-(2-methylallyl)phenyl)ethanone (4c) (Table 3, Entry 5)54
Procedure A was followed with 2-methylallyl acetate (2f) (114 mg, 1.00 mmol) and 4-iodoacetophenone (369 mg, 1.50 mmol). Purification by flash chromatography (9% ethyl acetate in hexanes) afforded 96 mg (55% yield) of the title compound as a pale yellow oil; analytical data matched those reported previously. 1H NMR (400 MHz; CDCl3) δ 7.93-7.91 (m, 2H), 7.30 (t, J = 7.8 Hz, 2H), 4.88 (s, 1H), 4.77 (s, 1H), 3.40 (s, 2H), 2.61 (s, 3H), 1.70 (s, 3H). 13C NMR (101 MHz; CDCl3) δ 197.8, 145.5, 144.1, 135.3, 129.1, 128.5, 112.7, 44.6, 26.6, 22.1. GC-MS (EI) m/z (% rel. int., ion): 174.10 (15.17, M+), 115 (100.00, M+ -CH3). HRMS (EI) [M+] calc. for C12H14O: 174.105; found: 174.105.
(2 E,4E)-hexa-2,4-dien-1-ylbenzene (4d) (Table 3, Entry 6)55
Procedure A was followed with (2E, 4E)-hexa-2,4-dien-1-yl acetate (2g) (140 mg, 1.00 mmol) and iodobenzene (167 μL, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 103 mg (65% yield) of the title compound as a colorless oil. Product was a mixture of olefin isomers: 6.9:1:1 [2 E,4E]:{2E,4Z]:[unidentified olefin isomer]. Olefin isomer ratio and identity were assigned based upon GC/MS fragmentation patterns and by comparison to previously reported NMR spectra (1H and 13C). We assigned one isomer as 2E, 4E in analogy to the other products. 1H NMR (400 MHz; CDCl3) Linear 2E, 4E isomer:56 δ 7.34-7.21 (m, 5H), 6.11-6.08 (m, 2H), 5.76-5.65 (m, 2H), 3.44 (d, J = 6.9 Hz, 2H), 1.78 (d, J = 6.2 Hz, 3H); linear 2E, 4Z isomer:57 δ 7.34-7.21 (m, 5H), 6.52-6.43 (dd, 1H), 6.11-6.08 (t, 1H), 5.76-5.65 (m, 1H), 5.51-5.46 (m, 1H), 3.50 (d, J = 7.0 Hz,), 1.78 (dd, 3H); unidentified olefin isomer: δ 7.34-7.21 (m, 5H), 6.11-6.08 (m, 2H), 5.76-5.65 (m, 2H), 3.56 (d, J = 7.4 Hz, 2H), 1.86-1.84 (d, 3H). 13C NMR (101 MHz; CDCl3) linear 2E, 4Z isomer: δ 140.5, 132.4, 129.2, 128.6, 128.4, 126.6, 125.0, 39.3, 13.4. Linear 2E, 4E and unidentified olefin isomer: δ 131.6, 131.4, 130.4, 130.1, 129.5, 128.5, 127.9, 127.4, 126.7, 126.1, 126.1, 126.0, 125.98, 125.95, 39.0, 33.9, 18.4, 18.1, 13.4 (no literature 13C NMR spectrum is available for linear 2E, 4E olefin isomer). GC-MS (EI) m/z (% rel. int., ion): 158.10 (52.92, M+), 129.10 (100.00, M+ -C2H4). HRMS (EI) [M+] calc. for C12H14: 158.120; found: 158.120.
(3,7-dimethylocta-2,6-dien-1-yl)benzene (4e) (Table 3, Entry 7)53a, 56, 57
Procedure A was followed with geranyl acetate (214 μL, 1.00 mmol) and iodobenzene (167 μL, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 171 mg (80% yield) of a mixture of the three isomers as a colorless oil. Isolated product has a 1.9:1 [linear]:[branched] ratio. The linear and branched products were identified by GC/MS fragmentation patterns and by comparison with literature NMR values. By NMR, the yield of linear products is 52% and the yield of branched product is 28%. Linear (E and Z) isomers:53a, 57 1H NMR (500 MHz; CDCl3): δ 7.36-7.21 (m, 10H), 5.40-5.36 (m, 2H), 5.19-5.07 (m, 2H), 3.40 (d, J = 7.2 Hz, 4H), 2.22-2.08 (m, 8H), 1.79 (s, 3H), 1.75 (s, 3H), 1.72 (s, 6H), 1.66 (s, 3H), 1.64 (s, 3H). 13C NMR (126 MHz; CDCl3): δ 142.0 (E), 141.9 (Z), 136.4 (E), 136.3 (Z), 131.9 (Z), 131.6 (E), 128.50 (E/Z), 128.47 (E/Z), 128.45 (E/Z), 125.83 (E/Z), 125.80 (E/Z), 124.4 (E), 124.3 (Z), 124.0 (Z), 123.2 (E), 39.9 (E), 34.33 (E/Z), 34.27 (E/Z), 32.1 (Z), 26.7 (E/Z), 25.9 (E), 25.8 (Z), 25.0 (Z), 23.6 (Z), 17.9 (E), 17.8 (Z), 16.3 (E). Branched isomer:56 1H NMR (500 MHz; CDCl3): δ 7.36-7.21 (m, 5H), 6.08 (dd, J = 17.5, 10.7 Hz, 1H), 5.19-5.07 (m, 2H), 1.91-1.82 (m, 4H), 1.69 (s, 3H), 1.55 (s, 3H), 1.42 (s, 3H). 13C NMR (126 MHz; CDCl3): δ 147.6, 147.0, 131.5, 128.2, 126.7, 125.9, 124.8, 111.9, 44.4, 41.3, 25.9, 23.4, 17.7. GC-MS (EI) m/z (% rel. int., ion): 214.15 (9.14, M+), 123.15 (100.00, M+ -C7H7). HRMS (EI) [M+] calc. for C16H22: 214.172; found: 214.173.
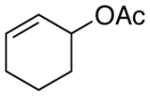
1-phenyl-2-cyclohexene (4f) (Table 3, Entry 8)53a
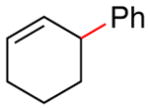
Procedure A was followed with cyclohex-2-en-1-yl acetate (2i) (140 mg, 1.00 mmol) and iodobenzene (167 μL, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 126 mg (80% yield) of the title compound as a colorless oil. 1H NMR (500 MHz; CDCl3) δ 7.35-7.22 (m, 5H), 5.95-5.91 (m, 1H), 5.78-5.74 (m, 1H), 3.45 (td, J = 5.1, 2.5 Hz, 1H), 2.15-2.03 (m, 3H), 1.81-1.77 (m, 1H), 1.69-1.55 (m, 2H). 13C NMR (101 MHz; CDCl3) δ 146.7, 130.2, 128.4, 128.3, 127.8, 126.0, 41.9, 32.7, 25.1, 21.2. GC-MS (EI) m/z (% rel. int., ion): 158.10 (100.00, M+). HRMS (EI) [M+] calc. for C12H14: 158.120; found: 158.110. NMR data matched those previously reported.
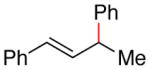
(E)-but-1-ene-1,3-diyldibenzene (4g) (Table 3, Entry 9)53a
Procedure A was followed with but-1-ene-1,3-diyldibenzene (2j) (190 mg, 1.00 mmol) and iodobenzene (167 μL, 1.50 mmol). Purification by flash chromatography (100% hexanes) afforded 152 mg (73% yield) of the title compound as a colorless oil; 1H NMR (400 MHz; CDCl3) δ 7.41-7.23 (m, 10H), 6.48-6.39 (m, 2H), 3.71-3.65 (m, 1H), 1.51 (d, J = 7.0 Hz, 3H).13C NMR (101 MHz; CDCl3) δ 145.8, 137.7, 135.4, 128.7, 128.5, 127.4, 127.2, 126.4, 126.3, 42.7, 21.4 GC-MS (EI) m/z (% rel. int., ion): 208.10 (66.46, M+), 115 (100.00, M+ -C7H8). HRMS (EI) [M+] calc. for C16H16: 208.125; found: 208.125. NMR data matched those previously reported.
Procedure D: Allylic acetates with alkyl halides in a nitrogen glove box
A 1 dram vial was charged with NiCl2(dme) (10.6 mg, 0.0483 mmol), 4,4′,4″-tri-tert-butyl-2,2′:6′,2″-terpyridine (20.0 mg, (0.0498 mmol), DMA (500 μL), a teflon coated magnetic stirbar was then added, and the reaction mixture was stirred for 5–10 minutes before the sequential addition of allylic acetate (1.3 mmol), secondary alkyl halide (1.0 mmol), THF (1500 μL), and manganese powder (109.6 mg, 2.00 mmol). The vial was then capped with a screw cap with a PTFE-faced silicone septum. The reaction was then treated as in Procedure A (vide supra).
Procedure E: Allylic acetates with alkyl halides on the bench top under argon
As in procedure D, but set up on the bench, without care to exclude air or moisture. Once the reaction was capped, the reaction mixture was sparged with Ar gas for 1 min, followed by a purge of the vial head space for 1 min. The needles were then removed and the vial placed in a heating block at 40 °C in the usual manner.
(E)-(4-methylnon-1-en-1-yl)benzene (5a) (Table 4, Entry 1)
Run 1 (1.00 mmol): General procedure D was followed with cinnamyl acetate (2a) (216.7 μL, 1.30 mmol) and 2-bromoheptane (172.9 μL, 1.00 mmol, distilled purity was 90.7% so amount was scaled from 156.8 μL). Purification by flash chromatography (100% hexanes) afforded 151.7 mg (70% yield) of the title compound as a clear oil. 1H NMR (400 MHz; CDCl3): δ 7.38-7.18 (m, 5H), 6.39 (d, J = 15.8 Hz, 1H), 6.23 (dt, J = 15.4, 7.5 Hz, 1H), 2.27-2.02 (m, 2H), 1.62-1.56 (m, 1H), 1.37-1.28 (m, 7H), 1.20-1.14 (m, 1H), 0.95-0.90 (m, 6H). 13C NMR (101 MHz; CDCl3): δ 138.1, 131.0, 130.0, 128.6, 126.9, 126.1, 40.8, 36.8, 33.5, 32.3, 27.0, 22.9, 19.8, 14.3. GC-MS (EI) m/z (% rel. int., ion): 216.15 (18.02, M+), 117.10 (100.00, M+-C7H15). HRMS (EI) [M+] calc. for C16H24: 216.188; found: 216.187. Run 2 (0.50 mmol): General procedure D was followed with cinnamyl acetate (83.4 μL, 0.65 mmol) and 2-bromoheptane (86.2 μL, 0.50 mmol, Aldrich Lot# 12798PJ was 90.7% so amount was scaled from 78.4 μL). Purification by flash chromatography (100% hexanes) afforded 95.6 mg (88% yield) of the title compound as a clear oil. 1H NMR matched those reported in run 1.
(E)-(3-cyclohexylprop-1-enyl)benzene (5b) (Table 4, Entry 2)58
General procedure D was followed with cinnamyl acetate (2a) (216.7 μL, 1.30 mmol) and bromocyclohexane (123.2 μL, 1.00 mmol). Purification by flash chromatography (100% hexanes) afforded 180.6 mg (90% yield) on Run 1 and 171.7 mg (86% yield) on Run 2 of the title compound as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 7.39-7.30 (m, 4H), 7.23-7.20 (m, 1H), 6.39 (d, J = 15.8 Hz, 1H), 6.25 (dt, J = 15.4, 7.5 Hz, 1H), 2.16-2.12 (m, 2H), 1.82-1.68 (m, 5H), 1.43 (ddtd, J = 14.5, 10.8, 7.2, 3.6 Hz, 1H), 1.32-1.14 (m, 3H), 1.04-0.94 (m, 2H). 13C NMR (101 MHz; CDCl3): δ 138.1, 130.8, 129.9, 128.6, 126.9, 126.0, 41.2, 38.3, 33.4, 26.7, 26.5. GC-MS (EI) m/z (% rel. int., ion): 200.15 (23.74, M+), 104.10 (100.00, M+-C7H13+H). HRMS (EI) [M+] calc. for C15H20: 200.157; found: 200.156.
(E)-(3-cyclohexylprop-1-enyl)benzene (5b) (Table 4, Entry 3)58
General procedure E was followed with cinnamyl acetate (2a) (216.7 μL, 1.30 mmol) and bromocyclohexane (123.2 μL, 1.00 mmol). Purification by flash chromatography (100% hexanes) afforded 179.4 mg (90% yield) of the title compound as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 7.39-7.30 (m, 4H), 7.23-7.19 (m, 1H), 6.38 (d, J = 15.8 Hz, 1H), 6.25 (dt, J = 15.7, 7.2 Hz, 1H), 2.15-2.12 (m, 2H), 1.81-1.67 (m, 5H), 1.43 (ddtd, J = 14.5, 10.9, 7.2, 3.6 Hz, 1H), 1.32-1.13 (m, 3H), 1.04-0.90 (m, 2H). 13C NMR (101 MHz; CDCl3): δ 138.1, 130.8, 129.9, 128.6, 126.9, 126.0, 41.2, 38.3, 33.4, 26.7, 26.5. GC-MS (EI) m/z (% rel. int., ion): 200.15 (22.48, M+), 104.10 (100.00, M+-C7H13+H). HRMS (EI) [M+] calc. for C15H20: 200.157; found: 200.156.
(E)-(3-cyclopentyl-1-propenyl)-benzene (5c) (Table 4, Entry 4)59
General procedure D was followed with cinnamyl acetate (2a) (216.7 μL, 1.30 mmol) and bromocyclopentane (107.2 μL, 1.00 mmol). Purification by flash chromatography (100% hexanes) afforded 129.1 mg (69% yield) on Run 1 and 123.7 mg (66% yield) on Run 2 of the title compound as a clear oil; analytical data matched those reported in the literature. 1H NMR (400 MHz; CDCl3): δ 7.37-7.29 (m, 4H), 7.20 (td, J = 7.2, 1.8 Hz, 1H), 6.39 (d, J = 15.8 Hz, 1H), 6.25 (dt, J = 15.8, 7.0 Hz, 1H), 2.26-2.22 (m, 2H), 1.97 (dquintet, J = 15.0, 7.5 Hz, 1H), 1.84-1.76 (m, 2H), 1.69-1.51 (m, 4H), 1.27-1.18 (m, 2H). 13C NMR (101 MHz; CDCl3): δ 138.1, 130.7, 130.3, 128.6, 126.9, 126.1, 40.2, 39.6, 32.5, 25.3. GC-MS (EI) m/z (% rel. int., ion): 186.10 (25.68, M+), 117.10 (100.00, M+-C5H9). HRMS (EI) [M+] calc. for C14H18: 186.141; found: 186.141.
(E)-(1S,2R,4R)-2-cinnamylbicyclo[2.2.1]heptane (5d) (Table 4, Entry 5)
General procedure D was followed with cinnamyl acetate (2a) (216.7 μL, 1.30 mmol) and exo-2-bromonorborane (128.4 μL, 1.00 mmol). Purification by flash chromatography (100% hexanes) afforded 179.0 mg (84% yield) on Run 1 and 150.2 mg (71% yield) on Run 2 of the title compound as a clear oil. 1H NMR (400 MHz; CDCl3): δ 7.38-7.29 (m, 4H), 7.23-7.18 (m, 1H), 6.38 (d, J = 15.8 Hz, 1H), 6.20 (dt, J = 15.8, 7.0 Hz, 1H), 2.24-2.14 (m, 2H), 2.07-2.00 (m, 2H), 1.61-1.42 (m, 4H), 1.42-1.32 (m, 1H), 1.31-1.10 (m, 4H). 13C NMR (101 MHz; CDCl3): δ 138.1, 130.4, 130.3, 128.6, 126.9, 126.1, 42.2, 40.9, 40.4, 38.0, 36.9, 35.3, 30.2, 29.0. GC-MS (EI) m/z (% rel. int., ion): 212.10 (12.51, M+), 95.10 (100.00, M+-C9H9). HRMS (EI) [M+] calc. for C16H20: 212.157; found: 212.157.
(E)-1-methoxy-4-(4-methylnon-1-en-1-yl)benzene (5e) (Table 4, Entry 6)
General procedure D was followed with (E)-3-(4-methoxyphenyl)allyl acetate (2k) (268.1 mg, 1.30 mmol) and 2-bromoheptane (172.9 μL, 1.10 mmol because material was 90.7% pure). Purification by flash chromatography (2% ethyl acetate in hexanes) afforded 209.8 mg (85% yield) on Run 1 and 178.2 mg (72% yield) on Run 2 of the title compound as a clear oil. 1H NMR (400 MHz; CDCl3): δ 7.31-7.27 (m, 2H), 6.87-6.83 (m, 2H), 6.32 (d, J = 15.7 Hz, 1H), 6.08 (dt, J = 15.7, 7.3 Hz, 1H), 3.81 (s, 3H), 2.24-1.99 (m, 2H), 1.61-1.53 (m, 1H), 1.39-1.27 (m, 7H), 1.20-1.13 (m, 1H), 0.93-0.89 (m, 6H). 13C NMR (101 MHz; CDCl3): δ 158.7, 131.0, 130.3, 127.8, 127.1, 114.0, 55.4, 40.7, 36.8, 33.5, 32.3, 27.0, 22.9, 19.8, 14.3. GC-MS (EI) m/z (% rel. int., ion): 246.20 (15.13, M+), 147.10 (100.00, M+-C7H15). HRMS (EI) [M+] calc. for C17H26O: 246.198; found: 246.198.
(E)-(4-methylnon-1-en-1-yl)-4-(trifluoromethyl)benzene (5f) (Table 4, Entry 7)
Run 1 (1.00 mmol): General procedure D was followed with (E)-3-(4-trifluoromethyl)phenyl)allyl acetate (2l) (317.5 mg, 1.30 mmol) and 2-bromoheptane (172.9 μL, 1.10 mmol because material was 90.7% pure). Purification by flash chromatography (100% hexanes) afforded 202.1 mg (71% yield) of the title compound as a clear oil. 1H and 13C NMR spectra match those reported for run 2. Run 2 (0.50 mmol): General procedure D was followed with (E)-3-(4-trifluoromethyl)phenyl)allyl acetate (2l) (175.2 mg, 0.65 mmol, 2l was 90.6% pure by GC so amount was scaled from 158.5 mg) and 2-bromoheptane (86.5 μL, 0.50 mmol, distilled purity was 90.7% so amount was scaled from 78.4 μL). Purification by flash chromatography (100% hexanes) afforded 85.6 mg (60% yield) of the title compound as a clear oil. 1H NMR (400 MHz; CDCl3): δ 7.54 (d, J = 8.3 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 6.42-6.28 (m, 2H), 2.28-2.03 (m, 2H), 1.64-1.54 (m, 1H), 1.37-1.25 (m, 7H), 1.19-1.14 (m, 1H), 0.93-0.88 (m, 6H). 13C NMR (101 MHz; CDCl3): δ 141.5, 132.9, 129.8, 126.2, 125.54, 125.51, 40.8, 36.8, 33.4, 32.3, 26.9, 22.9, 19.8, 14.3. We were unable to resolve one of the expected carbon atoms. We presume the missing carbon to be the CF3 quartet based upon literature data for similar compounds. 19F NMR (376 MHz; CDCl3): δ −62.8. GC-MS (EI) m/z (% rel. int., ion): 284.20 (8.51, M+), 57.10 (100.00, M+-C13H14F3). HRMS (EI) [M+] calc. for C17H23F3: 284.175; found: 284.176.
Procedure F: Allylic acetates and carbonates with a vinyl bromide in a nitrogen glove box: A
1 dram vial was charged with NiCl2(dme) (10.6 mg, 0.0483 mmol), 2,9-dimethyl-1,10-phenanthroline (10.4 mg, (0.0499 mmol), and DMA (500 μL) followed by a teflon coated magnetic stirbar. The reaction mixture was stirred for 5–10 minutes. In a separate 1 dram vial the branched allylic starting material (1.0 mmol) and 2-bromocyclohex-2-en-1-one (1.0 mmol) were massed. The catalyst solution was the added to the substrates, followed by THF (1500 μL), and manganese powder (−325 mesh, 109.6 mg, 2.00 mmol), sequentially. The remainder of the procedure followed proceure A (vide supra).
2-cinnamylcyclohex-2-enone (7a) (Table 5, Entry 3)
General procedure F was followed with 1-phenyl-2-propenyl acetate (2c) (176.2 mg, 1.00 mmol) and 2-bromocyclohex-2-en-1-one (6) (175.0 mg, 1.00 mmol). Purification by flash chromatography (5% ethyl acetate in hexanes) afforded 165.3 mg (78% yield) of the title compound as a pale yellow oil. 1H NMR (400 MHz; CDCl3): δ 7.36-7.18 (m, 5H), 6.78 (t, J = 4.2 Hz, 1H), 6.41 (d, J = 15.8 Hz, 1H), 6.22 (dt, J = 15.8, 7.0 Hz, 1H), 3.12-3.09 (m, 2H), 2.48-2.44 (m, 2H), 2.36 (tdt, J = 6.0, 4.0, 1.9 Hz, 2H), 2.01 (dq, J = 12.9, 6.3 Hz, 2H). 13C NMR (101 MHz; CDCl3): δ 199.1, 146.1, 138.4, 137.6, 131.7, 128.6, 127.7, 127.2, 126.2, 38.6, 32.9, 26.2, 23.2. GC-MS (EI) m/z (% rel. int., ion): 212.00 (62.05, M+), 121.05 (100.00). HRMS (EI) [M+] calc. for C15H16O: 212.120; found: 212.119.
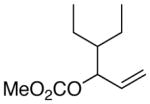
(E)-2-(4-ethylhex-2-en-1-yl)cyclohex-2-enone (7b) (Table 6, Entry 3)
General procedure F was followed with 4-ethyl-hex-1-en-3-yl methyl carbonate (2m) (186.3 mg, 1.00 mmol) and 2-bromocyclohex-2-en-1-one (6) (175.0 mg, 1.00 mmol). Purification by flash chromatography (5% ethyl acetate in hexanes) afforded 104.6 mg (51% yield) of the title compound as a dark yellow oil. 1H NMR (400 MHz; CDCl3): δ 6.70 (t, J = 4.2 Hz, 1H), 5.32 (dt, J = 14.8, 7.2 Hz, 1H), 5.14 (dd, J = 15.2, 8.7 Hz, 1H), 2.89 (d, J = 6.8 Hz, 2H), 2.42 (t, J = 6.8 Hz, 2H), 2.34 (tdt, J = 5.9, 4.0, 1.9 Hz, 2H), 2.03-1.94 (m, 2H), 1.73 (dtd, J = 13.3, 8.7, 4.7 Hz, 1H), 1.43-1.33 (m, 2H), 1.23-1.14 (m, 2H), 0.82 (t, J = 7.4 Hz, 6H). 13C NMR (101 MHz; CDCl3): δ 199.3, 145.2, 139.2, 137.1, 127.0, 46.4, 38.7, 32.3, 27.8, 26.2, 23.3, 11.9. GC-MS (EI) m/z (% rel. int., ion): 206.10 (3.97, M+), 135.05 (100.00, M+-C5H11). HRMS (EI) [M+] calc. for C14H22O: 206.167; found: 206.166.
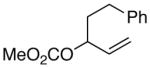
(E)-2-(5-phenylpent-2-en-1-yl)cyclohex-2-enone (7c) (Table 6, Entry 4)
General procedure F was followed with 5-phenyl-pent-1-en-3-yl methyl carbonate (2n) (220.3 mg, 1.00 mmol) and 2-bromocyclohex-2-en-1-one (6) (175.0 mg, 1.00 mmol). Purification by flash chromatography (5% ethyl acetate in hexanes) afforded 104.6 mg (51% yield) of the title compound as a pale yellow oil. 1H NMR (400 MHz; CDCl3): δ 7.29-7.16 (m, 5H), 6.52 (t, J = 4.2 Hz, 1H), 5.51-5.35 (m, 2H), 2.86 (d, J = 6.3 Hz, 2H), 2.69 (t, J = 7.7 Hz, 2H), 2.42 (td, J = 6.8, 3.0 Hz, 2H), 2.37-2.28 (m, 4H), 1.96 (dt, J = 13.0, 6.4 Hz, 2H). 13C NMR (101 MHz; CDCl3): δ 199.3, 145.6, 142.1, 138.7, 131.7, 128.7, 128.3, 127.9, 125.8, 38.6, 36.0, 34.4, 32.2, 26.2, 23.2. GC-MS (EI) m/z (% rel. int., ion): 240.10 (14.85, M+), 91.00 (100.00, M+-C10H13O). HRMS (EI) [M+] calc. for C17H20O: 240.151; found: 240.151.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Science of the National Institutes of Health under award number R01 GM097243. Analytical data were obtained from the CENTC Elemental Analysis Facility at the University of Rochester, funded by NSF CHE-0650456. Mass spectrometry data were obtained from the Univeristy of Illinois, with instrumentation funded in part by the NIH (RR 04648).
Footnotes
Supporting Information. Full selectivity data (Tables S1–S5), Table S6, details on methods and starting materials, and copies of 1H and 13C NMR spectra are available in the Supplementary Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Boron For a review, see Pigge FC. Synthesis. 2010:1745–1762.Miyaura N, Yamada K, Suginome H, Suzuki A. J Am Chem Soc. 1985;107:972–980.Trost BM, Spagnol MD. Perkin Trans 1. 1995:2083–2097.Kobayashi Y, Mizojiri R, Ikeda E. J Org Chem. 1996;61:5391–5399.Kabalka GW, Dong G, Venkataiah B. Org Lett. 2003;5:893–895. doi: 10.1021/ol034067d.bismuth Rao MLN, Banerjee D, Giri S. J Organomet Chem. 2010;695:1518–1525.magnesium Mayer M, Czaplik WM, Jacobi von Wangelin A. Adv Synth Catal. 2010;352:2147–2152.Rao Volla CM, Dubbaka SR, Vogel P. Tetrahedron. 2009;65:504–511.zinc Dunet G, Knochel P. Synlett. 2007:1383–1386.
- 2.Silicon Hatanaka Y, Ebina Y, Hiyama T. J Am Chem Soc. 1991;113:7075–7076.Denmark S, Werner N. J Am Chem Soc. 2008;130:16382–16393. doi: 10.1021/ja805951j.indium Seomoon D, Lee K, Kim H, Lee PH. Chem–Eur J. 2007;13:5197–5206. doi: 10.1002/chem.200601338.Lee K, Kim H, Mo J, Lee PH. Chem–Asian J. 2011;6:2147–2157. doi: 10.1002/asia.201000890.tin Kosugi M, Sasazawa K, Shimizu Y, Migita T. Chem Lett. 1977;6:301–302.Farina V, Krishnamurthy V, Scott WJ. Org React. 1997;50:1–652.boron Kalinin VN, Denisov FS, Bubnov YN. Mendeleev Commun. 1996;6:206–207.Fürstner A, Seidel G. Synlett. 1998;1998:161–162.Yamamoto Y, Takada S, Miyaura N. Chem Lett. 2006;35:704–705.Sebelius S, Olsson VJ, Wallner OA, Szabó KJ. J Am Chem Soc. 2006;128:8150–8151. doi: 10.1021/ja062585o.Glasspoole BW, Ghozati K, Moir JW, Crudden CM. Chem Commun. 2012;48:1230–1232. doi: 10.1039/c2cc16076e.Doucet H. Eur J Org Chem. 2008:2013–2030.
- 3.(a) Heck RF. Org React. 1982;27:345–390. [Google Scholar]; (b) Meijere A, Meyer FE. Angew Chem, Int Ed. 1994;33:2379–2411. [Google Scholar]
- 4.Typically, a mixture of isomers is obtained, for example: Demont EH, Faller A, MacPherson DT, Milner PH, Naylor A, Redshaw S, Stanway SJ, Vesey DR, Walter DS. WO2004050619A1. Preparation of hydroxyethylamine derivatives for the treatment of Alzheimer’s disease. 2004
- 5.For an overview: Sigman MS, Werner EW. Acc Chem Res. 2012;45:874–884. doi: 10.1021/ar200236v.
- 6.(a) Gomes P, Gosmini C, Périchon J. J Org Chem. 2003;68:1142–1145. doi: 10.1021/jo026421b. [DOI] [PubMed] [Google Scholar]; (b) Gomes P, Buriez O, Labbe E, Gosmini C, Périchon J. J Electroanal Chem. 2004;562:255–260. [Google Scholar]
- 7.Gomes P, Gosmini C, Périchon J. Org Lett. 2003;5:1043–1045. doi: 10.1021/ol0340641. [DOI] [PubMed] [Google Scholar]
- 8.(a) Durandetti M, Nédélec JY, Périchon J. J Org Chem. 1996;61:1748–1755. doi: 10.1021/jo9518314. [DOI] [PubMed] [Google Scholar]; (b) Wang S, Qian Q, Gong H. Org Lett. 2012;14:3352–3355. doi: 10.1021/ol3013342. [DOI] [PubMed] [Google Scholar]
- 9.(a) Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920–921. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; (b) Everson DA, Jones BA, Weix DJ. J Am Chem Soc. 2012;134:6146–6159. doi: 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prinsell MR, Everson DA, Weix DJ. Chem Commun. 2010;46:5743–5745. doi: 10.1039/c0cc01716g. and unpublished studies. [DOI] [PubMed] [Google Scholar]
- 11.Semmelhack MF. Org React. 1972;19:115–198. [Google Scholar]
- 12.Qian X, Auffrant A, Felouat A, Gosmini C. Angew Chem, Int Ed. 2011;50:10402–10405. doi: 10.1002/anie.201104390. [DOI] [PubMed] [Google Scholar]
- 13.Dai Y, Wu F, Zang Z, You H, Gong H. Chem–Eur J. 2012;18:808–812. doi: 10.1002/chem.201102984. [DOI] [PubMed] [Google Scholar]
- 14.Shi L, Fan CA, Tu YQ, Wang M, Zhang FM. Tetrahedron. 2004;60:2851–2855. [Google Scholar]
- 15.Durandetti M, Gosmini C, Périchon J. Tetrahedron. 2007;63:1146–1153. [Google Scholar]
- 16.See Table 1 and Tables S1–S5 in the Supporting Information.
- 17.(a) Majid TN, Knochel P. Tetrahedron Lett. 1990;31:4413–4416. [Google Scholar]; (b) Ikegami R, Koresawa A, Shibata T, Takagi K. J Org Chem. 2003;68:2195–2199. doi: 10.1021/jo026746s. [DOI] [PubMed] [Google Scholar]; (c) Krasovskiy A, Malakhov V, Gavryushin A, Knochel P. Angew Chem, Int Ed. 2006;45:6040–6044. doi: 10.1002/anie.200601450. [DOI] [PubMed] [Google Scholar]; (d) Sibille S, Ratovelomanana V, Périchon J. Chem Commun. 1992:283–284. [Google Scholar]
- 18.Similarly, Mn is slow to insert into alkyl and vinyl bromides: Peng Z, Knochel P. Org Lett. 2011;13:3198–3201. doi: 10.1021/ol201109g.Cahiez G, Duplais C, Buendia J. Chem Rev. 2009;109:1434–1476. doi: 10.1021/cr800341a.
- 19.Ylijoki KEO, Lavy S, Fretzen A, Kündig EP, Berclaz T, Bernardinelli G, Besnard C. Organometallics. 2012;31:5396–5404. [Google Scholar]
- 20.A test reaction run in a THF/DMF mixture gave a GC yield of 73%, compared to 90% in THF/NEP.
- 21.Hydrogen atom abstraction from starting materials or product remains possible, for example.
- 22.Oishi S, Hatano K, Tsubouchi A, Takeda T. Chem Commun. 2011;47:11639–11640. doi: 10.1039/c1cc14765j. and references therein. [DOI] [PubMed] [Google Scholar]
- 23.Peng J, Liu X, Kishi Y. Tetrahedron Lett. 2011;52:2172–2175. [Google Scholar]
- 24.Shrestha R, Dorn SCM, Weix DJ. Submitted. [Google Scholar]
- 25.Kusuda S, Watanabe Y, Ueno Y, Toru T. J Org Chem. 1992;57:3145–3152. [Google Scholar]
- 26.Majetich G, Hull K, Casares AM, Khetani V. J Org Chem. 1991;56:3958–3973. [Google Scholar]
- 27.Takahashi T, Hori K, Tsuji J. Tetrahedron Lett. 1981;22:119–122. [Google Scholar]
- 28.Yu B, Jiang T, Quan W, Li J, Pan X, She X. Org Lett. 2009;11:629–632. doi: 10.1021/ol8026895. [DOI] [PubMed] [Google Scholar]
- 29.Nickel-catalyzed dimerization reactions of alkyl halides and allylic acetates were rapid with L1, but dimerization of aryl halides was much slower.
- 30.(a) Corey EJ, Semmelhack MF, Hegedus LS. J Am Chem Soc. 1968;90:2416–2417. [Google Scholar]; (b) Hegedus LS, Miller LL. J Am Chem Soc. 1975;97:459–460. [Google Scholar]; (c) Tsou T, Kochi J. J Am Chem Soc. 1979;101:7547–7560. [Google Scholar]; (d) Hegedus LS, Thompson DHP. J Am Chem Soc. 1985;107:5663–5669. [Google Scholar]
- 31.Ikeda S-i, Suzuki K, Odashima K. Chem Commun. 2006:457–459. doi: 10.1039/b515508h. [DOI] [PubMed] [Google Scholar]
- 32.Ward LGL, Pipal JR. Inorg Synth. 1971;13:154–164. [Google Scholar]
- 33.Hadda TB, Le Bozec H. Polyhedron. 1988;7:575–577. [Google Scholar]
- 34.Orita A, Miyamoto K, Nakashima M, Ye F, Otera J. Adv Synth Catal. 2004;346:767–776. [Google Scholar]
- 35.Wilhelm T, Lautens M. Org Lett. 2005;7:4053–4056. doi: 10.1021/ol051628n. [DOI] [PubMed] [Google Scholar]
- 36.Melissaris AP, Litt MH. J Org Chem. 1994;59:5818–5821. [Google Scholar]
- 37.Toumi M, Couty F, Evano G. J Org Chem. 2007;72:9003–9009. doi: 10.1021/jo070517u. [DOI] [PubMed] [Google Scholar]
- 38.Monnereau C, Blart E, Odobel F. Tetrahedron Lett. 2005;46:5421–5423. [Google Scholar]
- 39.Ueno S, Hartwig JF. Angew Chem, Int Ed. 2008;47:1928–1931. doi: 10.1002/anie.200705267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim IS, Dong GR, Jung YH. J Org Chem. 2007;72:5424–5426. doi: 10.1021/jo0705263. [DOI] [PubMed] [Google Scholar]
- 41.(a) Nishizawa R, Nishiyama T, Hisaichi K, Minamoto C, Murota M, Takaoka Y, Nakai H, Tada H, Sagawa K, Shibayama S, Fukushima D, Maeda K, Mitsuya H. Bioorg Med Chem. 2011;19:4028–4042. doi: 10.1016/j.bmc.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pontikis R, Benhida R, Aubertin AM, Grierson DS, Monneret C. J Med Chem. 1997;40:1845–1854. doi: 10.1021/jm960765a. [DOI] [PubMed] [Google Scholar]
- 42.Bricout H, Carpentier JF, Mortreux A. Tetrahedron. 1998;54:1073–1084. [Google Scholar]
- 43.(a) Luche JL. J Am Chem Soc. 1978;100:2226–2227. [Google Scholar]; (b) Hayashi T, Kawatsura M, Uozumi Y. J Am Chem Soc. 1998;120:1681–1687. [Google Scholar]
- 44.Hou J, Feng C, Li Z, Fang Q, Wang H, Gu G, Shi Y, Liu P, Xu F, Yin Z, Shen J, Wang P. Eur J Med Chem. 2011;46:3190–3200. doi: 10.1016/j.ejmech.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 45.Fischer DF, Barakat A, Xin Z-q, Weiss ME, Peters R. Chem–Eur J. 2009;15:8722–8741. doi: 10.1002/chem.200900712. [DOI] [PubMed] [Google Scholar]
- 46.(a) Kawatsura M, Ata F, Hayase S, Itoh T. Chem Commun. 2007:4283–4285. doi: 10.1039/b709218k. [DOI] [PubMed] [Google Scholar]; (b) Lölsberg W, Ye S, Schmalz HG. Adv Synth Catal. 2010;352:2023–2031. [Google Scholar]
- 47.Easton CJ, Heath GA, Hughes CMM, Lee CKY, Savage GP, Simpson GW, Tiekink ERT, Vuckovic GJ, Webster RD. J Chem Soc, Perkin Trans 1. 2001:1168–1174. [Google Scholar]
- 48.Commandeur M, Commandeur C, Cossy J. Org Lett. 2011;13:6018–6021. doi: 10.1021/ol202483u. [DOI] [PubMed] [Google Scholar]
- 49.(a) Ito H, Kawakami C, Sawamura M. J Am Chem Soc. 2005;127:16034–16035. doi: 10.1021/ja056099x. [DOI] [PubMed] [Google Scholar]; (b) Hon YS, Devulapally R. Tetrahedron Lett. 2009;50:2831–2834. [Google Scholar]; (c) Evans PA, Clizbe EA. J Am Chem Soc. 2009;131:8722–8723. doi: 10.1021/ja9041302. [DOI] [PubMed] [Google Scholar]
- 50.Li MB, Wang Y, Tian SK. Angew Chem, Int Ed. 2012;51:2968–2971. doi: 10.1002/anie.201109171. [DOI] [PubMed] [Google Scholar]
- 51.Tsukamoto H, Uchiyama T, Suzuki T, Kondo Y. Org Biomol Chem. 2008;6:3005–3013. doi: 10.1039/b804991b. [DOI] [PubMed] [Google Scholar]
- 52.Sharma R, Bulger PG, McNevin M, Dormer PG, Ball RG, Streckfuss E, Cuff JF, Yin J, Chen C-y. Org Lett. 2009;11:3194–3197. doi: 10.1021/ol9010147. [DOI] [PubMed] [Google Scholar]
- 53.(a) Yamada YMA, Sarkar SM, Uozumi Y. J Am Chem Soc. 2012;134:3190–3198. doi: 10.1021/ja210772v. [DOI] [PubMed] [Google Scholar]; (b) Latham CM, Blake AJ, Lewis W, Lawrence M, Woodward S. Eur J Org Chem. 2012;2012:699–707. [Google Scholar]
- 54.Hayashi S, Hirano K, Yorimitsu H, Oshima K. J Am Chem Soc. 2006;128:2210–2211. doi: 10.1021/ja058055u. [DOI] [PubMed] [Google Scholar]
- 55.(a) Pünner F, Schmidt A, Hilt G. Angew Chem, Int Ed. 2012;51:1270–1273. doi: 10.1002/anie.201107512. [DOI] [PubMed] [Google Scholar]; (b) Narasaka K, Kusama H, Hayashi Y. Tetrahedron. 1992;48:2059–2068. [Google Scholar]
- 56.Rodriguez D, Sestelo JP, Sarandeses LA. J Org Chem. 2003;68:2518–2520. doi: 10.1021/jo0265939. [DOI] [PubMed] [Google Scholar]
- 57.Sarkar SM, Uozumi Y, Yamada YMA. Angew Chem, Int Ed. 2011;50:9437–9441. doi: 10.1002/anie.201103799. [DOI] [PubMed] [Google Scholar]
- 58.Phapale VB, Guisán-Ceinos M, Buñuel E, Cárdenas DJ. Chem–Eur J. 2009;15:12681–12688. doi: 10.1002/chem.200901913. [DOI] [PubMed] [Google Scholar]
- 59.Yus M, Ortiz R. Eur J Org Chem. 2004:3833–3841. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.