Abstract
Adenosine deaminases that act on RNA (ADARs) deaminate adenosines in dsRNA to produce inosines. ADARs are essential in mammals and are particularly important in the nervous system. Altered levels of adenosine-to-inosine (A-to-I) editing are observed in several diseases. The extent to which an adenosine is edited depends on sequence context. Human ADAR2 (hADAR2) has 5′ and 3′ neighbor preferences, but which amino acids mediate these preferences, and by what mechanism, is unknown. We performed a screen in yeast to identify mutations in the hADAR2 catalytic domain that allow editing of an adenosine within a disfavored triplet. Binding affinity, catalytic rate, base flipping, and preferences were monitored to understand the effects of the mutations on ADAR reactivity. Our data provide information on the amino acids that affect preferences and point to a conserved loop as being of key importance. Unexpectedly, our data suggest that hADAR2’s preferences derive from differential base flipping rather than from direct recognition of neighboring bases. Our studies set the stage for understanding the basis of altered editing levels in disease and for developing therapeutic reagents.
Keywords: 2-aminopurine, RNA editing
Adenosine deaminases that act on RNA (ADARs) target double-stranded regions of precursor mRNAs (pre-mRNAs), noncoding RNAs, and viral RNAs, deaminating adenosines to create inosines (1–3). Inosine is recognized as guanosine; thus, adenosine-to-inosine (A-to-I) editing in a pre-mRNA can alter codons and splice-forms, leading to multiple protein isoforms from a single gene. ADARs also alter microRNA and endogenous siRNA biogenesis and targeting (2–4). A-to-I editing of viral RNAs can reduce virus growth as well as enhance it (5).
ADARs are found in most metazoans, and often more than one ADAR exists in an organism. For example, there are three mammalian ADAR genes: ADAR1, ADAR2, and ADAR3, and each has two or three N-terminal dsRNA-binding motifs (dsRBMs) and a highly conserved C-terminal deaminase domain. ADAR1 and ADAR2 are active deaminases, but enzymatic activity has not been observed with ADAR3 (6, 7).
Two of the most studied ADAR substrates are the pre-mRNAs of glutamate receptor, ionotropic, AMPA 2 (GRIA2) and the 5-HT2C serotonin receptor. GRIA2 pre-mRNA has two editing sites, one that recodes glutamine into arginine (Q/R), and another that recodes an arginine into glycine (R/G). Aberrant A-to-I editing is correlated with several diseases (8). For example, underediting of the Q/R site of GRIA2 pre-mRNA is implicated in amyotrophic lateral sclerosis, overediting of the R/G site is observed in epilepsy patients, and an increase in editing of the 5-HT2C serotonin receptor pre-mRNA is observed in patients with depression and in suicide victims. In addition, the locus for dyschromatosis symmetrica hereditaria (DSH), a pigmentary genodermatosis, maps to the ADAR1 gene. The mechanistic basis for altered levels of editing in various diseases is entirely unclear.
ADARs specifically edit certain adenosines over others, and the extent of editing also varies. There are two determinants of specificity: selectivity and preferences. The fraction of sites edited in a dsRNA, referred to as “selectivity,” depends on its length and whether it contains mismatches, bulges, and internal loops (3). In vitro studies show that nonselective editing occurs in completely base-paired dsRNA of 50 bp or more, whereas adenosines in shorter dsRNA or in dsRNA containing mismatches, bulges, and loops are edited more selectively (9–11). ADARs’ dsRBMs are believed to play a large role in selectivity (12).
The extent of A-to-I editing at a particular site depends on sequence context, and these rules are referred to as “preferences” (11, 13). Human ADAR1 (hADAR1) and human ADAR2 (hADAR2) have a 5′ nearest-neighbor preference of U > A > C > G and a 3′ nearest-neighbor preference of G > C ∼A > U and G > C > U ∼A, respectively (14). Truncated forms of hADAR1 and hADAR2 comprising only the catalytic domain have the same 5′ preference as the full-length proteins and similar but distinct 3′ preferences (G > C > A > U and C ∼G ∼A > U, respectively) (14). The 3′ preferences of the truncated forms indicate that the dsRBMs play a role in the 3′ neighbor preference, as is consistent with NMR solution structures of mammalian ADAR2 dsRBMs bound to the R/G hairpin of GRIA2 that show a hydrogen bond from S258 in the second dsRBM to the amino group of the 3′ G (15). In another study, when the deaminase domains of hADAR1 and hADAR2 are switched, substrate specificity of the chimeric protein tracks with its deaminase domain (16). These studies suggest that preferences derive mainly from the catalytic domain, but which amino acids in the catalytic domain mediate preferences is not known.
To identify the amino acids that mediate preferences, we performed a screen for mutations within the hADAR2 catalytic domain that allow editing of an adenosine in a poor sequence context. Collectively, the hADAR2 variants we identified point to a conserved loop near the active site as important for preferences. Unexpectedly, our data suggest that hADAR2’s preferences derive from differential base flipping rather than from direct recognition of the neighboring bases. These studies offer insight into the correlation of altered editing levels with disease and set the stage for developing therapeutic reagents.
Results
Screen Identifies Residues in the hADAR2 Catalytic Domain That Affect Preferences.
For both hADAR1 and hADAR2, nearest-neighbor preferences derive mainly from the catalytic domain (14, 16). A crystal structure of the catalytic domain of hADAR2 has been solved (17); therefore, to facilitate our analysis, we focused on this enzyme. We adapted a previously reported screen in Saccharomyces cerevisiae (18) to identify mutations in the hADAR2 catalytic domain that allow editing of an adenosine in the context of a disfavored triplet, GAC, where the underline indicates the targeted adenosine.
The screen relied on a hairpin-reporter that was introduced into the chromosome of the haploid yeast strain, W303α, under the control of a constitutive promoter, ADH1. The hairpin (shown in red in Fig. 1A) contained an ADAR editing site within a stop codon (bold) either in a disfavored context, UGAC (Fig. 1B), or in a favored context, UAG (Fig. 1C), and its sequence was based on the R/G editing site of GRIA2 pre-mRNA. Editing of either stop codon created a tryptophan codon, allowing expression of the downstream α-galactosidase reporter and turning yeast colonies green on 5-bromo-4-chloro-3-indolyl-α-d-galactopyranoside (X-α-Gal) plates. The sequence upstream of the RNA hairpin (shown in blue in Fig. 1A) was the signal sequence for secretion of α-galactosidase.
Fig. 1.

Mutants that edit the disfavored GAC hairpin were identified from a screen in yeast. (A) Schematic of the hairpin-reporter used in the screen. The hairpin, in red, is an ADAR substrate with the target adenosine in context of a stop codon that must be edited for expression of the downstream α-galactosidase reporter. (B and C) Sequences of the GAC (B) and UAG (C) hairpins (hp) with the target adenosine within a disfavored and favored triplet, respectively. (D) Control experiments showing CM −URA plates with yeast colonies that have a GAC (Left) or UAG (Right) hairpin-reporter integrated into a chromosome and transformed with WT hADAR2. (E) Mutants identified from the screen, listed from left to right in terms of decreasing green intensity of yeast colonies, an indication of decreasing in vivo editing efficiency. Green intensity of yeast colonies with the control UAG hairpin-reporter is indicated also. “++++” indicates that yeast colonies started turning green in ≤4 d; “+++” and “++” indicate 5–7 d, and “+” indicates low levels of editing taking 2–5 wk to turn faint green. (F) Mutated residues (yellow sticks) mapped onto the crystal structure of the catalytic domain of hADAR2 (Protein Data Bank ID code 1ZY7) (17) with Zn (pink sphere), IP6 (orange and red stick), and modeled in AMP (pink stick). (G) Alignment of hADAR1, hADAR2, hADAR3, and ADAR2 from different species. Mutants that were characterized further are indicated. The highly conserved loop that includes two β-strands and comprises 14 residues is highlighted in yellow.
S. cerevisiae lack an ADAR gene, an essential part of the screen. hADAR2 was introduced in the low-copy CEN vector to ensure uniform protein expression. Expression was under the control of an inducible GAL promoter to facilitate induction by galactose after replica plating. Control experiments established that introduction of WT hADAR2 into strains containing the hairpin-reporter with the favored UAG editing site (the UAG yeast strain) allowed expression of α-galactosidase, but introduction into the strain containing the hairpin-reporter with the disfavored GAC editing site (the GAC yeast strain) did not (Fig. 1D).
The hADAR2 catalytic domain was mutagenized randomly by error-prone PCR to attain a mutation rate of zero to four mutations per kilobase and was introduced into the GAC yeast strain in context of the full-length protein (Materials and Methods). Thirty-five thousand colonies were screened, and 24 positives were obtained that could edit GAC more than WT hADAR2. Seven positives had single mutations in the catalytic domain, and of these, E488Q exhibited the highest level of editing as judged by green intensity. (Fig. 1E). Four mutations appeared more than once, suggesting the screen was saturated. Plasmids also were rescued from some white colonies, representative of mutants that did not edit GAC, and sequencing verified that these plasmids were mostly WT hADAR2 or variants with stop codons in the ORF. One of these, T490A, was at an interesting location between two residues mutated in the positives, E488Q and V493A. All mutant forms of hADAR2 were introduced into the UAG yeast strain, and all seven positives retained the ability to edit UAG, although T490A edited UAG poorly (Fig. 1E). We did not identify any mutants showing a reversed preference that could edit an adenosine within GAC but not UAG.
When the identified mutations were mapped onto the crystal structure of the hADAR2 catalytic domain (17), most mapped onto the surface of the predicted RNA-binding site (17) (Fig. 1F). E488Q, T490A, and V493A were on a highly conserved loop that includes two β-strands and comprises 14 residues (shown in green in Fig. 1F) and amino acids 480–493 (shaded yellow in Fig. 1G). Other mutations, e.g., A589V, N597K, and S599T, mapped onto another loop and a β-strand on the protein surface (shown in blue in Fig. 1F), and N613K mapped nearby. Identification of two proximal asparagine-to-lysine mutations suggested that these mutants may have been selected because they improved RNA binding. Residues N597 and N613 are conserved in ADAR2 from different species but are negatively charged residues in ADAR1 (Fig. 1G).
T490A and the four mutants that showed maximal editing of GAC were selected for further characterization. Additional mutagenesis was performed to understand the properties required at these positions. PCR libraries encoding all possible amino acids at each of the five residues were created and introduced into either the GAC yeast strain (E488, V493, N597, and N613; Materials and Methods) or both the GAC and UAG yeast strains (T490). At least 15–30 positives and as many negatives were selected, sequenced, and retransformed into the GAC and UAG yeast strains. Most of the negatives had stop codons or frame shifts in the ORF.
At position 488, a variety of amino acids were able to substitute for glutamic acid to allow editing of UAG, but only glutamine and asparagine, polar, uncharged amino acids with an amide side chain, allowed editing of GAC (Table 1). Substituting E488 with large hydrophobic residues such as phenylalanine, tryptophan, and leucine resulted in loss of editing of both UAG and GAC. We did not identify any amino acids at position 490 that allowed editing at GAC. Furthermore, only serine and cysteine could replace threonine for editing UAG, suggesting that the predicted hydrogen bond (17) from the side chain of T490 to R481 is important (see Fig. 6B). At position 493, a less hydrophobic amino acid, alanine, and polar, uncharged amino acids with an hydroxyl side chain, threonine and serine, edited GAC more than the WT valine. At positions 597 and 613, only positively charged residues, lysine and arginine, allowed editing of GAC, further supporting the idea that these residues were selected in the screen because of improved RNA binding.
Table 1.
Mutational analysis of mutants identified from the screen
| Editing† |
|||
| Residue | Amino acid substitution* | UAG | GAC |
| 488 | Q (25), N (3) | ++++ | +++ |
| E (2), A (1), S (1), M (1), R (1) | ++++ | − | |
| F (1), L (3), W (1) | − | − | |
| 490 | T (28), C (8), S (8) | ++++ | − |
| A (3) | ++ | − | |
| F (1), Y (1) | + | − | |
| R (2), K (1), P (3), E (2) | − | − | |
| 493 | T (4), S (13), A (4) | ++++ | + |
| V (1), R (1), D (1), P (1), G (1) | ++++ | − | |
| 597 | K (19), R (13) | ++++ | + |
| N (3), A (1), E (1), H (3), G (1), Y (1) | ++++ | − | |
| F (2) | − | − | |
| 613 | K (6), R (7) | ++++ | + |
| N (1), A (1), E (1) | ++++ | − | |
*WT residues are in bold. The number beside each amino acid substitution indicates number of clones isolated.
†Extent of editing of UAG and GAC hairpin-reporters in vivo as determined from green intensity of yeast colonies on X-α-Gal plates, as defined in the legend of Fig. 1E.
Fig. 6.

Model describing preference for adenosine in the context of UAG compared with GAC and the role of residues in the conserved loop. (A) Three steps—binding, base flipping, and editing—are shown for WT hADAR2 and E488Q with UAG and GAC substrates. Binding affinities, increase in FI upon mixing enzymes with 2-AP substituted UAG or GAC substrates, and catalytic rates are indicated. Binding affinities of WT hADAR2 and E488Q are similar for UAG and GAC substrates, indicating that discrimination is not derived from differences in binding affinity. In the second step, base flipping is more efficient for adenosine within UAG than within GAC (indicated by font size), correlating with the increased catalytic rate (third step) and suggesting that preferences derive from differences in base flipping. Additionally, E488Q facilitates base flipping, leading to a further increase in catalytic rate for E488Q compared with WT hADAR2. In our model, T490 is essential for the stability of the active conformation of the conserved loop, and a residue on this loop is important for base flipping. (B) A close view of the conserved loop that includes two β-strands and comprises 14 residues. Hydrogen bonds between the R481 side chain and T490 backbone carbonyl oxygen and side chain hydroxyl are shown. Residues E488 and V493 on this loop are indicated also (yellow sticks). (C) Cartoon showing all seven hydrogen bonds within the 14 residues as determined in the crystal structure (17). Red dots indicate a hydrogen bond from a side chain; other bonds involve a backbone carbonyl oxygen or amine hydrogen.
hADAR2 Catalytic Domain Mutants Separate into Two Classes Based on Binding Affinity.
WT and mutant hADAR2 proteins were purified to homogeneity (Materials and Methods) and subjected to in vitro characterization. We first performed gel mobility-shift assays comparing the proteins for binding to UAG or GAC hairpins that were chemically synthesized and 32P 5′-end–labeled. Representative gel shifts are shown for WT hADAR2 and the N597K variant (Fig. 2 A and B). WT hADAR2 and all mutant forms showed the formation of two protein–RNA complexes, as observed in previous studies (19). For all proteins tested, a mobility shift was first observed at a protein concentration of ≤1.5 nM, and a second, slower mobility shift, likely caused by a second binding event on the RNA, appeared at high protein concentrations of ∼50–100 nM. RNA was bound almost completely at a protein concentration of 500 nM. Kd values were determined for the complex represented by the first, fast mobility shift; binding isotherms are shown in Fig. 2 C and D.
Fig. 2.

Binding affinity for some mutants is similar to that of WT hADAR2 but is increased for others. (A and B) PhosphorImages showing representative gel-shift assays of WT hADAR2 (A) and the N597K mutant (B) with 20 pM of 32P 5′-end–labeled UAG (Left) and GAC (Right) hairpins (Fig. 1 B and C). Protein concentrations are indicated at the top of each gel. (C and D) UAG (C) and GAC (D) hairpin binding isotherms for WT hADAR2 and mutant enzymes. Radioactivity corresponding to RNAtotal and RNAfree was quantified to determine the fraction bound: Fraction bound = 1 − (RNAfree/RNATotal). All data points were fit using the Hill formalism. Error bars indicate SD; n ≥ 3.
WT hADAR2 had a Kd of ∼2.1 nM for both UAG and GAC hairpins, emphasizing that the editing preference for UAG over GAC is not derived from differences in binding affinity. Further, all mutants showed a similar binding affinity for UAG and GAC hairpins (Table 2). E488Q, T490A, and V493T showed binding affinity similar to that of WT hADAR2, indicating that these mutations do not affect the binding step. However, N597K and N613K showed an approximately twofold increase in binding affinity, compared with WT hADAR2, for both UAG and GAC hairpins; this difference was reproducible among different experiments and different protein preparations. Possibly, these mutants were selected in the screen because of increased binding affinity. Consistent with this idea, both residues are on the surface of the protein (17). In the full-length protein used for the gel-shift assays, the dsRBMs likely contributed far more than the catalytic domain to the observed affinity, possibly masking the full impact of the mutations on interactions with the catalytic domain. In fact, a full-length protein containing both mutations still showed only an approximately twofold increase in binding affinity compared with WT hADAR2 (Fig. S1C). However, when we compared truncated proteins, one consisting of the WT catalytic domain (truncWT), and the other consisting of the catalytic domain containing both mutations (truncN597K/N613K), truncN597K/N613K had an approximately fourfold higher binding affinity than truncWT (Fig. S1 A and B).
Table 2.
Characterization of hADAR2 WT and mutants
|
Kd (nM) |
kdeam (min−1) |
FI (a.u.)* |
|||||
| hADAR2 proteins | UAG | GAC | UAG | GAC | UA2APG-28 | GA2APC-27 | Srel† |
| WT | 2.1 ± 0.3 | 2.1 ± 0.3 | 0.9 ± 0.1 | (4.4 ± 0.4) × 10−4 | 4.6 ± 0.4 | 1.9 ± 0.1 | 1.00 |
| E488Q | 1.8 ± 0.3 | 1.8 ± 0.3 | ∼ >2.5‡ | (2.6 ± 0.3) × 10−2 | 9.8 ± 0.4 | 2.6 ± 0.2 | 0.85 |
| T490A | 1.8 ± 0.2 | 1.8 ± 0.2 | (1.2 ± 0.1) × 10−2 | UN | 3.9 ± 0.3 | 2.0 ± 0.1 | 1.20 |
| E488Q/T490A | 2.1 ± 0.4 | 1.9 ± 0.4 | 1.0 ± 0.1 | (6.3 ± 0.4) × 10−3 | 3.9 ± 0.2 | 2.0 ± 0.1 | ND |
| V493T | 2.2 ± 0.1 | 2.6 ± 0.3 | ∼ >2.5‡ | (2.6 ± 0.3) × 10−3 | 4.2 ± 0.2 | 1.7 ± 0.1 | ND§ |
| N597K | 0.9 ± 0.1 | 1.2 ± 0.1 | 2.3 ± 0.5 | (2.3 ± 0.3) × 10−3 | NR | NR | 0.67 |
| N613K | 1.0 ± 0.2 | 1.2 ± 0.1 | 1.2 ± 0.2 | (1.6 ± 0.2) × 10−3 | 2.6 ± 0.1 | 1.6 ± 0.1 | 0.79 |
ND, not determined; NR, not relevant; UN, undetectable.
*FI, increase in fluorescence intensity (arbitrary units) on addition of protein to UA2APG-28 or GA2APC-27. FI of controls: ssUA2APG-28 = 10.2 ± 0.3, duplex UA2APG-28 = 3.7 ± 0.1, ssGA2APC-27 = 9.9 ± 0.3, duplex GA2APC-27 = 3.4 ± 0.2.
†Srel, Relative nearest-neighbor specificity for WT and mutants (Srel = Sprotein/SWT). Nearest-neighbor specificity for proteins, for example, WT hADAR2, was determined from the equation:
SWT = [∑|Average percent edited for a triplet -20|]WT
which is summation of the absolute values obtained by subtracting 20 from the average % edited for each triplet. Total percent editing was normalized to 20% (see text and Fig. 5).
‡Values measured for E488Q and V493T with UAG were actually 7.3 ± 0.3 min−1 and 3.4 ± 0.3 min−1, respectively. However, in our experience the manual pipetting method we used is inaccurate for values above 2.5 min−1, and a more accurate value must await measurement by rapid quench protocols.
§Srel = 0.80 for V493A, a different mutation at this position.
Catalytic Rate of E488Q Is Increased and That of T490A Is Decreased Compared with WT hADAR2.
WT hADAR2 and variants showed similar binding affinity for the UAG and GAC hairpins, indicating that discrimination between UAG and GAC occurs after the initial binding step. To understand further the basis of preferences, we determined the deamination rate (kdeam) of WT hADAR2 and mutants under single-turnover conditions. Slow turnover rate and substrate inhibition of ADARs makes steady-state measurements challenging (20). To determine kdeam, we used the same UAG and GAC hairpins used for binding-affinity studies, except that the editing site adenosine was 32P-labeled at its 5′ phosphate using a splint-ligation technique (21). Hairpins were incubated with enzyme, and then the RNA was treated with nuclease P1 to produce nucleoside 5′-monophosphates, which were separated by TLC. The kdeam was determined by monitoring the amount of 5′-AMP converted to 5′-IMP over time.
Representative deamination assays for WT and E488Q mutant hADAR2 using the UAG hairpin are shown in Fig. 3 A and B, and quantitation of multiple assays for both UAG and GAC hairpins is plotted in Fig. 3 C and D. Compared with WT hADAR2, the E488Q mutant showed an increase in kdeam for the UAG hairpin (Table 2 and Fig. 3C). V493T and N597K also showed an increase in kdeam for UAG, albeit to a lesser extent than observed with E488Q (Table 2 and Fig. 3C). Dramatically, the E488Q mutant showed an ∼60-fold increase in kdeam for the GAC hairpin compared with WT hADAR2, whereas V493T, N597K, and N613K showed only a slight increase in kdeam for this hairpin (Table 2 and Fig. 3D). These data correlated with in vivo data obtained from the screen, which showed that E488Q edited GAC most efficiently. T490A edited the UAG hairpin poorly and did not edit the GAC hairpin, again correlating with in vivo data. These data indicated that T490 is important for WT levels of editing and is involved in a step after the initial binding.
Fig. 3.

The kdeam values for some mutant enzymes are similar to that of WT hADAR2, but others differ. (A and B) PhosphorImages showing representative TLC plates used in the kdeam assay with 250 nM WT hADAR2 (A) or E488Q mutant (B) and 0.5 nM UAG hairpin with the target adenosine labeled at its 5′ phosphate. Time points are indicated at the top of the TLC plate, positions of origin (O), 5′ AMP (pA), and 5′ IMP (pI) are indicated on the left. Control experiments using less protein or twice the amount of RNA confirmed single-turnover conditions (Fig. S2) and also established that WT and mutant hADAR2 were stable for the duration of the experiment (Fig. S3). (C and D) Plots showing the fraction of inosine produced as a function of time for WT hADAR2 and mutants with UAG (C) and GAC (D) hairpins. Data points were fitted to the equation, Ft = Fend (1 − e−kt), where Ft is the fraction of inosine at time t, Fend is the fitted fraction of inosine at end point, and k is the fitted rate constant. Error bars indicate SD; n ≥ 3. Insets expand the x-axis for reactions with the UAG hairpin and the y-axis for reactions with the GAC hairpin. Although the overall fit to this equation was good, late time points showed a continued increase in inosine for the UAG hairpin. This continued increase could indicate a double-exponential rate, but the kdeam values obtained on excluding the late time points by fitting the data points up to 30 min were similar to that obtained from 60-min time points. The small increase at later time points possibly was caused by slow editing of contaminating 32P 5′-end–labeled 54-nt RNA used as starting material for preparing the 60-nt UAG hairpin by splint ligation.
Assays of 2-Aminopurine Fluorescence Suggest Certain Mutants Alter Base Flipping.
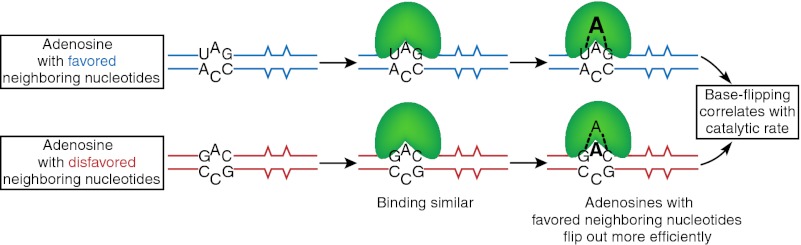
Because the E488Q mutant bound RNA with an affinity similar to that of WT hADAR2, its large increase in kdeam was likely caused by a subsequent step. Like other enzymes that modify bases within a double helix, ADARs are thought to use a base-flipping mechanism (20). In another base-flipping enzyme, the cytosine-specific DNA methyl transferase M.HhaI, the position occupied by the target cytosine in the DNA duplex is assumed by a glutamine when cytosine flips out (22). This glutamine is flanked by glycine residues proposed to be crucial for positioning the glutamine side chain for deep penetration into the helix. E488Q also has flanking glycine residues and is on a loop proximal to the active site. Thus, we investigated the base-flipping ability of mutants on this conserved loop (E488Q, T490A, and V493T) by substituting the target adenine with 2-aminopurine (2-AP), a fluorescent adenine analog previously used to probe base flipping (23), including in studies of hADAR2 (20).
The fluorescence of 2-AP is dependent on its molecular environment. When present in single-stranded oligonucleotides or free in aqueous solution, 2-AP fluoresces. However, when 2-AP is incorporated into a double helix, its fluorescence is quenched by base-stacking interactions. Two ADAR substrates were synthesized with 2-AP in the context of favored (UA2APG-28) or disfavored (GA2APC-27) neighbors (Fig. 4A). These substrates were similar to those used for determining binding affinity and kdeam, but the editing site adenine was replaced with 2-AP, and intermolecular duplexes were used instead of a hairpin to enable control experiments with single-stranded RNA (ssRNA). As expected, in control experiments, ssRNA with 2-AP showed a dramatic increase in fluorescence intensity (FI) compared with duplex RNA with 2-AP (gray dashed and solid lines, respectively, in Fig. 4 B and C). However, the 2-AP fluorescence in the duplex was not quenched completely, possibly because the fluorescence of 2-AP in a mismatch is quenched less effectively than that of 2-AP in a base pair (23). In the absence of protein, UA2APG-28 and GA2APC-27 duplexes showed similar FI, as did single-stranded UA2APG-28 and GA2APC-27 (Fig. 4D).
Fig. 4.

2-AP fluorescence assays suggest certain mutants alter base flipping. (A) Sequences of constructs used for base-flipping studies, with target adenosines replaced by 2-AP (red). UA2APG-28 and GA2APC-27 are intermolecular duplexes made from complementary strands of 28 and 27 nt, respectively. (B and C) Plots showing FI in arbitrary units (a.u.) as a function of wavelength for samples containing only RNA (0.6 μM) or both protein (2.4 μM) and RNA (0.6 μM) for UA2APG-28 and GA2APC-27 (see legend Insets). Control experiments confirmed protein was saturating (Fig. S4). Excitation was at 320 nm to minimize background fluorescence from excitation of protein residues, and emission was scanned from 335–430 nm. Each spectrum is the average of multiple analyses (n ≥ 3). (D) Mean FI at emission maximum is plotted; error bars indicate SD. Dotted lines indicate observed FI of WT hADAR2 with UA2APG-28 and GA2APC-27 for reference. *P = 0.02, **P = 0.007, ***P = 0.00002, mutants compared with WT. (E) Plot of Pearson product-moment correlation coefficient (r) measuring correlation between catalytic rate and FI increase observed with UAG (blue squares) or GAC substrates (red squares) for WT hADAR2 and mutants. Correlation coefficient (r) and P value are indicated. The correlation coefficients for UAG substrate data only or GAC substrate data only also were greater than 0.91. (F and G) Plot (F) and bar graph (G) showing FI with UA2APG hairpin, analyzed similarly to UA2APG -28 duplex.
All fluorescence measurements were made at saturating protein concentrations. In agreement with previous studies (20), when WT hADAR2 was added to the UA2APG-28 duplex, FI increased compared with that observed with duplex UA2APG-28 alone (P = 0.02) (compare solid red and gray lines in Fig. 4B). Most notably, E488Q showed a dramatic increase in FI compared with WT hADAR2 (P = 0.00002) (compare solid green versus red lines in Fig. 4B), suggesting that a glutamine at residue 488 enhances base flipping. Our binding studies indicated that T490A has a WT affinity for dsRNA (Table 2), but the FI observed when this protein was added to UA2APG-28 was indistinguishable from that of the duplex alone (solid blue and gray lines, respectively, in Fig. 4B). This result suggests that T490 might be required for base flipping or in a step upstream of base flipping. V493T did not show a statistically significant difference in FI compared with WT hADAR2 (compare solid pink and red lines in Fig. 4B).
Surprisingly, when added to duplex GA2APC-27 (gray solid line in Fig. 4C), WT hADAR2 and all mutants showed a decrease in FI compared with duplex GA2APC-27 alone. However, the FI observed with E488Q was higher than that observed with WT hADAR2 (P = 0.007) (compare green and red lines in Fig. 4C), suggesting that E488Q also enhances base flipping of adenosine within GAC. These data suggest that the net FI observed in our steady-state fluorescence measurements reflects both quenching caused by protein binding at the mismatched 2-AP, as well as base flipping. According to this hypothesis, for WT hADAR2, E488Q, and V493T, base flipping with the UA2APG-28 duplex is more robust than that occurring with GA2APC-27 duplex, leading to an increase in FI that counterbalances the quenching caused by protein binding and producing a net increase in FI (Fig. 4B). Accounting for both binding and base flipping in the net FI also might explain the relatively small increase in FI when WT hADAR2 was added to the UA2APG-28 duplex and the results with N613K, a mutant with an approximately twofold higher binding affinity and a catalytic rate similar to WT hADAR2. Compared with WT hADAR2, N613K showed an approximately twofold decrease in FI with UA2APG-28 duplex (compare red and dashed orange lines in Fig. 4B and see Table 2). Finally, given that T490A exhibits low levels of editing with UAG substrates, it likely is capable of base flipping, albeit inefficiently. We assume that the effects of this base flipping on FI are counterbalanced by quenching, so that the FI with T490A and the UA2APG-28 duplex is indistinguishable from that of the duplex alone.
Although our FI measurements likely reflect contributions from both binding and base flipping, for proteins with similar affinity an increase in FI should correlate with increased base flipping. Consistent with this expectation, we observed a positive correlation between an increase in FI and the catalytic rate for WT and hADAR2 variants with similar affinity (Pearson’s product moment correlation coefficient (r) = 0.9248, P = 0.0004) (Fig. 4E). To confirm that fluorescence experiments performed with duplexes can be compared with experiments using hairpins, we incorporated 2-AP at the editing site of the UAG hairpin used in binding and deamination assays. When the UA2APG hairpin was mixed with WT hADAR2 and E488Q, we observed an increase in FI comparable to that observed with the UA2APG-28 duplex (Fig. 4 F and G).
Characterization of the Double Mutant E488Q/T490A.
Compared with WT hADAR2, the E488Q mutant showed a dramatic increase in FI when added to the UA2APG-28 duplex, suggesting that glutamine at residue 488 affects base flipping. In contrast, when added to this duplex, the T490A mutant exhibited an FI that was slightly less than that observed with WT hADAR2 (difference significant at P = 0.02), suggesting that T490 is required for efficient base flipping or in a step before base flipping. We hypothesized that if T490 were essential for efficient base flipping or in a step before base flipping, then the double mutant, E488Q/T490A, would not exhibit the dramatic increase in FI observed with the E488Q mutant.
We first confirmed that binding affinity of E488Q/T490A was similar to that of WT hADAR2 for both UAG and GAC hairpins (Fig. 2 C and D and Table 2). Although the catalytic rate of the T490A mutant was extremely low for both hairpins, in the context of the E488Q/T490A double mutant, kdeam increased for both hairpins but was far less than the high kdeam exhibited by the E488Q single mutant (Fig. 3 C and D and Table 2). Further, when the E488Q/T490A mutant was mixed with the UA2APG-28 or GA2APC-27 duplex, the increase in FI was similar to that of T490A, indicating that T490 is essential for the increase in FI observed with E488Q (Fig. 4 B–D and Table 2). The E488Q mutation could not enhance base flipping of the T490A mutation, but the increased kdeam of the double mutant compared with the T490A mutant suggested that E488Q has an additional role in catalysis.
hADAR2 Specificity Is Affected by the E488Q and T490A Mutations.
With WT hADAR2, a protein-induced increase in FI was observed with the UA2APG-28 duplex compared with the GA2APC-27 duplex, suggesting that hADAR2’s preference for UAG over GAC is derived from differences in base flipping. Are there other examples in which the base-flipping efficiency of the enzyme affects specificity? In M.EcoRI, an N6-adenine DNA methyl transferase that uses a bending, base-flipping, and intercalation mechanism, a bending-deficient mutant decreases base flipping and increases specificity (24, 25). For noncognate substrates, M.EcoRI specificity arises from partitioning the enzyme/DNA intermediate into the unbent form (25, 26). We investigated whether E488Q and T490A, mutants that showed differences in 2-AP FI compared with WT hADAR2, also showed differences in substrate specificity. We determined nearest-neighbor preferences for all possible 16 triplet contexts in which the target adenosine can occur using a long, synthetic, perfectly base-paired dsRNA (14). Each protein was incubated with non-radiolabeled 418-bp dsRNA to achieve ∼20% overall editing, thus ensuring that well-edited sites were not saturated to 100% editing (saturation could result in loss of information) and also that the majority of editing sites with a 5′ adenosine were not 5′ inosine (14), which could skew preference determinations.
If hADAR2 lacked preferences, each of the graphs in Fig. 5 would show a horizontal line at 20% editing (represented by the dotted line in the graphs). However, as illustrated for the WT protein, hADAR2, like all ADARs, exhibits preferences, and the line graph is plotted with the most preferred triplets to the right of the graph. Consistent with the idea that increased base flipping correlates with a decrease in specificity, most points on the line graph for E488Q moved closer to the dotted line representing 20% editing (Fig. 5A). Overall, the E488Q mutant showed more editing for the least-preferred triplets (left half of the graph) and less editing for the most-preferred triplets (right half of the graph), although some triplets (CAA, CAC, AAU, and UAG) did not follow this pattern. To facilitate comparison among enzyme variants, we defined a relative nearest-neighbor specificity, Srel; compared with the WT enzyme, the E488Q enzyme exhibited an Srel of 0.85 (Table 2). As expected, the E488Q mutant showed a slight-to-moderate increase in editing of all four triplets that had a 5′ G. However, compared with WT hADAR2, only a 1.6-fold increase in editing of GAC was observed with E488Q in the 418-bp dsRNA, in contrast to the approximately 60-fold increase in kdeam observed in the GAC hairpin. Possibly this disparity indicates that the mismatched adenosine of the GAC hairpin is more amenable to base flipping than adenosines within the completely base-paired 418-bp dsRNA.
Fig. 5.

hADAR2 mutations affect specificity. (A) Plot showing average percent editing of adenosine in each of the 16 possible triplet contexts, determined from analysis of editing in a 418-bp dsRNA, for E488Q compared with WT hADAR2. One hundred ninety-six adenosines were used to calculate average percent editing of adenosine in 16 triplet contexts. Triplets are ordered in terms of 5′ nearest neighbor, with 5′ G followed by C, A, and U. Mean percent editing across 196 adenosines was 19.9% for WT hADAR2 and 21.3% for E488Q and was normalized to 20% as indicated by the dotted line. Error bars indicate SD; n ≥ 3. (B) Similar plot to A comparing T490A with WT hADAR2, but incubations were at 30 °C to facilitate a 20% editing level by the T490A mutant. Control experiments established that WT hADAR2 preferences were almost identical at both temperatures. Sequencing for a portion of the antisense strand was not clean for the T490A reactions, so only 153 adenosines were used to calculate average percent editing of adenosine in 16 triplet contexts for both T490A and WT hADAR2. Mean percent editing across 153 adenosines was 18.7% for WT hADAR2 and 19.2% for T490A and was normalized to 20% as in A. Error bars represent SD; n ≥ 2. (C) As in A, comparing V493A, N597K, and N613K with WT hADAR2. Mean percent editing across 196 adenosines was 19.6% for V493A, 19.0% for N597K, and 20.6% for N613K and was normalized to 20% as in A. Error bars indicate SD; n ≥ 3.
The T490A mutant, on the other hand, showed an overall increase in specificity, with Srel = 1.20 (Table 2); triplets poorly edited by WT hADAR2 were edited to a lower percentage, and triplets well edited by WT hADAR2 were edited to a greater percentage (Fig. 5B). Mutants V493A, N597K, and N613K showed a decrease in specificity (Table 2); triplets poorly edited by WT hADAR2 were edited more, and triplets well edited by WT hADAR2 were edited less (Fig. 5C). The decreased specificity observed with N597K and N613K could be caused by their higher binding affinity compared with WT hADAR2.
Discussion
ADARs exhibit nearest-neighbor preferences in choosing adenosines for deamination. Preferences derive mainly from the catalytic domain (14, 16), but the amino acids that mediate preferences and the mechanism involved are unclear. We investigated these issues by performing a screen for mutations within the catalytic domain that allowed editing of an adenosine within a disfavored nearest-neighbor context, GAC. We identified seven mutations, most of which mapped onto two distinct regions on the surface of the predicted RNA-binding site (17). We characterized effects of these mutations by performing in vitro assays on mutant hADAR2 enzymes to determine binding affinity, catalytic rate, and nearest-neighbor preferences. Using a hADAR2 substrate with the fluorescent analog 2-AP incorporated at the editing site, we also compared changes in fluorescence that occurred upon addition of WT or mutant enzymes. These data support the idea that ADARs use a base-flipping mechanism to access the target adenosine and, unexpectedly, suggest that preferences derive mainly from nearest-neighbor effects on base flipping rather than from direct recognition of neighboring bases. Our studies point to a conserved loop on the surface of hADAR2 and close to the active site, as important for preferences.
Fig. 6 presents a model that incorporates the results of our studies. The model compares WT hADAR2 with the E488Q mutant but is consistent with properties of all characterized mutations. As illustrated, we observed that WT hADAR2 and the E488Q mutant bound the favored UAG and disfavored GAC substrates with identical affinities, a trend that was consistent for all mutant enzymes. Even for the two mutants N597K and N613K found to have an affinity higher than that of the WT enzyme, no differences were observed between UAG and GAC substrates. Thus, our data indicate that preferences do not derive from differential binding. Our model posits that a base-flipping step follows binding, and here our experiments with 2-AP suggest a clear difference between UAG and GAC substrates. For WT hADAR2 and all studied mutants, a greater increase in fluorescence was observed when the protein was mixed with UAG substrates than with GAC substrates, and in all cases this increase in fluorescence correlated with a higher kdeam (Fig. 4 D and E). The change in fluorescence was most dramatic with the E488Q mutant, suggesting that this mutant has enhanced base-flipping properties. Our data suggest that preferences are based on the effects of nearest neighbors on the base-flipping step of the ADAR reaction.
We also characterized residue T490, because it was on the conserved loop, proximal to other residues identified in the screen (Figs. 1 F and G and 6B). The mutant T490A exhibited a greatly reduced catalytic rate (Table 2) and minimal base flipping that was not enhanced in the E488Q/T490A double mutant. These data suggest that T490 is required for efficient base flipping, and our favored model is that it is important for maintaining a conformation of the conserved loop that promotes base flipping. Indeed, the hADAR2 crystal structure shows the side chain hydroxyl and backbone carbonyl of T490 hydrogen bonding with the side chain of R481 (Fig. 6 B and C) (17). Correspondingly, R481A did not show editing of adenosine within UAG or GAC hairpins in the in vivo α-galactosidase reporter assay (Fig. S5). Further, in the mutational analysis of residue T490 (Table 1), only serine and cysteine could replace threonine for editing UAG, suggesting that the predicted hydrogen bond (17) from the side chain of T490 to R481 is important (Fig. 6B).
The double mutant E488Q/T490A showed a minimal level of base flipping similar to that of T490A but higher kdeam than T490A. This result suggests that a glutamine at residue 488 has effects in addition to enhancing base flipping. Similar to another base-flipping enzyme, M.EcoRI, in which enhanced base flipping compromises specificity (24), the enhanced base flipping of the E488Q mutant correlated with a decreased specificity for most triplets (Fig. 5A); however, some triplets did not conform, again raising the possibility that the E488Q mutation has other effects. Compared with WT hADAR2, only glutamine or asparagine at residue 488 enhanced GAC editing (Table 1), emphasizing the importance of an amide side chain for GAC editing. Possibly a hydrogen bond from the amide side chain to the RNA promotes this specificity. With E488Q, a small increase in base flipping of adenosine within GAC (FI ∼2.6) resulted in an ∼60-fold increase in editing. This increase also might indicate that a glutamine at residue 488 has effects beyond base flipping, but it also is possible that base flipping is rate limiting for GAC and that a small increase in base flipping results in a large increase in catalytic rate.
Our studies suggest that preferences are determined mainly by the propensity of the target adenosine for base flipping rather than by direct recognition of the neighboring bases. This idea is supported by the fact that we did not identify any mutants showing a reversed preference, i.e., that could edit an adenosine within GAC but not within UAG. However, we did identify a mutation that increased base flipping and thereby increased the efficiency for editing adenosines in unfavorable contexts (Figs. 4 and 5). Possibly ADAR preferences arose from the intrinsic base-flipping properties of an RNA double helix, and ADARs evolved to attain a balance between editing efficiency and specificity. The latter possibility is suggested by our observation that the T490A mutant has a dramatically reduced catalytic rate but increased specificity, whereas the E488Q mutant has an increased catalytic rate but decreased specificity (Figs. 3 C and D and 5 A and B).
2-AP fluorescence is affected by changes in the immediate environment, such as alterations in base pairing or protein interactions (27). The analog is used frequently to report on base flipping (23), including with hADAR2 (20), and in limited cases the observed changes in fluorescence have been correlated with a crystal structure with 2-AP in a flipped-out position (28). Although we cannot be certain our assays with 2-AP are measuring base flipping, this explanation seems to be the most likely. The C6 position of adenine that undergoes nucleophilic attack during the deamination reaction lies deep in the major groove of dsRNA, and, as previously proposed (11, 20), it is difficult to imagine anything but base flipping that would allow ADAR access to the C6 atom. Further, the crystal structure of the hADAR2 catalytic domain shows a large basic patch on the surface, which likely facilitates binding to dsRNA, with a deep pocket that contains the active site zinc ion (17), a perfect arrangement for interacting with a helix with a flipped-out adenosine.
Proven base-flipping enzymes use various mechanisms to gain access to a base. For example, in the cytosine-specific DNA methyl transferase M.HhaI, a glutamine side chain directly pushes the target cytosine out of the helix, and Gly residues flanking the glutamine are proposed to be crucial for positioning its side chain for penetration into the helix (22, 29). Residue E488 also has flanking glycine residues and is on a loop proximal to the active site, and it is enticing to speculate that ADARs use a similar mechanism for base flipping the target adenosine. However, definitive proof of this mechanism awaits further experimentation. Other mechanisms used by base-flipping enzymes include the serine-mediated pinch-pull-push mechanism (30) and the helix-bending, base-flipping, and intercalation mechanism (26). An alternative passive mechanism also has been proposed in which the protein simply traps a transiently flipped-out base (31). Our studies suggest that hADAR2 base flips an adenosine more efficiently within a UAG context than within a GAC context, but further studies will be necessary to determine mechanistic details. If ADARs use the passive mechanism, then by implication an adenosine with a 5′ U intrinsically flips more readily than an adenosine with a 5′ G. Indeed, NMR experiments and theoretical calculations indicate that the probability of base-pair opening is affected by nearest neighbors (32, 33), and data from such analyses are consistent with ADAR preferences. For example, the opening probability of an A•T base pair with a 5′ G is less than that with a 5′ C or 5′ T, possibly because of the stronger stacking interaction of adenosine with a 5′ G (33). Alternatively, base flipping may be protein induced, but the extent of flipping might be influenced by the sequence context. Molecular dynamics simulations suggest that, of the two A•C mismatches in the R/G hairpin, adenosine of the target A•C pair is more prone to base flipping than that in the second A•C pair, which is not edited (34). Interestingly, the target adenosine has a 5′ A, whereas the adenosine in the second A•C pair has a 5′ G.
The locus for DSH, a pigmentary genodermatosis, maps to the human ADAR1 gene (8). Of more than 100 mutations reported in the human ADAR1 gene of DSH patients, at least 47 are missense mutations in the catalytic domain (8, 35). DSH is not usually associated with other diseases, but in the two reported cases in which DSH was accompanied by dystonia, brain calcification, and mental deterioration, the same mutation, G1007R, was identified (36, 37). The equivalent residue in hADAR2 is G487, one of the two glycine residues flanking E488 on the conserved loop. It will be informative to study this mutant further to determine if disease symptoms result from altered hADAR1 specificity.
Materials and Methods
Construction of hADAR2 Plasmids for the Screen.
The hADAR2 construct includes an N-terminal 12-histidine tag followed by a TEV protease-recognition site in a yeast expression plasmid, YEpTOP2PGAL1 (38). We modified this construct to include a restriction site before the catalytic domain (YEpTOP2PGAL1-RSinADAR2) (SI Materials and Methods). Both constructs were cloned into yeast centromere plasmid (YCp) vector with a GAL promoter and URA3 marker (YCp-ADAR2 and YCp-RSinADAR). See SI Materials and Methods for details.
Construction of Hairpin-Reporter Strains.
The plasmid encoding UAG hairpin upstream of the α-galactosidase reporter pR/GαGal has been described (18). We constructed the plasmid encoding the GAC hairpin-reporter by performing sewing PCR and incorporated the favored six-nucleotide loop (SI Materials and Methods) (18). Both hairpin-reporter constructs were cloned separately into the YCp vector with the ADH1 promoter and TRP1 marker and then were cloned into the corresponding yeast-integrating plasmid (YIp) vector (SI Materials and Methods). YIp vectors containing hairpin-reporters were linearized and integrated into the W303α chromosome by lithium acetate transformation (SI Materials and Methods).
Screen.
YCp-RSinADAR2 was restriction digested to produce a gapped vector lacking the catalytic domain (SI Materials and Methods). The hADAR2 catalytic domain was PCR amplified from YCp-ADAR2 (SI Materials and Methods) and was used as a template for random mutagenesis by error-prone PCR using the GeneMorph II Random Mutagenesis Kit (Stratagene). The amount of DNA template and the number of PCR cycles were optimized to get zero to four mutations per kilobase. One hundred nanograms of the gapped vector and 300 ng of random mutagenized PCR product were transformed simultaneously with lithium acetate into the GAC hairpin-reporter yeast strain, plated on complete minimal minus uracil (CM −URA) agar plates, and incubated at 30 °C for 2 d. Plates were replica plated onto agar plates containing CM −URA, 3% (wt/vol) galactose, 2% (wt/vol) raffinose, and 0.06 mg/mL X-α-Gal, and were incubated at 20 °C for 4–5 wk. Plasmids were rescued from selected colonies, retransformed into fresh GAC hairpin-reporter yeast strains to confirm hADAR2 dependence, and sequenced to identify mutations.
Additional mutational analysis for selected residues used the same hADAR2 catalytic domain template used for random mutagenesis. We performed sewing PCR using two outside primers (CDRanMutP1 and CDRanMutP3) and two inside primers specific for each mutant (Table S1). Inside primers were degenerate, with the three nucleotides coding for the relevant amino acid randomized. Mutagenized PCR product and gapped vector were transformed into hairpin-reporter yeast strains, as for the screen, and were analyzed similarly.
Protein Purification.
Mutants in the YCp vector were cloned back into the yeast-expression plasmid YEpTOP2PGAL1 (SI Materials and Methods). WT hADAR2 and all mutants (in YEpTOP2PGAL1) were purified as described (38) to greater than 97% purity, as determined by Coomassie staining. Identity of purified proteins was confirmed by mass spectrometry.
Preparation of RNA for in Vitro Studies.
RNA for in vitro studies was chemically synthesized and gel purified after denaturing PAGE (SI Materials and Methods). The 5′-end–labeled hairpins for binding experiments were prepared as described in ref. 21 (SI Materials and Methods). Internally radiolabeled hairpins for rate determination were prepared by splint ligation as described in ref. 20 (SI Materials and Methods). Duplexes or hairpins with the target adenine replaced with 2-AP were prepared as described (20) with modifications. Purified top and bottom strands were dissolved in hybridization buffer [10 mM Tris⋅HCl (pH 7.5), 0.1 mM EDTA, 50 mM KCl], heated to 95 °C for 4 min, slowly cooled to room temperature for 2 h, and ethanol precipitated and gel purified after 15% native PAGE. Internally radiolabeled and non-radiolabeled 418-bp RNA were synthesized as described in ref. 14 (SI Materials and Methods). This dsRNA has a 21-nt overhang at each 5′ terminus. On the 3′ termini, sense strands had a 12-nt overhang, and antisense strands had a 13-nt overhang.
Gel Mobility-Shift Assay.
Gel-shift assays were as described in ref. 19, with modifications. Assays were performed with 20 pM RNA and varying protein concentrations, incubated at 4 °C for 20 min in buffer containing 14 mM Tris⋅HCl (pH 8), 130 mM KCl, 10% (vol/vol) glycerol, 1 mM DTT, 100 μg/mL BSA, 0.2 mM β-mercaptoethanol, and 20 mM NaCl. Reactions were stopped by loading 10 μL of the reaction directly onto a 6% (37.5:1 acrylamide/bis-acrylamide) native gel running at 150 V, at 4 °C in 0.5× Tris/borate/EDTA. Gels were electrophoresed for an additional 2 h, dried or frozen, and autoradiographed. For truncated proteins, gel shifts were performed as for full-length proteins, except that 35 mM salt and 29:1 acrylamide/bis-acrylamide were used.
Deamination Assay.
Deamination assays were performed under single-turnover conditions as described in ref. 21, using 0.5 nM RNA and 250 nM protein. Initial experiments with WT hADAR2 and UAG substrate at 30 °C resulted in ∼60% editing at 20 s, so that discerning differences was challenging. Thus, for UAG substrate, all incubations were at 20 °C to slow the reaction. For the GAC substrate, incubations were at 30 °C.
2-AP Fluorescence Assay.
2-AP fluorescence experiments were performed on a Perkin-Elmer LS 50 Luminescence Spectrometer at room temperature. Excitation was at 320 nm, emission was scanned from 335–430 nm, and a 10-nm slit width was used for excitation and emission. Fluorescence was measured using a ultra-micro cuvettete from Hellma (30 μL) with 2.4 μM protein and 0.6 μM RNA in buffer containing 16 mM Tris⋅HCl (pH 7.5), 8 mM Tris (pH 8), 20 mM KCl, 40 mM NaCl, 8% (vol/vol) glycerol, 1 mM DTT (Roche), 0.01% Nonidet P-40, and 0.4 mM β-mercaptoethanol (Sigma). Control experiments used 0.6 μM duplex or ssRNA. Corrections were made to emission spectra to account for fluorescence from protein and buffer; for RNA-only samples, the spectrum of the buffer was subtracted, and for protein-RNA samples, the spectrum of the protein in buffer was subtracted. To ascertain that saturating protein concentrations were used, we performed control experiments using 3.0 and 1.8 μM protein instead of 2.4 μM (Fig. S4).
Preference Assay.
An initial time course was performed as described in ref. 14 with internally radiolabeled 418-bp dsRNA to determine time required for ∼20% editing. Preference assays were as described in ref. 14 by incubating 0.25 nM non-radiolabeled 418-bp dsRNA and 250 nM protein at 20 °C for the time required to achieve ∼20% editing (SI Materials and Methods). For T490A, incubations were at 30 °C, because 20% editing was not achieved at 20 °C by 1 h. For comparison, WT hADAR2 preferences were determined at 30 °C and were found to be almost identical to those at 20 °C.
Supplementary Material
Acknowledgments
We thank S. Pokharel and P. Beal for the plasmid pR/GαGal; D. Stillman for the W303α yeast strain, YCp and YIp vectors, and essential advice, guidance, and patience; D. Winge for access to the Perkin Elmer LS 50 Luminescence Spectrometer; B. Schackmann and M. Hanson at the University of Utah DNA/Peptide Core Facility for RNA and primer synthesis; C. Nelson and K. Parsawar at the University of Utah Mass Spectrometry and Proteomics Core Facility for mass spectrometry of mutant ADARs; and A. A. Krauchuk and P. J. Aruscavage for technical assistance. Shared core resources were supported by P30CA042014 from the National Cancer Institute. This work was supported by Grant R01GM044073 from the National Institute of General Medical Sciences (to B.L.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 19521 (volume 109, number 48).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212548109/-/DCSupplemental.
References
- 1.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hundley HA, Bass BL. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem Sci. 2010;35(7):377–383. doi: 10.1016/j.tibs.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warf MB, Shepherd BA, Johnson WE, Bass BL. Effects of ADARs on small RNA processing pathways in C. elegans. Genome Res. 2012;22(8):1488–1498. doi: 10.1101/gr.134841.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411(2):180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen CX, et al. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6(5):755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melcher T, et al. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996;271(50):31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 8.Maas S, Kawahara Y, Tamburro KM, Nishikura K. A-to-I RNA editing and human disease. RNA Biol. 2006;3(1):1–9. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann KA, Bass BL. The importance of internal loops within RNA substrates of ADAR1. J Mol Biol. 1999;291(1):1–13. doi: 10.1006/jmbi.1999.2914. [DOI] [PubMed] [Google Scholar]
- 10.Nishikura K, et al. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J. 1991;10(11):3523–3532. doi: 10.1002/j.1460-2075.1991.tb04916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polson AG, Bass BL. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13(23):5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stephens OM, Haudenschild BL, Beal PA. The binding selectivity of ADAR2’s dsRBMs contributes to RNA-editing selectivity. Chem Biol. 2004;11(9):1239–1250. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39(42):12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 14.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stefl R, et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143(2):225–237. doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong SK, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7(6):846–858. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macbeth MR, et al. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309(5740):1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pokharel S, Beal PA. High-throughput screening for functional adenosine to inosine RNA editing systems. ACS Chem Biol. 2006;1(12):761–765. doi: 10.1021/cb6003838. [DOI] [PubMed] [Google Scholar]
- 19.Ohman M, Källman AM, Bass BL. In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA. 2000;6(5):687–697. doi: 10.1017/s1355838200000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stephens OM, Yi-Brunozzi HY, Beal PA. Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry. 2000;39(40):12243–12251. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 21.Macbeth MR, Lingam AT, Bass BL. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA. 2004;10(10):1563–1571. doi: 10.1261/rna.7920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76(2):357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 23.Holz B, Klimasauskas S, Serva S, Weinhold E. 2-Aminopurine as a fluorescent probe for DNA base flipping by methyltransferases. Nucleic Acids Res. 1998;26(4):1076–1083. doi: 10.1093/nar/26.4.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allan BW, et al. DNA bending by EcoRI DNA methyltransferase accelerates base flipping but compromises specificity. J Biol Chem. 1999;274(27):19269–19275. doi: 10.1074/jbc.274.27.19269. [DOI] [PubMed] [Google Scholar]
- 25.Youngblood B, Bonnist E, Dryden DT, Jones AC, Reich NO. Differential stabilization of reaction intermediates: Specificity checkpoints for M.EcoRI revealed by transient fluorescence and fluorescence lifetime studies. Nucleic Acids Res. 2008;36(9):2917–2925. doi: 10.1093/nar/gkn131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Youngblood B, Reich NO. Conformational transitions as determinants of specificity for the DNA methyltransferase EcoRI. J Biol Chem. 2006;281(37):26821–26831. doi: 10.1074/jbc.M603388200. [DOI] [PubMed] [Google Scholar]
- 27.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29(18):3784–3795. doi: 10.1093/nar/29.18.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenz T, et al. 2-Aminopurine flipped into the active site of the adenine-specific DNA methyltransferase M.TaqI: Crystal structures and time-resolved fluorescence. J Am Chem Soc. 2007;129(19):6240–6248. doi: 10.1021/ja069366n. [DOI] [PubMed] [Google Scholar]
- 29.Daujotyte D, et al. HhaI DNA methyltransferase uses the protruding Gln237 for active flipping of its target cytosine. Structure. 2004;12(6):1047–1055. doi: 10.1016/j.str.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Wong I, Lundquist AJ, Bernards AS, Mosbaugh DW. Presteady-state analysis of a single catalytic turnover by Escherichia coli uracil-DNA glycosylase reveals a “pinch-pull-push” mechanism. J Biol Chem. 2002;277(22):19424–19432. doi: 10.1074/jbc.M201198200. [DOI] [PubMed] [Google Scholar]
- 31.Roberts RJ, Cheng X. Base flipping. Annu Rev Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 32.Coman D, Russu IM. A nuclear magnetic resonance investigation of the energetics of basepair opening pathways in DNA. Biophys J. 2005;89(5):3285–3292. doi: 10.1529/biophysj.105.065763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krueger A, Protozanova E, Frank-Kamenetskii MD. Sequence-dependent base pair opening in DNA double helix. Biophys J. 2006;90(9):3091–3099. doi: 10.1529/biophysj.105.078774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hart K, Nyström B, Ohman M, Nilsson L. Molecular dynamics simulations and free energy calculations of base flipping in dsRNA. RNA. 2005;11(5):609–618. doi: 10.1261/rna.7147805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, et al. Mutational spectrum of the ADAR1 gene in dyschromatosis symmetrica hereditaria. Arch Dermatol Res. 2010;302(6):469–476. doi: 10.1007/s00403-010-1039-2. [DOI] [PubMed] [Google Scholar]
- 36.Tojo K, et al. Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation. Mov Disord. 2006;21(9):1510–1513. doi: 10.1002/mds.21011. [DOI] [PubMed] [Google Scholar]
- 37.Kondo T, et al. Dyschromatosis symmetrica hereditaria associated with neurological disorders. J Dermatol. 2008;35(10):662–666. doi: 10.1111/j.1346-8138.2008.00540.x. [DOI] [PubMed] [Google Scholar]
- 38.Macbeth MR, Bass BL. Large-scale overexpression and purification of ADARs from Saccharomyces cerevisiae for biophysical and biochemical studies. Methods Enzymol. 2007;424:319–331. doi: 10.1016/S0076-6879(07)24015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]