

Abstract
A doubly substituted form of the nitrogenase MoFe protein (α-70Val→Ala, α-195His→Gln) has the capacity to catalyze the reduction of carbon dioxide (CO2) to yield methane (CH4). Under optimized conditions, 1 nmol of the substituted MoFe protein catalyzes the formation of 21 nmol of CH4 within 20 min. The catalytic rate depends on the partial pressure of CO2 (or concentration of HCO3−) and the electron flux through nitrogenase. The doubly substituted MoFe protein also has the capacity to catalyze the unprecedented formation of propylene (H2C = CH-CH3) through the reductive coupling of CO2 and acetylene (HC≡CH). In light of these observations, we suggest that an emerging understanding of the mechanistic features of nitrogenase could be relevant to the design of synthetic catalysts for CO2 sequestration and formation of olefins.
Keywords: metalloenyzme, multi-electron reduction, hydrocarbon
Carbon dioxide (CO2) is an abundant and stable form of carbon that is the product of respiration and burning of fossil fuels. As a result of these activities, the atmospheric concentration of CO2, a greenhouse gas, has been rising over the last century and contributing to global warming (1). There is strong interest in developing methods for sequestering CO2 either by capturing it or by chemically converting it to valuable chemicals (2–5). Of particular interest are possible routes to reduction of CO2 by multiple electrons to yield methanol (CH3OH) and methane (CH4), which are renewable fuels (2). The reduction of CO2 is difficult, with a limited number of reports of metal-based compounds able to catalyze these reactions (6–13). In biology, only a few enzymes are known to reduce CO2 (14–18), and none of these can catalyze the eight electron reduction to CH4.
The bacterial Mo-dependent nitrogenase enzyme catalyzes the multielectron/proton reduction of dinitrogen (N2) to two ammonia (NH3) at a metal cluster designated FeMo-cofactor [7Fe-9S-1Mo-1C-R-homocitrate] (Fig. 1) in a reaction that requires ATP hydrolysis and evolution of H2, with a minimal reaction stoichiometry shown in Eq. 1 (19–22).
Fig. 1.

The FeMo cofactor with some key amino acid residues. Colors are Mo in magenta, Fe in rust, S in yellow, C in gray, O in red, and N in blue. The central atom is C (gray), and the structure and numbering of Fe atoms are based on the protein database file PDB: ID code 1M1N.
Given that nitrogenase is effective at catalyzing the difficult multielectron reduction of N2, it was of interest to determine whether this enzyme might also catalyze the reduction of CO2 to the level of CH4. Nitrogenase is known to have the capacity to reduce a variety of other small, relatively inert, doubly or triply bonded compounds, such as acetylene (HC≡CH) (19, 23). It has been shown that an alternative form of nitrogenase, which contains V in place of Mo in the active site cofactor, has the remarkable capacity to reduce CO and couple multiple CO molecules, yielding short chain alkenes and alkanes such as ethylene (C2H4), ethane (C2H6), propylene (C3H6), and propane (C3H8) (24, 25). In contrast, the Mo-nitrogenase is only able to reduce CO at exceedingly low rates (24). However, we have found that the MoFe protein can be remodeled by substitution of amino acid residues that provide the first shell of noncovalent interactions with the active site FeMo cofactor such that CO becomes a much more robust substrate with catalytic formation of methane and short chain alkenes and alkanes (26). Related to these observations, we have also reported that the native nitrogenase has the capacity to reduce CO2 at relatively low rates to yield CO (27). Here, we report that a remodeled nitrogenase MoFe protein can achieve the eight-electron reduction of CO2 to CH4. Further, it is shown that CO2 reduction can be coupled to the reduction of other substrates (e.g., acetylene, C2H2) to form longer chain, high value hydrocarbons (e.g., propylene).
Results
CO2 Reduction to CH4 by Remodeled Nitrogenase.
When the wild-type nitrogenase was tested for reduction of CO2 to yield CH4, no CH4 above background could be detected over the course of 20 min (Fig. 2). Earlier work has demonstrated that several amino acids having side chains that approach FeMo cofactor play an important role in controlling substrate binding and reduction (20, 28). Among these residues are α-70Val, α-195His, and α-191Gln (Fig. 1). Variant forms of the MoFe protein having α-70 substituted by Ala, α-195 substituted by Gln, or α-70 and α-191 both substituted by Ala showed no appreciable capacity for reduction of CO2 to yield CH4. In contrast, a doubly substituted MoFe protein, α-70Ala/α-195Gln, was found to catalyze the formation of CH4 from CO2, forming up to 16 nmol CH4/nmol MoFe protein over 20 min (Fig. 2). The formation of CH4 depended on the presence of CO2, Fe protein, MoFe protein, and MgATP . The rate of CH4 production was found to increase with increasing partial pressure of CO2 up to 0.45 atm (Fig. S1). A fit of these data to the Michaelis–Menten equation gave a Km for CO2 of 0.23 atm and a Vmax of 21 nmol CH4/nmol MoFe protein over 20 min. In a Bis-Tris buffer at pH 6.7, sodium bicarbonate (NaHCO3) could also serve as a substrate for CH4 formation, with a determined Km of 16 mM for NaHCO3 and Vmax of 14 nmol CH4/nmol MoFe protein over 20 min (Fig. S2).
Fig. 2.

CH4 formation as a function of time for different MoFe proteins. CO2 reduction to CH4 is shown as a function of time for the wild-type (○), α-70Ala (◇), α-195Gln (△), α-70Ala/α-191Ala (□), and α-70Ala/α-195Gln (■) MoFe proteins. The partial pressure of CO2 was 0.45 atm, the concentration of MoFe protein was 0.5 mg/mL, and Fe protein was 3 mg/mL The reaction temperature was 30 °C. The complete assay for α-70Ala/α-195Gln MoFe protein was done in triplicate, with SE bars shown.
The rate of electron flow through nitrogenase (called electron flux) can be regulated by altering the ratio of Fe protein to MoFe protein (Fe protein:MoFe protein), with a low ratio corresponding to low electron flux and a high ratio corresponding to high electron flux. Under all conditions, the majority of electrons passing through nitrogenase in the presence of saturating CO2 were found to reduce protons to make H2 with relatively low rates of associated CH4 formation (Fig. 3). However, as the electron flux increased, the proportion of electrons passing to CO2 reduction increased, reaching a maximum at a molar ratio of approximately 50 Fe protein per MoFe protein. At this highest flux, the molar ratio of H2 formed per CH4 formed was ∼250:1. Given that proton reduction is a two-electron reduction and CO2 reduction to CH4 is an eight-electron reduction, up to 2% of the total electron flux passing through nitrogenase goes to CO2 reduction to methane under these conditions.
Fig. 3.

Electron flux dependence for CO2 reduction to CH4. The ratio CH4 formed to H2 formed is shown as a function of the electron flux through the α-70Ala/α-195Gln MoFe protein for assays quenched after 20 min at 30 °C. The partial pressure of CO2 was 0.45 atm, the concentration of MoFe protein was 0.5 mg/mL, and Fe protein was varied from 0.5 to 6 mg/mL
The use of 12C- or 13C-enriched bicarbonate (HCO3−) as substrate and product analysis by gas chromatography-mass spectrometry (GC-MS) confirmed that CH4 formation was derived from added CO2. When H12CO3− was the added substrate, a peak having the same retention time as methane showed a mass over charge (m/z) peak of 16, whereas no peak with m/z of 17 was observed (Fig. S3). When H13CO3− was the substrate, a peak having the same retention time was found to have a m/z of 17, which can be ascribed to the molecular mass of 13CH4. This result demonstrates that HCO3− or CO2 is the substrate for CH4 formation rather than some other component in the reaction mixture.
When the CO2 reduction reaction catalyzed by the remodeled nitrogenase was performed in the presence of 0.30 mg/mL deoxyhemoglobin, the amount of CH4 formed was lowered by ∼25% (Fig. S4). Deoxyhemoglobin binds CO very rapidly (rate constant k ∼ 2 × 105 M−1⋅s−1) and with a high affinity (dissociation constant Kd ∼ 50 nM) and would, therefore, bind any CO released into solution during CO2 reduction by nitrogenase (27). Although CO is expected to be an intermediate along the reaction pathway from CO2 to CH4, a relatively small lowering of the rate of formation of CH4 from CO2 when deoxyhemoglobin is included in reaction mixture indicates that CO2 reduction follows primarily a nondissociative mechanism.
Several earlier studies revealed multiple inhibitor and substrate binding sites on FeMo-cofactor, including at least two binding sites for CO (29–33) and acetylene (31, 34, 35). Two adjacent binding sites can explain the earlier reports that two or three CO molecules can be reduced and coupled to form C2 and C3 hydrocarbon products (24, 26). It was therefore of interest to test whether the doubly substituted MoFe protein could couple two or more CO2 molecules to yield short chain hydrocarbons. Under the assay conditions examined, no C2 or C3 hydrocarbon products were detected above the background when CO2 was the sole C substrate. However, when a small amount of acetylene was added when CO2 was used as substrate, the C3 hydrocarbon propylene (H2C = CH-CH3) was detected as the major product and propane (H3C-CH2-CH3) as a minor product. Propylene formation only occurred in a reaction with all components for a complete nitrogenase assay, revealing that propylene is formed by nitrogenase turnover. Interestingly, it was found that propylene formation was favored under relatively low electron flux conditions (4 Fe protein:1 MoFe protein), with higher electron flux favoring CH4 formation at the expense of propylene formation under 0.45 atm of CO2 and 0.014 atm of acetylene (Fig. 4). Under the optimal electron flux condition, the amount of propylene formed increased with increasing acetylene partial pressure up to 0.027 atm and then decreased rapidly at higher acetylene concentrations (Fig. 5) likely due to inhibition of CO2 reduction by acetylene. The results indicate there is an optimal concentration ratio between CO2 and acetylene to achieve reductive coupling of the two molecules at a given electron flux. All possible combinations of electron flux and acetylene and CO2 concentration have not been examined.
Fig. 4.

Propylene formation from CO2 and acetylene as a function of electron flux. The amount of propylene formed as a function of electron flux is shown for the α-70Ala/α-195Gln MoFe protein. The partial pressure of CO2 was 0.45 atm and C2H2 was 0.014 atm. The concentration of MoFe protein was 0.5 mg/mL, and Fe protein was varied from 0.25 to 3 mg/mL. The reactions were incubated at 30 °C for 60 min. Inset shows the CH4 production as a function of electron flux.
Fig. 5.

Propylene formation from CO2 and acetylene. The dependence of propylene formation on the partial pressure of acetylene at a partial pressure of CO2 of 0.40–0.45 atm is shown for the α-70Ala/α-195Gln MoFe protein. The concentration of MoFe protein was 0.5 mg/mL, and Fe protein was 0.5 mg/mL. The reactions were incubated at 30 °C for 60 min.
Formation of propylene from the reductive coupling of one CO2 and one C2H2 was confirmed by GC-MS analysis. When H12CO3− and 12C2H2 were used as substrates, the propylene elution peak displayed a molecular ion peak with m/z of 42, which is ascribed to 12C3H6. Trace amounts of a fragment with a m/z of 43 was observed because of natural abundance of 13C and 2H. When H13CO3− was used together with 12C2H2, a molecular ion peak with m/z of 43 was detected at the same retention time as propylene, consistent with the coupling of one 13CO2 with one 12C2H2 to form 13CH3-12CH=12CH2 (Fig. S5).
Discussion
The discovery reported here that remodeled nitrogenase is able to reduce CO2 by eight electrons to CH4 (Eq. 2) makes it unique among known enzyme catalyzed reactions. Further, the ability to reduce CO2 and couple it to acetylene to form propylene (Eq. 3) makes nitrogenase unique among all reported catalysts (5).
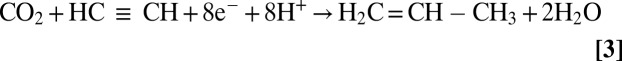 |
Propylene is an especially important hydrocarbon, being the starting point for the synthesis of a variety of polymers (36). A limited number of metal-based catalysts have been shown to reduce CO2 to yield different reduction products including formate (HCOO−), carbon monoxide (CO), formaldehyde (CH2O), methanol (CH3OH), and methane (CH4) (2, 6–13). Common features of homogeneous catalysts for CO2 reduction to CH4 are low reaction rates (e.g., turnover frequencies) and limited number of turnovers (e.g., turnover number) before inactivation of the catalyst (37, 38). Further, for electrochemical reductions, a high overpotential is required, with production of H2 as a waste of electron flux (39, 40). The nitrogenase catalyzed reduction of CO2 to CH4 reported here is comparable in turnover frequency (approximately 1 min−1) and turnover number, with notable slowing of the reaction beyond 20 min. Nitrogenase also diverts most of its electron flux to H2 formation, with only a small percentage going to CO2 reduction. In contrast to the electrochemical catalysts, nitrogenase catalyzes these reactions at modest electrochemical potentials (dithionite is the reductant used in these experiments), however, it does require considerable energy input from the obligate hydrolysis of ATP.
No other known single enzyme can catalyze CO2 reduction to CH4. Methanogenic bacteria convert CO2 to CH4, but this is accomplished by the action of a consortium of enzymes functioning as part of a metabolic pathway (16). In acetogenic bacteria, CO2 is converted to acetate by the action of several enzymes including CO dehydrogenase, which catalyzes the reversible interconversion of CO2 and CO (41). Like nitrogenase, CO dehydrogenase also uses a complex metal cluster to achieve this reaction. Other enzymes have been shown to reduce CO2 to formate or methanol (17, 18), but none to CH4 as reported here for nitrogenase.
The reduction of CO2 catalyzed by nitrogenase can be considered in the context of our current understanding of the mechanism for the reduction of the physiological substrate N2 by six electrons to two ammonia molecules with two additional electrons being used to evolve H2 (20, 28, 42). Several recent studies have added to earlier work in building a probable mechanism for how N2 might be reduced at the active site FeMo cofactor. One important insight relevant to the current discussion is the observation of metal-bound hydrides (determined as two hydrides bridging between Fe atoms) as an integral part of the FeMo-cofactor reactivity toward N2 (43–45). These metal bound hydrides (M-H−) have been proposed to participate in the initial reduction of N2 to the proposed intermediate diazene (HN = NH). Further reduction of the metal bound diazene to two ammonia molecules is proposed to involve successive addition of electrons and protons. An important observation regarding this mechanistic feature is that during N2 reduction, the proposed reaction intermediates (diazene HN = NH or hydrazine H2N-NH2) are not detected in appreciable quantities, indicating that intermediates remain bound to the active site until the final products are released. This phenomenon is likely explained by stabilization of key intermediates along the reaction pathway through appropriate functional groups, thereby minimizing kinetic barriers in going from N2 to two ammonias. The observation reported here that nitrogenase can achieve the multielectron reduction of CO2 to CH4 suggests that nitrogenase can also stabilize key intermediates along this reaction pathway through appropriate functional groups. Metal hydrides have been suggested to be involved in the initial steps of CO2 reduction catalyzed by metal complexes (39), suggesting that nitrogenase might also achieve the two electron reduction of CO2 by hydride insertion, in a process parallel to the one proposed for the initial steps in N2 reduction. Whether partial reduction intermediates (e.g., formate or formaldehyde) are leaked from nitrogenase during CO2 reduction is technically challenging to determine because accurate measurement of formate or formaldehyde in solutions containing dithionite is complicated by interference from dithionite.
Another key finding reported here is the need to remodel the protein environment around FeMo cofactor to activate the reduction of CO2 to CH4. Earlier studies have illustrated that the protein environment immediately surrounding FeMo cofactor control both the size of compounds that can be substrates and the reactivity of FeMo cofactor toward those compounds (46). We earlier found that CO2 could be reduced to CO by the wild-type MoFe protein (27), but very little further reduction products are detected, suggesting that CO2 had limited access to the active part of FeMo cofactor in the wild-type enzyme and that the reaction cannot go forward beyond CO. The inability to go beyond CO could be due to steric constraints imposed by the active site on subsequent intermediates in the reaction pathway or the lack of functional groups to stabilize reaction intermediates. Such possibilities are supported by the requirement for amino acid substitutions to achieve CO2 reduction beyond CO all of the way to CH4 and the fact that the unsubstituted MoFe protein has an exceedingly poor capacity to reduce CO (24, 26).
A final notable observation from the current work is the capacity for reductive coupling of CO2 with acetylene to yield the 3C olefin propylene by the remodeled MoFe protein. Several earlier studies have suggested two binding sites on FeMo cofactor. For example, it has been proposed that two CO molecules bind to FeMo cofactor in the high CO concentration inhibited state (29–33). Likewise, two acetylene binding sites have been implicated from studies combining kinetics and amino acid substitutions (31, 34, 35). Finally, the finding that CO can be reduced and coupled to make C2 and C3 hydrocarbons is consistent with two adjacent binding sites (24, 26). Here, we report that CO2 can be reduced to the level of CH4 and coupled to acetylene, yielding predominately propylene. Up to 8% of the C3 product during CO2 and acetylene reduction catalyzed by nitrogenase is the C3 product propane. Addition of ethylene to a reaction with CO2 did not yield propane. This result clearly suggests that both CO2 and acetylene are binding to the active site and both are being activated within the active site during the coupling reaction. This observation is best explained by two adjacent substrate activation sites, which can be populated to varying extents by changing the electron flux through nitrogenase and the partial pressures of CO2 and acetylene. Reductive coupling of CO2 to alkynes yielding oxygenated hydrocarbons (e.g., carboxylic acids) has been reported for metal catalysts (10, 47) but, to our knowledge, the production of olefins is unique to the reactions reported here.
In summary, the findings presented here initiate the understanding of how nitrogenase can reduce and couple CO2 by multiple electrons to the industrially interesting CH4 and propylene. The findings presented here begin to shed light on these reactions and provide insights into the broader context of how N2 is reduced to ammonia. Future studies should look toward understanding how reaction barriers are lowered through stabilization of key reaction intermediates, which should provide guiding insights that can be used in the design of more robust catalysts for the reduction of CO2 to various hydrocarbons.
Materials and Methods
Reagents and Protein Purification.
All reagents were obtained from Sigma-Aldrich or Fisher Scientific and were used without further purification, unless specified otherwise. Sodium bicarbonate (NaHCO3) was from Avantor Performance Materials. Sodium dithionite was purified to approximately 99% purity according to a published procedure (48). Gases were purchased from Air Liquide: CO2 and acetylene. Methane gas was obtained from household natural gas line with an estimated purity of 97%. Propane gas was obtained from a propane fuel tank with an estimated purity of 86%. All other gases were purchased from Air Liquide. Azotobacter vinelandii strains DJ1260 (wild-type, WT, or α-70Val), DJ997 (α-195Gln), DJ1310 (α-70Ala), DJ1316 (α-70Ala/α-195Gln), and DJ1495 (α-70Ala/α-191Ala) were grown, and the corresponding nitrogenase MoFe proteins were expressed and purified as described (49). All MoFe proteins in this study contain a seven-His tag addition near the carboxyl-terminal end of the α-subunit. The purification of these proteins was accomplished according to a published purification protocol (50). Protein concentrations were determined by the Biuret assay using BSA as standard. The purities of these proteins were >95% based on SDS/PAGE analysis with Coomassie staining. Manipulation of proteins and buffers was done in septum-sealed serum vials under an argon atmosphere or on a Schlenk vacuum line. All gases and liquid transfers used gas-tight syringes.
Carbon Dioxide Reduction Assays.
Using CO2 as substrate, assays were conducted in 9.4-mL serum vials containing 2 mL of an assay buffer consisting of approximately 100 mM sodium dithionite, a MgATP regenerating system (13.4 mM MgCl2, 10 mM ATP, 60 mM phosphocreatine, 0.6 mg/mL BSA, and 0.4 mg/mL creatine phosphokinase) in a combination buffer of 33.3 mM Mops, 33.3 mM Mes, and 33.3 mM TAPS at pH 8.0 except for the CO2 partial pressure dependence study, which was done in 100 mM Bis-Tris buffer, pH 6.7. After making the solution anaerobic, addition of CO2 and equilibration between the gas phase and liquid phase for approximately 20 min, the MoFe protein were added. Then the assay vials were ventilated to atmospheric pressure. Reactions were initiated by the addition of Fe protein and incubated at 30 °C. Reactions were quenched by the addition of 400 µL of 400 mM EDTA at pH 8.0 solution. When using NaHCO3 as the substrate for reduction or coupling assays, the reaction mixture was made by mixing a 100 mM Bis-Tris buffer (pH 6.7) containing all components as described above and a stock solution of NaHCO3 dissolved in 100 mM Bis-Tris buffer (pH 6.7). For coupling reactions between CO2 and C2H2, the three-component buffer system (33.3 mM Mops, 33.3 mM Mes, and 33.3 mM TAPS at pH 8.0) was used. All pH values in this work were nominal values before mixing and/or equilibration with CO2 gas or NaHCO3 solution. Methane (CH4) and propylene (C3H6) were quantified by gas chromatography by injection of 500 µL of the gas phase of the reaction vial into a Shimadzu GC-8A equipped with a flame ionization detector fitted with a 30 cm × 0.3 cm Porapak N column with nitrogen as the carrier gas. The injection/detection temperature was set to 180 °C, and the column temperature was set to 110 °C. The standard curves with high linearity were created by using methane, and propane gases diluted with argon in 9.4-mL serum vials.
GC-MS Analysis.
The production of CH4 from CO2 reduction and C3H6 from the reductive coupling between CO2 and C2H2 was confirmed on a Shimadzu GC-2010 gas chromatograph equipped with a programmed temperature vaporization (PTV) injector and a Shimadzu GCMS-QP2010S mass spectrometer by using 12/13C-enriched NaHCO3 as CO2 source. Separation of methane was achieved with a GC-CARBONPLOT column [30 m, 0.32 mm inner diameter (ID), and 3.0-µm film thickness] (Agilent Technologies), and separation of propylene and propane was achieved with a Rt-Alumina BOND/KCl column (30 m, 0.32 mm ID, and 5.0 µm film thickness) (Restek). The injector and column temperatures were set to 35 °C. Ultrapure helium was used as the carrier gas set at a linear velocity of 50 cm/s for methane separation and 60 cm/s for propylene separation. For separation of methane and propylene, 25 µL and 500 µL of headspace gases were directly injected into the PTV injector, respectively. The mass spectrometer was operated in electron ionization and selected ion monitoring mode.
Supplementary Material
Acknowledgments
This work has been supported by National Institutes of Health Grant GM 59087 (to D.R.D. and L.C.S.). V.R.M was the recipient of Brazilian Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) Mobility Fellowship 0388/11-4.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213159109/-/DCSupplemental.
References
- 1.Mann ME. The Hockey Stick and the Climate Wars: Dispatches from the Front Lines. New York: Columbia Univ Press; 2012. [Google Scholar]
- 2.Olah GA, Prakash GKS, Goeppert A. Anthropogenic chemical carbon cycle for a sustainable future. J Am Chem Soc. 2011;133(33):12881–12898. doi: 10.1021/ja202642y. [DOI] [PubMed] [Google Scholar]
- 3.Lackner KS. Climate change. A guide to CO2 sequestration. Science. 2003;300(5626):1677–1678. doi: 10.1126/science.1079033. [DOI] [PubMed] [Google Scholar]
- 4.Szulczewski ML, MacMinn CW, Herzog HJ, Juanes R. Lifetime of carbon capture and storage as a climate-change mitigation technology. Proc Natl Acad Sci USA. 2012;109(14):5185–5189. doi: 10.1073/pnas.1115347109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Omae I. Recent developments in carbon dioxide utilization for the production of organic chemicals. Coord Chem Rev. 2012;256(13-14):1384–1405. [Google Scholar]
- 6.Rakowski DuBois M, DuBois DL. Development of molecular electrocatalysts for CO2 reduction and H2 production/oxidation. Acc Chem Res. 2009;42(12):1974–1982. doi: 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]
- 7.Morris AJ, Meyer GJ, Fujita E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc Chem Res. 2009;42(12):1983–1994. doi: 10.1021/ar9001679. [DOI] [PubMed] [Google Scholar]
- 8.Olah GA. Beyond oil and gas: The methanol economy. Angew Chem Int Ed Engl. 2005;44(18):2636–2639. doi: 10.1002/anie.200462121. [DOI] [PubMed] [Google Scholar]
- 9.Cokoja M, Bruckmeier C, Rieger B, Herrmann WA, Kühn FE. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew Chem Int Ed Engl. 2011;50(37):8510–8537. doi: 10.1002/anie.201102010. [DOI] [PubMed] [Google Scholar]
- 10.Schaub T, Paciello RA. A process for the synthesis of formic acid by CO2 hydrogenation: Thermodynamic aspects and the role of CO. Angew Chem Int Ed Engl. 2011;50(32):7278–7282. doi: 10.1002/anie.201101292. [DOI] [PubMed] [Google Scholar]
- 11.Sakakura T, Choi J-C, Yasuda H. Transformation of carbon dioxide. Chem Rev. 2007;107(6):2365–2387. doi: 10.1021/cr068357u. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Wang S, Ma X, Gong J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev. 2011;40(7):3703–3727. doi: 10.1039/c1cs15008a. [DOI] [PubMed] [Google Scholar]
- 13.Darensbourg DJ. Chemistry of carbon dioxide relevant to its utilization: A personal perspective. Inorg Chem. 2010;49(23):10765–10780. doi: 10.1021/ic101800d. [DOI] [PubMed] [Google Scholar]
- 14.Reda T, Plugge CM, Abram NJ, Hirst J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc Natl Acad Sci USA. 2008;105(31):10654–10658. doi: 10.1073/pnas.0801290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ragsdale SW, Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO(2) fixation. Biochim Biophys Acta. 2008;1784(12):1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6(8):579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 17.Heo J, Skjeldal L, Staples CR, Ludden PW. Carbon monoxide dehydrogenase from Rhodospirillum rubrum produces formate. J Biol Inorg Chem. 2002;7(7-8):810–814. doi: 10.1007/s00775-002-0365-z. [DOI] [PubMed] [Google Scholar]
- 18.Obert R, Dave BC. Enzymatic conversion of carbon dioxide to methanol: Enhanced methanol production in silica sol−gel matrices. J Am Chem Soc. 1999;121(51):12192–12193. [Google Scholar]
- 19.Burgess BK, Lowe DJ. Mechanism of molybdenum nitrogenase. Chem Rev. 1996;96(7):2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 20.Seefeldt LC, Hoffman BM, Dean DR. Mechanism of Mo-dependent nitrogenase. Annu Rev Biochem. 2009;78:701–722. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spatzal T, et al. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science. 2011;334(6058):940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lancaster KM, et al. X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science. 2011;334(6058):974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera-Ortiz JM, Burris RH. Interactions among substrates and inhibitors of nitrogenase. J Bacteriol. 1975;123(2):537–545. doi: 10.1128/jb.123.2.537-545.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CC, Hu Y, Ribbe MW. Vanadium nitrogenase reduces CO. Science. 2010;329(5992):642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Y, Lee CC, Ribbe MW. Extending the carbon chain: Hydrocarbon formation catalyzed by vanadium/molybdenum nitrogenases. Science. 2011;333(6043):753–755. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z-Y, Dean DR, Seefeldt LC. Molybdenum nitrogenase catalyzes the reduction and coupling of CO to form hydrocarbons. J Biol Chem. 2011;286(22):19417–19421. doi: 10.1074/jbc.M111.229344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seefeldt LC, Rasche ME, Ensign SA. Carbonyl sulfide and carbon dioxide as new substrates, and carbon disulfide as a new inhibitor, of nitrogenase. Biochemistry. 1995;34(16):5382–5389. doi: 10.1021/bi00016a009. [DOI] [PubMed] [Google Scholar]
- 28.Seefeldt LC, Hoffman BM, Dean DR. Electron transfer in nitrogenase catalysis. Curr Opin Chem Biol. 2012;16(1-2):19–25. doi: 10.1016/j.cbpa.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H-I, Cameron LM, Hales BJ, Hoffman BM. CO binding to the FeMo cofactor of CO-inhibited nitrogenase: 13CO and 1H Q-band ENDOR investigation. J Am Chem Soc. 1997;119(42):10121–10126. [Google Scholar]
- 30.Pollock RC, et al. Investigation of CO bound to inhibited forms of nitrogenase MoFe protein by 13C ENDOR. J Am Chem Soc. 1995;117(33):8686–8687. [Google Scholar]
- 31.Davis LC, Henzl MT, Burris RH, Orme-Johnson WH. Iron-sulfur clusters in the molybdenum-iron protein component of nitrogenase. Electron paramagnetic resonance of the carbon monoxide inhibited state. Biochemistry. 1979;18(22):4860–4869. doi: 10.1021/bi00589a014. [DOI] [PubMed] [Google Scholar]
- 32.Maskos Z, Fisher K, Sørlie M, Newton WE, Hales BJ. Variant MoFe proteins of Azotobacter vinelandii: Effects of carbon monoxide on electron paramagnetic resonance spectra generated during enzyme turnover. J Biol Inorg Chem. 2005;10(4):394–406. doi: 10.1007/s00775-005-0648-2. [DOI] [PubMed] [Google Scholar]
- 33.Christie PD, et al. Identification of the CO-binding cluster in nitrogenase MoFe protein by ENDOR of 57Fe isotopomers. J Am Chem Soc. 1996;118(36):8707–8709. [Google Scholar]
- 34.Shen J, Dean DR, Newton WE. Evidence for multiple substrate-reduction sites and distinct inhibitor-binding sites from an altered Azotobacter vinelandii nitrogenase MoFe protein. Biochemistry. 1997;36(16):4884–4894. doi: 10.1021/bi9628578. [DOI] [PubMed] [Google Scholar]
- 35.Hwang JC, Chen CH, Burris RH. Inhibition of nitrogenase-catalyzed reductions. Biochim Biophys Acta. 1973;292(1):256–270. doi: 10.1016/0005-2728(73)90270-3. [DOI] [PubMed] [Google Scholar]
- 36.Olah GA, Molnár Á. Hydrocarbon Chemistry. 2nd Ed. Hoboken, NJ: Wiley; 2003. [Google Scholar]
- 37.Matsuo T, Kawaguchi H. From carbon dioxide to methane: Homogeneous reduction of carbon dioxide with hydrosilanes catalyzed by zirconium-borane complexes. J Am Chem Soc. 2006;128(38):12362–12363. doi: 10.1021/ja0647250. [DOI] [PubMed] [Google Scholar]
- 38.Khandelwal M, Wehmschulte RJ. Deoxygenative reduction of carbon dioxide to methane, toluene, and diphenylmethane with [Et2Al]+ as catalyst. Angew Chem Int Ed Engl. 2012;51(29):7323–7326. doi: 10.1002/anie.201201282. [DOI] [PubMed] [Google Scholar]
- 39.Benson EE, Kubiak CP, Sathrum AJ, Smieja JM. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem Soc Rev. 2009;38(1):89–99. doi: 10.1039/b804323j. [DOI] [PubMed] [Google Scholar]
- 40.Cook TR, et al. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem Rev. 2010;110(11):6474–6502. doi: 10.1021/cr100246c. [DOI] [PubMed] [Google Scholar]
- 41.Ragsdale SW. Enzymology of the Wood-Ljungdahl pathway of acetogenesis. Ann N Y Acad Sci. 2008;1125:129–136. doi: 10.1196/annals.1419.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman BM, Dean DR, Seefeldt LC. Climbing nitrogenase: Toward a mechanism of enzymatic nitrogen fixation. Acc Chem Res. 2009;42(5):609–619. doi: 10.1021/ar8002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lukoyanov D, et al. Unification of reaction pathway and kinetic scheme for N2 reduction catalyzed by nitrogenase. Proc Natl Acad Sci USA. 2012;109(15):5583–5587. doi: 10.1073/pnas.1202197109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM. Is Mo involved in hydride binding by the four-electron reduced (E4) intermediate of the nitrogenase MoFe protein? J Am Chem Soc. 2010;132(8):2526–2527. doi: 10.1021/ja910613m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Igarashi RY, et al. Trapping H- bound to the nitrogenase FeMo-cofactor active site during H2 evolution: Characterization by ENDOR spectroscopy. J Am Chem Soc. 2005;127(17):6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- 46.Dos Santos PC, et al. Substrate interactions with the nitrogenase active site. Acc Chem Res. 2005;38(3):208–214. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Riduan SN. Catalytic hydrocarboxylation of alkenes and alkynes with CO2. Angew Chem Int Ed Engl. 2011;50(28):6210–6212. doi: 10.1002/anie.201101341. [DOI] [PubMed] [Google Scholar]
- 48.McKenna CE, Gutheil WG, Song W. A method for preparing analytically pure sodium dithionite. Dithionite quality and observed nitrogenase-specific activities. Biochim Biophys Acta. 1991;1075(1):109–117. doi: 10.1016/0304-4165(91)90082-r. [DOI] [PubMed] [Google Scholar]
- 49.Barney BM, et al. Diazene (HN=NH) is a substrate for nitrogenase: Insights into the pathway of N2 reduction. Biochemistry. 2007;46(23):6784–6794. doi: 10.1021/bi062294s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR. Catalytic and biophysical properties of a nitrogenase Apo-MoFe protein produced by a nifB-deletion mutant of Azotobacter vinelandii. Biochemistry. 1998;37(36):12611–12623. doi: 10.1021/bi981165b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.