

Abstract
Genetic studies in the last 5 years have greatly facilitated our understanding of how the dysregulation of diverse components of the innate immune system contributes to pathophysiology of SLE. A role for macrophages in the pathogenesis of SLE was first proposed as early as the 1980s following the discovery that SLE macrophages were defective in their ability to clear apoptotic cell debris, thus prolonging exposure of potential autoantigens to the adaptive immune response. More recently, there is an emerging appreciation of the contribution both monocytes and macrophages play in orchestrating immune responses with perturbations in their activation or regulation leading to immune dysregulation. This paper will focus on understanding the relevance of genes identified as being associated with innate immune function of monocytes and macrophages and development of SLE, particularly with respect to their role in (1) immune complex (IC) recognition and clearance, (2) nucleic acid recognition via toll-like receptors (TLRs) and downstream signalling, and (3) interferon signalling. Particular attention will be paid to the functional consequences these genetic associations have for disease susceptibility or pathogenesis.
1. Macrophages in Disease: SLE Candidate Genes and Functional Relevance
Systemic lupus erythematosus (SLE) is a multisystem chronic autoimmune disease, which affects approximately 0.1% of the population, with women being approximately nine times more likely to develop the disease than men [1]. SLE is a complex disease encompassing a broad spectrum of clinical symptoms, particular combinations of which can result in varying disease severity. To date the majority of work undertaken with respect to understanding the pathophysiology of this condition has focused on the autoreactive B and T lymphocytes [2]. However, recently attention has shifted to the role of the innate immune system and particularly myeloid cells in disease. Both monocytes and macrophages are phenotypically altered in SLE, with SLE macrophages demonstrated to have reduced uptake of apoptotic cells, enhanced activatory status, an altered skew of proinflammatory and anti-inflammatory macrophages and an overproduction of inflammatory cytokines such as tumour necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), interleukin-10 (IL-10), and antiviral type I interferons (IFNs) (Figure 1) [3–5]. As such, SLE monocytes and macrophages present self-antigens to autoreactive T cells in an inflammatory context, rather than the immuno-silent presentation normally associated with material from apoptotic cells [3]. In addition to this, the overproduction of type I IFNs by myeloid cells (including dendritic cells) also contributes to the breaking of immune tolerance due to their ability to stimulate antibody production and class switching from B cells [4]. The inadequate regulation of these processes in myeloid cells may be as a result of the influence of variants within SLE susceptibility genes.
Figure 1.
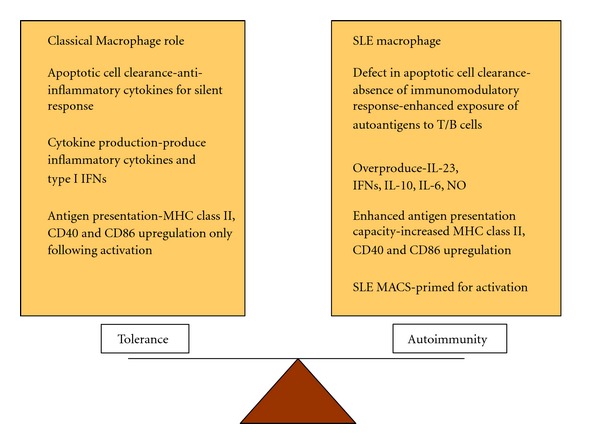
Dysregulation of macrophage function in SLE. The ability of the immune system to regulate macrophage function is altered in patients suffering from SLE. SLE macrophages have a defect in apoptotic cell clearance, overproduce IL-21, IFNs, IL-10, IL-6, and NO, have enhanced antigen presentation capacity and are primed for activation, leading to a skew towards autoimmunity.
Genetic analysis in human and murine studies indicate that susceptibility to SLE is heritable and that a number of different genetic loci are associated with disease risk [5]. Both candidate gene studies and Genome-wide association studies (GWAS) have unearthed many genes whose function can be clustered into 3 different categories, each clearly rooted in innate immune cell signalling and function (Table 1): (1) immune complex (IC) recognition and clearance such as the complement components and the Fc gamma receptors [6–8]; (2) nucleic acid recognition via toll-like receptors (TLRs) [9–11] and downstream signalling components such as TNF receptor-associated factor-6 (TRAF6) [12] and interferon regulatory factors (IRFs) [13] and (3) interferon signalling [14]. Characterisation of the molecular involvement of many of these genes in the function of SLE monocytes and macrophages has placed these cells as key orchestrators of SLE pathogenesis. Whilst the focus of this paper is the involvement of these candidate genes in macrophage function and their contribution to SLE pathology, it must be stressed that many of the candidate genes discussed below, particularly those that regulate type I IFN production, also play an important role in dendritic cell-driven autoimmune pathology [15, 16].
Table 1.
Polymorphisms of genes associated with SLE outlined in this review.
| Category | Gene | SNP | Ethnicity |
|---|---|---|---|
| Immune complex recognition |
C1q [8, 17, 18] |
||
| C1r [19] | |||
| C1s [20] | Rs292001 [21] |
Turkish [17], Mexican [18] |
|
| C2 [22] | |||
| C4 [8, 22–24] | |||
|
| |||
| Nucleic acid recognition |
rs3853839 [25] |
Chinese, Japanese [25] |
|
| rs179019 [26] | Japanese [26] | ||
| TLR7 [25] | rs179010 [26] | ||
| rs179008 [27] | Brazilian [27] |
||
| rs5743836 [27] | Brazilian [27] | ||
| TLR9 | rs352139 [28] | Japanese [28] | |
| rs352140 [29] | Chinese [29] | ||
| Asian [30], | |||
| IRF7 | rs1131665 [30] |
European American [30], | |
| African American [30] | |||
| rs5030437 [12] | |||
| TRAF6 | rs4755453 [12] | African American [12] | |
|
rs540386 [12] |
|||
| rs13192841 [31] | European [31] | ||
| rs2230926 [31, 32] | European [31], | ||
| TNFAIP3 | rs6922466 [31] | Chinese Han [33] | |
| rs5029939 [34] | European [31] | ||
| rs7708392 [35, 36] | Caucasian, Chinese [35, 36] | ||
| TNIP1 | rs10036748 [35, 36] | Japanese [26] | |
| Caucasian, Chinese [35, 36] | |||
|
| |||
| Interferon signalling |
rs7582694 [37] | Caucasian [37] | |
| STAT4 | rs7601754 [38] | Caucasian [38] | |
| rs7574865 [38] | Caucasian [38], | ||
| Northern Han Chinese [39] | |||
|
|
rs7582694 [40] | Caucasian [40] | |
| TYK2 | rs280519 [41] | UK, Swedish [41] | |
| rs2304256 [13] | Scandinavian [13] | ||
| IRF5 | rs12720270 [42] | UK [42] | |
| rs10488631 [43] | |||
2. Immune Complex Recognition and Uptake
2.1. The Complement System and Its Association with SLE
The principal function of activated components of the complement system include production of inflammatory and chemotactic proteins (C3a and C5a), cell lysis through the formation of the membrane attack complex (complex of C5b-9 proteins), and most importantly in the context of SLE, recognition and clearance of immune complexes and apoptotic cells (C1–C4) [6–8, 44]. Although genetic deficiencies in individual loci are rare, homozygous deficiency of each of the classical pathway components (C1q, C1r, C1s, C4, and C2) has been shown to be associated with SLE in humans [22]. A hierarchy of susceptibility and severity of disease is present where association is greatest with homozygous C1q deficiency followed by homozygous C4 and C2 deficiency [8]. Hereditary deficiencies of C1s and C1r are rarer than that of C1q and, in the majority of cases, deficiencies of both these components are inherited together [19, 20]. Both C1q and C4 are important in clearance of apoptotic cells and immune complexes, thereby preventing inappropriate activation of autoreactive B and T cells. Thus, reduced functioning of this important housekeeping function of complement proteins is strongly associated with increased risk of developing SLE [13–15, 19, 45].
C1q functions in facilitating clearance of immune complexes and apoptotic cells, thus protecting against autoimmunity. In addition, recent work has demonstrated that C1q can protect against SLE by preventing the production of type I IFN by dendritic cells [45, 46]. Individuals with a congenital deficiency of C1q gene (C1qD) develop SLE-like symptoms at more than 90% prevalence [47, 48]. Interestingly, the importance of ethnicity and the possible influence of haplotypes is highlighted by the observation that although C1q deficiency has been reported in Turkish [17] and Mexican [18] individuals affected by SLE, no association has been in Malaysian patients [49].
Homozygous deficiency of complement C4 is one of the strongest genetic risk factors for SLE and results in lupus-like disease in approximately 80% of the 28 known affected individuals [23, 24, 47]. To date, 28 individuals with complete C4 deficiency from 19 families have been reported, among these 15 individuals developed SLE, 7 developed lupus-like disease and four of the remaining subjects had kidney disease [24]. Through a five nucleotide substitution in exon 26, the C4 gene can encode either a C4A or a C4B protein [50], both of which have differential functions. C4A preferentially binds to amino groups in immune complexes and is the preferential ligand for complement receptor 1 (CR1) [51] whereas C4B is thought to be a more potent initiator of the complement activation cascade. The complement C4 gene located in the class III region of the major histocompatibility complex (MHC) on chromosome 6p21.3 and exhibits significant interindividual copy number variation (CNV). Boteva et al. demonstrated low C4A genome copy number significantly predisposed to SLE in UK and Spanish populations (P < 0.001) however, high C4A genome copy number was not associated with disease in either case (P = 0.63 and P = 0.76, resp.) [52]. Interestingly, C4B genome copy number demonstrated no association in the UK SLE group but was significantly associated with the Spanish cohort (P = 0.001). The discrepancies reported across different patient populations with respect to C4 copy number suggests that partial C4 deficiency states secondary to low C4A or C4B copy number are not independent genetic risk factors for susceptibility to disease [52, 53].
In addition to the rare inherited immunodeficiencies observed, many SLE patients have reduced levels of circulating C1q or C4 as a result of autoantibodies against these proteins, thus resulting in loss of their protective functions. Thus combined, mutations or decreased function of the early complement components has a profound effect on an individual's susceptibility to developing SLE.
2.2. Fc Gamma Receptors
Studies have investigated the contribution of the Fc-gamma family of receptors (FcγRs) to the pathogenesis of SLE given their role in the recognition of the Fc portion of IgG and subsequent responses to circulating and deposited immune complexes. Recent work in animal models indicates that the development of many human autoimmune diseases might be caused by impairment of the FcγR regulatory system (reviewed in [54]). FcγRs bind IgG, and can be further classified as activatory (FcγRI, IIA, IIIA, IIIB, and IV) or inhibitory (FcγRIIB) following IgG binding [55]. Additionally they can be subcategorised by relative affinity for IgG, with FcγRI having highest affinity, while FcγRII and III display lower affinity [56]. Currently there are no known polymorphisms in the FcγRI gene reported in humans and the rare individuals lacking this gene are healthy with no signs of autoimmune immune pathology [57]. However, polymorphisms in the activatory receptors FcγRIIA and FcγRIIIA have been identified [58–64].
FcγRII and III are encoded by two families of genes (FCGR2, FCGR3) clustered on chromosome 1q23-24, each containing multiple distinct genes [58]. FcγRIIA is a low-affinity receptor, comprised of multiple isoforms, which is expressed by B cells, monocytes, macrophages, and dendritic cells (DCs). It has two codominantly expressed alleles, R131 and H131, which differ in their affinity for IgG subclasses. Substitition of arginine to histidine at position 131 at the membrane proximal portion of the receptor results in enhanced affinity of FcγRIIA for binding of IgG2 and IgG3 by the H131 variant and increased levels of phagocytosis [59]. The allelic variant of FcγRIIA (R131) has been found to be strongly associated with lupus nephritis and renal failure in Brazilian lupus patients (P = 0.06) [60]. Interestingly meta analysis of European, African, and Asian populations demonstrated a significant association between the homozygous RR genotype and the development of SLE (P = 0.0016). This polymorphism was shown to increase the risk of devolving SLE 1.3-fold [61]. However analysis of this polymorphism in a Malaysian population found no significant association with disease [62].
FcγRIII encodes an activatory FcγR which is expressed on NK cells and monocytes, and has two isoforms: FcγRIIIA and FcγRIIIB. The wild-type sequence at position 176 encodes a phenylalanine (176-F) while the polymorphic variant is 176-valine (176-V) resulting in increased binding of IgG1 and IgG3 [63]. Recent studies in Japanese and Chinese patient cohorts found that positivity for the 176F allele was significantly increased in patients (P = 0.02 and P = 0.05 resp.), indicating a significant association of this allele with SLE [64, 65]. Additionally a significant association with this polymorphism and the development of lupus nephritis was observed among the Japanese patient cohort (P = 0.03) [64].
FcγRIIIB is an alternative membrane form of FcγRIII that is predominantly expressed on neutrophils and preferentially binds IgG1 and IgG3. The FcγRIIIB gene has three polymorphic forms known as HNA-1a, HNA-1b, and HNA-1c, encoded by the alleles FCGR3B*01, FCGR3B*02, and FCGR3B*03 (also referred to NA1, NA2, and SH) [66]. These different isoforms of FcγRIII exhibit differential function with increased levels of phagocytosis reported for FCGR3B*01 homozygotes compared to cells from FCGR3B*02 homozygotes, despite similar levels of receptor expression [67]. Reduced function of the FCGR3B*02 allele has been associated with impaired IC clearance in Caucasian populations [68] and has been strongly associated with disease susceptibility in Japanese and Thai populations (P = 0.008 and P = 0.02, resp.) [69, 70] and significantly associated with the development of lupus nephritis among the Japanese patient cohort (P = 0.007), whereas as no association was found in other population studies [62, 71].
As an inhibitory FcγR, loss of FcγRIIB not surprisingly results in development of lupus-like symptoms in mice, with the development of autoantibodies and autoimmune glomerulonephritis, consistent with a lack of inhibitory mechanisms on the development of autoreactive B cells [72]. Subsequent studies have demonstrated that increasing the expression of FcγRIIB in B cells derived from autoimmune-prone mice restored tolerance and prevented autoimmune disease [73]. With respect to the role of FcγRIIB in human autoimmune disease, reduced expression of FcγRIIb has been reported for memory B cells and plasma cells from SLE patients [74]. Interestingly, a polymorphism of FcγRIIb which changes the threonine at position 232 to an isoleucine (I232T) was found to be associated with SLE as positivity for the 232I allele was significantly decreased in SLE patients suggesting a significant association of the 232T/T genotype with SLE [64]. This study also found that the odds ratios (ORs) for the development of SLE among individuals with the T/T and I/T genotypes versus the I/I genotype were 2.3 and 1.1, respectively. A further comparison of genotype frequencies with patient clinical data revealed that FCGR2B polymorphismsstrongly associated with lupus nephritis (P = 0.01). This amino acid is in the transmembrane domain of FcγRIIb, and the polymorphism reduces the signalling capability of FcγRIIb due to its exclusion from lipid rafts [75]. Thus balanced signalling through activatory and inhibitory FcγRs regulates the activity of various cells in the immune system and genetic evidence in both mice and humans strongly supports the role of this receptor family in preventing the development of autoimmunity.
2.3. CD11b/ITGAM
ITGAM encodes integrin alpha-M (also commonly known as CD11b or complement receptor 3), the alpha chain of αMβ2 integrin which binds the cleavage fragment of complement component C3b, an opsonin and facilitates uptake of C3b-coated particles/pathogens into phagocytic cells (reviewed in [76]). Genetic association of ITGAM with SLE was found independently in 2 European GWAS [77, 78], with a non-synonomous functional variant being identified in a subsequent study [79]. Functionally this variant encodes an arginine to histine mutation at amino acid 77 which alters both the structure and function of integrin αM, thus reducing its ability to clear immune complexes [80].
3. Toll-Like Receptor Signalling and IFN Induction in SLE
Our increased awareness of the role played by cells of the innate immune system in disease has stemmed from the discovery of families of innate immune receptors, such as the TLRs, which have evolved to recognise and discriminate between different classes of pathogenesis reviewed in [81]. A link between antiviral pathogen recognition receptors and SLE is now well established, thus giving credence perhaps to the long-held view point that viral infection plays an important role in either the etiology of SLE or in driving flares in affected individuals [82]. With respect to SLE, receptors that can recognise viral nucleic acids, such as the endosomally located antiviral TLRs (TLR3, 7/8, and 9) [9–11] the intracellular RIG-I-like receptors (RLRs) [83] and AIM2-like receptors (ALRs) receptor families [84], have been implicated in SLE. There now exists strong genetic and functional evidence that RNA/DNA immune-complexes found in lupus patients can drive IFN-α production through the activation of TLR7 or TLR9 [85], respectively, indicating that TLR7/9 activation may be an important primary trigger for the generation of autoimmune disease (reviewed in [5, 86]). Plasmacytoid dendritic cells have been identified as the primary interferon-producing cells [87], however immature monocytes have also been demonstrated to produce significant levels of IFN-α in a mouse model of lupus and also in human SLE monocytes in response to immune complex activation [88, 89]. In addition to the viral TLRs themselves playing a role in the pathogenesis of this condition, downstream signalling components of these and their products may also contribute to the progression of this condition. Firstly, it is well documented that roughly half of all SLE patients overexpress IFN-α, thus giving rise to changes of gene expression that can be detected in peripheral blood monocytes, termed the IFN gene signature [90–93]. More recently, the activity and expression of certain members of the IRF family of transcription factors which regulate IFN production and mediate its effects, specifically IRF3 and IRF5, have been shown to be enhanced in SLE monocytes, resulting in increased expression of a subset of IRF-dependent genes [89, 94, 95]. For example, recent studies have shown that the levels of IRF3 bound to the promoter of a key pathogenic cytokine in SLE, IL-23, are enhanced in monocytes from SLE patients, thus resulting in increased basal production of this cytokine in SLE monocytes [95]. Likewise, monocytes from SLE patients present increased basal levels of nuclear IRF5 thus potentially contributing to enhanced production of the cytokines IFN-α, TNF-α, and IL-6 [96]. Thus not only are the triggers for activating SLE monocytes or macrophages in abundance due to impaired apoptotic cell clearance but also key downstream transcription factors such as the IRF family appear to be hyperresponsive in SLE monocytes [96], a finding inspired by genetic evidence linking IRF5 to disease [97].
3.1. Genetic Association of Antiviral Toll-Like Receptors TLR7 and TLR9 with SLE
With respect to the initial recognition of self-RNA and self-DNA by the antiviral TLRs, there have been several genetic studies in both human and murine models that further implicate these receptors in the pathogenesis of this condition in particular TLRs 7 and 9 [25–27, 88–96, 98–102].
3.1.1. Toll Like Receptor 7 (TLR7)
Mice lacking the TLR7 gene (located at Xp22.2) exhibit ameliorated disease, decreased lymphocyte activation and a marked reduction in the levels of RNA-containing antigens [98]. Interestingly, BBXSB/MpJ (BXSB) mice bearing the Yaa gene (Y chromosome-linked autoimmune acceleration gene) spontaneously develop a lupus-like autoimmunity, with males being affected much earlier and to a greater extent than their female counterparts. These Yaa containing mice were found to have increased expression of TLR7 due to the translocation of approximately 17 genes, including TLR7, onto the pseudoautosomal region of the Y chromosome [99, 100]. Deane et al. (2007) demonstrated that this duplication of the TLR7 gene and as a result, increased TLR7 expression, promoted the production of RNA-containing autoantibodies and development of lupus nephritis [101]. Although murine studies have indicated associations between TLR7 gene variations and SLE, there is controversy regarding human association studies. Using candidate gene approaches, Shen et al. (2010) investigated a role for TLR7 in SLE in Eastern Asian populations in which they identified a functional polymorphism in 3′ UTR of the TLR7 gene. This common variant (rs3853839G/C) was found to be robustly associated with SLE (P = 0.016), with a stronger effect seen in male subjects compared to their female counterparts [25]. The elevated levels of TLR7 transcripts and as a result, the enhanced IFN signature in patients with the G-allele of this single nucleotide polymorphism (SNP), have supported a functional role for this polymorphism in SLE. However when this SNP was studied in a non-Asian population, there was no evidence for this SNP as a risk factor for SLE in males with only females of non-Asian descent showing this association [102]. Following on from this multicentre study, Kawasaki et al. (2011), observed two additional variants located within the intron (rs179019A/C and rs179010T/C) that were also associated with SLE in a Japanese cohort (P = 0.016 and 0.018, resp.) thus further supporting the role of TLR7 as a risk factor for the development of this autoimmune condition [26]. Further studies into TLR7 polymorphisms in a Brazilian population also suggested the TLR7 SNP rs179008A/T as an SLE susceptibility factor in women of European descent (P = 0.020); however, this was not replicated in a Spanish population [27, 103]. Moreover an additional study into the role of copy number variants of TLR7 in SLE identified that increased TLR7 copy number was also a risk factor for the onset of juvenile SLE [104].
3.1.2. Toll-Like Receptor 9 (TLR9)
In addition to enhanced TLR7 expression, TLR9 has also been demonstrated to be upregulated in SLE B cells [105], further implicating a role for these viral TLRs in B cell tolerance and as result in the progression of SLE. In murine models, a role for TLR9 in disease susceptibility has also been examined with varying results. Christensen et al. (2005) demonstrated that TLR9 knockout mice crossed with lupus-prone mice exhibit decreased levels of anti-DNA antibodies implicating this gene as important in the progression of this condition [11]. However, in contrast the genetic ablation study carried out by Wu and Peng investigating the role of TLR9 in SLE, demonstrated that MRL mice lacking TLR9 developed more severe lupus than MRL controls, demonstrating a protective role for this gene in the pathogenesis of this condition [106]. Although numerous SNPs have been identified in the TLR9 locus (chromosome 3p21.3), which falls into the SLE susceptibility region, there is very little correlation between these variants and the onset of SLE and again this is an area of major controversy within the literature. A number of these common SNPs (rs187084, rs5743836, rs352139, and rs352140) were investigated in a Hong Kong Chinese population [107], however although overrepresented in SLE showed no significant association. When the rs5743836 SNP was further analysed in Caucasian American individuals, again no functional association was identified with this polymorphism [108]. In contrast, studies in Brazilian patients replicated these results reporting this SNP as an SLE susceptibility factor (P = 0.045) [27]. Consistent with results seen by Tao et al. investigating TLR9, the exonic region rs352139A/G SNP has been mildly associated with SLE (P = 0.040), with genetic analysis in a Japanese population indicating that carrying the G allele of this polymorphism predisposes individuals to an increased risk of SLE through the downregulation of TLR9 expression levels in reporter gene assays [28]. In addition to this, when the rs352140C/T in exon 2, was examined using a family-based association in China it was also reported that this SNP was also mildly associated with disease susceptibility (P = 0.045) [29].
The divergent roles played by TLR7 and 9 in autoimmunity are reflected in the variation seen in TLR7/9 polymorphisms and their subsequent effect on disease progression. TLR7 polymorphisms appear to increase expression of this gene leading to the enhanced recognition of autoantibodies culminating in an enhanced IFN signature thus predisposing these individuals to SLE [99]. On the other hand, TLR9 polymorphism associations, whilst controversial, particularly with respect to the different genetic backgrounds of the populations examined, suggest that TLR9 SNPs downregulate its expression and in doing so increase disease susceptibility [107]. Although the exact mechanism for this is not yet known, it has been suggested that lower levels of TLR9 expression lead to defective T regulatory cell activation which contributes to the decrease in number and immunosuppressive function of these cells in the MRL model of murine lupus [106]. Despite some controversy surrounding individual TLR7/9 SNPs in SLE, there is a growing body of evidence emerging to suggest that polymorphisms in these receptors play a role in genetic predisposition to this condition.
3.2. TLR Signalling Components
Activation of TLR7 and TLR9 by self nucleic acids and immune complexes has been demonstrated to contribute to the pathogenic production of IFN-α and proinflammatory cytokines such as TNF-α, IL-6 and IL-12 [109, 110]. A number of genes involved in type I IFN production and signalling have been linked to SLE [111–113]. Proteins directly activated downstream of TLR7 and TLR9, such as TRAF6 [12] and the IRF family of transcription factors [13], have known genetic association with SLE. In addition, proteins that negatively regulate TLR-induced activation of transcription factors IRF7 and NF-κB such as A20, have also been shown to contribute to lupus susceptibility in a combination of either GWAS or candidate gene approaches [31, 33, 114].
3.2.1. TRAF6
TNF-receptor-associated factor 6 (TRAF6) plays an important role in many signalling pathways that are important for immune regulation. TRAF6 was firstly identified in 1996 as a signal transducer in the NF-κB pathway which associates with interleukin-1 receptor-associated kinase (IRAK) [115]. Recent studies have suggested that polymorphisms within TRAF6 may be associated with the development of SLE, with SNPs in the TRAF6 gene giving nominal signals of association with SLE in an extended family Swedish cohort [116]. A more recent study showed a direct correlation between TRAF6 SNPs and SLE, supporting the notion that TRAF6 is potentially involved in the pathogenesis of autoimmune conditions [12]. In this study, fifteen SNPs across TRAF6 were evaluated in 7,490 SLE patients and 6,780 control subjects from different ancestries. Evidence of associations was detected in multiple SNPs, with rs5030437 and rs4755453 showing the strongest association [12].
3.2.2. TREX1
TREX1 encodes the most abundant 3′-5′ exonuclease in mammalian cells and has also been implicated in the cell death process, recognising and degrading genomic DNA and endogenous retroviral elements to minimize potential immune activation by persistent immunostimulatory DNA in the cytoplasm [117] (reviewed in [118]). Various genetic studies have identified a number of loss-of-function mutations in TREX1 that give rise to SLE, familial chilblain lupus (FCL), or Aicardi-Gautieres syndrome (AGS), an autoimmune disorder that presents as early onset encephalopathy resulting in severe intellectual and physical handicap [119–122]. Functional studies into these loss-of-function mutants of TREX1 demonstrate that they result in enhanced levels of immunostimulatory DNA resulting in enahnced type I IFN production. For example, TREX1D18N and TREX1D200N heterozygous mutants have been identified in FCL and AGS, respectively, and functionally are completely deficient at degrading dsDNA and demonstrate a lower rate of degradation of ssDNA than wild-type TREX1. The TREX1R114H homozygous mutation identified in AGS patients is found as a heterozygous mutation in SLE. As a homodimer TREX1R114H shows defects in its ability to degrade both ds- and ss-DNA, indicating that loss of function of TREX1 results in enhanced levels of immunostimulatory DNA which in turn results in enhanced levels of type I IFNs observed in both SLE and AGS [123, 124]. These findings clearly implicate TREX1 as an important endogenous DNA sensor that works to prevent inappropriate immune activation.
3.2.3. IRF5
Association of IRF5 genetic variants with SLE susceptibility has been first reported following a screening of genes involved in type I IFN production and response in Swedish, Icelandic, and Finnish patients with SLE [13]. Since then, the evidence of a link between IRF5 and SLE has been replicated in a number of case-control linkage studies in different populations [125–128] and GWAS analyses (reviewed in [129]). Association of IRF5 with SLE is complex, and a number of genetic studies have allowed defining risk, neutral, and protective haplotypes. Initially, 3 common polymorphisms in the IRF5 gene (SNPs rs2004640 in the 5′ UTR and rs10954213 in the 3′ UTR and a 30 nucleotides insertion in exon 6) [97] were proposed to alter the function or levels of IRF5 mRNA and proteins, thus explaining the association of risk alleles of these polymorphisms with SLE. A subsequent study by Sigurdsson et al. [130] identified two IRF5 polymorphisms independently and strongly associated with SLE: a 5 bp CGGGG insertion located 64 base pairs upstream of IRF5 exon 1a (P = 4.6 × 10−9) and a SNP (rs10488631) downstream of the IRF5 gene (P = 9.4 × 10−10). The presence of the insertion creates an additional binding site for the transcription factor Sp1, leading to increased transcription of IRF5 [130]. Interestingly, the CGGGG insertion is in linkage disequilibrium with SNPs rs2004640 and rs10954213, thus accounting for the association previously observed between these two SNPs and SLE. Interestingly, the CGGGG insertion in IRF5 promoter has been associated with a number of autoimmune conditions, such as primary Sjögren's syndrome [131], Multiple sclerosis [132], inflammatory bowel disease and Crohn's disease [133], while the haplotype tagged by rs10488631 seems to be specific in conferring SLE susceptibility [130]. A recent study by Hedl and Abraham [134] has found that monocyte-derived cells from healthy individuals carrying the risk alleles of SNP rs2004640 and the CGGGG insertion secreted elevated levels of proinflammatory cytokines following stimulation with Nod2 and TLRs ligands, thus suggesting a correlation between IRF5 genetic variants and transcriptional activity. In keeping with this, it has been shown that patients carrying IRF5-risk haplotypes have increased levels of circulating IFNα in the serum compared to patients carrying neutral or protective haplotypes. Of note, such correlation was observed only in patients positive for either anti-dsDNA or anti-RBP autoantibodies [135], and the study was sub sequentially expanded to show that different classes of autoantibodies are linked to different IRF5 haplotypes. Since autoantibodies can deliver self nucleic acids to endosomal TLRs [136], thus activating IRF5, the authors proposed that distinct classes of autoantibodies could activate specific IRF5 variants, leading to dysregulation of IFNα production and increased transcription of interferon-stimulated genes [137].
3.2.4. IRF7
IRF7 is considered the master regulator of IFNα production downstream the antiviral TLRs [138], and polymorphisms in this gene could therefore be an ideal candidate for genetic susceptibility to SLE. Together with IRF5, IRF7 has been shown to be necessary for murine DCs-mediated production of IL-6 and IFNα induced by immune complexes isolated from SLE patients' sera, again indicating a central role for these transcription factors in the disease context [85]. SNPs in the genetic region spanning the IRF7 gene (adjacent to the PHRF1 locus, also known as KIAA1542) have been identified, and different groups have attempted to associate common genetic variants at this site with SLE susceptibility. A GWAS in women affected by SLE has found a correlation between SNP rs4963128 in KIAA1542 and lupus (P = 3 × 10−10). Since this SNP is in strong linkage disequilibrium with SNP rs702966 located within 0.6 kb of IRF7, it was thought that variability at this site could represent the signal deriving from IRF7 [78]. Association of these two SNPs with lupus susceptibility has been replicated in populations of different ancestries by Salloum et al. [139]. Interestingly, this study demonstrated a correlation between the risk alleles of these SNPs and increased levels of IFNα, but only in patients with autoantibodies. Similar to what has been suggested for IRF5, potential autoantibodies might cooperate with SLE-associated IRF7 variants through TLR activation, resulting in increased type I IFN production which leads to breaking of tolerance and the onset of disease. In keeping with this, SNP rs4963128 was correlated with nephritis and anti-Ro/anti-La autoantibodies in a Chinese population, although no association of this SNP with lupus susceptibility was observed in this genetic background [39]. To date, the only known functional polymorphism in IRF7 is the nonsynonymous SNP rs1131665 which encodes a protein carrying a Q to R mutation at position 412 [30]. This variant has been shown to be associated with SLE patients of Asian and European American ancestry (P = 6.18 × 10−6), and functional analysis of the mutated protein revealed its enhanced transcriptional activation of an ISRE-dependent promoter. This is in keeping with the hypothesis that SLE-associated IRF7 polymorphisms may lead to the expression of proteins with increased activity downstream of the TLRs, thus leading to overproduction of type I IFN characteristic of the disease.
3.2.5. TNFAIP3
Tumour necrosis factor α-induced protein 3 (TNFAIP3), the gene product of which is the ubiquitin editing protein A20, is an essential negative regulator of pathways regulating NF-κB [140–142]. Recently TNFAIP3/A20 has been shown to interact with and negatively regulate IRF7, thus potentially explaining its molecular involvement in SLE [143]. Polymorphisms within the TNFAIP3 genomic locus, located at 6q23, have been associated with autoimmune disorders such as SLE [31, 35, 114, 144, 145] in Caucasian, Asian and Japanese populations. In particular, three independent SNPs in the TNFAIP3 gene (rs13192841, rs2230926 and rs6922466) are thought to be associated with SLE patients of European ancestry [31]. More recently the results of a meta-analysis of genome-wide association scans and replication in independent sets for TNFAIP3 polymorphism and SLE showed another TNFAIP3 SNP (rs2230926) to have an association with SLE [32]. This sample set contained 12,416 subjects with SLE from multiple ethnic groups and so suggested that this particular SNP may be conserved throughout diverse populations. In order to fully characterise the TNFAIP3 risk haplotype, fine mapping and genomic resequencing in ethnically diverse populations were carried out [146]. Results suggested a TT>A variant to be the most likely functional polymorphism responsible for the association between TNFAIP3 and SLE in subjects of both European and Korean ancestry [146]. This variant displayed a reduced binding avidity for NF-κB subunits. In addition, the haplotype carrying this variant resulted in reduced TNFAIP3 mRNA and A20 protein expression [146]. These findings underscore the crucial role of NF-κB regulation in the pathogenesis of SLE.
The TNFAIP3 interacting protein 1, (TNIP1), also known as ABIN1, interacts with TNFAIP3/A20 and promotes inhibition of NF-κB activity [147, 148]. TNIP1 has also been shown to be associated with SLE in a wide range of ethnic groups. Two individual GWAS revealed association of TNIP1 intronic SNPs, rs7708392, and rs10036748, with SLE in both Caucasian and Chinese populations [35, 36]. Subsequently a study was carried out in a Japanese population which confirmed the association of TNIP1 rs7708392 with SLE [148]. Interestingly in this study, this SNP showed a tendency of stronger association with SLE patients with renal disorder than in all SLE patients. Overall these studies highlight the important role that both TNFAIP3 and TNIP1 play in genetic predisposition to autoimmune disorders such as SLE.
3.3. Interferon Signalling Components
Serum levels of type I IFN correlate with disease activity and clinical manifestation [14], and interestingly lupus-like disorders can be induced during type I IFN therapy, again highlighting the pivotal role of these cytokines in disease development [149, 150]. Secreted type I IFN can then signal through the type I IFN receptor and kinases; tyrosine kinase 2 (TYK2) and janus kinase 1 (JAK1) [151]. Activation of the type I IFN receptor triggers phosphorylation of the transcription factors signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2) and assembly of the interferon stimulated gene factor 3 (ISGF3) complex, which then translocates to the nucleus where it regulates production of IFN-stimulated genes necessary to establish the antiviral state (Figure 2) [152]. Polymorphisms in genes such as TYK2 and STAT4, involved in signalling downstream of the type I IFN receptor and a number of other cytokines, have been identified that might instead alter responses to type I IFN in SLE [37, 38, 41, 42, 153–159].
Figure 2.
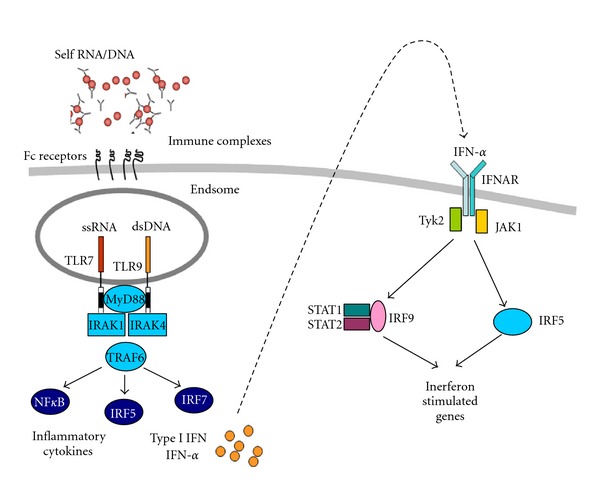
TLR induced IFN production and signalling in SLE. A brief outline of the signalling pathways involved in the production of type I IFNs in SLE. Activation of the transcription factors downstream of endosomal TLRs and Fc Receptors leads to the production of type I IFNs. These IFNs are secreted and further detected by IFN receptors, further activating interferon stimulated genes.
3.3.1. STAT4
STAT4, the signal transducer and activator of transcription 4 gene, encodes a transcription factor that mediates the effect of several cytokines, including IL-12, the type I interferons, and IL-23 in T cells and monocytes [153]. Thus, STAT4 has a role in T-cell differentiation, monocyte activation, and IFN-γ production. STAT4 was confirmed in 2003 by Jacob et al. to play a key role in the pathogenesis of a lupus-like disease in mice [154]. They showed that loss of STAT4 led to accelerated renal disease and increased mortality. A number of genetic studies have identified STAT4 SNPs with links to SLE in Caucasian populations for example, rs7582694 [37], rs7601754 and rs7574865 [38], and rs7582694 [155], in addition to rs7574865 and SLE in a Northern Han Chinese population [39]. Using transmission disequilibrium test analysis the rs7582694 SNP was found to have a strong association with SLE (P = 0.002, OR = 2.57) in a Finnish family cohort [37]. Using meta-analysis the SNPs rs7601754 and rs7574865 were found to have a significant association with SLE (P < 0.001) in populations of European and African origin [38]. Sigurdsson et al. (2008), in using a candidate gene study, also identified the SNP rs758294 as part of a common-risk haplotype for SLE (P = 1.7 × 10−5) in Swedish patients with SLE [155]. Li et al. (2011), using a candidate gene study in a Northern Han Chinese population, found a strong association between the SNP rs7574865 and SLE (P = 1.57 × 10−6) [39]. These SNPs are located within introns and are therefore suggested to play a role in the regulation of the expression level or splicing of the gene [155].
3.3.2. TYK2
TYK2 binds to the type I IFN receptor (IFNAR), thus initiating the JAK-STAT signalling cascade, culminating in the transcription of further type I IFN and IFN inducible genes [156]. A number of SNPs in TYK2 have been recently reported to be associated with SLE in Caucasian populations, namely, rs280519, rs2304256, and rs12720270 [13, 41, 42]. The TYK2 SNP rs280519 was found to be associated with SLE across a genome-wide association combined between a UK and Swedish cohort (P = 3.88 × 10−8) [41]. The TYK2 SNP rs2304256, was found to be associated with SLE in a Scandinavian cohort (P = 5.6 × 10−6) [13], but not associated with SLE in a UK cohort [42], however this same UK study also found another TYK2 SNP, rs12720270, associated with SLE that was not found within the Scandinavian cohort (P = 0.004). This SNP, rs12720270, however, was not found to be associated with SLE by Lee et al., when conducting meta-analysis on associations between SLE susceptibility and this SNP of TYK2 [157]. The rs2304256 is located in exon 8, and the rare A allele of the SNP causes a substitution of Val to Phe at residue 362 in the Jak-homology 4 (JH4) region of TYK2. This region is important for the interaction of TYK2 with IFNAR1, its function [158], as well as for maintaining the expression of IFNAR1 on the cell surface [159], suggesting that this SNP may reduce the function of TYK2 and thus susceptibility to autoimmune diseases.
4. Conclusion
Evidence from GWAS and candidate gene approaches have uncovered an array of genes that have functional consequences for how monocytes and macrophages respond to immune challenge during the course of disease. Many of these genes regulate either phagocytic, TLR, or IFN systems—three areas now well recognised to contribute to disease pathology. And as we become increasingly aware of the growing role of macrophages in disease pathology, it is interesting to note that cross-regulation of dendritic cells, the other major innate immune cell player in SLE pathology, by macrophages has an important role in driving disease. For example, C1q deficiency not only results in reduced uptake of immune complexes by macrophages and dendritic cells but it also is a negative regulator of IFN production by dendritic cells, thus its loss negatively impacts both macrophage and dendritic cell function in the context of disease pathology—exacerbating type I IFN production and contributing to a vicious cycle of reduced immune tolerance [160]. With respect to many of the genes discussed above, the functional relevance of their genetic variation has yet to be determined—do they contribute to pathogenic splice variants, altered transcript, or protein stability, or indeed introduce functional mutations that contribute to either over- or underactivation of the gene product? For others however, such as Trex1, not only is the molecular involvement of these variants in disease known, but research into their involvement in disease has uncovered novel functions for these proteins in innate immunity. However, where genetic associations uncovered have yet to conclusively demonstrate functional relevance for immune function in the context of SLE, we must be aware that many of the SNPs uncovered in SLE susceptibility regions may in fact have no true role in genetic susceptibility but instead, through linkage disequilibrium, act as a tag or marker for the real susceptibility gene. As researchers continue unravelling the functionality of genetic variability within SLE and translating these findings functionally to their contribution to immune dysregulation in SLE then we can undoubtedly expect this knowledge to contribute to greater insight into the molecular workings of disease. Already there are indications that certain SNPs appear to stratify with different disease manifestations and autoantibody profiles in SLE [161, 162], indicating the utility of screening to better inform and manage disease.
References
- 1.Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States—part I. Arthritis & Rheumatism. 2008;58(1):15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 2.Peng SL. Altered T and B lymphocyte signaling pathways in lupus. Autoimmunity Reviews. 2009;8(3):179–183. doi: 10.1016/j.autrev.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 3.Kavai M, Szegedi G. Immune complex clearance by monocytes and macrophages in systemic Lupus erythematosus. Autoimmunity Reviews. 2007;6(7):497–502. doi: 10.1016/j.autrev.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 4.Rönnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic Lupus erythematosus. Arthritis & Rheumatism. 2006;54(2):408–420. doi: 10.1002/art.21571. [DOI] [PubMed] [Google Scholar]
- 5.Sestak AL, Fürnrohr BG, Harley JB, Merrill JT, Namjou B. The genetics of systemic Lupus erythematosus and implications for targeted therapy. Annals of the Rheumatic Diseases. 2011;70(1, supplement):i37–i43. doi: 10.1136/ard.2010.138057. [DOI] [PubMed] [Google Scholar]
- 6.Davies KA, Schifferli JA, Walport MJ. Complement deficiency and immune complex disease. Springer Seminars in Immunopathology. 1994;15(4):397–416. doi: 10.1007/BF01837367. [DOI] [PubMed] [Google Scholar]
- 7.Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic Lupus erythematosus revisited. Journal of Immunology. 1997;158(10):4525–4528. [PubMed] [Google Scholar]
- 8.Pickering MC, Walport MJ. Links between complement abnormalities and systemic Lupus erythematosus. Rheumatology. 2000;39(2):133–141. doi: 10.1093/rheumatology/39.2.133. [DOI] [PubMed] [Google Scholar]
- 9.Patole PS, Gröne HJ, Segerer S, et al. Viral double-stranded RNA aggravates lupus nephritis through toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. Journal of the American Society of Nephrology. 2005;16(5):1326–1338. doi: 10.1681/ASN.2004100820. [DOI] [PubMed] [Google Scholar]
- 10.Lee PY, Kumagai Y, Li Y, et al. TLR7-dependent and FcγR-independent production of type I interferon in experimental mouse lupus. Journal of Experimental Medicine. 2008;205(13):2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. Journal of Experimental Medicine. 2005;202(2):321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Namjou B, Choi CB, Harley IT, et al. Evaluation of TRAF6 in a large multiancestral lupus cohort. Arthritis & Rheumatism. 2012;64(6):1960–1969. doi: 10.1002/art.34361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigurdsson S, Nordmark G, Göring HHH, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic Lupus erythematosus. American Journal of Human Genetics. 2005;76(3):528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dall’Era MC, Cardarelli PM, Preston BT, Witte A, Davis JC. Type I interferon correlates with serological and clinical manifestations of SLE. Annals of the Rheumatic Diseases. 2005;64(12):1692–1697. doi: 10.1136/ard.2004.033753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto M, Hirota K, Yoshitomi H, et al. Complement drives Th17 cell differentiation and triggers autoimmune arthritis. Journal of Experimental Medicine. 2010;207(6):1135–1143. doi: 10.1084/jem.20092301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Liu S, Yu Y, et al. Immune complex enhances tolerogenecity of immature dendritic cells via FcγRIIb and promotes FcγRIIb-overexpressing dendritic cells to attenuate lupus. European Journal of Immunology. 2011;41(4):1154–1164. doi: 10.1002/eji.201040767. [DOI] [PubMed] [Google Scholar]
- 17.Topaloglu R, Bakkaloglu A, Slingsby JH, et al. Molecular basis of hereditary C1q deficiency associated with SLE and IgA nephropathy in a Turkish family. Kidney International. 1996;50(2):635–642. doi: 10.1038/ki.1996.359. [DOI] [PubMed] [Google Scholar]
- 18.Petry F. Molecular basis of hereditary C1q deficiency. Immunobiology. 1998;199(2):286–294. doi: 10.1016/S0171-2985(98)80033-8. [DOI] [PubMed] [Google Scholar]
- 19.Loos M, Heinz HP. Component deficiencies—1. The first component: C1q, C1r, C1s. Progress in Allergy. 1986;39:212–231. [PubMed] [Google Scholar]
- 20.Chevailler A, Drouet C, Ponard D, et al. Non-coordinated biosynthesis of early complement components in a deficiency of complement proteins C1r and C1s. Scandinavian Journal of Immunology. 1994;40(4):383–388. doi: 10.1111/j.1365-3083.1994.tb03478.x. [DOI] [PubMed] [Google Scholar]
- 21.Zervou MI, Vazgiourakis VM, Yilmaz N, et al. TRAF1/C5, eNOS, C1q, but not STAT4 and PTPN22 gene polymorphisms are associated with genetic susceptibility to systemic lupus erythematosus in Turkey. Human Immunology. 2011;72(12):1210–1213. doi: 10.1016/j.humimm.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Morgan BP, Walport MJ. Complement deficiency and disease. Immunology Today. 1991;12(9):301–306. doi: 10.1016/0167-5699(91)90003-C. [DOI] [PubMed] [Google Scholar]
- 23.Wu YL, Hauptmann G, Viguier M, Yu CY. Molecular basis of complete complement C4 deficiency in two North-African families with systemic Lupus erythematosus. Genes and Immunity. 2009;10(5):433–445. doi: 10.1038/gene.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Chung EK, Zhou B, et al. The intricate role of complement component C4 in human systemic Lupus erythematosus. Current Directions in Autoimmunity. 2004;7:98–132. doi: 10.1159/000075689. [DOI] [PubMed] [Google Scholar]
- 25.Shen N, Fu Q, Deng Y, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic Lupus erythematosus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15838–15843. doi: 10.1073/pnas.1001337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawasaki A, Furukawa H, Kondo Y, et al. TLR7 single-nucleotide polymorphisms in the 3’ untranslated region and intron 2 independently contribute to systemic Lupus erythematosus in Japanese women: a case-control association study. Arthritis Research and Therapy. 2011;13(2, article R41) doi: 10.1186/ar3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.dos Santos BP, Valverde JV, Rohr P, et al. TLR7/8/9 polymorphisms and their associations in systemic Lupus erythematosus patients from southern Brazil. Lupus. 2012;21(3):302–309. doi: 10.1177/0961203311425522. [DOI] [PubMed] [Google Scholar]
- 28.Tao K, Fujii M, Tsukumo SI, et al. Genetic variations of Toll-like receptor 9 predispose to systemic Lupus erythematosus in Japanese population. Annals of the Rheumatic Diseases. 2007;66(7):905–909. doi: 10.1136/ard.2006.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu CJ, Zhang WH, Pan HF, Li XP, Xu JH, Ye DQ. Association study of a single nucleotide polymorphism in the exon 2 region of toll-like receptor 9 (TLR9) gene with susceptibility to systemic Lupus erythematosus among Chinese. Molecular Biology Reports. 2009;36(8):2245–2248. doi: 10.1007/s11033-008-9440-z. [DOI] [PubMed] [Google Scholar]
- 30.Fu Q, Zhao J, Qian X, et al. Association of a functional IRF7 variant with systemic Lupus erythematosus. Arthritis & Rheumatism. 2011;63(3):749–754. doi: 10.1002/art.30193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic Lupus erythematosus. Nature Genetics. 2008;40(9):1062–1064. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan Y, Tao JH, Zhang LP, Li LH, Ye DQ. The association between BANK1 and TNFAIP3 gene polymorphisms and systemic Lupus erythematosus: a meta-analysis. International Journal of Immunogenetics. 2011;38(2):151–159. doi: 10.1111/j.1744-313X.2010.00990.x. [DOI] [PubMed] [Google Scholar]
- 33.Cai LQ, Wang ZX, Lu WS, et al. A single-nucleotide polymorphism of the TNFAIP3 gene is associated with systemic Lupus erythematosus in Chinese Han population. Molecular Biology Reports. 2010;37(1):389–394. doi: 10.1007/s11033-009-9818-6. [DOI] [PubMed] [Google Scholar]
- 34.Bates JS, Lessard CJ, Leon JM, et al. Meta-analysis and imputation identifies a 109kb risk haplotype spanning TNFAIP3 associated with lupus nephritis and hematologic manifestations. Genes and Immunity. 2009;10(5):470–477. doi: 10.1038/gene.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han JW, Zheng HF, Cui Y, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic Lupus erythematosus. Nature Genetics. 2009;41(11):1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 36.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic Lupus erythematosus. Nature Genetics. 2009;41(11):1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hellquist A, Sandling JK, Zucchelli M, et al. Variation in STAT4 is associated with systemic Lupus erythematosus in a Finnish family cohort. Annals of the Rheumatic Diseases. 2010;69(5):883–886. doi: 10.1136/ard.2009.112284. [DOI] [PubMed] [Google Scholar]
- 38.Yuan H, Jin-Bao F, Hai-Feng P, et al. A meta-analysis of the association of STAT4 polymorphism with systemic Lupus erythematosus. Modern Rheumatology. 2010;20(3):257–262. doi: 10.1007/s10165-010-0275-9. [DOI] [PubMed] [Google Scholar]
- 39.Li P, Cao C, Luan H, et al. Association of genetic variations in the STAT4 and IRF7/KIAA1542 regions with systemic Lupus erythematosus in a Northern Han Chinese population. Human Immunology. 2011;72(3):249–255. doi: 10.1016/j.humimm.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 40.Sigurdsson S, Nordmark G, Garnier S, et al. A risk haplotype of STAT4 for systemic Lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Human Molecular Genetics. 2008;17(18):2868–2876. doi: 10.1093/hmg/ddn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunninghame Graham DS, Morris DL, Bhangale TR, et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic Lupus erythematosus. PLoS Genet. 2011;7(10) doi: 10.1371/journal.pgen.1002341.e1002341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham DSC, Akil M, Vyse TJ. Association of polymorphisms across the tyrosine kinase gene, TYK2 in UK SLE families. Rheumatology. 2007;46(6):927–930. doi: 10.1093/rheumatology/kel449. [DOI] [PubMed] [Google Scholar]
- 43.Ferreiro-Neira I, Calaza M, Alonso-Perez E, et al. Opposed independent effects and epistasis in the complex association of IRF5 to SLE. Genes and Immunity. 2007;8(5):429–438. doi: 10.1038/sj.gene.6364407. [DOI] [PubMed] [Google Scholar]
- 44.Taylor PR, Carugati A, Fadok VA, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. Journal of Experimental Medicine. 2000;192(3):359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santer DM, Wiedeman AE, Teal TH, Ghosh P, Elkon KB. Plasmacytoid dendritic cells and C1q differentially regulate inflammatory gene induction by lupus immune complexes. Journal of Immunology. 2012;188(2):902–915. doi: 10.4049/jimmunol.1102797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santer DM, Hall BE, George TC, et al. C1q deficiency leads to the defective suppression of IFN-α in response to nucleoprotein containing immune complexes. Journal of Immunology. 2010;185(8):4738–4749. doi: 10.4049/jimmunol.1001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic Lupus erythematosus, complement deficiency, and apoptosis. Advances in Immunology. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- 48.Walport MJ, Davies KA, Botto M. C1q and systemic Lupus erythematosus. Immunobiology. 1998;199(2):265–285. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]
- 49.Chew CH, Chua KH, Lian LH, Puah SM, Tan SY. PCR-RFLP genotyping of C1q mutations and single nucleotide polymorphisms in Malaysian patients with systemic Lupus erythematosus. Human Biology. 2008;80(1):83–93. doi: 10.3378/1534-6617(2008)80[83:PGOCMA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 50.Yung Yu C, Chung EK, Yang Y, et al. Dancing with Complement C4 and the RP-C4-CYP21-TNX (RCCX) Modules of the Major Histocompatibility Complex. Progress in Nucleic Acid Research and Molecular Biology. 2003;75:217–292. doi: 10.1016/s0079-6603(03)75007-7. [DOI] [PubMed] [Google Scholar]
- 51.Schifferli JA, Hauptmann G, Paccaud JP. Complement-mediated adherence of immune complexes to human erythrocytes. Difference in the requirements for C4A and C4B. FEBS Letters. 1987;213(2):415–418. doi: 10.1016/0014-5793(87)81533-8. [DOI] [PubMed] [Google Scholar]
- 52.Boteva L, Morris DL, Cortés-Hernández J, Martin J, Vyse TJ, Fernando MM. Genetically determined partial complement C4 deficiency states are not independent risk factors for SLE in UK and Spanish populations. The American Journal of Human Genetics. 2012;90(3):445–456. doi: 10.1016/j.ajhg.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y, Chung EK, Yee LW, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic Lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. American Journal of Human Genetics. 2007;80(6):1037–1054. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2(8):580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- 55.Nimmerjahn F. Activating and inhibitory FcγRs in autoimmune disorders. Springer Seminars in Immunopathology. 2006;28(4):305–319. doi: 10.1007/s00281-006-0052-1. [DOI] [PubMed] [Google Scholar]
- 56.Sondermann P, Kaiser J, Jacob U. Molecular basis for immune complex recognition: a comparison of Fc-receptor structures. Journal of Molecular Biology. 2001;309(3):737–749. doi: 10.1006/jmbi.2001.4670. [DOI] [PubMed] [Google Scholar]
- 57.Ceuppens JL, Baroja ML, Van Vaeck F, Anderson CL. Defect in the membrane expression of high affinity 72-kD Fc γ receptors on phagocytic cells in four healthy subjects. Journal of Clinical Investigation. 1988;82(2):571–578. doi: 10.1172/JCI113634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dijstelbloem HM, Kallenberg CGM, Van De Winkel JGJ. Inflammation in autoimmunity: receptors for IgG revisited. Trends in Immunology. 2001;22(9):510–516. doi: 10.1016/s1471-4906(01)02014-2. [DOI] [PubMed] [Google Scholar]
- 59.Warmerdam PAM, Van de Winkel JGJ, Vlug A, Westerdaal NAC, Capel PJA. A single amino acid in the second Ig-like domain of the human Fcγ receptor II is critical for human IgG2 binding. Journal of Immunology. 1991;147(4):1338–1343. [PubMed] [Google Scholar]
- 60.Bazilio AP, Viana VST, Toledo R, Woronik V, Bonfá E, Monteiro RC. FcγRIIa polymorphismml: a susceptibility factor for immune complex-mediated lupus nephritis in Brazilian patients. Nephrology Dialysis Transplantation. 2004;19(6):1427–1431. doi: 10.1093/ndt/gfh121. [DOI] [PubMed] [Google Scholar]
- 61.Karassa FB, Trikalinos TA, Ioannidis JPA. Role of the Fcγ receptor IIa polymorphism in susceptibility to systemic Lupus erythematosus and lupus nephritis: a meta-analysis. Arthritis & Rheumatism. 2002;46(6):1563–1571. doi: 10.1002/art.10306. [DOI] [PubMed] [Google Scholar]
- 62.Yap SN, Phipps ME, Manivasagar M, Tan SY, Bosco JJ. Human Fc gamma receptor IIA (FcγRIIA) genotyping and association with systemic Lupus erythematosus (SLE) in Chinese and Malays in Malaysia. Lupus. 1999;8(4):305–310. doi: 10.1191/096120399678847876. [DOI] [PubMed] [Google Scholar]
- 63.Karassa FB, Trikalinos TA, Ioannidis JPA, et al. The FcγRIIIA-F158 allele is a risk factor for the development of lupus nephritis: a meta-analysis. Kidney International. 2003;63(4):1475–1482. doi: 10.1046/j.1523-1755.2003.00873.x. [DOI] [PubMed] [Google Scholar]
- 64.Kyogoku C, Dijstelbloem HM, Tsuchiya N, et al. Fcγ receptor gene polymorphisms in Japanese patients with systemic Lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis & Rheumatism. 2002;46(5):1242–1254. doi: 10.1002/art.10257. [DOI] [PubMed] [Google Scholar]
- 65.Chu ZT, Tsuchiya N, Kyogoku C, et al. Association of Fcγ receptor IIb polymorphism with susceptibility to systemic Lupus erythematosus in Chinese: a common susceptibility gene in the Asian populations. Tissue Antigens. 2004;63(1):21–27. doi: 10.1111/j.1399-0039.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 66.Koene HR, Kleijer M, Roos D, De Haas M, Von Dem Borne AEGK. FcγRIIIB gene duplication: evidence for presence and expression of three distinct FcγRIIIB genes in NA(1+,2+)SH(+) individuals. Blood. 1998;91(2):673–679. [PubMed] [Google Scholar]
- 67.Bredius RGM, Fijen CAP, De Haas M, et al. Role of neutrophil FcγRIIa (CD32) and FcγRIIIb (CD16) polymorphic forms in phagocytosis of human IgG1- and IgG3-opsonized bacteria and erythrocytes. Immunology. 1994;83(4):624–630. [PMC free article] [PubMed] [Google Scholar]
- 68.Dijstelbloem HM, Bijl M, Fijnheer R, et al. Fcgamma receptor polymorphisms in systemic Lupus erythematosus: association with disease and in vivo clearance of immune complexes. Arthritis & Rheumatism. 2000;43(12):2793–2800. doi: 10.1002/1529-0131(200012)43:12<2793::AID-ANR20>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 69.Hatta Y, Tsuchiya N, Ohashi J, et al. Association of Fcγ receptor IIIB, but not of Fcγ receptor IIA and IIIA, polymorphisms with systemic Lupus erythematosus in Japanese. Genes and Immunity. 1999;1(1):53–60. doi: 10.1038/sj.gene.6363639. [DOI] [PubMed] [Google Scholar]
- 70.Siriboonrit U, Tsuchiya N, Sirikong M, et al. Association of Fcγ receptor IIb and IIIb polymorphisms with susceptibility to systemic Lupus erythematosus in Thais. Tissue Antigens. 2003;61(5):374–383. doi: 10.1034/j.1399-0039.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 71.Hong CH. The association between fcgammaRIIIB polymorphisms and systemic Lupus erythematosus in Korea. Lupus. 2005;14(5):346–350. doi: 10.1191/0961203305lu2086oa. [DOI] [PubMed] [Google Scholar]
- 72.Bolland S, Ravetch JV. Spontaneous autoimmune disease in FcγRIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13(2):277–285. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 73.McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. 2005;307(5709):590–593. doi: 10.1126/science.1105160. [DOI] [PubMed] [Google Scholar]
- 74.Mackay M, Stanevsky A, Wang T. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. Journal of Experimental Medicine. 2006;203(9):2157–2164. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Floto RA. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nature Medicine. 2005;11(10):1056–1058. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 76.Bernet J, Mullick J, Singh AK, Sahu A. Viral mimicry of the complement system. Journal of Biosciences. 2003;28(3):249–264. doi: 10.1007/BF02970145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hom G, Graham RR, Modrek B, et al. Association of systemic Lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. New England Journal of Medicine. 2008;358(9):900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 78.Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic Lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature Genetics. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nath SK, Han S, Kim-Howard X, et al. A nonsynonymous functional variant in integrin-αM (encoded by ITGAM) is associated with systemic Lupus erythematosus. Nature Genetics. 2008;40(2):152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 80.Rhodes B, et al. The rs1143679 (R77H) lupus associated variant of ITGAM (CD11b) impairs complement receptor 3 mediated functions in human monocytes. doi: 10.1136/annrheumdis-2012-201390. Annals of the Rheumatic Diseases, 2012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nature Immunology. 2010;11(5):373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 82.Kono DH, Haraldsson MK, Lawson BR, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(29):12061–12066. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: an interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(2):637–642. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kimkong I, Avihingsanon Y, Hirankarn N. Expression profile of HIN200 in leukocytes and renal biopsy of SLE patients by real-time RT-PCR. Lupus. 2009;18(12):1066–1072. doi: 10.1177/0961203309106699. [DOI] [PubMed] [Google Scholar]
- 85.Yasuda K, Richez C, Maciaszek JW, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes Is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. Journal of Immunology. 2007;178(11):6876–6885. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 86.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunological Reviews. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 87.Lövgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Rönnblom L. Induction of interferon-α by immune complexes or liposomes containing systemic Lupus erythematosus autoantigen-and Sjögren’s syndrome auto antigen-associated RNA. Arthritis & Rheumatism. 2006;54(6):1917–1927. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 88.Lee PY, Weinstein JS, Nacionales DC, et al. A novel type i IFN-producing cell subset in murine lupus. Journal of Immunology. 2008;180(7):5101–5108. doi: 10.4049/jimmunol.180.7.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feng D, Stone RC, Eloranta ML, et al. Genetic variants and disease-associated factors contribute to enhanced interferon regulatory factor 5 expression in blood cells of patients with systemic Lupus erythematosus. Arthritis & Rheumatism. 2010;62(2):562–573. doi: 10.1002/art.27223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Obermoser G. Lupus erythematosus and the skin: a journey at times perplexing, usually complex, often challenging, and evermore exhilarating. Lupus. 2010;19(9):1009–1011. doi: 10.1177/0961203310373108. [DOI] [PubMed] [Google Scholar]
- 91.Rönnblom L, Alm GV, Eloranta ML. The type I interferon system in the development of lupus. Seminars in Immunology. 2011;23(2):113–121. doi: 10.1016/j.smim.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 92.Crow MK. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Research & Therapy. 2010;12:p. S5. doi: 10.1186/ar2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kozyrev SV, Alarcon-Riquelme ME. The genetics and biology of Irf5-mediated signaling in lupus. Autoimmunity. 2007;40(8):591–601. doi: 10.1080/08916930701510905. [DOI] [PubMed] [Google Scholar]
- 95.Smith S, Gabhann JN, Higgs R, et al. Enhanced interferon regulatory factor 3 binding to the interleukin-23p19 promoter correlates with enhanced interleukin-23 expression in systemic Lupus erythematosus. Arthritis & Rheumatism. 2012;64(5):1601–1609. doi: 10.1002/art.33494. [DOI] [PubMed] [Google Scholar]
- 96.Stone RC. Interferon regulatory factor 5 activation in monocytes of systemic Lupus erythematosus patients is triggered by circulating autoantigens independent of type I interferons. Arthritis & Rheumatism. 2012;64(3):788–798. doi: 10.1002/art.33395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(16):6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell R, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of Lupus. Immunity. 2006;25(3):417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 99.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312(5780):1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 100.Subramanian S, Tus K, Li QZ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(26):9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deane JA, Pisitkun P, Barrett RS, et al. Control of Toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27(5):801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deng Y, Zhao J, Tan W, et al. Association of variants in the TLR7-TLR8 region with systemic Lupus erythematosus in non-asian populations. Arthritis & Rheumatism. 2010;62, supplement 10:p. 1584. [Google Scholar]
- 103.Sánchez E, Callejas-Rubio JL, Sabio JM, et al. Investigation of TLR5 and TLR7 as candidate genes for susceptibility to systemic Lupus erythematosus. Clinical and Experimental Rheumatology. 2009;27(2):267–271. [PubMed] [Google Scholar]
- 104.García-Ortiz H, Velázquez-Cruz R, Espinosa-Rosales F, Jiménez-Morales S, Baca V, Orozco L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic Lupus erythematosus in Mexican population. Annals of the Rheumatic Diseases. 2010;69(10):1861–1865. doi: 10.1136/ard.2009.124313. [DOI] [PubMed] [Google Scholar]
- 105.Papadimitraki ED, Choulaki C, Koutala E, et al. Expansion of toll-like receptor 9-expressing B cells in active systemic Lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis & Rheumatism. 2006;54(11):3601–3611. doi: 10.1002/art.22197. [DOI] [PubMed] [Google Scholar]
- 106.Wu X, Peng SL. Toll-like receptor 9 signaling protects against murine lupus. Arthritis & Rheumatism. 2006;54(1):336–342. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- 107.Ng MW, Lau CS, Chan TM, Wong WHS, Lau YL. Polymorphisms of the toll-like receptor 9 (TLR9) gene with systemic Lupus erythematosus in Chinese [1] Rheumatology. 2005;44(11):1456–1457. doi: 10.1093/rheumatology/kei120. [DOI] [PubMed] [Google Scholar]
- 108.Demirci FYK, Manzi S, Ramsey-Goldman R, et al. Association study of Toll-like Receptor 5 (TLR5) and Toll-like Receptor 9 (TLR9) polymorphisms in systemic Lupus erythematosus. Journal of Rheumatology. 2007;34(8):1708–1711. [PubMed] [Google Scholar]
- 109.Ganguly D, Chamilos G, Lande R, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. Journal of Experimental Medicine. 2009;206(9):1983–1994. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lamphier MS, Sirois CM, Verma A, Golenbock DT, Latz E. TLR9 and the recognition of self and non-self nucleic acids. Annals of the New York Academy of Sciences. 2006;1082:31–43. doi: 10.1196/annals.1348.005. [DOI] [PubMed] [Google Scholar]
- 111.Gaffney PM, Kearns GM, Shark KB, et al. A genome-wide search for susceptibility genes in human systemic Lupus erythematosus sib-pair families. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(25):14875–14879. doi: 10.1073/pnas.95.25.14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shai R, Quismorio FP, Li L, et al. Genome-wide screen for systemic Lupus erythematosus susceptibility genes in multiplex families. Human Molecular Genetics. 1999;8(4):639–644. doi: 10.1093/hmg/8.4.639. [DOI] [PubMed] [Google Scholar]
- 113.Kyogoku C, Tsuchiya N. A compass that points to lupus: genetic studies on type I interferon pathway. Genes and Immunity. 2007;8(6):445–455. doi: 10.1038/sj.gene.6364409. [DOI] [PubMed] [Google Scholar]
- 114.Kawasaki A, Ito I, Ito S, et al. Association of TNFAIP3 polymorphism with susceptibility to systemic Lupus erythematosus in a Japanese population. Journal of Biomedicine and Biotechnology. 2010;2010 doi: 10.1155/2010/207578.207578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383(6599):443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 116.Sandling JK, Garnier S, Sigurdsson S, et al. A candidate gene study of the type i interferon pathway implicates IKBKE and IL8 as risk loci for SLE. European Journal of Human Genetics. 2011;19(4):479–484. doi: 10.1038/ejhg.2010.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134(4):587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Human Molecular Genetics. 2009;18(2):R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Aicardi J, Goutieres F. Systemic Lupus erythematosus or Aicardi-Goutieres syndrome? Neuropediatrics. 2000;31(3):p. 113. doi: 10.1055/s-2000-7533. [DOI] [PubMed] [Google Scholar]
- 120.Rice G, Newman WG, Dean J, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutières syndrome. American Journal of Human Genetics. 2007;80(4):811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee-Kirsch MA, Gong M, Chowdhury D, et al. Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 are associated with systemic Lupus erythematosus. Nature Genetics. 2007;39(9):1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 122.Crow YJ, Hayward BE, Parmar R, et al. Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nature Genetics. 2006;38(8):917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 123.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. Journal of Biological Chemistry. 2008;283(46):31649–31656. doi: 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fye JM, Orebaugh CD, Coffin SR, et al. Dominant mutation of the TREX1 exonuclease gene in lupus and Aicardi-Goutieres syndrome. Journal of Biological Chemistry. 2011;286(37):32373–32382. doi: 10.1074/jbc.M111.276287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Demirci FYK, Manzi S, Ramsey-Goldman R, et al. Association of a Common Interferon Regulatory Factor 5 (IRF5) variant with increased risk of Systemic Lupus erythematosus (SLE) Annals of Human Genetics. 2007;71(3):308–311. doi: 10.1111/j.1469-1809.2006.00336.x. [DOI] [PubMed] [Google Scholar]
- 126.Graham DSC, Manku H, Wagner S, et al. Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Human Molecular Genetics. 2007;16(6):579–591. doi: 10.1093/hmg/ddl469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kawasaki A, Kyogoku C, Ohashi J, et al. Association of IRF5 polymorphisms with systemic Lupus erythematosus in a Japanese population: support for a crucial role of intron 1 polymorphisms. Arthritis & Rheumatism. 2008;58(3):826–834. doi: 10.1002/art.23216. [DOI] [PubMed] [Google Scholar]
- 128.Siu HO, Yang W, Lau CS, et al. Association of a haplotype of IRF5 gene with systemic Lupus erythematosus in Chinese. Journal of Rheumatology. 2008;35(2):360–362. [PubMed] [Google Scholar]
- 129.Graham RR, Hom G, Ortmann W, Behrens TW. Review of recent genome-wide association scans in lupus. Journal of Internal Medicine. 2009;265(6):680–688. doi: 10.1111/j.1365-2796.2009.02096.x. [DOI] [PubMed] [Google Scholar]
- 130.Sigurdsson S, Göring HHH, Kristjansdottir G, et al. Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (IRF5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic Lupus erythematosus. Human Molecular Genetics. 2008;17(6):872–881. doi: 10.1093/hmg/ddm359. [DOI] [PubMed] [Google Scholar]
- 131.Miceli-Richard C, Gestermann N, Ittah M, et al. The CGGGG insertion/deletion polymorphism of the IRF5 promoter is a strong risk factor for primary Sjögren’s syndrome. Arthritis & Rheumatism. 2009;60(7):1991–1997. doi: 10.1002/art.24662. [DOI] [PubMed] [Google Scholar]
- 132.Kristjansdottir G, Sandling JK, Bonetti A, et al. Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. Journal of Medical Genetics. 2008;45(6):362–369. doi: 10.1136/jmg.2007.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dideberg V, Kristjansdottir G, Milani L, et al. An insertion-deletion polymorphism in the Interferon Regulatory Factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Human Molecular Genetics. 2007;16(24):3008–3016. doi: 10.1093/hmg/ddm259. [DOI] [PubMed] [Google Scholar]
- 134.Hedl M, Abraham C. IRF5 risk polymorphisms contribute to interindividual variance in pattern recognition receptor-mediated cytokine secretion in human monocyte-derived cells. The Journal of Immunology. 2012;188(11):5348–5356. doi: 10.4049/jimmunol.1103319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-α activity in systemic Lupus erythematosus patients. Arthritis & Rheumatism. 2008;58(8):2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lövgren T, Eloranta ML, Båve U, Alm GV, Rönnblom L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis & Rheumatism. 2004;50(6):1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 137.Niewold TB, Kelly JA, Kariuki SN, et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic Lupus erythematosus. Annals of the Rheumatic Diseases. 2012;71(3):463–469. doi: 10.1136/annrheumdis-2011-200463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 139.Salloum R, Franek BS, Kariuki SN, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-α activity in lupus patients. Arthritis & Rheumatism. 2010;62(2):553–561. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dixit VM, Green S, Sarma V, et al. Tumor necrosis factor-α induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. Journal of Biological Chemistry. 1990;265(5):2973–2978. [PubMed] [Google Scholar]
- 142.Opipari AW, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor α encodes a novel type of zinc finger protein. Journal of Biological Chemistry. 1990;265(25):14705–14708. [PubMed] [Google Scholar]
- 143.Ning S, Pagano JS. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. Journal of Virology. 2010;84(12):6130–6138. doi: 10.1128/JVI.00364-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic Lupus erythematosus. Nature Genetics. 2008;40(9):1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shimane K, Kochi Y, Horita T, et al. The association of a nonsynonymous single-nucleotide polymorphism in TNFAIP3 with systemic Lupus erythematosus and rheumatoid arthritis in the japanese population. Arthritis & Rheumatism. 2010;62(2):574–579. doi: 10.1002/art.27190. [DOI] [PubMed] [Google Scholar]
- 146.Adrianto I, Wen F, Templeton A, et al. Association of a functional variant downstream of TNFAIP3 with systemic Lupus erythematosus. Nature Genetics. 2011;43(3):253–258. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mauro C, Pacifico F, Lavorgna A, et al. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. Journal of Biological Chemistry. 2006;281(27):18482–18488. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 148.Kawasaki A, Ito S, Furukawa H, et al. Association of TNFAIP3 interacting protein 1, TNIP1 with systemic Lupus erythematosus in a Japanese population: a case-control association study. Arthritis research & therapy. 2010;12(5):p. R174. doi: 10.1186/ar3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ronnblom LE, Alm GV, Oberg KE. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Annals of Internal Medicine. 1991;115(3):178–183. doi: 10.7326/0003-4819-115-3-178. [DOI] [PubMed] [Google Scholar]
- 150.Ronnblom LE, Alm GV, Oberg K. Autoimmune phenomena in patients with malignant carcinoid tumors during interferon-α treatment. Acta Oncologica. 1991;30(4):537–540. doi: 10.3109/02841869109092414. [DOI] [PubMed] [Google Scholar]
- 151.O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clinical Cancer Research. 2011;17(9):2619–2627. doi: 10.1158/1078-0432.CCR-10-1114. [DOI] [PubMed] [Google Scholar]
- 153.Korman BD, Kastner DL, Gregersen PK, Remmers EF. STAT4: genetics, mechanisms, and implications for autoimmunity. Current allergy and asthma reports. 2008;8(5):398–403. doi: 10.1007/s11882-008-0077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jacob CO, Zang S, Li L, et al. Pivotal role of Stat4 and Stat6 in the pathogenesis of the lupus-like disease in the New Zealand mixed 2328 mice. Journal of Immunology. 2003;171(3):1564–1571. doi: 10.4049/jimmunol.171.3.1564. [DOI] [PubMed] [Google Scholar]
- 155.Sigurdsson S, Nordmark G, Garnier S, et al. A risk haplotype of STAT4 for systemic Lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Human Molecular Genetics. 2008;17(18):2868–2876. doi: 10.1093/hmg/ddn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. International Immunology. 2005;17(11):1367–1378. doi: 10.1093/intimm/dxh318. [DOI] [PubMed] [Google Scholar]
- 157.Lee YH, Choi SJ, Ji JD, Song GG. Associations between PXK and TYK2 polymorphisms and systemic lupus erythematosus: a meta-analysis. Inflammation Research. 2012;61(9):949–954. doi: 10.1007/s00011-012-0486-y. [DOI] [PubMed] [Google Scholar]
- 158.Richter MF, Duménil G, Uzé G, Fellous M, Pellegrini S. Specific contribution of Tyk2 JH regions to the binding and the expression of the interferon α/β receptor component IFNAR1. Journal of Biological Chemistry. 1998;273(38):24723–24729. doi: 10.1074/jbc.273.38.24723. [DOI] [PubMed] [Google Scholar]
- 159.Ragimbeau J, Dondi E, Alcover A, Eid P, Uzé G, Pellegrini S. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO Journal. 2003;22(3):537–547. doi: 10.1093/emboj/cdg038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Teh BK, Yeo JG, Chern LM, Lu J. C1q regulation of dendritic cell development from monocytes with distinct cytokine production and T cell stimulation. Molecular Immunology. 2011;48(9-10):1128–1138. doi: 10.1016/j.molimm.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 161.Kariuki SN, Franek BS, Kumar AA, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic Lupus erythematosus. Arthritis Research and Therapy. 2010;12(4, article R151) doi: 10.1186/ar3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Koldobskaya Y, Ko K, Kumar AA, et al. Gene-expression-guided selection of candidate Loci and molecular phenotype analyses enhance genetic discovery in systemic lupus erythematosus. Clinical and Developmental Immunology. 2012;2012:9 pages. doi: 10.1155/2012/682018.682018 [DOI] [PMC free article] [PubMed] [Google Scholar]