

1. Introduction
The cells of the kidney contain many specialized ion channels and transporters, which act in concert to regulate volume and ionic concentration by absorption or secretion of ions into the urine. Each region of the kidney involved in filtration and concentration of ions expresses a particular subset of ion channels. Together, these ion channels ensure appropriate electrolyte homeostasis. However, a number of hereditary and genetic mutations render these channels mis-or non-functional. Mutations to one or more of these ion channels are associated with a variety of symptoms including proteinuria, progressive loss of renal function, and renal hypertension. The progressive loss of renal function, culminating in end-stage renal disease is typically treated by dialysis or transplantation. End-stage renal disease is an increasing health problem, both in terms of prevalence and economic burden. The scope of this review is to first provide a general overview of the kidney and function, and then specifically address the ion channels that, when mutated, lead to kidney disease.
1.1. Physiology of renal ion handling
The basic unit of the kidney is the nephron, and its function is to balance the ionic composition of the blood by filtering the blood, retrieving the necessary ions, secreting excess ions, and conserving water to concentrate the urine. Renal disease can be a manifestation of genetic mutations to renal channels (the focus of this review) or transporters (not discussed here, but there are many excellent reviews1). The correlation between distribution of a particular ion channel and its function for the kidney is a critical factor in the localization of disease. Most of these ion channels are tightly regulated and linked to a particular region of the nephron. Malfunctions in these channels can lead to impaired absorption of ions, and ultimately alter the osmotic balance in the kidney, with consequences on the ionic balance of the blood and tissues of the body. Specifically, mutations to a particular ion channel can have large effects beyond the kidney, as the ionic balance regulates a plethora of cotransporters required for transport of additional ions and other nutrients as well.
The nephron can be divided into the renal corpuscle, responsible for initial filtration, and the renal tubule, responsible for secretion and reabsorption of ions. The outline below describes the path of fluid filtration and concentration through the kidney, and identifies the ion channels that will be the subject of further discussion in Section 2. The order in which the ion channels are discussed in Section 2 reflects the fluid path through the kidney (Table 1). Figure 1 provides a schematic of the kidney filtration and concentration apparatus, and localizes the ion channels that will be addressed in this review.
Table 1.
Summary of the renal ion channels discussed in this review.
| Protein (gene) name | Distribution in kidney | Ion affected | Type of mutation | Disease associated | Section | References for mutations |
|---|---|---|---|---|---|---|
| TRPC6 (Trpc6) | Glomerulus | Ca2+ | Gain-of-function | Focal Segmental Glomerulosclerosis | 2.1 | 17–19 |
| TRPM6 (Trpm6) | Distal convoluted tubule | Mg2+ | Loss-of-function | Hypomagnesemia | 2.1 | 12,21 |
| ClC-5 (CLCN5) | Convoluted proximal tubule | Cl−/H+ | Loss-of-function | Dent’s disease | 2.2 | 33–36 |
| ClC-Kb (CLCNKB) | Thick ascending loop of Henle | Cl− | Loss-of-function | Bartter syndrome | 2.2 | 41,44,46–49 |
| ROMK (KCNJ1) | Thick ascending loop of Henle; Distal nephron | K+ | Loss-of-function | Bartter syndrome | 2.3 | 59,66–67 |
| Kir4.1 (KCNJ10) | Collecting duct | K+ | Loss-of-function | EAST syndrome | 2.3 | 60 |
| ENaC (Scnn1a) | Collecting duct | Na+ | Loss-of-function | Pseudohypoaldosteronism | 2.4 | 82b,83,85–86 |
| ENaC (Scnn1a) | Collecting duct | Na+ | Gain-of-function | Liddle’s syndrome | 2.4 | 87–88,91–92 |
| Polycystin 2 (PKD2) | Convoluted tubule | Ca2+ | Loss-of-function | Polycystic kidney disease | 2.5 | 101,121 |
Figure 1.

Overview of the kidney nephron and the distribution of ion channels discussed in this review. Fluid enters the glomerulus, then down the convoluted proximal tubule. After passing through the loop of Henle, the fluid is further concentrated in the distal convoluted tubule, and then the fluid reaches the collecting duct. See text for details.
The renal corpuscle is comprised of the glomerulus, which filters the blood, and the Bowman’s capsule. The Bowman’s capsule is composed of an inner layer of podocytes and an outer single layer of epithelial cells. Podocytes are specialized glomerular epithelial cells that surround the glomerular capillaries. Fluids from blood in the glomerulus are filtered through gaps between the podocytes, and the resulting fluid passed to the renal tubule. The concentration of the major ions, sodium (Na+), potassium (K+), chloride (Cl−), carbonate (HCO3−), calcium (Ca2+), through the Bowman’s space is the same as in whole blood2. Mutations to the transient receptor potential (TRP) canonical channel TRPC6 (See Section2.1) found in this region result in Focal Segmental Glomerulosclerosis.
After exiting the Bowman’s capsule, fluid enters the proximal convoluted tubule. Through this region, up to 67% of filtered Na+ and K+ is reabsorbed. The loop of Henle is comprised of the descending and ascending limbs. The ascending limb of the loop of Henle consists of the thin ascending limb, and a distal portion known as the thick ascending limb of the loop of Henle. Defects in the normal function of the ion channels within the thick ascending loop of Henle are relevant to Na+, K+, and Cl− imbalances, and impair the absorption of these ions. In the thick ascending limb of the loop of Henle, NaCl enters the cell via the bumetanide- sensitive Na+-K+-2Cl−-cotransporter (NKCC2 or BSC1, a transporter which will not be discussed further here), whereas K+ is recycled into the lumen via an adenosine triphosphate (ATP)-sensitive K channel (ROMK, see Section 2.3); this channel, ROMK, provides the K+ necessary for NKCC2 activity. Cl− leaves the cell by the basolateral membrane through either the chloride channel (ClC-Kb, see Section 2.2.3.1) or is cotransported with K+ using NKCC2 or other transporters. Na+, on the other hand, exits the cell through the Na+-K+-ATPase (an ATP driven pump which will not be dealt with in this review). Recirculation of K+ to the lumen together with the exit of Cl− across the basolateral membrane provides the lumen-positive transepithelial voltage gradient that drives Na+, K+, Ca2+, and Mg2+ reabsorption (see Section 2.1). In the thick ascending limb of the loop of Henle, 20% of filtered Na+ and K+ is reabsorbed. Together, the upstream proximal tubule and thick ascending limb of loop of Henle reabsorb 90% of filtered Ca2+.
After leaving the proximal tubule, the fluid enters the distal tubule. In the early distal convoluted tubule, NaCl reabsorption is mediated by the luminal NaCl cotransporter (another transporter which will not be dealt with in this review) and leaves the cell through ClC-Kb associated with barttin (mutations of which are discussed in Section 2.2), and through the Na+-K+-ATPase as well.
Finally, the modified filtrate enters the collecting system before it passes to the urinary bladder. This part of the nephron is comprised of connecting tubules, cortical collecting ducts, and medullary collecting ducts. Na+ in this region is reabsorbed via the epithelial Na+ channel (ENaC, see Section 2.4) on the luminal side and again exits the cell by the Na+-K+-ATPase. The inwardly rectifying potassium channel Kir4.1 (see Section 2.3.3), found on the basolateral membrane is believed to provide the K+ that drives the Na+-K+-ATPase pump. At the same time as Na+ absorption, K+ is extruded by the Ca2+-activated big conductance K+ channel (BK, not further discussed here, but see the following reviews3) and ROMK (see Section 2.3). The activity of the channels can be enhanced with aldosterone, which stimulates the mineralocorticoid receptor, and increases ENaC, the Na+-K+-ATPase, and ROMK channel expression and activity. Furthermore, mineralocorticoids increase the serum-and glucocorticoid-inducible kinase transcription, which also activates ENaC, the Na+-K+-ATPase, and ROMK.
In addition to the plasma membrane ion channels, one important intracellular ion channel, polycystin 2 will also be discussed (see Section 2.5). As a member of the TRP family, this channel primarily resides in the endoplasmic reticulum, where it can act as a Ca2+ release channel. Due to the widespread distribution of polycystin 2, mutations can result in the development cysts in any of the nephron segments (Figures 1 and 2).
Figure 2.

Distribution of the transient receptor potential (TRP) channels in the kidney. Note that the distribution of TRP channels is different depending on the region. For example, polycystin 2 is most prominently expressed in the distal convoluted tubule.
1.2. Treatment Strategies
As alluded to above, most patients with end-stage renal disease are given kidney dialysis treatment, or, if available, a transplant. Other treatment strategies include angiotensin-converting enzyme inhibitors that act to prevent proteinuria, and slow or halt the progression of proteinuric nephropathies. Frontline treatment of renal hypertension is through treatment with Ca2+ channel blockers primarily targeting the voltage-gated Ca2+ channels4 found on vascular smooth muscle cells in the peripheral resistance vessels. Additionally, the drugs alter the degree of constriction of the renal afferent arterioles. The voltage-gated Ca2+ channels are not included here, as this review is mainly addressing the genetic diseases associated with ion channels of the kidney, specifically with renal channelopathies. For more detailed information about these vascular voltage-gated Ca2+ channels, the reader is directed to reviews addressing the pharmacology of Ca2+ channel blockers, primarily the dihydropyridine blockers4. Although these treatment strategies are currently used today, it is anticipated that the identification of the ion channel genes involved in specific kidney disease and the consequential modifications to channel and kidney function will result in novel and more kidney specific therapeutic approaches to delay or even prevent dialysis or kidney transplantation.
1.3. Tools used to identify genes mutated in renal disease
Many of the ion channels and associated mutations discussed below have been identified using modern genetic tools including gene expression arrays, linkage analysis and association studies. In turn, these genetic tools can be used to screen and find new therapeutic strategies. Genetic arrays are a powerful and widely used approach for the analysis of gene transcription and protein expression, whereas linkage analysis and association studies link disease susceptibility to particular genetic regions. The identification of specific mutations has allowed for screening by in vitro models. These investigations are complemented by the use of transgenic animal models, which allows for an assessment of a mutant channel in disease progression. A more thorough treatment of this topic can be found in the following reviews5. Whereas the tools used to identify genes in renal disease have been ultimately successful, screening approaches have also been employed. However, what is fundamental to the application of these treatment strategies is an understanding of how each of the ion channels function, and how a particular mutation to the channel causes disease. As expanded below, many of the genes associated with renal ion channel disease have a multitude of pathogenic mutations, with no hot spot or particular region mutated. Thus, in order to specifically target each mutation, an approach combining structure and function studies is necessary.
2. Renal Channelopathies
2.1. Non-Selective Transient Receptor Channels: TRPC and TRPM
The Transient Receptor Potential (TRP) channels are non-selective ionic channels, allowing a variety of ions through the cell. Like other transmembrane proteins such as the voltage gated K+ or Ca2+ channels, they are comprised of 6 transmembrane helices. In order to form a channel, TRPs are believed to oligomerize in either hetero or homotetramers, with transmembrane domains 5 and 6 to form the pore. There are eight main families of TRP channels, with eight members found within the kidney, each with a specific regional distribution (Figure 2). Of the eight types of TRP channel, three of these channels (TRPC6, TRPM6 and TRPP2) are associated with channelopathies within the kidney. A discussion on TRPP2 can be found in Section 2.5.
The TRPC (C for canonical) family is a group of Ca2+-permeable cation channels that are important for the increase in intracellular Ca2+ concentration upon stimulation of G protein–coupled receptors and receptor tyrosine kinases6. Two members of this family, TRPC3 and 6 are of particular importance in the kidney. TRPC3 and 6 are detected along the glomerulus and the collecting duct. Gain-of-function mutations in TRPC6 lead to late-onset and childhood7 Focal Segmental Glomerulosclerosis, due to loss of integrity of the glomerular filter. Although mutation to TRPC3 have not been associated with renal disease, TRPC3 is compensated in the absence of TRPC68.
The TRPM family (M for melastatin) is so called due to their expression in melanocytes and activation by melastatin. Of the eight members, two are prominent in the kidney, TRPM6 and TRPM7. Like other TRP channels, TRPMs can conduct Ca2+ currents. However, TRPM6 and 7 are unique in that they preferentially conduct Mg2+ 9. Loss-of-function mutations to TRPM6 prevent Mg2+ reabsorption into the renal epithelial cells, and thus result in excessive Mg2+ excretion (hypomagnesemia) with secondary hypocalcemia (loss of Ca2+).
2.1.1. Structure of TRPC6 and TRPM6
Few studies have addressed the generic structure of TRP channels. Indeed, most use either the voltage gated K+ or Ca2+ channels as a model for how TRP channels fold. TRPC6 is a 936 amino acid protein with an extensive amino (N)-terminus, including several ankyrin repeats, where mutations result in channel dysfunction, and a coiled-coil domain. Motifs of interest in the carboxy (C)-terminal domain include the TRP box, thought to be involved with gating and a calmodulin binding site (enabling regulation by Ca2+). No known structures exist for TRPC6. However, studies on yeast TRPC310, which has roughly 75% homology to TRPC6 and 7, have been carried out and a low resolution (15 Å), cryo-electron microscopy structure of yeast TRPC3 has been described11.
TRPM6 consists of 2,022 amino acids, with extensive N- and C- termini (predicted to be 872 and 955 residues respectively)12. Structural studies on TRPM6, like TRPC6 have not been carried out, but atomic force microscopy suggest that the family member TRPM813 is a homotetramer. Interestingly, computerized modeling of TRMP8 compared with the protein importin, which has a homologous N-terminus to TRPM8, suggested that the N-terminus could form a tetramer. Less provocatively, the same modeling also suggested that the C-terminus of TRPM8 could form a tetramer through the coiled-coil region14, which had been previously described by X-ray crystallography15. These studies provide a starting point in understanding how TRPM proteins oligomerize to form channels.
The Mg2+ selectivity of TRPM6 and 7 is due to two conserved acidic residues between helices 5 and 6 in TRPM6 and 7, not present in other TRPM channels. Mutations to these residues were also found to alter Ca2+ sensitivity as well as the pH sensitivity of the pore16. Although TRPM7 homotetramers can also conduct Mg2+, and this channel localizes to the apical membrane like TRPM6 in the kidney, the resultant hypomagnesemia in patients with TRPM6 mutations suggests that the deficiency is not compensated by TRPM7 (Figure 3). This finding may indicate that the gating of TRPM6 heterotetramers versus TRPM7 homotetramers is different, or that compensation by TRPM7 is not adequate. As yet, no studies have been done to explore these possibilities. Moreover, a comparison alignment (Figure 4) between the two proteins reveals several regions with dissimilarity, particularly the C-terminus, although the functional consequences of these regions are unknown.
Figure 3.



Sequence Alignment of TRPM6 and TRPM7. Yellow highlighted residues correspond to known mutations in TRPM6. The putative transmembrane domains (TM) are underlined. Alignments were carried out using MultAlin140. Symbols represent conserved amino acids, where: ! can either be I or V, $ can be L or M, % can be F or Y and # can be B, D, E, N, Q, and Z.
Figure 4.

TRPM6 and 7 on the apical membrane permits Mg2+ influx into the cell. The identity of the basolateral Mg2+ channel is not know, although Mg2+ flux is believed to be coupled to Na+ moving in the opposite direction141. Mutations to TRPM6 lead to low blood Mg2+.
2.1.2. TRPC6 and Focal Segmental Glomerulosclerosis
The filter of the glomerulus is composed of three components- the capillary endothelium, the glomerular basement membrane and the slit diaphragm between the podocyte foot processes. Diseases associated with a loss of podocyte function, such as Focal Segmental Glomerulosclerosis, typified by partial scarring of the glomerulus, result in proteinuria. If left untreated, Focal Segmental Glomerulosclerosis leads to end stage renal disease. TRPC6 (Figure 5) was first described to be associated with Focal Segmental Glomerulosclerosis in 200517, where an N-terminal missense mutation was found in an ankyrin domain at P112. The mutation P112Q increased surface expression of TRPC6, resulting in an increase in Ca2+ influx, as measured by Ca2+ imaging. A subsequent study identified five other mutations in TRPC6. Patch clamp analysis demonstrated that two mutations, R895C and E897K, both in the C-terminus, lead to enhanced Ca2+ current amplitude18. Interestingly, the other three mutations, two in conserved ankyrin repeat regions in the N-termini (N143S, S270T), and a third being a truncation (K874X in the C-terminus), did not affect current amplitude nor protein trafficking to the plasma membrane18. This finding suggests that these mutations affect accessory down stream proteins, and are not necessarily coupled to Ca2+ channel function per se. More recently, three more mutations have been identified, two in the N-terminus, and one in the C-terminus, surprisingly associated with both childhood and adult onset Focal Segmental Glomerulosclerosis19. Of particular interest is a mutation in an N-terminus ankyrin repeat, M132T, which results in a dramatic increase in non-selective current, including a 10-fold increase in Ca2+ influx. This larger influx may contribute to the faster onset of disease with the M132T mutation, compared to the other previously described TRPC6 mutants. What is particularly striking about all known mutations of TRPC6 is that they have been mapped to either the N or C-terminal domains of the protein, with no known mutations found in the transmembrane domains. This distribution of mutations suggests that alterations to the transmembrane region would be lethal.
Figure 5.

Depiction of TRPC6 on the podocyte. Ca2+ entering through TRPC6 cases the reorganization of actin, and closure of the slit diaphragm by the zippering action of nephrin.
The surprising finding that childhood as well as adult onset Focal Segmental Glomerulosclerosis are consequences of mutations in TRPC6 suggests that the mutations to TRPC6 correlated to childhood onset are more deleterious than the adult onset mutations19. Indeed, evidence of a gene dosage effect of TRPC6 was observed in mice overexpressing either wild type TRPC6 or the originally described missense mutant P112Q TRPC6. Both types of mice developed glomerular lesions, indicating that either increased protein, or mutations that increase Ca2+ influx, is sufficient to cause disease. Moreover, when these findings are taken together with the M132T mutation, it appears that the glomerular lesions are a downstream consequence of elevated Ca2+ influx. An added complexity for understanding and treating Focal Segmental Glomerulosclerosis exists, as mutations have been found in a number of other proteins that either directly interact with TRPC6 or are involved in the structural integrity of the glomerular filter. These associated proteins include nephrin, podocin, α-actinin-4, phospholipase C and laminin 2 (for a further discussion of these proteins, see this review20).
Consistent with the idea that disease causing mutations to TRPC6 arise from a gain-of-function is the phenotype of the TRPC6 knockout mice. These mice were found to be viable, without kidney defects and no gross phenotype8. Although an elevated blood pressure and increased vascular contractility was reported, this was found to be due to a compensatory mechanism, the upregulation of TRPC38.
2.1.3. TRPM and Hypomagnesemia
Mutations in TRPM6 were found to be associated with abnormal Mg2+ excretion, indicating a disruption in the TRPM6’s ability to conduct Mg2+ from the extracellular environment into the cell12. Three mutations were in splice donor or acceptor sites, whereas the other two mutations resulted in premature stops (R484X, R56X). Additional mutations in a Turkish pedigree reveal splice site mutations21. Interestingly, TRPM6 is unable to form a channel in its own right, and forms heterotetramers with TRPM7 to form plasma membrane channels in Xenopus oocytes and HEK cells22. Even though the two proteins form a complex, no human mutations of TRPM7 associated with hypomagnesemia have been reported. However, modulation of TRPM7 expression in zebrafish studies indicate that the phenotype is similar to TRPM6 deficiency, in that Mg2+ and Ca2+ uptake is impaired23. Although no known mutations within TRPM7 have been described, both TRPM6 and TRPM7 expression and activity levels were found to be decreased in studies where hyperaldosterone studies lead to hypomagnesemia24.
2.1.4. Pharmacology and treatment
Finding agonists or antagonists for TRPs remains an elusive task (see this recent review25 for an overview of TRP pharmacology). At present, no known specific inhibitors or agonists of TRPC6 or TRPM6 exist.
2.2. Chloride Channels- ClCs
Chloride is the major anion found within the human body. Most of the Cl− is regulated via voltage gated Cl− channels (ClCs). Of the nine known ClCs, eight of these are found within the kidney, and mutations to two of these, ClC-5 and ClC-Kb are associated with the kidney disorders Dent’s disease and Bartter syndrome, respectively. Although ClC-5 was originally considered to be a Cl− channel, it has now been reclassified as a Cl−/H+ antiporter26.
2.2.1. Structure of ClCs
Structural understanding of ClC channels has largely been carried out by studying the bacterial homologue of mammalian ClC, although the mammalian cloned channel was first described in 199327. All the ClCs have similar structure, comprised of a homodimer28 of α subunits, though some isoforms are coupled with a β subunit. ClCs are multi-transmembrane proteins, with the mammalian ClCs containing between 10–12 transmembranes (for an excellent review on ClC structure, see29 (Figure 6)). Although the bacterial homolog of ClC has been important in studying ClC function, the mammalian ClCs contain two cystathionine β-synthetase binding sites in the C-terminus, not present in the bacterial ClC. The cystathionine β-synthetase binding sites are common regulatory nucleotide binding domains that bind ATP, ADP or AMP.
Figure 6.

ClC structure and function. A). Schematic arrangement of the α helices that are in mammalian ClC. It is believed that there are 10–12 transmembranes in ClC, however, not all helices span the membrane. The first 9 α helices (shaded blue) and the second 9 α helices (shaded pink) are similar in structure, though there is little homology at the amino acid level. Note the presence of the two cystathionine β-synthetase binding sites domains in the C-terminal tail. B). Structure of the cystathionine β-synthetase binding sites by X-ray crystallography30. A cleft between the two CBS sites binds the nucleotides. PDB accession code: 2J9L. Structure was modified the UCSF Chimera package142. C). ClC-5 acts to transport H+ and Cl− in endosomes, to counteract the activity of vesicular ATPase in the convoluted proximal tubule. Loss of ClC-5 function leads to the acidification of the endosome.
The cystathionine β-synthetase binding sites of ClC-5, containing 185 amino acids, was recently solved in complex with ADP by X-ray crystallography at 3.05Å30 (Figure 6B). This structure revealed that the nucleotide binds in a cleft, or interface region between the two cystathionine β-synthetase binding sites30. Binding of adenine nucleotides to the intracellular side of ClC-5 results in potentiated current. The residues important in this facilitation included Y617 and D727, but not S61831, a site previously shown to bind nucleotides in the high micromolar range with the α and γ phosphate groups of ATP30. The similar level of potentiation provided by all three adenine nucleotides, however, raises additional questions as to the specific role these nucleotides play in vivo. In contrast to ClC-5, ClC-Ka does not bind nucleotides, although like ClC-5, the cystathionine β-synthetase binding sites forms a dimeric domain32.
2.2.2. ClC-5 and Dent’s disease
Dent’s disease is a type of Fanconi syndrome (diseases that occur in the proximal renal tubule) with typical symptoms including tubular proteinuria (high urinary protein), hypercalciuria (excessive urinary Ca2+ excretion), Ca2+ nephrolithiasis (kidney stones), nephrocalcinosis (deposition of Ca2+ in the kidney), ultimately culminating in chronic renal failure. It is an X-linked genetic disease, due to mutations in the gene CLCN5 (that encodes ClC-5) residing on the X chromosome.
A number of mutations (over 45) in ClC-5 result in Dent’s disease, with the vast majority being truncations33. Analysis of the mutations in ClC-5 demonstrates that there are no hot spots across this channel. However, the majority of the missense (12 out of 15) mutations that abolish all current occur at the interface lining the pore region, indicating the importance of functional Cl− currents33. The remaining three mutations were positioned on helices that are external to the pore helices and are correlated with a reduction of 70% of the current33.
The means by which Dent’s disease arises can be for a number of reasons. For example, the version of ClC-5 with the mutation G212A trafficked normally to the cell surface and to early endosomes, underwent N-linked glycosylation at the cell surface like wild-type ClC-5, but exhibited significant reductions in outwardly rectifying ion currents (Figure 6C). In contrast, other mutations in ClC-5 including G179D, L200R, S203L, C219R, C221R, L469P, and R718X were improperly N-glycosylated and were non-functional due to retention in the endoplasmic reticulum34. Similar finding were observed in another study that found that four mutations (S270R, G513E, R516W, I524K) were unable to traffic from the endoplasmic reticulum35. An E527D mutation was found to have defects in endosomal acidification, and finally, two milder phenotypes (G57V, R280P) had altered endosomal distribution35. Of the known truncation mutations, two have extensive amino acid deletions over 300 residues. More surprisingly, there is another non-functional truncation mutation that deletes only 28 amino acids in the C-terminal region36. The latter mutation suggests that the whole protein, including the C-terminal tail, is essential for proper channel function.
An important finding was made using the knockout ClC-5 mouse model. Jentsch and colleagues showed that a major symptom of Dent’s disease, proteinuria, was caused by strongly reduced endocytosis in the proximal tubule37. This finding was later verified in an in vitro setting as well38. However, the means by which ClC-5 causes the downstream effects remain to be fully elucidated. Indeed, a recent genetic screen of the same ClC-5 knockout animal and wild type mice has demonstrated that over 700 genes are significantly altered in the proximal tubule39. Although this type of screen provides a fingerprint of the genes altered in Dent’s disease, it also demonstrates the complexity and downstream effects of mutating this one important Cl− channel.
2.2.3. Bartter Syndrome
Bartter syndrome and the closely related Gitelman syndrome are due to mutations in ion channels in the thick ascending limb within the loop of Henle. The five types of Bartter syndrome (each type traditionally associated with a different gene) and Gitelman syndrome (caused by mutations to the NaCl cotransporter40) are typified by hypokalaemic metabolic alkalosis, renal salt loss, hyper-reninaemic hyperaldosteronism and normal blood pressure. One type of Bartter syndrome, type 3 is due to defects within the CLCNKB gene that encodes the CLC-K channel41. Bartter syndrome types 4 and type 2 will also be discussed in the following sections, affecting the ClC accessory protein barttin and the potassium channel ROMK, respectively.
2.2.3.1. ClC-Kb
There are two known isoforms of ClC-K, variously known as ClC-Ka and b in humans, or ClC-K1 and 2 in rodents. Over 90% of the two proteins are homologous to each other; however, the two differ in distribution within the kidney. ClC-Ka is expressed in the thin ascending limb of Henle’s loop of the nephron, whereas ClC-Kb is restricted to the basolateral membranes of epithelial cells in the thick ascending limb, connecting tubule, distal convoluted tubule, and intercalated cells (Figure 7). Primarily mutations in ClC-Kb are associated with Bartter syndrome. In comparison to expression in the kidney, both ClC-K isoforms have similar distributions in the ear, thus the loss of function of ClC-Kb can be compensated for by ClC-Ka. However, mutation of both ClC-K genes results in sensorineural hearing loss (i.e: hearing loss due to damage in the inner ear)42.
Figure 7.

In the thick ascending loop of Henle, ClC-Kb on the basement membrane together with barttin enable the efflux of Cl−. Mutations lead to Bartter syndrome.
ClC-K is unique among the ClCs as being modulated by both extracellular Ca2+ and pH. Although extracellular Ca2+ is tightly controlled in most cellular environments (around 2.5 mM), in the kidney, extracellular Ca2+ concentrations can vary +/− 1 mM43. For ClC-K, the activity of the channel can vary 2.5 fold between a Ca2+ range of 1–10 mM44. Unlike proton gating for the other ClCs, which are gated by a glutamate residue, the residue at the equivalent site for ClC-K is a valine. The gating of ClC-K is complex, with the channel inhibited by both high and low pH45. The mutations in ClC-Kb were identified by linkage analysis on chromosome 141, and led to the discovery of missense mutations spread throughout the channel, and nonsense mutations leading to truncations. Unlike other channel mutations, no known mutations result in trafficking discrepancies. The mutations with known functional consequences will be discussed below. A recent study identified and characterized three mutations in ClC-Kb with electrophysiology studies46. Unsurprisingly, the truncated R30X mutation did not form a channel. The two missense mutations, A210V and R351W, were both found to significantly reduce the current amplitude. A210V, which is located in the third transmembrane domain, was almost as sensitive as the wild type to extracellular pH and Ca2+. In contrast, R351W, which is located in the extracellular loop between transmembrane domains 5 and 6 removed extracellular Ca2+ activation and markedly reduced alkaline pH activation of ClC-Kb. These results indicate that this loop contributed to gating. Another mutation in the C-terminus tail R538P is also linked with Bartter syndrome44. Whereas pH sensitivity was unchanged, the R538P mutation was found to activate at hyperpolarizing potentials, rather than inactivate, and surprisingly, there was abolition in Ca2+ sensitivity. Moreover, when the same mutation was made in ClC-Ka channels, no alteration of ClC-Ka currents was observed, suggesting that in spite of the 90% homology, the two channels also have specific residues that gate and activate the channel44. A severe clinical manifestation of Bartter syndrome in a young boy was associated with a missense mutation in ClC-Kb of G470E47. Although this mutation has not been tested in vitro, it is predicted that the current will be decreased, as the mutation is located in a transmembrane region.
Another example of the broad phenotype associated with ClC-Kb was demonstrated in an analysis of two family members, both with mutations at the same site in CLC-Kb but with vastly different symptom severity48. Moreover, a patient with symptoms associated with Gitelman syndrome was found to have a novel mutation A204T in ClC-Kb, but no defects in the NaCl transporter49, SLC12A3 gene, which is traditionally associated with Gitelman syndrome. Similarly, other patients with mixed Gitelman and Bartter syndrome symptoms were found to have mutations in ClC-Kb, including missense mutation R438H, but surprisingly no mutations in SLC12A350. Taken together, these reports provide evidence that associated proteins or other factors alters the spectrum of disease observed with ClC-Kb mutations. So far, no known animal models of ClC-K2 have been described. However, deletion of the ClC-K1 encoding gene in mice lead to a diabetic phenotype, an observation not reported in humans51.
2.2.3.2. Bartter syndrome with hearing loss- Barttin defects
Barttin, encoded by the gene BSND, is an important β subunit of ClC-Kb. Barttin acts to both traffic the ClC to the plasma membrane, as well as act as a regulatory subunit52. It is interesting to note that ClC-Ka is functional without co-expression of barttin, although currents are enhanced with barttin52. First described in 200153 barttin was found to be present in both the kidney and ear. Barttin is a small protein of 320 amino acids, consisting of two transmembrane helices formed by the amino acids between 9 and 54 and a cytoplasmic C-terminus of 266 amino acids. Three distinct functional roles for barttin have been described54. Deletion analysis demonstrated that the transmembrane core was necessary and sufficient to traffic the ClC-K/barttin complex from the endoplasmic reticulum to the surface membrane54. The transmembrane region and the start of the C-terminus affected channel opening and current amplitude in human and rat ClC-K, respectively. Finally, the C-terminal tail was found to be necessary for setting the absolute channel open probability54. A subsequent study demonstrated that barttin was associated with the outer lateral side of ClC. However, as barttin was able to bind to two different transmembrane domains of ClC, corresponding to transmembrane domains 1 and 8, which have 40% homology, it is hypothesized that a hydrophobic region, rather than specific residues per se are involved in the binding55. The verification of the requirement of the N-terminal region for trafficking was demonstrated by the development of a mutant (R8L) barttin. When this mutated barttin was genetically inserted in mice, there was reduced ClC-K plasma membrane expression and the mouse had the disease phenotype of Bartter syndrome56.
2.2.4. Pharmacology and treatment
The classical blocker of ClC channels is 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS). Although it is of interest to develop similar compounds as diuretics, this compound does not treat ClC related disease. Specifically, there is no known pharmacological treatment for Bartter syndrome, where activators of ClCs would be required. However, elucidating the sites at which blockers bind to either ClC-Ka or Kb may also reveal critical residues that may be the target of activating compounds. A study revealed that there was a five-fold difference between ClC-Ka and Kb in sensitivity to DIDs and another ClC blocker, 2-(p-chlorophenoxy)-3-phenylpropionic acid57, with ClC-Kb less sensitive. In spite of the high homology between ClC-Ka and ClC-Kb, two amino acids with side chains exposed to the pore, N68D and G72E in ClC-Ka and Kb respectively, were found to confer the difference in drug sensitivity57. Mutation of the D68 and E72 sites in ClC-Kb from negatively charged to neutral residues increased the sensitivity of ClC-Kb to DIDs, to that of ClC-Ka, thus these two amino acids may define the drug binding site57. A more recent strategy has been to modulate activation sites on ClC-Ka, leading to the discovery of molecules that can act as inhibitors with high affinities (less that 10 μM)58. However, it is also apparent that this type of pharmacological development needs to be applied more generally, to create compounds that activate ClC-Kb.
2.3. Potassium Channels- ROMK and Kir4.1
One of the more severe forms of Bartter syndrome is clinically manifested with childhood renal salt wasting and water loss, linked to defects in absorption in the thick ascending limb of the loop of Henle. The first association of channels other than ClC that would cause Bartter syndrome was in 1996, with the discovery that genetic mutations in ROMK caused childhood Bartter syndrome59 (Figure 8A). A second potassium channel that has been linked to renal tubulopathy is the inwardly rectifying potassium channel Kir4.1, encoded by the gene KCNJ1060 (Figure 8B).
Figure 8.

The role of the K+ channels. A, top). Schematic diagram of ROMK on the apical membrane of the thick ascending loop of Henle. Na+, K+ and Cl− are taken up by NKCC2, however, as luminal K+ is low, K+ must be recycled back to the lumen by ROMK. The removal K together with the activity of ClC-Kb ensures that the cell maintains a transcellular electrical potential, that in turn enables Na absorption by the paracellular pathway. Bottom, Disruption of ROMK impairs both the activity of NKCC2 and also disrupts the transcellular extracellular electrical potential. Thus, Na+ absorbtion is impaired. Note, however, that the phenotype of the ROMK knockout mice is not as severe as NKCC2 mutations, suggesting that alternative K+ influx pathways are still available68. B, top). Schematic diagram of the role of Kir4.1 on the basolateral membrane in the distal convoluted tubule. Bottom, Mutations to Kir4.1 impair moment of K+ out of the cell
2.3.1. Structure of ROMK
The ROMK channel (also known as KIR 1.1) was first cloned in 199361, and initial studies revealed several regulatory domains, including a pH sensor domain, an ATP binding domain, and other regulatory domains. Like other KIR channels, it is believed that four homologous units come together to form a central pore. The pH sensor domain is of particular importance in the kidney, where the channel is inhibited by H+ ions. As the channel is exquisitely pH dependent62, mutations to this site have been linked to disease, in particular Bartter syndrome. This conserved pH dependent region is found in the N-terminal cytoplasmic region of the protein, just upstream of the first transmembrane segment. Although a lysine residue (amino acid 80 in ROMK1; 61 in ROMK2) was initially believed to be responsible for the pH dependence63, later studies identified additional critical residues, including a hydrophobic region 10 amino acids downstream of the initial site64. This latter site was predicted using the hypothesis that the pKa of lysine was too high (pH 10.5), and suggested that a combination of regions were responsible for the pH dependence. Structural conformation of this site is lacking, although Sackin and colleagues have used the homology model of bacterial KIR1.1 coupled with a mutation analysis of the pH site of ROMK to suggest that the pH site acts as a gate for the channel, with the channel open at higher pHs65.
2.3.2. ROMK and Bartter syndrome
Mutations associated with ROMK include one that locks the channel in a closed state66. Others include truncations to the C-terminal tail, which also renders the channel non-functional, suggesting that a regulatory or gating domain may reside in the C-terminal tail. Additionally, the introduction of even one mutant unit combined with three WT units also diminishes all channel activity66, indicating that all four components must be fully functional. This study also demonstrated that the defects were not due to a trafficking or an oligomerization defect. What the regulatory element is and how this region interacts is still unknown. Interestingly, this region appears to be conserved across other ion channels, yet it is still unidentified66. Another study identified 17 novel mutations distributed throughout ROMK causing classic Bartter syndrome67. As no functional studies were done on the mutated residues, the underlying basis for ROMK dysfunction is not known.
In 2002, creation of a ROMK null mouse was published68. Surprisingly, this mouse was not embryonic lethal, although over 95% of the affected mice died within 3 weeks of birth. Of the surviving mice, impaired renal function was documented. Further studies using this mouse demonstrated that the loss of ROMK function was compensated by an upregulation in maxi-K+ channels as well as an upregulation in Na-Cl- cotransport activity and water transporting proteins69.
2.3.3. Kir4.1
Recently, mutations in the Kir4.1 have been found to be associated with epilepsy, ataxia, sensorineural deafness, and a renal salt-losing tubulopathy in several families, defined as EAST syndrome60b. Kir4.1 is an inwardly rectifying potassium channel comprising of two transmembrane domains, and is located on the basolateral membrane of the distal convoluted tubule, the connecting tubule, and the early cortical collecting duct. Kir4.1 knockout mice died shortly after birth but also have renal salt-losing tubulopathy typified with a lower urinary creatinine concentration, an elevated urinary sodium concentration and reduced Ca2+ excretion60b. It is posited by Bockenhauer and collegues60b that the role of the Kir4.1 channel on the basolateral membrane may be to provide potassium recycling from the Na+,K+-ATPase pump, and thus impairment of this channel may result in reduced Na+,K+-ATPase function, and consequently, a lack of sodium absorption by the cell, and therefore elevated sodium in the urine. The mutations that result in the EAST syndrome are a combination of missense mutations and truncations that all reduce or inhibit function. Known mutations include the missense mutations R65P and G77R, both of which lie within the first transmembrane domain, and result in a dramatic reduction of current60b, a truncation R199X, which results in loss of channel function, and an R175Q mutation, that has a substantially changed pH sensitivity in the alkaline range and reduced channel function60a. Additional mutations have been identified including the missense mutations R65C and F75L, and a truncation mutation V259X, that all result in reduced potassium currents70.
2.3.4. Pharmacology and treatment
So far, no direct treatment of either of the potassium channelopathies that have been discussed in the preceding paragraphs (ROMK and Kir4.1) are established. Little is understood structurally about the ROMK channel, in spite of its important function in the nephron. Additional studies on the other regulatory elements of ROMK, coupled with structural studies may complement the current findings. Compounds that inhibit ROMK are of interest, as they may act as a diuretic. A recent study using a small molecule approach has identified a compound (Figure 9) that blocks ROMK with ~300 nM affinity, although this compound also blocks KIR 7.1 with micromolar affinity71. The mode of block appears to be through the ion permeation pathway, though this block can be alleviated by hyperpolarization and high extracellular K+ concentrations. It is important to point out, however, that this compound is not a useful drug yet, as it blocks ROMK intracellularly, and to be clinically effective, it will need to be membrane permeant.
Figure 9.
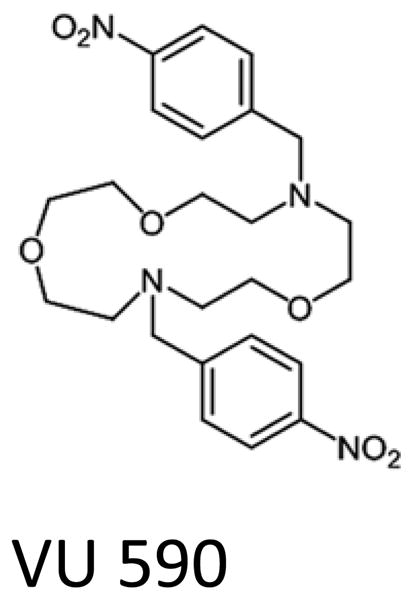
Structure of a ROMK inhibitor with 300 nM affinity, identified by small molecule screening71. This inhibitor binds the channel from the intracellular side.
2.4. Sodium channels-ENaC
The epithelial sodium/degenerin channel (ENaC/Deg) family includes a number of proteins including acid sensing ion channel 1 (ASIC)72. ENaCs are non-voltage sensitive and this channel is the primary limiting Na+ reabsorption channel in the epithelium. Na+ reabsorption at the channel level can be increased in two key ways-first by an increase in channel open probability and gating, and second, by increasing the population of ENaC channels on the plasma membrane. Mutations to ENaC are associated with two genetic disorders, Pseudohypoaldosteronism type 1 (PHA1) which is a monogenic disorder of mineralocorticoid resistance characterized by salt wasting, hyperkalemia, high aldosterone levels, and failure to thrive. An autosomal recessive form of PHA1 (AR-PHA1) is caused by loss-of-function mutations in ENaC with usually severe and persisting multiorgan symptoms. Conversely, gain-of-function mutations in ENaC are associated with Liddle’s syndrome, which is typified by hypertension, low plasma renin activity and hypokalemia and hypoaldosteronism. Examining the structure of ENaC can help in understanding how mutations in ENaC result in two differing phenotypes. As this section will only be addressing the more recent efforts into understanding ENaC structure, the reader is directed to other references for a fuller discussion of ENaCs72.
2.4.1. Structure of ENaC-Insights from chicken ASIC 1
ENaCs are comprised of a trio of subunits, α, β and γ, which together form the channel (Figure 10A). The three subunits share ~35% homology by amino acid sequence. Each subunit consists of two transmembrane domains, with short N- and C-termini, and an extensive extracellular loop73. The first inroads into ENaC structural understanding was made in 2007 with a truncated, non-active crystal structure of chicken ASIC (cASIC1) at 1.9 Å resolution74. In spite of the observation that there is little overall homology between cASIC1 and ENaC (20%), modeling suggests that the structure of these two channels overlap well75. The structure included a fist like hand, with a negatively charged pocket between the thumb and palm which, when mutated (D356N and D350N), diminished proton sensitivity74. However, there is still uncertainty as to the residues that confer acid sensitivity. A comprehensive mutagenesis study covering 40 conserved charged amino acids, identified four residues, E63, H72, H73, and D78 which, when mutated, abolished proton sensitivity76. A subsequent study later suggested that the initial residues identified were involved in stabilizing the interaction between the thumb and finger, with an enhancing effect on acid sensing77. What is apparent is that the relationship between acid sensitivity and gating is complex, and it is likely that multiple sites78 interact to contribute to gating and acid sensitivity. The initial ASIC structure was followed by another one that extended into transmembrane domain 2, however, when tested for function, this version of ASIC had reduced activity79. The structure of the extracellular and pore regions provides insight into channel assembly, processing, and the ability of these channels to sense the external environment. However, absence of intracellular structures precludes insight into important interactions with intracellular factors that regulate trafficking and function. Moreover, a striking difference that remains to be resolved is that ASIC is acid sensing whereas ENaC is not gated by pH.
Figure 10.

A). Schematic diagram of the arrangement of the α, β and γ subunits of ENaC. Each ENaC subunit consists of a short N and C-termini, and an extensive extracellular loop. The C-terminus contains a PPXY motif that interacts with the Nedd-4 WW domains. See text for details. B). ENaC is localized to the apical membrane. Loss-of-function mutations result in non-functional ENaCs either being mistrafficked, or unable to conduct. These mutations lead to impaired Na+ transport. C). Gain-of-function mutations are a result of mutations to the binding site of Nedd-4, a protein required for the ubiqination of ENaC, leading to an accumulation of ENaC, and increased Na+ cellular absorption.
2.4.2. Loss-of-function mutations in ENaC- Pseudohypoaldosteronism
The link between ENaC and pseudohypoaldosteronism (PHA) was first made by homozygosity mapping in 199680. PHA is characterized by the inability to respond to aldosterone, resulting in sodium wasting (Figure 10B). Aldosterone is linked to ENaC as it regulates the gene expression of ENaC81. Since 1996, several mutations have been described which result in loss or reduced ENaC function82. A missense mutation G37S in the β subunit, and conserved across all three subunits was found to significantly reduce currents by altering channel gating83. Moreover, when the same mutation was made to the α and γ subunits, currents were also reduced, without affecting channel trafficking to the membrane83. Further analysis of G37S mutation revealed that the voltage sensitivity of the channel was altered at hyperpolarized potentials84. A stop mutation in the α subunit at R508X was surprisingly found to still form channels, with similar Na+ conductance with β and γ, although, it was apparent that the channel plasma density was decreased, implying a trafficking role for the C-terminus of the α subunit85. In contrast, another stop mutation, R492X α demonstrated decreased currents, though a trafficking defect was not reported86.
2.4.3. Gain-of-function mutations in ENaC– Liddle’s syndrome
The mutations in ENaC that are associated with Liddle’s syndrome all appear to lead to gain-of-function (Figure 10C). These mutants still respond to stimuli, including aldosterone and vasopressin87. Moreover, these mutants have a longer half life in the cell, as a result of no longer being targets of ubiquination. These gain-of-function mutations correlated with truncations and missense mutations in the C-terminal regions of β and γ, and are associated with a proline rich region88. Site directed mutagenesis of the proline rich sequence with the C-terminal tail revealed a PPXY sequence present in α, γ and β subunits, which bind WW domains. A WW domain is a 38 amino acid residue region that folds into a three stranded β sheet, and has been given its name due to two conserved tryptophan (W) residues. When PPXY mutants are expressed in Xenopus oocyte expression systems, the resultant channels have increased activity89. The PPXY motif may therefore provide a target for small molecule screening studies90.
In addition to mutations in the PPXY site, mutations have also been observed at other sites, including α ENaC C618F91, A663T and A334T, all of which result in a human phenotype of essential hypertension. The two mutations C618F and A663T resulted in increased activity, and this perceived enhanced activity was through an increase in the number of channels at the membrane92. Of particular note is that although the cysteine residue at 618 is conserved across mammalian species, this is not the case in amphibian or avian ENaC, thus hindering the application of ENaC structure and functional correlation across species.
2.4.3.1. Nedd-4
One important protein modulator of ENaC is Nedd-4, a ubiquitin protein ligase. The protein consists of an N-terminal C2 domain, multiple WW domains and a C-terminal ubiquitin ligase domain. The interface of interaction is through WW domains on Nedd-492, which bind to the PPXY motifs present in each of the ENaC subunits, and are believed to suppress ENaC activity at the plasma membrane by ubiquination93. Whereas mutations to the PPXY region of ENaC are associated with Liddle’s syndrome, mutations in Nedd-4 mimic Liddle’s syndrome. Less is known structurally about this protein, though the solution structure of ENaC with Nedd-4 has been published94. The WW domains 2–4 of Nedd-4 interact with each of the PPXY regions of all three ENaC subunits, with domain 3 necessary and sufficient for binding95. Mutations to domains 2 and 4 decreased the inhibitory effect of Nedd-4, indicating a regulatory role for the other WW domains95.
A number of animal models incorporating various knockouts of ENaC have been described. The first transgenic knockout mouse in was described in 199796. Phenotypically, this mouse had metabolic acidosis, urinary salt-wasting, growth retardation, and 50% mortality by days 5–9, primarily due to fluid collection in the lungs. By contrast, the transgenic knockout β ENaC mouse mimicked the human condition of PHA, without the pulmonary phenotype97. Another β ENaC mutant mouse incorporated a stop codon in the C-terminus to mimic the first reported mutation by Liddle98. The mouse recapitulated many of the symptoms observed in the human form of Liddle’s disease. Finally, a cre-flox mouse model resulted in targeted deletion of α ENaC in the connecting tubule and collection duct had high Na+ and a higher volume of urine99. Although there are already a number of mouse models, future mouse models with mutations in ENaC will enable researchers to determine the functional importance of each of the different subunits individually, and in a particular region of the kidney.
2.4.4. Pharmacology and treatment
One striking pharmacological characteristic about the ENaC channels is their inhibition by amiloride, a guanidium group compound. Indeed, the channel was previously identified as being amiloride sensitive. In addition to the pore blocking compound, amiloride, and its derivatives, other compounds are actively being investigated as inhibitors of ENaC. As mentioned above, the PPXY motif may provide a useful target for small molecule screening studies90. However, activators of ENaC, which would be therapeutically applicable for PHA patients, have not yet been identified. One promising compound, S3969, was found to increase open probability of ENaC by an interaction with the extracellular loop of the β subunit. Even more promising was the rescue of activity for the G37S mutation described above100. Unfortunately, this compound could only be used when all three ENaC subunits were expressed, thus possibly limiting its benefit on patients with truncated ENaC subunits.
2.5. A special case of TRPs-Polycystin 2
Autosomal dominant polycystic kidney disease (ADPKD) is a common (prevelence of 1:500–1:800) adult onset disease typified by cysts in the kidney, pancreas and bile duct. In addition, numerous vascular complaints including mitral valve prolapse, aneurysms and hypertension. Mutations in TRPP2101 also known as polycystin 2, account for at least 15% of all cases of ADPKD. Mutations in polycystin 1, a non-TRP protein comprise the remaining 85%102. Like the other channelopathies discussed in this review, there is no known cure for polycystic kidney disease, with renal transplant and dialysis being the main means of treatment to circumvent renal failure. How mutations of the polycystin genes lead to cystic development is still an open question, although it is known from animal models that mutation of both copies of either polycystin gene is embryonic lethal103. The late onset of disease in humans (usually age 40 for mutations in polycystin 1 and later for polycystin 2) is believed to be due to a two hit hypothesis, where the good copy of the gene is mutated, leading to the increased secretion of fluid by the renal epithelial cells, and the subsequent development of fluid-filled cysts and finally renal failure104. However, in the transgenic rat model expressing a truncated form of polycystin 2, cysts could still develop even though a wild-type full-length copy could still be detected105.
2.5.1. Structural insights on polycystin 2
Like other TRP channel proteins, polycystin 2 is believed to form a tetramer with each subunit contributing 6 transmembrane spanning domains, with both the N- and C-termini cytoplasmic106. Unlike the other channels discussed in this review, polycystin 2 is unique in that it primarily resides in the endoplasmic reticulum of renal epithelial cells, where it acts as a Ca2+ release channel107. One particular site thought to be involved in activation and gating of the channel is at the EF hand of the C-terminus108. EF hands comprise a helix-loop-helix motif, with six well-conserved residues within the loop to coordinate Ca2+. EF hands are generally found as pairs of helix-loop helix motifs, with the two sites usually showing cooperative binding of Ca2+109. However, the EF hand of polycystin 2 is particularly interesting, as it has one non-cannoical, non-Ca2+ binding site, due to the loss of the Ca2+ binding loop, whereas the other EF hand binds Ca2+110 (Figure 7). In addition to the EF hand motif, there are believed to be other Ca2+ interacting sites within the C-terminus of PC2111.
Little is known about the transmembrane configuration of polycystin 2, however, the pore size of polycystin 2 is large (11 Å), as based on experiments using organic cation permeation112. A recent focus has been on the structural and functional features of the C-terminus tail (amino acids 704 to 968). In addition to the EF hand described above, there is also a linker region that has several serine residues that can be phosphorylated. One phosphorylation site, S812 has been shown to be constitutively phosphorylated in the cell, and when mutated, the channel has a decreased Ca2+ sensitivity113. Another site, S829 is phosphorylated by Aurora A, and reduces Ca2+ release114. Downstream of the phosphorylation sites is the coiled-coil domain, which has been extensively studied115. In recent studies, the region of the coiled-coil has been further refined, with initial prediction studies indicating a more extensive region. Moreover, modeling indicates that there is a kink in the coil, one third of the way down108. In isolation, the coiled-coil regions can exist as a monomer, dimer, but also as a trimer, and most recently as a tetramer116 in the presence and absence of Ca2+111.
Truncation of the channel at L703, a site that leads to the loss of the C-terminal tail but preserves the remaining channel forming regions, results in a protein that can traffic to the plasma membrane. The truncated L703 protein, however, can still function as a channel, albeit with openings only observed with depolarized holding potentials107, which could be functionally relevant when the truncated proteins moves to the plasma membrane. These results are consistent with other truncated version of polycystin 2, which have also demonstrated channel activity117. However, at 0 mV, the resting membrane potential of the endoplasmic reticulum, no currents are observed, thus it is unlikely that truncated polycystin 2 in the endoplasmic reticulum functions as a Ca2+ channel107.
In addition to the coiled-coil, two other interaction domains that enable polycystin to interact with itself have been identified, including a N-terminal domain118 and a region between the 5–6 loop of the polycystin 2 channel119. The region between the 5–6 loop of polycystin 2 has also been described to interact with the protein Hax-1120.
2.5.2. Mutations to polycystin 2 and interacting proteins
Over 100 mutations for polycystin 2 have been documented, with the majority of these resulting in truncations to polycystin 2. Mutations in polycystin 2 are fairly evenly spread throughout the protein with no hotspots. One functionally validating mutation is D511V, located on the putative 5th transmembrane domain. Although this variant correctly localizes to the endoplasmic reticulum, the channel is functionally unable to conduct Ca2+107. For other known mutations, especially the C-termini truncations, it is unclear whether the downstream pathology occurs due to increased trafficking to the plasma membrane, or a loss of intracellular Ca2+ signaling121.
Polycystin 2 is known to bind and modulate other Ca2+ release channels within the endoplasmic reticulum, namely the inositol trisphosphate (InsP3) and ryanodine receptors. Polycystin 2 interacts with InsP3R type 1 through its C-terminal tail to sustain Ca2+ release122. A subsequent study demonstrated that polycystin 2 could also alter Ca2+ release through an interaction with InsP3R type 3 in a region of the C-terminal tail that is highly acidic123, between the EF-hand and the coiled-coil. The N-terminus of polycystin 2 also binds to the ryanodine receptor, however, only the C-terminus of polycystin 2 was found to modulate the ryanodine receptor currents by decreasing open probability124.
Although polycystin 2 is largely found within the endoplasmic reticulum, with over 99% expressed in this region, polycystin 2 also localizes to the primary cilia through an N-terminus RVXP motif125. In the cilia, polycystin 2 is believed to act in concert with polycystin 1 as a chemical and/or flow sensor126. A subpopulation of polycystin 2 is detectable in the primary cilium of epithelial cells, where polycystin 1 is also present126. It is believed that polycystin 2 present within the cilia contributes to Ca2+ influx upon sheer stress127. Indeed, cells deficient in polycystin 1 have impaired cillary mechanosenation, and reduced Ca2+ signaling in the cilia128.
Polycystin 2 interaction with polycystin 1 has been shown by multiple groups, and the interaction site is through the coiled-coil region of the C-termini tail129. The interaction site was verified using the R878130 variant, which had a single amino acid deleted from the KRRE cluster in the coiled-coil. This variant induces a rotation of the helix, and thus disrupts the downstream interaction site. In addition to an interaction with polycystin 1, other investigators have identified possible interactions with other TRP channels such as TRPC1131 and TRPV4132. In overexpression systems, polycystin 2 together with TRPC1 or TRPV4 produce enhanced currents131–132. Polycystin 2 has also been shown to form heterotetramers with an alternating 2:2 stoichiometry with TRPC1 and TRPV4, as assessed by atomic force microscopy133. However, what interaction these heterotetramers may have physiologically is not yet clear, as the majority of polycystin 2 resides on the endoplasmic reticulum, with little expression on the plasma membrane, where TRPC1 and TRPV4 predominantly reside. As the TRPC1 knockout mouse does have polycystic kidneys134, it is unlikely that the interaction between polycystin 2 and TRPC1 is important in the progression of polycystic kidney disease. However, the interaction between TRPV4 and polycystin 2 may be important in flow sensing, as both of these proteins co-localize to cilia132.
2.5.3. Pharmacology and treatment
Although no known specific inhibitors for polycystin 2 exist112, there have been exciting developments using triptolide, the active ingredient the traditional Chinese medicine Lei Gong Teng, which acts to induce Ca2+ release through polycystin 2135. Application of triptolide was found to reduce cyst growth136. Kidney cysts in animals with altered polycystin levels were also shown to reduce in size following treatment with the mTOR inhibitor, rapamycin137. However, results from clinical trials have been disappointing, as the dose required to observe cystic reduction was at a concentration where deleterious side effects occur138.
3. Conclusion and perspectives
The critical balance of electrolytes is largely controlled through a functioning kidney and the many ion channels localized to specific regions. Mutations in any one of these ion channels can cause an alteration in ion balance leading to renal disease. As mutations to ion channels can cause a variety of effects, including cell surface trafficking, increased activity or decreased activity, the interventions into preventing and treating ion channel related disease must be similarly varied. For diseases where the mutations result in a gain-of-function (eg: Liddle’s syndrome), pharmacological approaches finding specific antagonists will help. Conversely, a more complex approach will be warranted where a loss-of-function mutation results in disease (eg: Dent’s disease). In this case, the use of high throughput techniques such as LC-MS/MS is another exciting avenue in understanding and targeting the downstream pathways139.
The recent advances in genetic technology have enabled researches to identify the exact location of mutations within renal ion channels that lead to disease. Simultaneously, great advances in uncovering the underlying structural-functional relationships have been made. Understanding the molecular, structural and consequential effects of ion channel mutations in renal disease will therefore enable the development of new treatment strategies for the prevention and treatment of renal failure.
Figure 11.
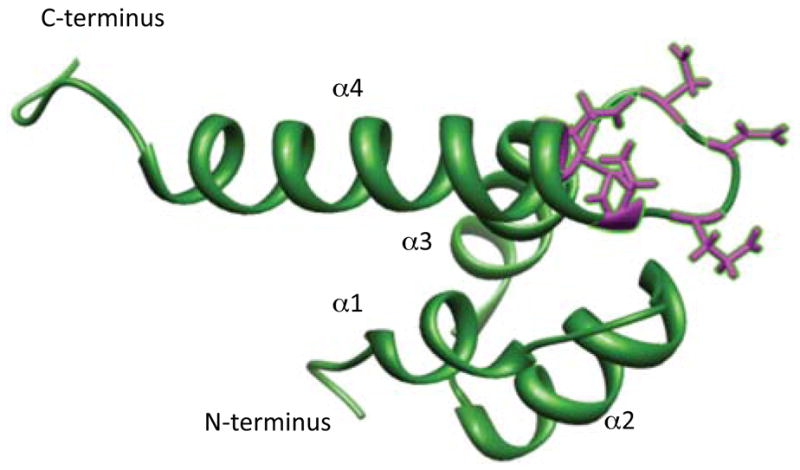
The EF hand domain of polycystin 2 (amino acids 720 to 797), found in the C-terminus. Polycystin 2 contains two EF hand motifs. The EF hand motif formed from α helices 1 and 2 is non-cannonical and cannot bind Ca2+ as critical residues required for Ca2+ binding are missing. The residues involved in Ca2+ coordination between a helices 3 and are highlighted in purple. PDB accession code: 2KQ6. Structure was modified the UCSF Chimera package142.
Acknowledgments
Research on polycystin 2 in the Ehrlich lab is funded by NIH grants P30 DK090744I, R01 DK61747 and R01 DK087844. I.Y.K. is the recipient of an American Heart Association Postdoctoral Fellowship R10682. We thank Dr A. Rachel Gallagher for comments.
Biographies

Dr. Ivana Y. Kuo is an AHA postdoctoral fellow with Dr Ehrlich, and has a keen interest in ion channels and calcium signaling. She received her PhD in Neuroscience, studying voltage-gated calcium channels in the cerebrovasculature at The Australian National University, under Dr. Caryl Hill in 2010.

Dr. Barbara E. Ehrlich is professor and director of the Molecular Hermeneutics group in the Department of Pharmacology, Yale University. She has a long-standing interest in intracellular calcium signaling and ion channel function. She obtained her PhD at UCLA with Dr. Jared Diamond, before doing postdoctoral studies with Dr. Alan Finkelstein at Albert Einstein College of Medicine.
References
- 1.(a) Miyamoto K, Haito-Sugino S, Kuwahara S, Ohi A, Nomura K, Ito M, Kuwahata M, Kido S, Tatsumi S, Kaneko I, Segawa H. J Pharm Sci. 2011;100:3719. doi: 10.1002/jps.22614. [DOI] [PubMed] [Google Scholar]; (b) Saito H. Pharmacol Ther. 2010;125:79. doi: 10.1016/j.pharmthera.2009.09.008. [DOI] [PubMed] [Google Scholar]; (c) Jentsch TJ. Crit Rev Biochem Mol Biol. 2008;43:3. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]; (d) Lang F, Capasso G, Schwab M, Waldegger S. Clin Exp Nephrol. 2005;9:91. doi: 10.1007/s10157-005-0355-x. [DOI] [PubMed] [Google Scholar]
- 2.Amirlak I, Dawson KP. QJM. 2000;93:207. doi: 10.1093/qjmed/93.4.207. [DOI] [PubMed] [Google Scholar]
- 3.(a) Holtzclaw JD, Grimm PR, Sansom SC. Curr Opin Nephrol Hypertens. 2011;20:512. doi: 10.1097/MNH.0b013e3283488889. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pluznick JL, Sansom SC. Am J Physiol Renal Physiol. 2006;291:F517. doi: 10.1152/ajprenal.00118.2006. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi K, Wakino S, Sugano N, Ozawa Y, Homma K, Saruta T. Circ Res. 2007;100:342. doi: 10.1161/01.RES.0000256155.31133.49. [DOI] [PubMed] [Google Scholar]
- 5.(a) de Borst MH, Benigni A, Remuzzi G. Nat Clin Pract Nephrol. 2008;4:265. doi: 10.1038/ncpneph0785. [DOI] [PubMed] [Google Scholar]; (b) Rumballe B, Georgas K, Wilkinson L, Little M. Pediatr Nephrol. 2010;25:1005. doi: 10.1007/s00467-009-1392-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nilius B. Biochim Biophys Acta. 2007;1772:805. doi: 10.1016/j.bbadis.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F. PLoS One. 2009;4:e7771. doi: 10.1371/journal.pone.0007771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dietrich A, Mederos YSM, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Mol Cell Biol. 2005;25:6980. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, Hoenderop JG. J Biol Chem. 2004;279:19. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]; (b) Voets T, Janssens A, Prenen J, Droogmans G, Nilius B. J Gen Physiol. 2003;121:245. doi: 10.1085/jgp.20028752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mio K, Ogura T, Hara Y, Mori Y, Sato C. Biochem Biophys Res Commun. 2005;333:768. doi: 10.1016/j.bbrc.2005.05.181. [DOI] [PubMed] [Google Scholar]
- 11.Mio K, Ogura T, Kiyonaka S, Hiroaki Y, Tanimura Y, Fujiyoshi Y, Mori Y, Sato C. J Mol Biol. 2007;367:373. doi: 10.1016/j.jmb.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 12.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC. Nat Genet. 2002;31:171. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 13.Stewart AP, Egressy K, Lim A, Edwardson JM. Biochem Biophys Res Commun. 2010;394:383. doi: 10.1016/j.bbrc.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 14.Pedretti A, Marconi C, Bettinelli I, Vistoli G. Biochim Biophys Acta. 2009;1788:973. doi: 10.1016/j.bbamem.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Fujiwara Y, Minor DL., Jr J Mol Biol. 2008;383:854. doi: 10.1016/j.jmb.2008.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Du J, Jiang J, Ratzan W, Su LT, Runnels LW, Yue L. J Biol Chem. 2007;282:25817. doi: 10.1074/jbc.M608972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. Science. 2005;308:1801. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 18.Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. Nat Genet. 2005;37:739. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santin S, Ars E, Rossetti S, Salido E, Silva I, Garcia-Maset R, Gimenez I, Ruiz P, Mendizabal S, Luciano Nieto J, Pena A, Camacho JA, Fraga G, Cobo MA, Bernis C, Ortiz A, de Pablos AL, Sanchez-Moreno A, Pintos G, Mirapeix E, Fernandez-Llama P, Ballarin J, Torra R, Zamora I, Lopez-Hellin J, Madrid A, Ventura C, Vilalta R, Espinosa L, Garcia C, Melgosa M, Navarro M, Gimenez A, Cots JV, Alexandra S, Caramelo C, Egido J, San Jose MD, de la Cerda F, Sala P, Raspall F, Vila A, Daza AM, Vazquez M, Ecija JL, Espinosa M, Justa ML, Poveda R, Aparicio C, Rosell J, Muley R, Montenegro J, Gonzalez D, Hidalgo E, de Frutos DB, Trillo E, Gracia S, de los Rios FJ. Nephrol Dial Transplant. 2009;24:3089. doi: 10.1093/ndt/gfp229. [DOI] [PubMed] [Google Scholar]
- 20.Lowik MM, Groenen PJ, Levtchenko EN, Monnens LA, van den Heuvel LP. Eur J Pediatr. 2009;168:1291. doi: 10.1007/s00431-009-1017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guran T, Akcay T, Bereket A, Atay Z, Turan S, Haisch L, Konrad M, Schlingmann KP. Nephrol Dial Transplant. 2011 doi: 10.1093/ndt/gfr300. [DOI] [PubMed] [Google Scholar]
- 22.(a) Chubanov V, Schlingmann KP, Waring J, Heinzinger J, Kaske S, Waldegger S, Mederosy Schnitzler M, Gudermann T. J Biol Chem. 2007;282:7656. doi: 10.1074/jbc.M611117200. [DOI] [PubMed] [Google Scholar]; (b) Chubanov V, Waldegger S, Mederosy Schnitzler M, Vitzthum H, Sassen MC, Seyberth HW, Konrad M, Gudermann T. Proc Natl Acad Sci USA. 2004;101:2894. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elizondo MR, Budi EH, Parichy DM. Endocrinology. 2010;151:5700. doi: 10.1210/en.2010-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Yogi A, Callera GE, O’Connor SE, He Y, Correa JW, Tostes RC, Mazur A, Touyz RM. J Hypertens. 2011;29:1400. doi: 10.1097/HJH.0b013e32834786d6. [DOI] [PubMed] [Google Scholar]; (b) Sontia B, Montezano AC, Paravicini T, Tabet F, Touyz RM. Hypertension. 2008;51:915. doi: 10.1161/HYPERTENSIONAHA.107.100339. [DOI] [PubMed] [Google Scholar]
- 25.Moran MM, McAlexander MA, Biro T, Szallasi A. Nat Rev Drug Discov. 2011;10:601. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- 26.(a) Picollo A, Pusch M. Nature. 2005;436:420. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]; (b) Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Nature. 2005;436:424. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- 27.Uchida S, Sasaki S, Furukawa T, Hiraoka M, Imai T, Hirata Y, Marumo F. J Biol Chem. 1993;268:3821. [PubMed] [Google Scholar]
- 28.(a) Ludewig U, Pusch M, Jentsch TJ. Nature. 1996;383:340. doi: 10.1038/383340a0. [DOI] [PubMed] [Google Scholar]; (b) Middleton RE, Pheasant DJ, Miller C. Nature. 1996;383:337. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- 29.Dutzler R. Curr Opin Struct Biol. 2006;16:439. doi: 10.1016/j.sbi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Meyer S, Savaresi S, Forster IC, Dutzler R. Nat Struct Mol Biol. 2007;14:60. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- 31.Zifarelli G, Pusch M. EMBO Rep. 2009;10:1111. doi: 10.1038/embor.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markovic S, Dutzler R. Structure. 2007;15:715. doi: 10.1016/j.str.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 33.Wu F, Roche P, Christie PT, Loh NY, Reed AA, Esnouf RM, Thakker RV. Kidney Int. 2003;63:1426. doi: 10.1046/j.1523-1755.2003.00859.x. [DOI] [PubMed] [Google Scholar]
- 34.Grand T, Mordasini D, L’Hoste S, Pennaforte T, Genete M, Biyeyeme MJ, Vargas-Poussou R, Blanchard A, Teulon J, Lourdel S. Kidney Int. 2009;76:999. doi: 10.1038/ki.2009.305. [DOI] [PubMed] [Google Scholar]
- 35.Smith AJ, Reed AA, Loh NY, Thakker RV, Lippiat JD. Am J Physiol Renal Physiol. 2009;296:F390. doi: 10.1152/ajprenal.90526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carballo-Trujillo I, Garcia-Nieto V, Moya-Angeler FJ, Anton-Gamero M, Loris C, Mendez-Alvarez S, Claverie-Martin F. Nephrol Dial Transplant. 2003;18:717. doi: 10.1093/ndt/gfg016. [DOI] [PubMed] [Google Scholar]
- 37.Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ. Nature. 2000;408:369. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- 38.Wang SS, Devuyst O, Courtoy PJ, Wang XT, Wang H, Wang Y, Thakker RV, Guggino S, Guggino WB. Hum Mol Genet. 2000;9:2937. doi: 10.1093/hmg/9.20.2937. [DOI] [PubMed] [Google Scholar]
- 39.Wright J, Morales MM, Sousa-Menzes J, Ornellas D, Sipes J, Cui Y, Cui I, Hulamm P, Cebotaru V, Cebotaru L, Guggino WB, Guggino SE. Physiol Genomics. 2008;33:341. doi: 10.1152/physiolgenomics.00024.2008. [DOI] [PubMed] [Google Scholar]
- 40.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Nat Genet. 1996;12:24. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 41.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Nat Genet. 1997;17:171. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 42.Schlingmann KP, Konrad M, Jeck N, Waldegger P, Reinalter SC, Holder M, Seyberth HW, Waldegger S. N Engl J Med. 2004;350:1314. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- 43.(a) Mupanomunda MM, Tian B, Ishioka N, Bukoski RD. Am J Physiol Renal Physiol. 2000;278:F644. doi: 10.1152/ajprenal.2000.278.4.F644. [DOI] [PubMed] [Google Scholar]; (b) Mupanomunda MM, Ishioka N, Bukoski RD. Am J Physiol. 1999;276:H1035. doi: 10.1152/ajpheart.1999.276.3.H1035. [DOI] [PubMed] [Google Scholar]
- 44.Martinez GQ, Maduke M. PLoS One. 2008;3:e2746. doi: 10.1371/journal.pone.0002746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.(a) Arreola J, Begenisich T, Melvin JE. J Physiol. 2002;541:103. doi: 10.1113/jphysiol.2002.016485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jordt SE, Jentsch TJ. The EMBO journal. 1997;16:1582. doi: 10.1093/emboj/16.7.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu Y, Xu C, Pan X, Ren H, Wang W, Meng X, Huang F, Chen N. Clin Genet. 2010;77:155. doi: 10.1111/j.1399-0004.2009.01288.x. [DOI] [PubMed] [Google Scholar]
- 47.Lin CM, Tsai JD, Lo YF, Yan MT, Yang SS, Lin SH. Eur J Pediatr. 2009;168:1129. doi: 10.1007/s00431-008-0883-y. [DOI] [PubMed] [Google Scholar]
- 48.Robitaille P, Merouani A, He N, Pei Y. Eur J Pediatr. 2011;170:1209. doi: 10.1007/s00431-011-1464-z. [DOI] [PubMed] [Google Scholar]
- 49.Enriquez R, Adam V, Sirvent AE, Garcia-Garcia AB, Millan I, Amoros F. Int Urol Nephrol. 2010;42:1099. doi: 10.1007/s11255-010-9850-4. [DOI] [PubMed] [Google Scholar]
- 50.(a) Jeck N, Konrad M, Peters M, Weber S, Bonzel KE, Seyberth HW. Pediatr Res. 2000;48:754. doi: 10.1203/00006450-200012000-00009. [DOI] [PubMed] [Google Scholar]; (b) Zelikovic I, Szargel R, Hawash A, Labay V, Hatib I, Cohen N, Nakhoul F. Kidney Int. 2003;63:24. doi: 10.1046/j.1523-1755.2003.00730.x. [DOI] [PubMed] [Google Scholar]
- 51.Matsumura Y, Uchida S, Kondo Y, Miyazaki H, Ko SB, Hayama A, Morimoto T, Liu W, Arisawa M, Sasaki S, Marumo F. Nat Genet. 1999;21:95. doi: 10.1038/5036. [DOI] [PubMed] [Google Scholar]
- 52.Waldegger S, Jeck N, Barth P, Peters M, Vitzthum H, Wolf K, Kurtz A, Konrad M, Seyberth HW. Pflugers Arch. 2002;444:411. doi: 10.1007/s00424-002-0819-8. [DOI] [PubMed] [Google Scholar]
- 53.(a) Birkenhager R, Otto E, Schurmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NV, Antignac C, Sudbrak R, Kispert A, Hildebrandt F. Nat Genet. 2001;29:310. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]; (b) Estevez R, Boettger T, Stein V, Birkenhager R, Otto E, Hildebrandt F, Jentsch TJ. Nature. 2001;414:558. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- 54.Scholl U, Hebeisen S, Janssen AG, Muller-Newen G, Alekov A, Fahlke C. Proc Natl Acad Sci USA. 2006;103:11411. doi: 10.1073/pnas.0601631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tajima M, Hayama A, Rai T, Sasaki S, Uchida S. Biochem Biophys Res Commun. 2007;362:858. doi: 10.1016/j.bbrc.2007.08.097. [DOI] [PubMed] [Google Scholar]
- 56.Nomura N, Tajima M, Sugawara N, Morimoto T, Kondo Y, Ohno M, Uchida K, Mutig K, Bachmann S, Soleimani M, Ohta E, Ohta A, Sohara E, Okado T, Rai T, Jentsch TJ, Sasaki S, Uchida S. Am J Physiol Renal Physiol. 2011;301:F297. doi: 10.1152/ajprenal.00604.2010. [DOI] [PubMed] [Google Scholar]
- 57.Picollo A, Liantonio A, Didonna MP, Elia L, Camerino DC, Pusch M. EMBO reports. 2004;5:584. doi: 10.1038/sj.embor.7400169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liantonio A, Picollo A, Carbonara G, Fracchiolla G, Tortorella P, Loiodice F, Laghezza A, Babini E, Zifarelli G, Pusch M, Camerino DC. Proc Natl Acad Sci USA. 2008;105:1369. doi: 10.1073/pnas.0708977105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP. Nat Genet. 1996;14:152. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 60.(a) Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R. Proc Natl Acad Sci USA. 2010;107:14490. doi: 10.1073/pnas.1003072107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’tHoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. N Engl J Med. 2009;360:1960. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Nature. 1993;362:31. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 62.Rapedius M, Haider S, Browne KF, Shang L, Sansom MS, Baukrowitz T, Tucker SJ. EMBO Rep. 2006;7:611. doi: 10.1038/sj.embor.7400678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.(a) Fakler B, Schultz JH, Yang J, Schulte U, Brandle U, Zenner HP, Jan LY, Ruppersberg JP. EMBO J. 1996;15:4093. [PMC free article] [PubMed] [Google Scholar]; (b) Doi T, Fakler B, Schultz JH, Schulte U, Brandle U, Weidemann S, Zenner HP, Lang F, Ruppersberg JP. J Biol Chem. 1996;271:17261. doi: 10.1074/jbc.271.29.17261. [DOI] [PubMed] [Google Scholar]
- 64.Choe H, Zhou H, Palmer LG, Sackin H. Am J Physiol. 1997;273:F516. doi: 10.1152/ajprenal.1997.273.4.F516. [DOI] [PubMed] [Google Scholar]
- 65.Sackin H, Nanazashvili M, Palmer LG, Krambis M, Walters DE. Biophys J. 2005;88:2597. doi: 10.1529/biophysj.104.051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flagg TP, Tate M, Merot J, Welling PA. J Gen Physiol. 1999;114:685. doi: 10.1085/jgp.114.5.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Konrad M, Vollmer M, Lemmink HH, van den Heuvel LP, Jeck N, Vargas-Poussou R, Lakings A, Ruf R, Deschenes G, Antignac C, Guay-Woodford L, Knoers NV, Seyberth HW, Feldmann D, Hildebrandt F. J Am Soc Nephrol. 2000;11:1449. doi: 10.1681/ASN.V1181449. [DOI] [PubMed] [Google Scholar]
- 68.Lorenz JN, Baird NR, Judd LM, Noonan WT, Andringa A, Doetschman T, Manning PA, Liu LH, Miller ML, Shull GE. J Biol Chem. 2002;277:37871. doi: 10.1074/jbc.M205627200. [DOI] [PubMed] [Google Scholar]
- 69.(a) Cantone A, Yang X, Yan Q, Giebisch G, Hebert SC, Wang T. Am J Physiol Renal Physiol. 2008;294:F1366. doi: 10.1152/ajprenal.00608.2007. [DOI] [PubMed] [Google Scholar]; (b) Wagner CA, Loffing-Cueni D, Yan Q, Schulz N, Fakitsas P, Carrel M, Wang T, Verrey F, Geibel JP, Giebisch G, Hebert SC, Loffing J. Am J Physiol Renal Physiol. 2008;294:F1373. doi: 10.1152/ajprenal.00613.2007. [DOI] [PubMed] [Google Scholar]
- 70.Freudenthal B, Kulaveerasingam D, Lingappa L, Shah MA, Brueton L, Wassmer E, Ognjanovic M, Dorison N, Reichold M, Bockenhauer D, Kleta R, Zdebik AA. Nephron Physiol. 2011;119:p40. doi: 10.1159/000330250. [DOI] [PubMed] [Google Scholar]
- 71.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. Mol Pharmacol. 2009;76:1094. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garty H, Palmer LG. Physiol Rev. 1997;77:359. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 73.(a) Renard S, Lingueglia E, Voilley N, Lazdunski M, Barbry P. J Biol Chem. 1994;269:12981. [PubMed] [Google Scholar]; (b) Snyder PM, Olson DR, Bucher DB. J Biol Chem. 1999;274:28484. doi: 10.1074/jbc.274.40.28484. [DOI] [PubMed] [Google Scholar]; (c) Snyder PM, McDonald FJ, Stokes JB, Welsh MJ. J Biol Chem. 1994;269:24379. [PubMed] [Google Scholar]; (d) Canessa CM, Horisberger JD, Rossier BC. Nature. 1993;361:467. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]; (e) Canessa CM, Merillat AM, Rossier BC. Am J Physiol. 1994;267:C1682. doi: 10.1152/ajpcell.1994.267.6.C1682. [DOI] [PubMed] [Google Scholar]
- 74.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Nature. 2007;449:316. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 75.Stockand JD, Staruschenko A, Pochynyuk O, Booth RE, Silverthorn DU. IUBMB Life. 2008;60:620. doi: 10.1002/iub.89. [DOI] [PubMed] [Google Scholar]
- 76.Paukert M, Chen X, Polleichtner G, Schindelin H, Grunder S. J Biol Chem. 2008;283:572. doi: 10.1074/jbc.M706811200. [DOI] [PubMed] [Google Scholar]
- 77.Yang H, Yu Y, Li WG, Yu F, Cao H, Xu TL, Jiang H. PLoS Biol. 2009;7:e1000151. doi: 10.1371/journal.pbio.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Y, Bianchi L. Biochemistry (Mosc) 2009;48:10005. doi: 10.1021/bi9014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gonzales EB, Kawate T, Gouaux E. Nature. 2009;460:599. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Strautnieks SS, Thompson RJ, Hanukoglu A, Dillon MJ, Hanukoglu I, Kuhnle U, Seckl J, Gardiner RM, Chung E. Hum Mol Genet. 1996;5:293. doi: 10.1093/hmg/5.2.293. [DOI] [PubMed] [Google Scholar]
- 81.Hamm LL, Feng Z, Hering-Smith KS. Curr Opin Nephrol Hypertens. 2010;19:98. doi: 10.1097/MNH.0b013e328332bda4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.(a) Thomas CP, Zhou J, Liu KZ, Mick VE, MacLaughlin E, Knowles M. Am J Respir Cell Mol Biol. 2002;27:314. doi: 10.1165/rcmb.2002-0029OC. [DOI] [PubMed] [Google Scholar]; (b) Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. Nat Genet. 1996;13:248. doi: 10.1038/ng0696-248. [DOI] [PubMed] [Google Scholar]
- 83.Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP, Rossier BC. EMBO J. 1997;16:899. doi: 10.1093/emboj/16.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kucher V, Boiko N, Pochynyuk O, Stockand JD. Biophys J. 2011;100:1930. doi: 10.1016/j.bpj.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bonny O, Chraibi A, Loffing J, Jaeger NF, Grunder S, Horisberger JD, Rossier BC. J Clin Invest. 1999;104:967. doi: 10.1172/JCI6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonny O, Knoers N, Monnens L, Rossier BC. Pediatr Nephrol. 2002;17:804. doi: 10.1007/s00467-002-0945-8. [DOI] [PubMed] [Google Scholar]
- 87.Auberson M, Hoffmann-Pochon N, Vandewalle A, Kellenberger S, Schild L. Am J Physiol Renal Physiol. 2003;285:F459. doi: 10.1152/ajprenal.00071.2003. [DOI] [PubMed] [Google Scholar]
- 88.(a) Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Nat Genet. 1995;11:76. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]; (b) Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. Proc Natl Acad Sci USA. 1995;92:11495. doi: 10.1073/pnas.92.25.11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. EMBO J. 1996;15:2381. [PMC free article] [PubMed] [Google Scholar]
- 90.Rotin D. BMC Biochem. 2008;9(Suppl 1):S5. doi: 10.1186/1471-2091-9-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Samaha FF, Rubenstein RC, Yan W, Ramkumar M, Levy DI, Ahn YJ, Sheng S, Kleyman TR. J Biol Chem. 2004;279:23900. doi: 10.1074/jbc.M401941200. [DOI] [PubMed] [Google Scholar]
- 92.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. EMBO J. 1996;15:2371. [PMC free article] [PubMed] [Google Scholar]
- 93.(a) Snyder PM, Olson DR, McDonald FJ, Bucher DB. J Biol Chem. 2001;276:28321. doi: 10.1074/jbc.M011487200. [DOI] [PubMed] [Google Scholar]; (b) Zhou R, Patel SV, Snyder PM. J Biol Chem. 2007;282:20207. doi: 10.1074/jbc.M611329200. [DOI] [PubMed] [Google Scholar]
- 94.Kanelis V, Rotin D, Forman-Kay JD. Nat Struct Biol. 2001;8:407. doi: 10.1038/87562. [DOI] [PubMed] [Google Scholar]
- 95.Tong Q, Menon AG, Stockand JD. Am J Physiol Renal Physiol. 2006;290:F821. doi: 10.1152/ajprenal.00312.2005. [DOI] [PubMed] [Google Scholar]
- 96.Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B, Gatzy J, Burnier M, Horisberger JD, Beermann F, Boucher R, Rossier BC. Proc Natl Acad Sci USA. 1997;94:11710. doi: 10.1073/pnas.94.21.11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB, Jr, Stokes JB, Welsh MJ, Williamson RA. Proc Natl Acad Sci USA. 1999;96:1727. doi: 10.1073/pnas.96.4.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, Hummler E, Rossier BC. J Am Soc Nephrol. 1999;10:2527. doi: 10.1681/ASN.V10122527. [DOI] [PubMed] [Google Scholar]
- 99.Christensen BM, Perrier R, Wang Q, Zuber AM, Maillard M, Mordasini D, Malsure S, Ronzaud C, Stehle JC, Rossier BC, Hummler E. J Am Soc Nephrol. 2010;21:1942. doi: 10.1681/ASN.2009101077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lu M, Echeverri F, Kalabat D, Laita B, Dahan DS, Smith RD, Xu H, Staszewski L, Yamamoto J, Ling J, Hwang N, Kimmich R, Li P, Patron E, Keung W, Patron A, Moyer BD. J Biol Chem. 2008;283:11981. doi: 10.1074/jbc.M708001200. [DOI] [PubMed] [Google Scholar]
- 101.(a) Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. Science. 1996;272:1339. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]; (b) Viribay M, Hayashi T, Telleria D, Mochizuki T, Reynolds DM, Alonso R, Lens XM, Moreno F, Harris PC, Somlo S, San Millan JL. Hum Genet. 1997;101:229. doi: 10.1007/s004390050621. [DOI] [PubMed] [Google Scholar]; (c) Xenophontos S, Constantinides R, Hayashi T, Mochizuki T, Somlo S, Pierides A, Deltas CC. Hum Mol Genet. 1997;6:949. doi: 10.1093/hmg/6.6.949. [DOI] [PubMed] [Google Scholar]
- 102.Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM, Walker DL, Bae K, Zhang QJ, Thompson PA, Miller JP, Harris PC. J Am Soc Nephrol. 2007;18:2143. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 103.(a) Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, Blum M, Dworniczak B. Curr Biol. 2002;12:938. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]; (b) Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R. Proc Natl Acad Sci USA. 2001;98:12174. doi: 10.1073/pnas.211191098. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu G, Markowitz GS, Li L, D’Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH, van Adelsberg J, Hou H, Jr, Kucherlapati R, Edelmann W, Somlo S. Nat Genet. 2000;24:75. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 104.(a) Qian F, Watnick TJ, Onuchic LF, Germino GG. Cell. 1996;87:979. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]; (b) Wu G, D’Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H, Jr, Kucherlapati R, Edelmann W, Somlo S. Cell. 1998;93:177. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 105.Gallagher AR, Hoffmann S, Brown N, Cedzich A, Meruvu S, Podlich D, Feng Y, Konecke V, de Vries U, Hammes HP, Gretz N, Witzgall R. J Am Soc Nephrol. 2006;17:2719. doi: 10.1681/ASN.2005090979. [DOI] [PubMed] [Google Scholar]
- 106.(a) Hoffmeister H, Gallagher AR, Rascle A, Witzgall R. Biochem J. 2011;433:285. doi: 10.1042/BJ20101141. [DOI] [PubMed] [Google Scholar]; (b) Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, Mochizuki T, Park JH, Witzgall R, Somlo S. J Biol Chem. 1999;274:28557. doi: 10.1074/jbc.274.40.28557. [DOI] [PubMed] [Google Scholar]
- 107.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S. Nat Cell Biol. 2002;4:191. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 108.Celic A, Petri ET, Demeler B, Ehrlich BE, Boggon TJ. J Biol Chem. 2008;283:28305. doi: 10.1074/jbc.M802743200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chazin WJ. Acc Chem Res. 2011;44:171. doi: 10.1021/ar100110d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Petri ET, Celic A, Kennedy SD, Ehrlich BE, Boggon TJ, Hodsdon ME. Proc Natl Acad Sci USA. 2010;107:9176. doi: 10.1073/pnas.0912295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schumann F, Hoffmeister H, Bader R, Schmidt M, Witzgall R, Kalbitzer HR. J Biol Chem. 2009;284:24372. doi: 10.1074/jbc.M109.025635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anyatonwu GI, Ehrlich BE. J Biol Chem. 2005;280:29488. doi: 10.1074/jbc.M504359200. [DOI] [PubMed] [Google Scholar]
- 113.Cai Y, Anyatonwu G, Okuhara D, Lee KB, Yu Z, Onoe T, Mei CL, Qian Q, Geng L, Wiztgall R, Ehrlich BE, Somlo S. J Biol Chem. 2004;279:19987. doi: 10.1074/jbc.M312031200. [DOI] [PubMed] [Google Scholar]
- 114.Plotnikova OV, Pugacheva EN, Golemis EA. J Cell Biol. 2011;193:1021. doi: 10.1083/jcb.201012061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Giamarchi A, Feng S, Rodat-Despoix L, Xu Y, Bubenshchikova E, Newby LJ, Hao J, Gaudioso C, Crest M, Lupas AN, Honore E, Williamson MP, Obara T, Ong AC, Delmas P. EMBO J. 2010;29:1176. doi: 10.1038/emboj.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ferreira FM, Oliveira LC, Germino GG, Onuchic JN, Onuchic LF. Proc Natl Acad Sci USA. 2011;108:9833. doi: 10.1073/pnas.1106766108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.(a) Chen XZ, Segal Y, Basora N, Guo L, Peng JB, Babakhanlou H, Vassilev PM, Brown EM, Hediger MA, Zhou J. Biochem Biophys Res Commun. 2001;282:1251. doi: 10.1006/bbrc.2001.4720. [DOI] [PubMed] [Google Scholar]; (b) Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, Babakhanlou H, Cruger G, Kanazirska M, Ye C, Brown EM, Hediger MA, Zhou J. Biochem Biophys Res Commun. 2001;282:341. doi: 10.1006/bbrc.2001.4554. [DOI] [PubMed] [Google Scholar]; (c) Delmas P, Nauli SM, Li X, Coste B, Osorio N, Crest M, Brown DA, Zhou J. FASEB J. 2004;18:740. doi: 10.1096/fj.03-0319fje. [DOI] [PubMed] [Google Scholar]; (d) Xu GM, Gonzalez-Perrett S, Essafi M, Timpanaro GA, Montalbetti N, Arnaout MA, Cantiello HF. J Biol Chem. 2003;278:1457. doi: 10.1074/jbc.M209996200. [DOI] [PubMed] [Google Scholar]
- 118.Feng S, Okenka GM, Bai CX, Streets AJ, Newby LJ, DeChant BT, Tsiokas L, Obara T, Ong AC. J Biol Chem. 2008;283:28471. doi: 10.1074/jbc.M803834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feng S, Rodat-Despoix L, Delmas P, Ong AC. J Biol Chem. 2011;286:18994. doi: 10.1074/jbc.M110.192286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gallagher AR, Cedzich A, Gretz N, Somlo S, Witzgall R. Proc Natl Acad Sci USA. 2000;97:4017. doi: 10.1073/pnas.97.8.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Miyagi K, Kiyonaka S, Yamada K, Miki T, Mori E, Kato K, Numata T, Sawaguchi Y, Numaga T, Kimura T, Kanai Y, Kawano M, Wakamori M, Nomura H, Koni I, Yamagishi M, Mori Y. J Biol Chem. 2009;284:34400. doi: 10.1074/jbc.M109.015149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Y, Wright JM, Qian F, Germino GG, Guggino WB. J Biol Chem. 2005;280:41298. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 123.Sammels E, Devogelaere B, Mekahli D, Bultynck G, Missiaen L, Parys JB, Cai Y, Somlo S, De Smedt H. J Biol Chem. 2010 doi: 10.1074/jbc.M109.090662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Proc Natl Acad Sci USA. 2007;104:6454. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Geng L, Okuhara D, Yu Z, Tian X, Cai Y, Shibazaki S, Somlo S. J Cell Sci. 2006;119:1383. doi: 10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- 126.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Nat Genet. 2003;33:129. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 127.Li Q, Montalbetti N, Wu Y, Ramos A, Raychowdhury MK, Chen XZ, Cantiello HF. J Biol Chem. 2006;281:37566. doi: 10.1074/jbc.M603643200. [DOI] [PubMed] [Google Scholar]
- 128.Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wandinger-Ness A, Bacallao R, Alper SL. Am J Phyiol Renal Physiol. 2007;292:F930. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.(a) Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. Nat Genet. 1997;16:179. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]; (b) Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Proc Natl Acad Sci USA. 1997;94:6965. doi: 10.1073/pnas.94.13.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Casuscelli J, Schmidt S, DeGray B, Petri ET, Celic A, Folta-Stogniew E, Ehrlich BE, Boggon TJ. Am J Phyiol Renal Physiol. 2009;297:F1310. doi: 10.1152/ajprenal.00412.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhu J, Yu Y, Ulbrich MH, Li MH, Isacoff EY, Honig B, Yang J. Proc Natl Acad Sci USA. 2011;108:10133. doi: 10.1073/pnas.1017669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P. EMBO Rep. 2008;9:472. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G. J Cell Biol. 2008;182:437. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.(a) Kobori T, Smith GD, Sandford R, Edwardson JM. J Biol Chem. 2009;284:35507. doi: 10.1074/jbc.M109.060228. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stewart AP, Smith GD, Sandford RN, Edwardson JM. Biophys J. 2010;99:790. doi: 10.1016/j.bpj.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dietrich A, Kalwa H, Storch U, Mederosy Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pflugers Arch. 2007;455:465. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- 135.Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, Crews CM. Proc Natl Acad Sci USA. 2007;104:4389. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM. J Am Soc Nephrol. 2008;19:1659. doi: 10.1681/ASN.2008030259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.(a) Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T. Proc Natl Acad Sci USA. 2006;103:5466. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu M, Wahl PR, Le Hir M, Wackerle-Men Y, Wuthrich RP, Serra AL. Kidney Blood Press Res. 2007;30:253. doi: 10.1159/000104818. [DOI] [PubMed] [Google Scholar]
- 138.(a) Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Horl WH, Obermuller N, Arns W, Pavenstadt H, Gaedeke J, Buchert M, May C, Gschaidmeier H, Kramer S, Eckardt KU. N Engl J Med. 2010;363:830. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]; (b) Perico N, Remuzzi G. Nature reviews Nephrology. 2010;6:696. doi: 10.1038/nrneph.2010.153. [DOI] [PubMed] [Google Scholar]; (c) Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, Wuthrich RP. N Engl J Med. 2010;363:820. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 139.(a) Sachs AN, Pisitkun T, Hoffert JD, Yu MJ, Knepper MA. Am J Physiol Renal Physiol. 2008;295:F1799. doi: 10.1152/ajprenal.90510.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pisitkun T, Bieniek J, Tchapyjnikov D, Wang G, Wu WW, Shen RF, Knepper MA. Physiol Genomics. 2006;25:263. doi: 10.1152/physiolgenomics.00214.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Corpet F. Nucleic Acids Res. 1988;16:10881. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dietrich A, Chubanov V, Gudermann T. J Am Soc Nephrol. 2010;21:736. doi: 10.1681/ASN.2009090948. [DOI] [PubMed] [Google Scholar]
- 142.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Journal of computational chemistry. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]