

Abstract
Mutations of the thyroid hormone receptor-β gene (THRB) cause resistance to thyroid hormone (RTH). A mouse model of RTH harboring a homozygous thyroid hormone receptor (TR)-β mutation known as PV (ThrbPV/PV mouse) spontaneously develops follicular thyroid cancer (FTC). Similar to RTH patients with mutations of two alleles of the THRB gene, the ThrbPV/PV mouse exhibits elevated thyroid hormones accompanied by highly nonsuppressible TSH. However, the heterozygous ThrbPV/+ mouse with mildly elevated TSH (∼2-fold) does not develop FTC. The present study examined whether the mutation of a single allele of the Thrb gene is sufficient to induce FTC in ThrbPV/+ mice under stimulation by high TSH. ThrbPV/+ mice and wild-type siblings were treated with propylthiouracil (PTU) to elevate serum TSH. ThrbPV/+mice treated with PTU (ThrbPV/+-PTU) spontaneously developed FTC similar to human thyroid cancer, but wild-type siblings treated with PTU did not. Interestingly, approximately 33% of ThrbPV/+-PTU mice developed asymmetrical thyroid tumors, as is frequently observed in human thyroid cancer. Molecular analyses showed activation of the cyclin 1-cyclin-dependent kinase-4-transcription factor E2F1 pathway to increase thyroid tumor cell proliferation of ThrbPV/+-PTU mice. Moreover, via extranuclear signaling, the PV also activated the integrin-Src-focal adhesion kinase-AKT-metalloproteinase pathway to increase migration and invasion of tumor cells. Therefore, mutation of a single allele of the Thrb gene is sufficient to drive the TSH-simulated hyperplastic thyroid follicular cells to undergo carcinogenesis. The present study suggests that the ThrbPV/+-PTU mouse model potentially could be used to gain insights into the molecular basis underlying the association between thyroid cancer and RTH seen in some affected patients.
Thyroid hormone receptors (TRs) are ligand-dependent transcription factors that mediate the biological activities of the thyroid hormone T3 in growth, development, differentiation, and metabolic homeostasis. Two genes, located on two different chromosomes, encode three major T3-binding TR isoforms: TR1, TRβ2, and TRα1. The critical role of TR is apparent from the deleterious effects of mutations of the THRB gene. One well-known consequence of TRβ mutations is exhibited in the syndrome of resistance to thyroid hormone (RTH). This disease is manifested by elevated levels of circulating thyroid hormones associated with normal or high levels of serum TSH (1). RTH can appear in a sporadic form, but most commonly it is a familial syndrome with autosomal dominant inheritance (1). Most patients are heterozygotes, with only one mutant THRB gene, and the clinical symptoms are mild. Reported clinical features include goiter, short stature, decreased weight, tachycardia, hearing loss, attention-deficit hyperactivity disorder, decreased intelligence quotient, and dyslexia (1, 2). To date, four patients with homozygous mutations of the THRB gene have been reported (3–5). These patients displayed an extraordinary and complex phenotype of extreme thyroid hormone resistance with very high levels of thyroid hormones and TSH. TRβ mutants derived from RTH patients have reduced or total loss of T3-binding affinities and transcriptional capacities. TRβ mutants act in a dominant-negative fashion to cause the clinical phenotype (2, 5, 6).
A mouse model of RTH created by knocking in a mutation (denoted as PV) into the Thrb gene has been extensively characterized (7, 8). PV was identified in an RTH patient that has a frame shift mutation in the C-terminal 14 amino acids (9). PV has completely lost T3 binding and transcription capacity. The heterozygous ThrbPV/+ mice faithfully recapitulate the mild resistance manifested in RTH patients with mutations of a single allele of the THRB gene. The homozygous ThrbPV/PV mice exhibit severe target tissue resistance as reflected by thyroid function tests [∼400-fold elevation in TSH and 9- to 15-fold increase in thyroid hormones as compared with wild-type mice (7)], similar to those RTH patients with mutations of both THRB alleles (3–5).
Remarkably, as the homozygous ThrbPV/PV mice age, they spontaneously develop follicular thyroid carcinoma similar to human thyroid cancer (10). Hyperplasia of follicular cells is evident beginning at approximately 3 months of age. Capsular and vascular invasion by thyroid tumor cells is frequent in mice at 5–7 months, followed by development of anaplasia and lung metastasis with a frequency similar to that of human thyroid follicular carcinoma (10). Moreover, as found in human follicular thyroid cancer, ThrbPV/PV mice exhibit aberrant signaling pathways that include constitutive activation of phosphatidyl inositol 3-kinase (PI3K)/AKT (11, 12), repression of peroxisome proliferator-activated receptor-γ signaling (13, 14), and aberrant accumulation of both the pituitary tumor-transforming gene (15, 16) and β-catenin (17). Recent studies have shown that thyroid cell proliferation stimulated by the elevated TSH is necessary but not sufficient to induce metastatic thyroid cancer in ThrbPV/PV mice (18). The oncogenic effect of a mutated TRβ is required to empower hyperplastic follicular cells to invade and to metastasize in ThrbPV/PV mice (18).
Taken together, these findings indicate that the deleterious effects of TRβ mutations go beyond RTH. This notion prompted us to ask whether mutation of a single allele of the THRB gene is sufficient to drive the TSH-stimulated hyperplastic thyroid follicular cells to undergo carcinogenesis. Because most RTH patients are heterozygous for the mutation of the THRB gene, this question is critical to understanding whether elevated TSH could potentially pose a risk of thyroid cancer in these patients. The importance of this question is exemplified by the recent reports that papillary thyroid cancer is associated with several RTH patients with single mutations of the THRB gene (19–21). The heterozygous ThrbPV/+ mouse provided an opportunity to address this question. Similar to RTH patients with mutation of a single allele of the THRB gene, the ThrbPV/+ mouse exhibits mild abnormality in the dysregulation of the pituitary-thyroid axis in that thyroid hormones are elevated about 2.5- to 2.8-fold, accompanied by a 2.1-fold increase in TSH. The thyroid weight is increased about 2-fold with only enlarged follicular cells but no hyperplasia of follicular cells (7). Thus, the ThrbPV/+ mouse provided an unprecedented opportunity to address the critical question as to whether hyperplastic follicular cells stimulated by highly elevated TSH is a prerequisite to collaborate with the mutations of a single allele of the Thrb gene to bring out thyroid carcinogenesis.
Materials and Methods
Animals
The animal protocols used in the present study were approved by the National Cancer Institute Animal Care and Use Committee. ThrbPV/+ mice were generated and genotyped as described previously (7). For the induction of highly elevated serum TSH levels, ThrbPV/+ mice (n = 12) and wild-type (WT) sibling cohorts (n = 19) were fed with an iodine-deficient diet supplemented with 0.15% propylthiouracil (PTU) (Harlan Teklab, Frederick, MD) ad libitum starting at the age of 2–3 months until the treated ThrbPV/+ mice became moribund and therefore were euthanized. The untreated ThrbPV/+ mice (n = 21) and WT siblings (n = 32) were killed at 12–13 months of age.
Hormone assays
Serum levels of total T3 (TT3) (WT, n = 21; ThrbPV/+, n = 16; WT-PTU, n = 6; and ThrbPV/+-PTU, n = 7) and total T4 (TT4) (WT, n = 21; ThrbPV/+, n = 32; WT-PTU, n = 7; and ThrbPV/+-PTU, n = 7) were determined by using a GammaCoat T4 or T3 assay RIA kit (DiaSolin, Stillwater, MN) according to the manufacturer's instructions. The serum TSH levels of the mice with two genotypes with or without PTU treatment (WT, n = 16; ThrbPV/+, n = 17; WT-PTU, n = 7; and ThrbPV/+-PTU, n = 10) were measured as previously described (7).
Western blot analysis and coimmunoprecipitation
Twenty-five micrograms of thyroid lysates prepared similarly as described previously (22) were analyzed for protein abundance by Western blot analysis. Antibodies used were cyclin D1 (sc-450), cyclin-dependent kinase 4 (CDK4) (sc-260), E2F1 (cs-251), fibronectin (sc-6952), metalloproteinase (MMP)-2 (sc-10736), MMP-9 (sc-6841-R), growth factor receptor binding protein 2 (Grb2) (sc-8034), all at 1:1000 dilution (from Santa Cruz Biotechnology, Santa Cruz, CA). The antibodies for total Src (no. 2109) and phosphorylated Src (Y416 no. 2101) were from Cell Signaling Technology, Inc. (Danvers, MA) and were used at 1:250 dilution: total focal adhesion kinase (FAK) (no. sc-557) at 1:250 dilution (purchased from Santa Cruz Biotechnology) or phosphorylated FAK (Y397) at 1:1000 dilution (from Invitrogen, Carlsbad, CA), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (14C10, no. 2118; Cell Signaling) at 1:1000 dilution. Western blotting analysis with three replicates was performed for the WT (n = 10), ThrbPV/+(n = 9), WT-PTU (n = 7), and ThrbPV/+-PTU (n = 6) mice. Representative results from four mice are shown in Results, with detailed information mentioned in the figure legends.
For coimmunoprecipitation, 1.5 mg of total thyroid lysates was immunoprecipitated with 8 μg monoclonal anti-TR antibody J53 (23) or polyclonal anti-TR antibodies that recognize the amino-terminal A/B domain of PV (8 μg per 600 μl: catalog no. 600-401-A96; Rockland Inc., Gilbertsville, PA), or with an irrelevant purified mouse monoclonal IgG (MOPC-141, M8894, Sigma, St. Louis, MO), 4 μg] as a negative control, followed by Western blot analysis using antibodies for integrin-β1(M-06; sc-8978), integrin-α5 (H-104; sc-10729), p85α (z-8; sc-423) (at 1:250 dilution; Santa Cruz Biotechnology), and p-AKT(Ser473) (no. 9271; at 1:1000 dilution; Cell Signaling).
Histopathological analysis and immunohistochemistry
Thyroid glands and lungs of the WT or ThrbPV/+ mice with or without PTU treatment (WT, n = 21; ThrbPV/+, n = 27; WT-PTU, n = 19; and ThrbPV/+-PTU, n = 9) were dissected, fixed in 10% neutral buffered formalin (Sigma-Aldrich, St. Louis, MO), and subsequently embedded in paraffin. Five-micrometer-thick sections were stained with hematoxylin and eosin (H & E) and analyzed.
Immunohistochemistry was performed as previously described (7) with modifications. For the antigen retrieval step, slides were heated in 0.05% citraconic anhydride (Sigma-Aldrich) solution (pH 7.4) at 98 C for 45 min, followed by treatment with rabbit anti-Ki67 (1:300 dilution; NeoMarker, Thermo Scientific, Rockford, IL) at 4 C overnight. The antigen signals were detected by treatment with peroxidase substrate diaminobenzidine, followed by counterstaining with Gill's hematoxylin.
Immunohistochemical analysis of bromodeoxyridine (BrdU; Sigma-Aldrich) incorporation was carried out first by ip injection of ThrbPV/+ mice and WT siblings with or without treatment of PTU with BrdU (50 μg/g of body weight). Euthanasia was performed 2 h after injection. Thyroid glands were dissected and fixed and paraffin blocks were prepared as described above. For the antigen retrieval step, slides were boiled in antigen unmasking solution (H-3300; Vector Laboratories Inc., Burlingame, CA) for 20 min, followed by incubation with mouse BrdU antibody [at 1:10 dilution; BD Biosciences (San Jose, CA); no. 347580] at 4 C overnight. The antigen signals were detected by treatment with peroxidase substrate diaminobenzidine, followed by counterstaining with Gill's hematoxylin. Cells were considered as positive when they displayed a yellow/brown-stained nucleus. Four representative fields of a thyroid section were photographed for each animal (n = 3–4 animals per group). The positively stained nuclei were counted after examining the total number of nuclei on the thyroid sections (500–1500 total nuclei were counted per microphotograph). We used National Institutes of Health IMAGE software (ImageJ 1.34s; Wayne Rasband, National Institutes of Health, Bethesda, MD) for counting.
Statistical analysis
All data are expressed as mean ± the sem. A Student's t test was used to determine the significant differences between two groups. The significant differences among the groups were determined by ANOVA. The Kaplan-Meier cumulative survival analysis was performed using GraphPad Prism version 5.0 for Mac OS X (GraphPad Software, San Diego, CA). Log rank (Mantel-Cox) tests were used to compare survival between groups. P < 0.05 is considered statistically significant.
Results
TSH is necessary but not sufficient to induce spontaneous development of follicular thyroid carcinoma in ThrbPV/+ mice
The effect of PTU treatment on thyroid function tests of mice is shown in Fig. 1. Without PTU treatment, the serum TSH levels of ThrbPV/+ mice were 2.4-fold higher than those of WT mice (bar 2 vs. bar 1; Fig. 1A). PTU treatment elevated serum TSH levels approximately 222-fold in ThrbPV/+-PTU mice (bar 4 vs. bar 2; Fig. 1A). PTU treatment also similarly elevated serum TSH levels in WT-PTU mice (bar 3 vs. bar 1; Fig. 1A). The elevated TSH levels of ThrbPV/+-PTU mice and WT-PTU mice did not differ significantly (Fig. 1A). As a result of elevated TSH by PTU treatment, both the serum TT4 and TT3 of ThrbPV/+-PTU mice and WT-PTU mice were lower than that of the corresponding untreated mice (bar 3 vs. bar 1; bar 4 vs. bar 2, Fig. 1, B and C). These thyroid function tests show similar effects of PTU on the hormonal profiles in WT and ThrbPV/+ mice.
Fig. 1.

Thyroid function tests of the ThrbPV/+ mice and WT siblings with or without treatment with PTU. A, Serum TSH levels for mice with genotypes are as marked (n = 4–17). No significant differences in TSH concentrations were detected between WT-PTU and ThrbPV/+-PTU mice. B, Serum TT4 for mice with genotypes are as marked (n = 7–32). No significant differences in TT4 were detected between WT-PTU and ThrbPV/+-PTU mice. C, Serum TT3 for mice with genotypes are as marked (n = 6–21). No significant differences in TT3 were detected between WT-PTU and ThrbPV/+-PTU mice. The significant differences among the four groups were determined by ANOVA. P values are shown. NS, Not significant.
However, ThrbPV/+-PTU mice had markedly reduced survival compared with WT-PTU mice after treatment with PTU for 12 months (Fig. 2). Five percent (one of 19 mice) and 16% (three of 19 mice) of WT-PTU mice showed signs of enlarged thyroids and difficulty in breathing. They had to be euthanized by the ages of 10 and 12 months, respectively. In contrast, 36% (five of 14 mice) and 100% (14 of 14 mice) of ThrbPV/+-PTU mice became moribund and had to be euthanized at those ages. During the same observation period, no death was observed for untreated WT and ThrbPV/+ mice (controls). Analysis of thyroid weight showed that PTU treatment led to an 8.5-fold increase in WT-PTU mice and a 21-fold increase in ThrbPV/+-PTU mice (Fig. 3A). Without PTU treatment, ThrbPV/+ mice had a 2-fold increase in thyroid weight due to the elevated TSH as compared with WT mice. The higher increase in thyroid weight of ThrbPV/+-PTU than that of WT-PTU mice reflected the effect of PV on thyroid growth of ThrbPV/+-PTU mice. Remarkably, about 33% of ThrbPV/+-PTU mice developed asymmetrical thyroid tumors as is frequently observed in human thyroid cancer (representative examples are shown in Fig. 3B, panels a, b, and c).
Fig. 2.
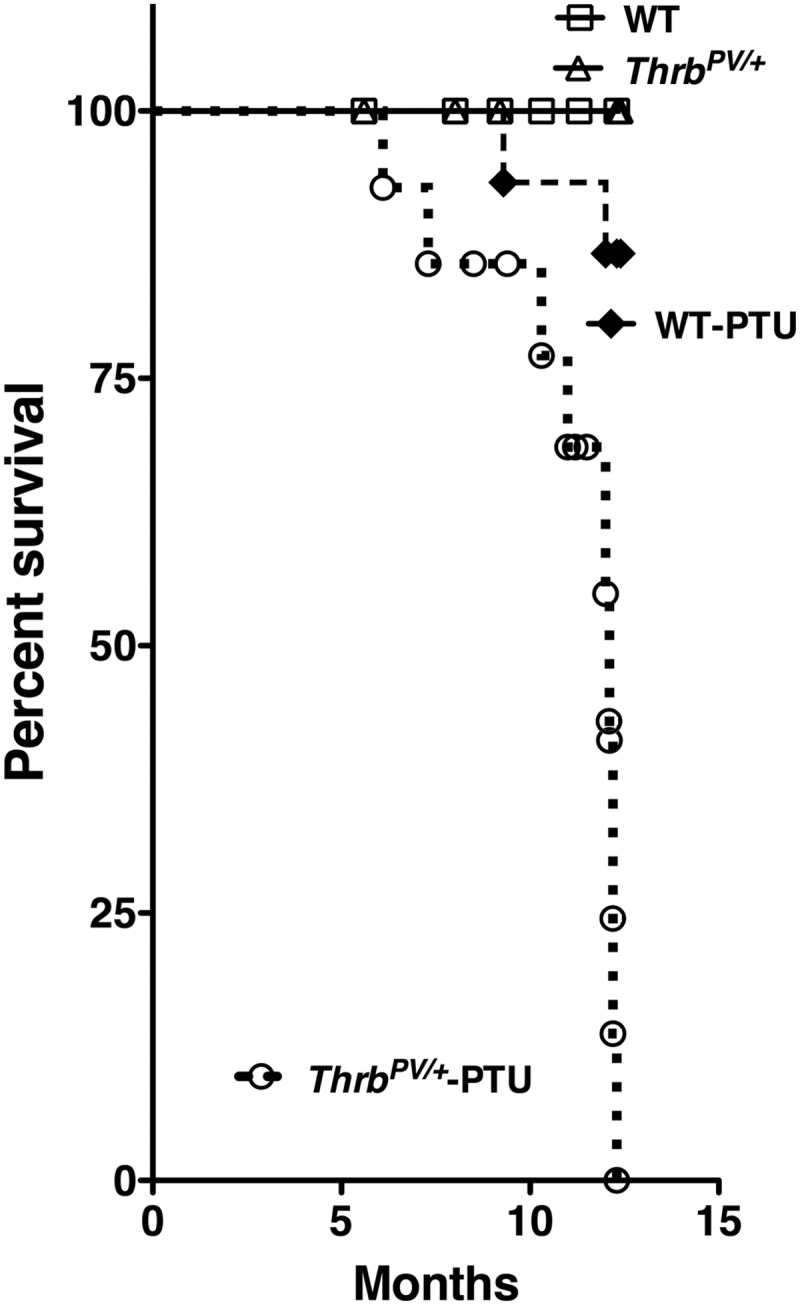
The Kaplan-Meier survival curves for the ThrbPV/+ mice and WT siblings with or without treatment with PTU for the observation period of 12 months. PTU treatment started at the ages of 2–3 months and continued for 12 months. The Kaplan-Meier cumulative survival analysis was performed using GraphPad Prism version 5.0. No deaths occurred for WT mice (n = 17) or untreated ThrbPV/+ mice (n = 12) during the observation period. Only three WT-PTU mice died (n = 19; three of 19, 16%) during the observation period. In contrast, all the ThrbPV/+-PTU mice (n = 14) died by 12 months of age. Percent survival of WT-PTU and ThrbPV/+-PTU mice differed significantly (P < 0.05) as determined by a Mantel-Cox test.
Fig. 3.
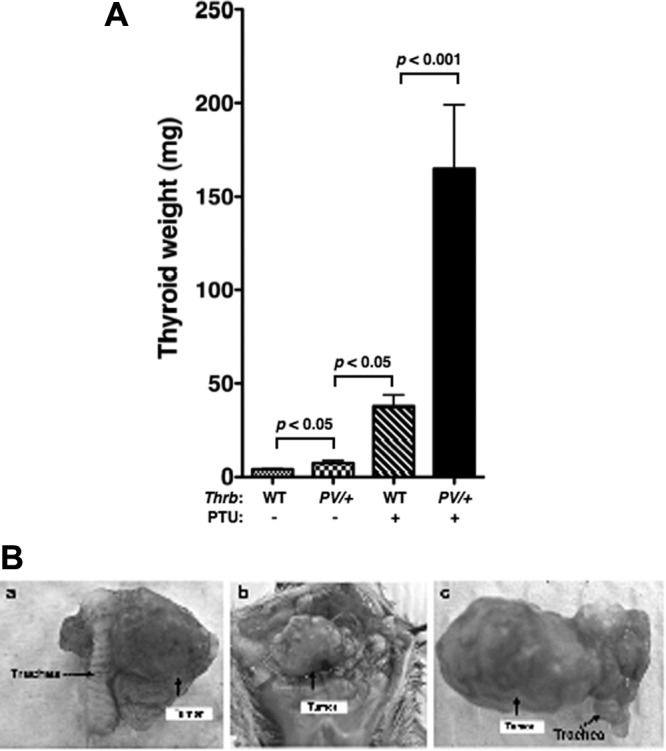
A, Comparison of thyroid growth in the ThrbPV/+ mice and WT siblings with or without treatment with PTU. Thyroids of mice with the genotypes indicated (n = 6–27) were dissected and their weights were compared. The thyroids of the ThrbPV/+-PTU mice were significantly larger than those of the WT-PTU mice and the untreated ThrbPV/+mice, as determined by ANOVA. The P values are indicated. B, Representative examples of anterior views of asymmetrical thyroid tumors spontaneously developed in the ThrbPV/+-PTU mice. Asymmetrical tumors are indicated by arrows.
Histopathological evaluation of the thyroids of ThrbPV/+-PTU mice demonstrated grossly enlarged glands, displaying diffuse adenomatous hyperplasia with dense nuclear chromatin and a glandular pattern characteristic of follicular carcinoma. Pathological changes progressed from hyperplasia (Fig. 4Ab) to capsular invasion (Fig. 4Ac), vascular invasion (Fig. 4Ad), anaplasia (Fig. 4Ae), and lung metastasis (Fig. 4Af), as compared with only enlarged follicles (Fig. 4Aa) and normal lung in untreated ThrbPV/+ mice, Fig. 4Ag). In contrast, WT-PTU mice did not exhibit vascular invasion, anaplasia, and metastasis (see Fig. 4B). Table 1 compares the occurrence of pathological changes in WT-PTU and ThrbPV/+-PTU mice. WT-PTU mice displayed hyperplasia (see also Fig. 4Ah for WT-PTU mice) and a lower frequency of occurrence of capsular invasion (16% for WT-PTU mice vs. 89% for ThrbPV/+-PTU mice; Fig. 4B), but no visible signs of vascular invasion, anaplasia, and metastasis were found for WT-PTU mice (Fig. 4B, panels b, c, and d). These results indicate that TSH is necessary but not sufficient for the development of thyroid cancer in ThrbPV/+-PTU mice.
Fig. 4.

Pathological progression during thyroid carcinogenesis of ThrbPV/+-PTU mice. A, Representative examples of hematoxylin and eosin (H and E)-stained thyroid cells (panels a, b, c, d, e, and h) and lung (panels f and g) of mice with genotypes and treatment indicated. The arrows indicate capsular invasion (panel c), vascular invasion (panel d), anaplasia (panel e), and lung metastasis (panel f) of the ThrbPV/+-PTU mice. B, Quantitative analysis of occurrence frequency (percentage) of capsular invasion (panel a), vascular invasion (panel b), anaplasia (panel c), and lung metastasis (panel d) of the WT (n = 6), ThrbPV/+(n = 6), WT-PTU (n = 19), and ThrbPV/+-PTU (n = 9) mice without or with PTU treatment for 6–12 months. Sections of thyroids and lungs from the WT, ThrbPV/+, WT-PTU, and ThrbPV/+-PTU mice were stained with H and E and analyzed for pathological progression. The data are expressed as the percentage of occurrence frequency of the mice examined. #, No occurrence frequency (percentage) during the observation period. All panels have the same magnification as shown in the bar in panel c.
Table 1.
Thyroid pathological features of WT-PTU and Thrb PV/+-PTU mice
| Pathological features | Hyperplasia | Capsular invasion | Vascular invasion | Anaplasia | Lung mets |
|---|---|---|---|---|---|
| WT-PTU | 19/19 (100%) | 3/19 (16%) | 0/19 (0%) | 0/19 (0%) | 0/19 (0%) |
| ThrbPV/+-PTU | 9/9 (100%) | 8/9 (89%) | 4/9 (44%) | 1/9 (11%) | 3/9 (33%) |
WT-PTU (n = 19) and ThrbPV/+-PTU (n = 9) mice were treated with PTU for 12 months. Sections of thyroids and lungs were stained with H & E and analyzed for pathological progression of hyperplasia, capsular invasion, vascular invasion, anaplasia, and metastases in the lung and heart. Mets, metastases.
Activation of proliferation signaling pathways in thyroids of ThrbPV/+-PTU mice
To understand whether thyroid tumor growth of ThrbPV/+-PTU mice was caused by increasing tumor cell proliferation, we immunostained the nuclear proliferation marker Ki-67 of thyroid cells. For control, Fig. 5Aa shows that only a few untreated WT cells were stained positively with Ki-67, whereas more Ki-67 positively stained thyroid tumor cells were detected in ThrbPV/+-PTU mice (Fig. 5Ad) than in WT-PTU (Fig. 5Ab) and untreated-ThrbPV/+ mice (Fig. 5Ac). The positively stained cells were counted and the quantitative data are graphed in panel e (Fig. 5A), indicating 10.5- and 2.3-fold increases in Ki-67 stained thyroid tumor cells of ThrbPV/+-PTU mice as compared with untreated ThrbPV/+ and WT-PTU mice, respectively. We further pulsed thyroid cells with BrdU for 2 h and used immunostaining to detect BrdU-incorporated cells (Fig. 5B). For control, virtually no BrdU incorporation was detected in the thyroid cells of WT mice (Fig. 5Ba) under the experimental conditions. However, more BrdU-incorporated cells were detected in thyroid of ThrbPV/+-PTU mice (Fig. 5Bd) than in WT-PTU (Fig. 5Bb) and untreated ThrbPV/+ mice (Fig. 5Bc). Figure 5Be shows positive controls using small intestine of WT mice, and Fig. 5Bf shows the negative control using the brain tissue of WT mice. The BrdU-containing cells were counted and the quantitative data are graphed in panel g (Fig. 5B), showing 8.8- and 3.9-fold increases in BrdU-incorporated thyroid cells of ThrbPV/+-PTU mice as compared with untreated ThrbPV/+ and WT-PTU mice, respectively. Taken together, these results indicate that PV mutant synergized with TSH to drive the proliferation of thyroid tumor cells of ThrbPV/+-PTU mice.
Fig. 5.

Increased tumor cell proliferation in the ThrbPV/+-PTU mice. A, The nuclear Ki-67 proliferation marker was immunohistochemically stained in the thyroid sections of untreated WT (panel a), WT-PTU (panel b), untreated ThrbPV/+ (panel c), and ThrbPV/+-PTU mice (panel d) as described in Materials and Methods. Arrows point to the representative positively stained cells. Quantitative analyses of positively stained cells were conducted. Significantly more positively Ki-67 stained cells were detected in the thyroid of the ThrbPV/+-PTU mice than in the WT-PTU, untreated ThrbPV/+, and WT mice (panel e). The significant differences among the four groups were determined by ANOVA. The P values are indicated. B, Thyroid sections were prepared after ip injection with BrdU as described in Materials and Methods. Nuclear BrdU incorporation was detected by the immunohistochemical staining of thyroid cells of the untreated WT (panel a), WT-PTU (panel b), untreated ThrbPV/+ (panel c), and ThrbPV/+-PTU mice (panel d) as described in Materials and Methods. Arrows point to the representative positively stained cells. For positive control, the small intestine cells of the WT mice were also similarly stained for BrdU (panel e). For negative control, brain cells of the WT mice were also similarly stained for BrdU, but no positively incorporated cells were detected under the experimental conditions (panel f). Quantitative analyses of positively stained cells are shown in panel g. Significantly more positively BrdU-incorporated cells were detected in the thyroid of ThrbPV/+-PTU mice than in the WT-PTU, untreated ThrbPV/+, and WT mice. The significant differences among the four groups were determined by ANOVA. The P values are indicated.
That proliferation of thyroid tumor cells of ThrbPV/+-PTU mice was increased prompted us to elucidate the underlying altered proliferation pathways. We first focused on the key cell cycle regulators. Figure 6 shows that the protein abundances of cyclin D1 (Fig. 6A, lanes 13–16, n = 4), CDK4 (Fig. 6B), and E2F1 (Fig. 6C) were markedly higher in thyroid tumors of ThrbPV/+-PTU mice than in WT (lanes 1–4), WT-PTU (lanes 5–8), and untreated ThrbPV/+ mice (lanes 9–12). These findings indicate the activation of the cell cycle progression of thyroid tumor cells of ThrbPV/+-PTU mice.
Fig. 6.

Activation of the cyclin D1-CDK4–E2F1 pathway to increase thyroid tumor cell proliferation of the ThrbPV/+-PTU mice. Thyroid extracts (25 μg) from mice with the genotypes indicated (n = 4 for each genotype) were analyzed for key cell cycle regulators as shown by Western blotting. Markedly elevated protein abundance of cyclin D1 (A), CDK4 (B), and E2F1 (C) were observed in the thyroids of ThrbPV/+-PTU mice (lanes 13–16) as compared with the WT-PTU mice (lanes 5–8). No apparent signals were observed in either the WT (lanes 1–4) or untreated ThrbPV/+mice (lanes 9–12). GAPDH (D) was used as the loading control.
Activation of integrin-Src-FAK-MMP signaling to increase tumor cell invasion and metastasis in thyroids of ThrbPV/+-PTU mice
Studies in the past decades indicated that Src proteins, complexed with FAK, affect cell adhesion, invasion, and motility to increase tumor progression and metastasis (24–26). Indeed, we showed previously that activation of Src-FAK-MMP signaling in the thyroid of ThrbPV/PV mice contributes to the migration, invasion, and metastasis of thyroid tumor cells (18). We therefore evaluated whether expression of a single allele of mutant PV is sufficient to activate Src-FAK-MMP signaling to contribute to the metastatic process of thyroid tumor cells in ThrbPV/+-PTU mice. Figure 7A shows more phosphorylated Src (Y416) was detected in the thyroid of ThrbPV/+-PTU mice (panel a; lanes 13–16) than in WT-PTU (lanes 5–8) or untreated ThrbPV/+ mice (lanes 9–12). Similarly, more phosphorylated FAK (Y397) was found in the thyroid of ThrbPV/+-PTU mice (panel c; lanes 13–16) than in WT-PTU (panel c, lanes 5–8) or untreated ThrbPV/+ mice (panel c, lanes 9–12). However, no apparent changes in total Src (panel b) and total FAK (panel d) were found. These results indicate the activation of Src-FAK signaling in the thyroid of ThrbPV/+-PTU mice. FAK, once activated, serves as a binding site for the Grb2 (27, 28). Figure 7Ae shows that Grb2 protein abundance was markedly elevated in ThrbPV/+-PTU mice (Fig. 7Ae, lanes 13–16) as compared with untreated ThrbPV/+ mice (Fig. 7Ae, lanes 9–12) and WT-PTU (Fig. 7Ae, lanes 5–8) mice. The consequence of the activated Src-FAK signaling was evident in the higher protein abundance of MMP-2 (Fig. 7Af) and MMP-9 (Fig. 7Ag, lanes 13–16) than in WT-PTU (Fig. 7A, f and g, lanes 5–8) or untreated ThrbPV/+ mice (Fig. 7A, f and g, lanes 9–12). These results indicate that the activation of the Src-FAK-Grb2-MMP signaling led to increased thyroid tumor cell invasion and metastasis in ThrbPV/+-PTU mice.
Fig. 7.

Activation of integrin-FN-Src-FAK signaling by PV. A, Thyroid extracts (25 μg) from mice with the genotypes indicated (n = 4 for each genotype) were analyzed for key cellular regulators (as marked) by Western blotting. Increased protein abundance of p-Src (panel a), total Src (panel b), p-FAK (panel c), total FAK (panel d), Grb2 (panel e), MMP-2 (panel f), and MMP-9 (panel g) were observed in the ThrbPV/+-PTU mice (lanes 13–16) as compared with the WT-PTU (lanes 5–8), WT (lanes 1–4), and untreated ThrbPV/+mice (lanes 9–12). No apparent changes were observed in total Src (panel b) and total FAK (panel d). GAPDH (panel h) was used as loading controls. B, Coimmunoprecipitation assays were carried out as described in Materials and Methods. PV physically interacted with integrin-β1 (panel a), integrin-α5 (panel b), FN (panel c), p-Src (panel d), p-FAK (panel e), p85α (panel f), and p-AKT (panel g). An increased association of PV with the indicated regulators was observed in the thyroid of the ThrbPV/+-PTU mice (lane 4) as compared with the WT-PTU mice (lane 3). No apparent association of PV with the indicated key regulators was apparent in the thyroid of the untreated ThrbPV/+(lane 2) mice. Lane 5 shows the negative control when an irrelevant antibody, mouse monoclonal IgG MOPC-141, was used in the immunoprecipitation.
Our observations prompted us to elucidate the molecular basis by which PV acted to stimulate Src-FAK-Grb2-MMP-2 signaling. It is known that FAK is localized to the specialized submembranous structures, known as focal adhesions (29). These are multiple protein complexes consisting of integrins, extracellular signals such as fibronectin (FN), and signaling proteins such FAK, Src, and Grb2 and their related downstream targets (30, 31). The focal adhesions serve to mediate strong cell-substrate adhesion and transduce extracellular signals into the cell (32). To test the hypothesis that PV could interact with key regulators in this complex to activate signaling, we carried out coimmunoprecipitation assays by first immunoprecipitating thyroid lysates with anti-PV antibodies, followed by Western blotting. Figure 7B shows that the integrins such as integrin-β1 (Fig. 7Ba) and integrin-α5 (Fig. 7Bb) and their ligand FN (Fig. 7Bc) were strongly associated with PV in the thyroid of ThrbPV/+-PTU mice (lane 4). Only background signals were detected in WT-PTU mice (lane 3). No association of PV was apparent in untreated ThrbPV/+ mice (lane 2). Importantly, more activated p-Src (Y416) (Fig. Bd) and p-FAK (Y397) (Fig. 7Be) were detected in the PV-associated complex (lane 4 vs. lane 3). Similarly, p85α, the regulatory subunit of PI3K (Fig. 7Bf), and its downstream effector p-AKT (Fig. 7Bg), were recruited to the PV-associated protein complexes (lane 4), but no such recruitment was visible in WT-PTU (lane 3) or untreated ThrbPV/+ mice (lane 2). As shown in lane 1 (Fig. 7B, a–g), no apparent association of PV with these regulators in the WT mice (lane 1, Fig. 7B, a–g) was observed. These results support our hypothesis in that PV acted to facilitate the recruitment of integrins, FN, Src, FAK, and p85α. The enhanced complex formation led to increased phosphorylation of Src, FAK, and AKT to promote signaling of integrin-FN-Src-FAK-AKT, leading to the activation of MMP-2/9 to increase cell migration and metastasis.
Discussion
The discovery that the mouse harboring the PV mutation in both alleles of the Thrb gene (ThrbPV/PV) spontaneously develops follicular thyroid cancer has led to the uncovering of altered signaling pathways during thyroid carcinogenesis, thereby identifying novel potential molecular targets for treatments and elucidating oncogenic actions of a mutated TRβ (i.e. PV). The highly elevated serum TSH exhibited by ThrbPV/PV mice was found to be necessary but not sufficient to induce thyroid carcinogenesis (18), suggesting the collaborating role of TSH with the oncogenic actions of PV. Similar to most RTH patients with mutations of a single allele of the THRB gene, ThrbPV/+ mice have only marginally elevated serum TSH levels (∼2-fold) with no occurrence of thyroid cancer. A critical question has arisen as to whether mutation of a single allele of PV would be sufficient to induce thyroid carcinogenesis in ThrbPV/+ mice if serum TSH levels were elevated to that of ThrbPV/PV mice. The present studies explored this question and showed that the mutation of a single allele of the Thrb gene was sufficient to collaborate with elevated TSH to induce spontaneous development of thyroid cancer. Thus, these findings further support the notion that loss of the normal TRβ suppressor functions caused by mutation of a single allele of the Thrb gene empowered the hyperplastic follicular cells to undergo carcinogenesis.
Molecular analyses showed that PV acted to stimulate cell proliferation in the thyroid of ThrbPV/+-PTU mice by increasing expression of cyclin D1 at the protein level (Fig. 6). Overexpression of cyclin D1 protein led to activation of the cyclin D1-CDK4-E2F pathway to drive cell proliferation in the thyroid of ThrbPV/+-PTU mice (Figs. 6 and 5, A and B). Previously we found that in addition to thyroid cancer, ThrbPV/PV mice also exhibit TSH-secreting tumors (33). Molecular analyses showed that the liganded wild-type TRβ represses Ccnd1 (cyclin D1) mRNA expression via tethering to the Ccnd1 promoter through binding to the cAMP response element-binding protein. That repression effect is lost in mutant PV, thereby resulting in constitutive activation of the Ccnd1 in the pituitary ThrbPV/PV mice (33). Such transcription regulation via TR-cAMP response element-binding protein protein-protein interaction was also observed in other cell types (34). Thus, it is reasonable to propose that a similar mechanism could also be operated by PV to increase the expression of the Ccnd1 gene in the thyroid of ThrbPV/+-PTU mice.
In addition to mediating its activity at the transcription level, PV also acted via extranuclear signaling in the thyroids of ThrbPV/+-PTU mice. PV physically interacted with integrins (integrin-β1 and integrin-α5)-Src-FAK-PI3K(p85α)-AKT complexes (Fig. 7B). In the PV-associated large protein complex, more phosphorylated Src, FAK, and AKT were found in the thyroid of ThrbPV/+-PTU mice than in WT-PTU mice, suggesting that interaction of PV with integrin-Src-FAK-PI3K-AKT complexes facilitated activation of downstream signaling by increasing phosphorylation cascades. The activated signaling was evidenced by increased MMP-2 and MMP-9 protein abundance (Fig. 7A), thereby promoting invasion and metastasis of thyroid tumor cells of ThrbPV/+-PTU mice (see Fig. 4, A and B).
Although the molecular oncogenic actions of PV in the thyroid of ThrbPV/+-PTU mice described here are similar to those reported for PV in ThrbPV/PV mice (18, 35, 36), there are differences in thyroid tumor phenotypes exhibited by ThrbPV/+-PTU and ThrbPV/PV mice. About 33% of the ThrbPV/+-PTU developed asymmetrical tumors (Fig. 3B), as is frequently observed in human thyroid cancers, whereas tumors in the ThrbPV/PV mice were exclusively bilateral tumors (10, 35). These observations suggested the possibility that there could be gene dosage dependency for the oncogenic actions of PV in thyroid carcinogenesis. Studies of the oncogenic actions of PV in ThrbPV/PV mice revealed that PV acts via multiple mechanisms. In addition to the transcription regulation exemplified by the up-regulation of the Ccnd1 gene and the extranuclear signaling via activation of PV-integrin-Src-FAK-PI3K(p85α)-AKT signaling pathways, PV also acts via gain of function in promoting thyroid carcinogenesis (37). Therefore, it is entirely possible that thyroid cancer exhibited asymmetrically in ThrbPV/+-PTU mice could reflect the oncogenic actions of PV from a lower gene dosage by mutation of a single allele of the Thrb gene. In the ThrbPV/PV mice, both alleles of the Thrb gene are mutated such that there is stronger and more extensive oncogenic action of PV, leading to exclusively bilateral follicular thyroid cancer. Currently the molecular basis underlying asymmetrical human thyroid cancer is not clear. It is possible that the gene-dosage effects of some oncogenes involved in thyroid carcinogenesis (e.g. Ras mutations) could account for the asymmetrical human thyroid tumors. However, this question is open for some additional exploration.
The role of TSH in thyroid carcinogenesis has been intensively studied in patients and mouse models. Increasing evidence has suggested a close association of TSH with risk of malignancy (38–41). Through the use of mouse models, it has been become possible to further elucidate the role of TSH in thyroid carcinogenesis at the molecular level. The ThrbPV/PV mouse with highly elevated TSH and mutations of two alleles of the Thrb gene spontaneously develops metastatic follicular thyroid cancer (10). However, in ThrbPV/PV mice with the proliferation signaling of TSH blocked by knocking out the TSH receptors (ThrbPV/PVTshr−/− mice), thyroids show only impaired growth with no occurrence of thyroid cancer (18). As also shown in the present studies, WT mice with TSH levels elevated by long-term treatment of PTU displayed only hyperplastic thyroids but no metastatic thyroid cancer (18). Thus, growth stimulated by TSH is a prerequisite but is not sufficient for metastatic thyroid cancer to occur. Additional genetic alterations (such as PV or other oncogenic changes), destined to alter focal adhesion and migration capacities, are required to empower hyperplastic follicular cells to become metastatic cancer cells.
This molecular model of growth and metastasis learned from the ThrbPV/PV mice is consistent with the clinical observations that higher TSH is associated with extrathyroidal extension of disease but not with distant metastases (38). The present study further strengthened this proposed molecular model of growth and metastasis learned from the ThrbPV/PV mice. ThrbPV/+ mice with only mildly elevated TSH (∼2-fold higher than in WT mice) and enlarged follicular cells, but with no apparent hyperplasia [see Fig. 4A and Kaneshige et al. (7)], were not at risk of developing metastatic thyroid cancer. Only upon stimulation by high TSH (as in ThrbPV/+-PTU mice) did the metastatic thyroid cancer occur. Recently several RTH patients with mutations of a single allele of the THRB gene were found to develop papillary thyroid cancer (19–21). At present, it is not possible to establish a cause-effect relationship between THRB mutations and thyroid cancer in these patients. Furthermore, with the very limited number of cases, it would be difficult to establish either the threshold of TSH concentrations or the duration of exposure to proliferative stimulation required to be at risk for thyroid cancer in RTH patients. However, these recent clinical observations support the lessons learned from the present studies of ThrbPV/+ and ThrbPV/+-PTU mice in that stimulation of follicular cells by TSH together with THRB mutations could be risk factors to develop thyroid cancer.
The ThrbPV/+ mouse was created to understand the molecular basis of RTH (7). Indeed, extensive molecular characterization of the ThrbPV/+ mouse has led to the understanding of the in vivo functions of TRβ mutants (i.e. PV) in mediating RTH (8, 42, 43). In the present study, the ThrbPV/+ mouse was further used to demonstrate that a single allele mutation of the Thrb gene was sufficient to collaborate with highly elevated TSH (as in ThrbPV/+-PTU mice) to transform hyperplastic follicular cells to thyroid cancer. The spontaneous development of asymmetrical thyroid cancer in ThrbPV/+-PTU mice could provide an opportunity for studies to shed new light on the genetic changes underlying asymmetrical thyroid carcinogenesis in humans.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Disclosure Summary: The authors declare no conflict of interest.
Footnotes
- BrdU
- Bromodeoxyridine
- CDK4
- cyclin-dependent kinase 4
- FAK
- focal adhesion kinase
- FN
- fibronectin
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- Grb2
- growth factor receptor binding protein 2
- H & E
- hematoxylin and eosin
- MMP
- metalloproteinase
- PI3K
- phosphatidyl inositol 3-kinase
- PTU
- propylthiouracil
- PV
- mouse model of RTH created by knocking in a mutation
- RTH
- resistance to thyroid hormone
- TR
- thyroid hormone receptor
- TT3
- total T3
- TT4
- total T4
- WT
- wild type.
References
- 1. Refetoff S, Weiss RE, Usala SJ. 1993. The syndromes of resistance to thyroid hormone. Endocr Rev 14:348–399 [DOI] [PubMed] [Google Scholar]
- 2. Brucker-Davis F, Skarulis MC, Grace MB, Benichou J, Hauser P, Wiggs E, Weintraub BD. 1995. Genetic and clinical features of 42 kindreds with resistance to thyroid hormone. The National Institutes of Health Prospective Study. Ann Intern Med 123:572–583 [DOI] [PubMed] [Google Scholar]
- 3. Usala SJ, Menke JB, Watson TL, Wondisford FE, Weintraub BD, Bérard J, Bradley WE, Ono S, Mueller OT, Bercu BB. 1991. A homozygous deletion in the c-erbA β thyroid hormone receptor gene in a patient with generalized thyroid hormone resistance: isolation and characterization of the mutant receptor. Mol Endocrinol 5:327–335 [DOI] [PubMed] [Google Scholar]
- 4. Ono S, Schwartz ID, Mueller OT, Root AW, Usala SJ, Bercu BB. 1991. Homozygosity for a dominant negative thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metab 73:990–994 [DOI] [PubMed] [Google Scholar]
- 5. Ferrara AM, Onigata K, Ercan O, Woodhead H, Weiss RE, Refetoff S. 2012. Homozygous thyroid hormone receptor beta-gene mutations in resistance to thyroid hormone: three new cases and review of the literature. J Clin Endocrinol Metab 97:1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yen PM, Chin WW. 1994. Molecular mechanisms of dominant negative activity by nuclear hormone receptors. Mol Endocrinol 8:1450–1454 [DOI] [PubMed] [Google Scholar]
- 7. Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S. 2000. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA 97:13209–13214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng SY. 2004. New development in thyroid hormone resistance. Hot Thyroidology No 1 [Google Scholar]
- 9. Parrilla R, Mixson AJ, McPherson JA, McClaskey JH, Weintraub BD. 1991. Characterization of seven novel mutations of the c-erbAβ gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest 88:2123–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki H, Willingham MC, Cheng SY. 2002. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid 12:963–969 [DOI] [PubMed] [Google Scholar]
- 11. Furuya F, Hanover JA, Cheng SY. 2006. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc Natl Acad Sci USA 103:1780–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, Bernet V, Burman KD, Kohn LD, Saji M. 2001. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res 61:6105–6111 [PubMed] [Google Scholar]
- 13. Ying H, Suzuki H, Zhao L, Willingham MC, Meltzer P, Cheng SY. 2003. Mutant thyroid hormone receptor beta represses the expression and transcriptional activity of peroxisome proliferator-activated receptor γ during thyroid carcinogenesis. Cancer Res 63:5274–5280 [PubMed] [Google Scholar]
- 14. Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, Cheng SY. 2006. PPARγ insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-κB signaling pathway. Oncogene 25:2736–2747 [DOI] [PubMed] [Google Scholar]
- 15. Ying H, Furuya F, Zhao L, Araki O, West BL, Hanover JA, Willingham MC, Cheng SY. 2006. Aberrant accumulation of PTTG1 induced by a mutated thyroid hormone β receptor inhibits mitotic progression. J Clin Invest 116:2972–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim CS, Ying H, Willingham MC, Cheng SY. 2007. The pituitary tumor-transforming gene promotes angiogenesis in a mouse model of follicular thyroid cancer. Carcinogenesis 28:932–939 [DOI] [PubMed] [Google Scholar]
- 17. Guigon CJ, Zhao L, Lu C, Willingham MC, Cheng SY. 2008. Regulation of β-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol Cell Biol 28:4598–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu C, Zhao L, Ying H, Willingham MC, Cheng SY. 2010. Growth activation alone is not sufficient to cause metastatic thyroid cancer in a mouse model of follicular thyroid carcinoma. Endocrinology 151:1929–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim HK, Kim D, Yoo EH, Lee JI, Jang HW, Tan AH, Hur KY, Kim JH, Kim KW, Chung JH, Kim SW. 2010. A case of resistance to thyroid hormone with thyroid cancer. J Korean Med Sci 25:1368–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paragliola RM, Lovicu RM, Locantore P, Senes P, Concolino P, Capoluongo E, Pontecorvi A, Corsello SM. 2011. Differentiated thyroid cancer in two patients with resistance to thyroid hormone. Thyroid 21:793–797 [DOI] [PubMed] [Google Scholar]
- 21. Weinert LS, Ceolin L, Romitti M, Camargo EG, Maia AL. 2012. Is there a role for inherited TRβ mutation in human carcinogenesis? Arq Bras Endocrinol Metab 56:67–71 [DOI] [PubMed] [Google Scholar]
- 22. Zhu XG, Zhao L, Willingham MC, Cheng SY. 2010. Thyroid hormone receptors are tumor suppressors in a mouse model of metastatic follicular thyroid carcinoma. Oncogene 29:1909–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin KH, Willingham MC, Liang CM, Cheng SY. 1991. Intracellular distribution of the endogenous and transfected β form of thyroid hormone nuclear receptor visualized by the use of domain-specific monoclonal antibodies. Endocrinology 128:2601–2609 [DOI] [PubMed] [Google Scholar]
- 24. Zhao X, Guan JL. 2011. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev 63:610–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guan JL. 2010. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life 62:268–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim LC, Song L, Haura EB. 2009. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol 6:587–595 [DOI] [PubMed] [Google Scholar]
- 27. Schlaepfer DD, Hunter T. 1996. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol 16:5623–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunton VG, Frame MC. 2008. Src and focal adhesion kinase as therapeutic targets in cancer. Curr Opin Pharmacol 8:427–432 [DOI] [PubMed] [Google Scholar]
- 29. Zachary I. 1997. Focal adhesion kinase. Int J Biochem Cell Biol 29:929–934 [DOI] [PubMed] [Google Scholar]
- 30. Cukierman E, Pankov R, Stevens DR, Yamada KM. 2001. Taking cell-matrix adhesions to the third dimension. Science 294:1708–1712 [DOI] [PubMed] [Google Scholar]
- 31. Hehlgans S, Haase M, Cordes N. 2007. Signalling via integrins: implications for cell survival and anticancer strategies. Biochim Biophys Acta 1775:163–180 [DOI] [PubMed] [Google Scholar]
- 32. Chatzizacharias NA, Kouraklis GP, Theocharis SE. 2008. Clinical significance of FAK expression in human neoplasia. Histol Histopathol 23:629–650 [DOI] [PubMed] [Google Scholar]
- 33. Furumoto H, Ying H, Chandramouli GV, Zhao L, Walker RL, Meltzer PS, Willingham MC, Cheng SY. 2005. An unliganded thyroid hormone β receptor activates the cyclin D1/cyclin-dependent kinase/retinoblastoma/E2F pathway and induces pituitary tumorigenesis. Mol Cell Biol 25:124–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Méndez-Pertuz M, Sánchez-Pacheco A, Aranda A. 2003. The thyroid hormone receptor antagonizes CREB-mediated transcription. EMBO J 22:3102–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ying H, Suzuki H, Furumoto H, Walker R, Meltzer P, Willingham MC, Cheng SY. 2003. Alterations in genomic profiles during tumor progression in a mouse model of follicular thyroid carcinoma. Carcinogenesis 24:1467–1479 [DOI] [PubMed] [Google Scholar]
- 36. Lu C, Cheng SY. 2011. Extranuclear signaling of mutated thyroid hormone receptors in promoting metastatic spread in thyroid carcinogenesis. Steroids 76:885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu C, Mishra A, Zhu YJ, Meltzer P, Cheng SY. 2011. Global expression profiling reveals gain-of-function oncogenic activity of a mutated thyroid hormone receptor in thyroid carcinogenesis. Am J Cancer Res 1:168–191 [PMC free article] [PubMed] [Google Scholar]
- 38. Haymart MR, Glinberg SL, Liu J, Sippel RS, Jaume JC, Chen H. 2009. Higher serum TSH in thyroid cancer patients occurs independent of age and correlates with extrathyroidal extension. Clin Endocrinol (Oxf) 71:434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boelaert K. 2009. The association between serum TSH concentration and thyroid cancer. Endocr Relat Cancer 16:1065–1072 [DOI] [PubMed] [Google Scholar]
- 40. Fiore E, Rago T, Provenzale MA, Scutari M, Ugolini C, Basolo F, Di Coscio G, Berti P, Grasso L, Elisei R, Pinchera A, Vitti P. 2009. Lower levels of TSH are associated with a lower risk of papillary thyroid cancer in patients with thyroid nodular disease: thyroid autonomy may play a protective role. Endocr Relat Cancer 16:1251–1260 [DOI] [PubMed] [Google Scholar]
- 41. Fiore E, Vitti P. 2012. Serum TSH and risk of papillary thyroid cancer in nodular thyroid disease. J Clin Endocrinol Metab 97:1134–1145 [DOI] [PubMed] [Google Scholar]
- 42. Cheng SY. 2007. Thyroid hormone receptor mutations and disease: insights from knock-in mouse models. Expert Rev Endocrinol Metab 2:47–57 [DOI] [PubMed] [Google Scholar]
- 43. Cheng SY. 2004. Multi-factorial regulation of in vivo action of TRβ mutants. Lessons learned from RTH mice with a targeted mutation in the TRβ gene. In: Beck-Peccoz P, ed. Syndromes of hormone resistance in the hypothalamic-pituitary-thyroid axis. London: Kluwer; Academic Publishers; 137–148 [Google Scholar]