

Abstract
Many forms of cellular motility are driven by the growth of branched networks of actin filaments, which push against a membrane. In the dendritic nucleation model, Arp2/3 complex is critical, binding to the side of an existing mother filament, nucleating a new daughter filament, and thus creating a branch. Spatial and temporal regulation of Arp2/3 activity is critical for efficient generation of force and movement. A diverse collection of Arp2/3 regulatory proteins has been identified. They bind to and/or activate Arp2/3 complex via an acidic motif with a conserved tryptophan residue. We tested this model for Arp2/3 regulator function in vivo, by examining the roles of multiple Arp2/3 regulators in endocytosis in living yeast cells. We measured the molecular composition of the actin network in cells with mutations that removed the acidic motifs of the four Arp2/3 regulators previously shown to influence the proper function of the actin network. Unexpectedly, we did not find a simple or direct correlation between defects in patch assembly and movement and changes in the composition and dynamics of dendritic nucleation proteins. Taken together our data does not support the simple hypothesis that the primary role for Arp2/3 regulators is to recruit and activate Arp2/3. Rather our data suggests that these regulators may be playing more subtle roles in establishing functional networks in vivo.
Introduction
Branched networks of growing actin filaments push against membranes and contribute force to a wide range of cellular movements (reviewed in (1)). The dendritic nucleation model proposes a molecular basis for how a branched actin filament network can form and generate a pushing force (2–4). In this model, Arp2/3 is recruited to the site of actin network formation. Once there, Arp2/3 binds to the side of an existing mother actin filament, and it is activated to nucleate a new daughter filament with a free barbed end, which establishes a branch point in the network. These steps are presumed to be under the control of Arp2/3 regulatory proteins that have been demonstrated in vitro to bind Arp2/3 complex and induce a conformational change that allows Arp2/3 to nucleate new actin filaments (reviewed in (5)).
Arp2/3 regulators are a diverse superfamily of multidomain proteins that bind to the Arp2/3 complex via an acidic motif containing a conserved tryptophan residue, referred to as an acidic/DDW region (see Fig. 1 D). The superfamily can be divided into two broad classes. The first class consists of proteins that bind simultaneously to Arp2/3 complex and to an actin monomer. These regulators are proposed to activate Arp2/3 and then provide the first actin monomer to create the new daughter filament. This class includes WASp (Wiskott-Aldrich Syndrome protein), N-WASp (neural WASp), WAVE (WASP family verprolin homology proteins), WASH (WASp and Scar homolog), WHAMM (WASP homolog associated with actin, membranes, and microtubules), and JMY (junction-mediating and regulatory protein), among others. The second class of Arp2/3 regulators binds simultaneously to Arp2/3 and to an actin filament. In vitro, the most prominent activity of these proteins is to stabilize Arp2/3-based filament branches. These proteins increase the nucleation activity of Arp2/3, but far less well than do the first class of regulators. This class of regulators includes cortactin, HS1, and Auxin binding protein 1 (Abp1) among others (reviewed in (1,5,6)).
Figure 1.
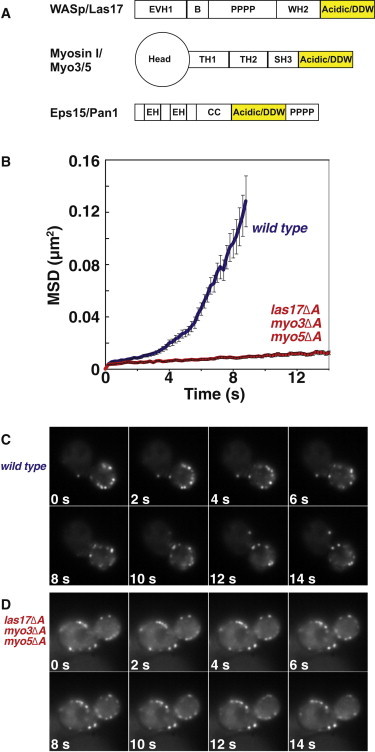
Movement of actin patches in Arp2/3 complex regulator mutants. (A) Schematics of the Arp2/3 regulators mutated in this study. The acidic/DDW motif, which was deleted in each protein, is indicated in yellow. EVH1 = Ena/VASP Homology domain 1; B = basic region; PPPP = Poly-proline; WH2 = WASp homology 1; Head = myosin head/motor domain; TH1 = Tail homology 1; TH2 = Tail homology 2; SH3 = Src homology 3; EH = Eps15 homology; CC = coiled coil. (B) MSD plots of WT (blue) and mutant (red) patches aligned at the start of their lifetimes. Error bars are ± one standard error. Patches in mutant cells are longer lived and do not undergo movement away from the cortex. Adapted with permission (23). (C and D) Time-lapse images of actin patches labeled with Abp1-GFP in WT and las17Δacidic myo3Δacidic myo5Δacidic cells.
In cells, the molecular mechanisms by which Arp2/3 regulators work are largely unknown. A large number of studies have documented that class-I and class-II Arp2/3 regulators promote assembly of branched actin networks and influence the cellular processes that depend on those networks (7–22). In terms of molecular mechanism, the regulator-Arp2/3 interaction has been shown to be critical for endocytosis, which is the primary Arp2/3-dependent process in budding yeast (23,24), but little evidence regarding the mechanism exists in other systems. Further obscuring our understanding of the function of these proteins is that only a few studies have examined loss of the interaction of an activator with Arp2/3 in cells under conditions where the mutant regulator is expressed at normal levels and the endogenous wild-type (WT) regulator is absent (18,25,26). No studies have tested molecular hypotheses for how disrupting the interaction between Arp2/3 regulators and Arp2/3 might affect the assembly and molecular composition of the actin filament network in cells.
In budding yeast, Arp2/3 is necessary for cortical actin patches to mediate endocytosis (27,28). First, endocytic proteins and Arp2/3 regulators are recruited to the site. Next, Arp2/3 is recruited and actin filament assembly begins. The patch then moves a short distance (∼200 nm) away from the membrane, which presumably corresponds to endocytic invagination. Next, endocytic proteins are lost from the patch, and fission of the vesicle from the plasma membrane occurs. Finally, the vesicle moves into the cytoplasm. Actin and Arp2/3 are required for these movements.
Actin patches in budding yeast provide an excellent model system to understand the formation of Arp2/3-based actin networks in cells, because actin patch assembly and movement requires Arp2/3, but Arp2/3 is not necessary for cell viability or for the assembly of other actin-based structures (29–31). The yeast actin patch contains one class-I Arp2/3 regulator, the WASp homolog Las17, and four class-II regulators, including two type-I myosins (Myo3 and Myo5), the eps15 homolog Pan1, and the actin-binding protein Abp1, which has a domain structure similar to cortactin. The acidic/DDW Arp2/3-binding regions of these proteins are known to have differing levels of importance for the assembly and movement of actin patches (23,24). These studies leave the question of molecular mechanism unanswered.
To investigate molecular mechanism, we have examined how mutations that disrupt interaction between Arp2/3 and a regulator affect the levels of dendritic nucleation proteins over time, using quantitative fluorescence microscopy of living cells. We quantitated the levels of Arp2/3 complex, barbed-end capping protein (CP), and Abp1, an actin filament side-binding protein, using an internal standard. Unexpectedly, mutations with large effects on patch movement produced relatively minor decreases in the accumulation of these molecular components. In some cases, the mutations with impaired movement even increased the levels of the molecular components. The results argue that simple notions of recruitment and activation are not sufficient to account for the cellular activity of the Arp2/3 regulators.
Materials and Methods
Yeast strains
The strains used in this study are listed in Table S1 in the Supporting Material. All strains are derived from YJC6494, an isogenic strain we generated by diploidizing BY4741. Green fluorescent protein (GFP) constructs were generated as described (32,33). Mutations were generated in this new isogenic background as described (23). We confirmed that the mutant proteins were expressed and localized to actin patches by constructing and expressing GFP-fusions of the mutant proteins. Fluorescence microscopy revealed that the fusions were expressed and that they localized to patches in cells (see Fig. S8).
Microscopy and image analysis
Strains were grown overnight in liquid yeast extract, peptone, adenine, and dextrose at 30°C to an optical density of ∼0.1–0.5. Cells were collected by centrifugation (82 × g, 5 min) and suspended in SD-complete medium or in a simplified nonfluorescent synthetic media (34).
To quantitate GFP fusion protein levels, we mixed cells expressing Cse4-GFP with cells expressing the fusion of interest and placed them onto a 1% agarose pad, made with the same nonfluorescent media and covered with a #1 coverslip. Fluorescence movies were collected with a widefield, inverted microscope (IX81, Olympus) with a 1.35 NA 100X oil immersion objective, and an EM-CCD camera (Hamamatsu). A 1.5X magnifier was placed in front of the camera, and a 1.6X magnifier was placed in the light path in the body of the microscope. GFP was excited with laser illumination at 488 nm. Images were collected from a single, equatorial focal plane with 900 ms exposures at 1 frame/s. For all experiments, 60 consecutive frames were collected. The temperature was maintained at 30°C for all observations.
Patch tracking was performed as described (23). Tracks were accepted for analysis if they satisfied all of the following criteria: 1), the patch was observed for at least 6 s. 2), The patch originated near the cortex. 3), The patch disappeared during the time course of the movie. 4), The patch could be distinguished from and was not immediately adjacent to another patch. 5), During subsequent intensity analysis a single assembly phase and a single disassembly phase were observed. All tracks were manually verified and corrected by the observer. The majority of the patches excluded by our criteria are excluded because we cannot resolve them from other patches sufficiently to measure them or because the entire life of the patch is not contained within the movie. For mutants exhibiting large increases in patch lifetime, our requirement that the entire lifetime of a patch be present in the movie may result in a slight bias in selecting the shortest lived patches. Intensity data were extracted using the X-Y and time coordinates from unmodified TIF images using a custom macro in ImageJ (Wayne Rasband, NIH, Bethesda, MD, http://imagej.nih.gov/ij/). The tracking software and the ImageJ macro are available from the authors. The integrated intensity of a 9 × 9 pixel square was collected, with background subtraction from pixels adjacent to the patch as described (35).
Cse4 at kinetochores was used as a standard, since the number of molecules at the kinetochore has been determined (36). During the preparation of this manuscript it became apparent that the original number of molecules of Cse4 at yeast kinetochores was underestimated (37,38). Instead of 32 molecules of Cse4 at one cluster of 16 kinetochores, the new studies found 122 and ∼80 molecules, and the investigators noted significant variation from strain to strain. We therefore determined the number of molecules of Cse4 in the kinetochores in our strain, using GFP::MotB in Escherichia coli as a reference (39). We found 109 molecules of Cse4 at each anaphase kinetochore cluster in our strain, within the range observed in the recent studies.
The intensity value of each Cse4 particle was determined in every frame of a 60-s movie. Intensity values for 6–8 Cse4 spots at each time point were averaged, giving Cse4 intensity for each time point during the movie. The value for fluorescence per Cse4 particle was divided by 109 to yield the fluorescence value corresponding to a single molecule of GFP at that time point. The fluorescence intensity value for each actin patch at every time point during the 60-s movie was divided by the value of fluorescence per GFP molecule at that time point. This process of normalizing at every time point corrected for photobleaching. The result was the number of molecules of GFP for each time point in a patch’s lifetime. The intensity versus time data for all patches from a given segregant was aligned at the time point of maximum fluorescence. At each time point, the mode of the distribution of numbers of molecules and the standard error of the mode (100 iterations) were determined as described (40). The rates of assembly and disassembly were determined from plots of number of molecules versus time for each individual patch, as the slope of the linear phase of assembly or disassembly. The time of the linear phase of assembly and disassembly were also determined for every patch.
Results
Actin assembly in an Arp2/3 activator mutant with a severe defect in patch motility
To begin our molecular analysis of Arp2/3-mediated actin assembly, we chose a triple mutant with a severe actin patch motility defect. We showed previously that cells lacking the acidic Arp2/3-binding regions of the activators WASp/Las17, Myo3, and Myo5 (diagrammed in Fig. 1 A) have severe defects in the motility of actin patches (23). For example, mean-square displacement (MSD) plots of Abp1-GFP-labeled patches in this mutant show an almost complete loss of movement of patches (Fig. 1 B). The prevailing model for Arp2/3 regulator function predicts that this motility defect should result from a decrease in the amount of actin filaments nucleated at patches. To the contrary, qualitative examination of fluorescence images of Abp1-GFP in patches did not show a noticeable decrease in the intensity of the patches (Fig. 1, C and D).
To confirm this qualitative observation, we quantitated the number of molecules of a given protein in a patch with a slightly modified version of the method employed by Joglekar and colleagues (36). From digital fluorescence images, we measured the fluorescence intensity of GFP-labeled Abp1 in a patch. We constructed an Abp1-GFP fusion by integration of GFP at the endogenous locus for ABP1. Thus, the cells expressed no unlabeled Abp1, and expression of Abp1-GFP was controlled by the endogenous elements of the locus. We converted the fluorescence value to the absolute number of molecules by comparison with the fluorescence intensity of the kinetochore in anaphase cells expressing the histone variant Cse4-GFP (see Materials and Methods). Cells expressing Abp1-GFP and Cse4-GFP strains were mixed and viewed on the same slide. Background subtraction was based on the intensity of pixels immediately adjacent to the region of interest, which helped control for local fluctuations in the background fluorescence (35).
Using this method, we collected data for the number of molecules of Abp1 per patch over the lifetime of a patch, in a large number of patches. Data from all the patches in cells of the same genotype were pooled. The data sets were aligned with each other by defining time zero as the time when the value of each patch reached its peak. Following alignment, the values for a given time point were not averaged; instead, the individual values for number of molecules were retained in the data set and plotted as histograms, as illustrated for a WT strain with the three-dimensional plot in Fig. 2 A. In this example, at each of the indicated times on the y axis, the graph presents a histogram of the number of molecules per patch on the x axis, with the number of patches having that value on the z axis. In this case, the total number of patches was 323. All the histograms, at every time point, display non-Gaussian distributions, with tails extended toward larger values. Because these distributions were not Gaussian, we calculated the mode of the distribution (see Materials and Methods). Throughout the study, we then present the mode ± one standard error of the mode, as a succinct way to display the data sets, unless otherwise indicated. A typical plot would be the mode of the number of Abp1 molecules versus time (Fig. 2 B). In the figures and supplements we have included histograms of the data of the critical parameters to allow for consideration of the entire population observed. In addition to the mode, we have also included values for the median, mean, and 90% confidence intervals for all the data sets in the supplemental tables. The full data sets will be available to other investigators.
Figure 2.
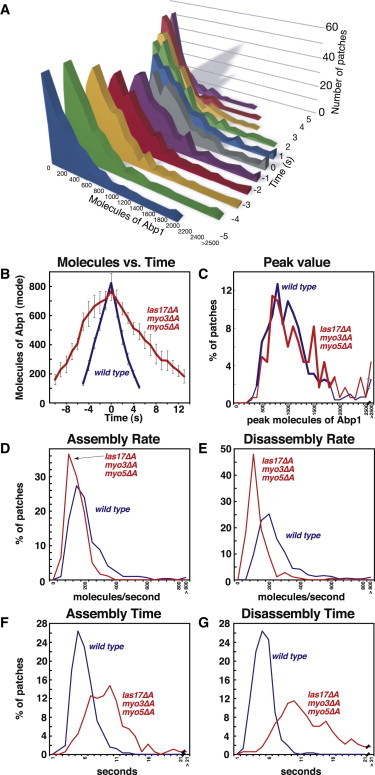
Assembly of Abp1, a marker for actin filaments, in actin patches over time. (A) Data for the absolute number of molecules of the Abp1 in patches over time. At each time point (y axis), the figure shows a histogram of the number of patches (z axis) that contain a certain number of Abp1 molecules (x axis). The data include 323 actin patches from 24 WT cells. (B–G) Analysis of Abp1 assembly in WT (blue) and las17Δacidic myo3Δacidic myo5Δacidic (red) cells. Data are from three segregants of each genotype. (B) Number of molecules of GFP-Abp1 per patch versus time. Plots were generated from data aligned at the maximal (peak) value for each patch. This time point was defined as zero, and the mode of the distribution at each time point was graphed. Error bars are ± one standard error of the mode. (C) Histogram of the peak number of molecules in patches. (D) Histogram of the rate of assembly. (E) Histogram of the rate of disassembly. (F) Histogram of the duration of Abp1 assembly. (G) Histogram of the duration of disassembly. Strain numbers and numbers of patches analyzed were as follows. Abp1-GFP strains: WT, YJC6718-20, N = 99, 114, 110; WASp/las17Δacidic myo3Δacidic myo5Δacidic, YJC7151-3, N = 75, 65, 44. Cse4-GFP: YJC6725-6.
The quantitative analysis of molecules of Abp1-GFP per patch in las17Δacidic myo3Δacidic myo5Δacidic cells confirmed the qualitative observation that the peak amount of Abp1-GFP per patch was not changed in the mutant (Fig. 2, B and C). Changes in the timing of accumulation and loss were apparent from the graphs of number of molecules versus time (Fig. 2 B). From the full original set of data, we calculated the rate and time for the assembly and disassembly phase for each individual patch. Those values are displayed as histograms in Fig. 2, D–G. In the mutant, the duration of the assembly phase was prolonged (Fig. 2 F, WT 5.2 ± 0.3 s, mutant 9.1 ± 0.6 s), with a decrease in the rate of accumulation (Fig. 2 D, WT 129 ± 14 molecules/s, mutant 85 ± 18 molecules/s). The disassembly phase was also prolonged (Fig. 2 G, WT 5.2 ± 0.4 s, mutant 10.0 ± 0.7 s), with a decreased rate (Fig. 2 E, WT 152 ± 9 molecule/s, mutant 69 ± 6 molecule/s).
The findings of normal peak levels and prolonged disassembly phases are not what one would expect if the role of these Arp2/3 regulators were simply to recruit and/or activate Arp2/3 at the patch. One would have expected large decreases in peak levels and/or greatly accelerated disassembly to account for the nearly complete loss of patch motility.
Molecular composition of actin patches in mutants with severe motility defects
Because these results did not support the simplest molecular model, we investigated the molecular composition of the actin network in these cells in more detail. The assembly of a branched actin network capable of producing force involves a balance among the rates of nucleation and elongation of actin filaments, the formation of branch points, and the termination of filament growth. To understand how this balance is perturbed in this mutant, we measured the content of molecular markers that reflect these activities. In addition to monitoring F-actin levels by quantitating Abp1-GFP, we also measured levels of Arp2/3 complex, which is proposed to be responsible for nucleation and branch formation, and for capping protein, which is proposed to be responsible for terminating filament growth at barbed ends.
For the Arp2/3 measurements, we quantitated the levels of two subunits, Arc35 and Arc15. As for Abp1, we used GFP fusions integrated at the endogenous locus. Unfortunately, triple-mutant haploid spores lacking the acidic domain of Las17, Myo3, and Myo5, which also expressed Arc35-GFP or Arc15-GFP failed to germinate. Instead, we examined the Arp2/3 levels in the double mutant las17Δacidic myo5Δacidic, for which Arc35-GFP haploid spores did germinate and cells grew normally. These cells have defects in patch motility almost as severe as those of las17Δacidic myo3Δacidic myo5Δacidic cells (23). In las17Δacidic myo5Δacidic cells, the amount of Arp2/3 accumulation was decreased, with a peak value about two-thirds that of WT. Within the distribution of values, many patches contained peak levels of Arp2/3 comparable to those seen in WT cells (Fig. 3, A and B). The rate at which Arp2/3 accumulated was decreased in the double mutant (WT 44.0 ± 3.2/s, mutant 18.1 ± 1.7/s, Fig. S1 A) and the duration of accumulation was increased (WT 5.8 ± 0.3 s, mutant 7.8 ± 0.6 s, Fig. S1 C). As we observed for Abp1, the disassembly process was also disrupted in these mutants. The rate of disassembly was decreased (WT 56.9 ± 4.0/s, mutant 20.7 ± 1.1/s, Fig. S1 B), and the duration was increased (WT 5.0 ± 0.1 s, mutant 9.1 ± 0.6 s).
Figure 3.
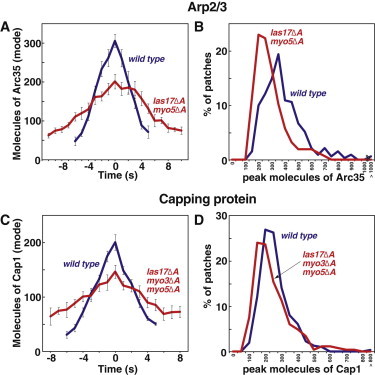
Assembly of Arp2/3 complex as GFP-Arc35 (A and B) and CP as GFP-Cap1 (C and D) in WT (blue), las17Δacidic myo5Δacidic (A and B, red) and las17Δacidic myo3Δacidic myo5Δacidic (C and D, red) cells. Data are from three segregants of each genotype. Panels A and C are plots of the number of molecules of the GFP-labeled protein per patch versus time. Plots were generated from data aligned at the maximal (peak) value for each patch. This time point was defined as zero, and the mode of the distribution at each time point was graphed. Error bars are ± one standard error of the mode. Panels B and D are histograms of the peak number of molecules in patches. Supplemental data related to these strains are presented in Fig. S1. Strain numbers and numbers of patches analyzed were as follows. Arc35-GFP: WT, YJC6756-YJC6758, N = 107, 126, 98. WASp/las17Δacidic myo5Δacidic, YJC7167-9, N = 60, 42, 55. Cap1-GFP: WT, YJC6804-6, N = 127, 96, 90. WASp/las17Δacidic myo3Δacidic myo5Δacidic, YJC7126-8. N = 77, 75, 115. Cse4-GFP: YJC6725-6.
If the Arp2/3 regulators function to activate Arp2/3 in patches, one would expect that patches in the mutant cells would contain a decreased number of barbed ends, reflecting decreased nucleation from Arp2/3. In cells, free barbed ends of actin filaments should be rapidly capped by CP. Therefore, we used CP-GFP as a reflection of the number of barbed ends. Triple mutant las17Δacidic myo3Δacidic myo5Δacidic cells were viable when expressing CP-GFP. The results for CP were very similar to what was observed for Arp2/3. In mutant cells, CP accumulated to a peak level of ∼2/3 of the WT level, and many patches contained WT amounts of CP at their peak (Fig. 3, C and D). The rates of CP assembly (WT 31.0 ± 1.9/s, mutant 17.3 ± 1.6/s, Fig. S1 E) and disassembly (WT 34.3 ± 3.2/s, mutant 16.4 ± 2.3/s, Fig. S1 F) were decreased, and the durations of assembly (WT 5.6 ± 0.3 s, mutant 6.3 ± 0.5 s, Fig. S1 G) and disassembly (WT 4.5 ± 0.3 s, mutant 7.5 ± 0.3 s, Fig. S1 H) was increased.
Addressing the complexity of multiple regulators
One challenge for understanding the regulation of Arp2/3 is that the various protein regulators are present in the same structure at the same time (23,24). In previous studies, WASP/Las17, Myo3, and Myo5 were found to have overlapping, as well as some distinct, functions in patch assembly and movement (23,24). Actin patches also contain another Arp2/3 regulator that can affect the assembly and movement of actin patches, the endocytic scaffold protein Pan1. To begin to address the complexity of the system, we tested how each of the Arp2/3 regulators, alone and in combination, influences actin patch assembly and motility. We quantitatively assessed the effects of mutations disrupting the acidic Arp2/3 binding region of Las17, Myo3, Myo5, and Pan1, individually and in all viable combinations. A fifth Arp2/3 regulator, Abp1 is also present in the actin patch. However, Abp1 does not have a major affect on patch assembly and movement; instead, its role appears to be in disassembling endocytic proteins, such as Sla1 and Sla2, from the patch (23,41). Abp1 was therefore not included in our analysis.
The WASp/las17 single mutant: unexpected increased accumulation
Among the single mutants, the las17Δacidic mutant has the strongest actin patch assembly and movement phenotype (23). Therefore, we hypothesized that the las17Δacidic mutant would display the greatest decrease in Arp2/3 recruitment or activation, among the single mutants. However, the peak level of Arp2/3 in patches was the same in WT and las17Δacidic mutant cells (Fig. 4, A and B). Unexpectedly, for CP and Abp1, peak levels of accumulation were increased in the mutant (Fig. 4, C–F), the opposite direction from what one would have predicted.
Figure 4.
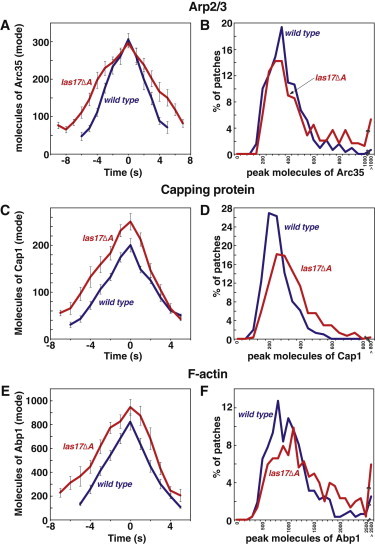
Assembly of Arp2/3 complex as GFP-Arc35 (A and B), CP as GFP-Cap1 (C and D) and F-actin as GFP-Abp1 (E and F) in WT (blue) and las17Δacidic (red) cells. Data are from three segregants of each genotype. Panels A, C, and E are plots of the number of molecules of the GFP-labeled protein per patch versus time. Plots were generated from data aligned at the maximal (peak) value for each patch. This time point was defined as zero, and the mode of the distribution at each time point was graphed. Error bars are ± one standard error of the mode. Panels B, D, and F are histograms of the peak number of molecules in patches. Supplemental data related to these strains are presented in Fig. S2. Strain numbers and numbers of patches analyzed were as follows. WT strains and data are the same as in Figs. 2 and 3. Arc35-GFP: WASp/las17Δacidic, YJC6759-YJC6761, N = 89, 68, 90. Cap1-GFP: WASp/las17Δacidic, YJC6807-9, N = 82, 96. 132. Abp1-GFP: WASp/las17Δacidic, YJC6721-3, N = 96, 93, 116. Cse4-GFP: YJC6725-6.
In the patch assembly phase, the rate of Arp2/3 assembly was slightly reduced in las17Δacidic cells (WT 44.0 ± 3.2/s, mutant 36.6 ± 2.5/s, Fig. S2 A), and the duration of assembly was increased (WT 5.8 ± 0.3 s, mutant 7.3 ± 0.3 s, Fig. S2 C). For both CP and Abp1, the rate was unaffected (Fig. S2 E and I), and the duration was increased (CP: WT 5.6 ± 0.3 s, mutant 7.2 ± 0.3 s, see Fig. 1 H, Fig. S2 G. Abp1: WT 5.2 ± 0.3 s, mutant 7.4 ± 0.3 s, Fig. S2 J).
Examining patch disassembly, we found that the rate of Arp2/3 disassembly was decreased in las17Δacidic cells (WT 56.9 ± 4.0/s, mutant 37.7 ± 3.4/s, Fig. S2 B). In contrast, the disassembly rates for CP and Abp1 were increased in the mutant (CP: WT 34.3 ± 3.2/s, mutant 44.7 ± 5.0/s, Fig. S2 F. Abp1: WT 152 ± 9 /s, mutant 179 ± 14/s, Fig. S2 J). The duration of disassembly was increased for Arp2/3 (WT 5.0 ± 0.1 s, mutant 6.2 ± 0.2 s, Fig. S2 D) and for CP (WT 4.5 ± 0.3 s, mutant 5.2 ± 0.2 s, Fig. S2 H), and it did not change for Abp1 (Fig. S2 L).
The most interesting and striking result in this section was the increased peak levels of CP and Arp2/3 for the mutant, contrasting with the decreased peak levels of CP and Arp2/3 in the previous triple mutant. Both mutants showed strong defects in actin patch movement; therefore, a simple molecular explanation is not sufficient to account for all the results.
The myosin-I Myo3/Myo5 double mutant
We next examined the consequences of loss of the acidic domains of the class-I myosins Myo3 and Myo5 on the accumulation of Arp2/3, CP, and Abp1. The myo3Δacidic myo5Δacidic cells have no defects in the movement of actin patches (23). As expected, no changes in the level of accumulation or the assembly parameters were observed for Arp2/3 and CP in the double-mutant cells (Fig. 5, A–D, Fig. S3, A, C, E, and G). However, the peak level of Abp1 accumulation was slightly reduced in mutant cells (Fig. 5, E and F); this was accompanied by a small decrease in the rate of assembly (WT 129 ± 14/s, mutant 101 ± 6/s, Fig. S3 I), but a normal duration (Fig. S3 K). We also found defects during disassembly. For CP, there was a slight increase in the rate of CP loss from the patch (Fig. S2 F). For Abp1, the rate was unaffected (Fig. S2 J), although the time of disassembly may have been slightly less (WT 5.2 ± 0.4 s, mutant 4.6 ± 0.3 s, Fig. S3 L).
Figure 5.
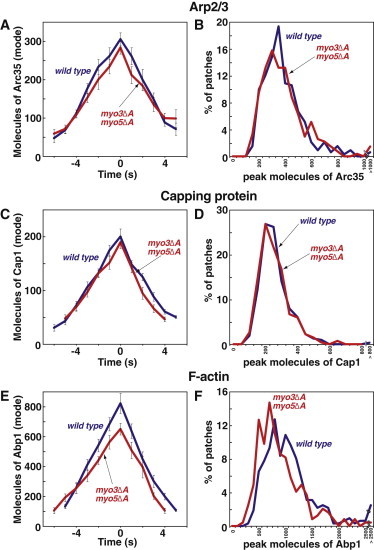
Assembly of Arp2/3 complex as GFP-Arc35 (A and B), CP as GFP-Cap1 (C and D) and F-actin as GFP-Abp1 (E and F) in WT (blue) and myo3Δacidic myo5Δacidic (red) cells. Data are from three segregants of each genotype. Panels as in Fig. 4. Supplemental data related to these strains are presented in Fig. S3. Strain numbers and numbers of patches analyzed were as follows. WT strains and data are the same as in Figs. 2 and 3. Arc35-GFP: myo3Δacidic myo5Δacidic, YJC7173-5, N = 94, 80, 99. Cap1-GFP: myo3Δacidic myo5Δacidic, YJC7123-5, N = 95, 89, 84. Abp1-GFP: myo3Δacidic myo5Δacidic, YJC7148-50, N = 91, 76, 98. Cse4-GFP: YJC6725-6.
In our view, the most interesting results from this section were that some differences in the molecular parameters were found, with magnitudes not terribly dissimilar from those for the triple Δacidic mutant and single las17 Δacidic mutant; however, the motility of the actin patches was unchanged. Therefore, small differences in the molecular composition are not sufficient to produce large changes in motility.
The Pan1 single mutant
In our previous studies, loss of the acidic domain of Pan1 had only a very minor effect on the motility of actin patches (23). Consistent with this observation, we found no effect on the levels or dynamics of any of the dendritic nucleation proteins we examined in this study (Fig. S4).
The Las17 Pan1 double mutant
The pan1Δacidic las17Δacidic double-mutant cells have a patch movement phenotype similar to that of las17Δacidic single-mutant cells, with the exception of showing a slight recovery in the very late movements (23). We expected, therefore, that las17Δacidic pan1Δacidic cells would have molecular parameters similar to those of las17Δacidic cells. Interestingly, this was not always the case.
For Arp2/3, the most striking difference was in the disassembly phase. Adding the pan1Δacidic mutation to the las17Δacidic resulted in a recovery to normal of both the rate (WT 56.9 ± 4.0 molecules/s, las17Δacidic 37.7 ± 3.4 molecules/s, las17Δacidic pan1Δacidic 60.1 ± 13.2 molecules/s; Fig. S2 B, Fig. S5 B) and time of disassembly (WT 5.0 ± 0.1 s, las17Δacidic 6.2 ± 0.2 s, las17Δacidic pan1Δacidic 5.6 ± 0.3 s; Fig. S2 D, Fig. S5 D).
The double mutant had a greater number of patches with high peak levels of Arp2/3 compared to the single mutant (Fig. 6 B vs. Fig. 4 B). Correspondingly, the duration of assembly was also increased by a greater amount in double-mutant cells compared to las17Δacidic cells (WT 5.8 ± 0.3 s, las17Δacidic 7.3 ± 0.3 s, las17Δacidic pan1Δacidic 9.1 ± 0.5 s; Fig. S2 C, Fig. S5 C). Assembly rates were closer to normal in the double mutant than in the single mutant (Fig. S2 A, Fig. S5 A).
Figure 6.
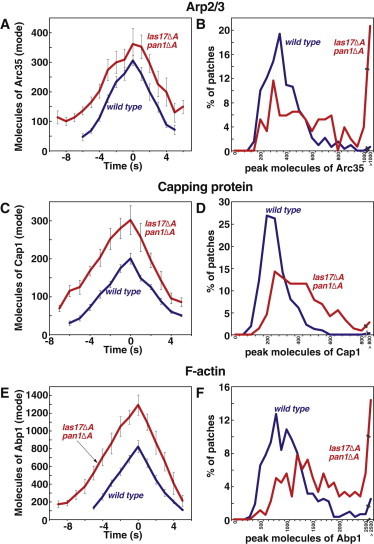
Assembly of Arp2/3 complex as GFP-Arc35 (A and B), CP as GFP-Cap1 (C and D) and F-actin as GFP-Abp1 (E and F) in WT (blue) and las17Δacidic pan1Δacidic (red) cells. Data are from three segregants of each genotype. Panels as in Fig. 4. Supplemental data related to these strains are presented in Fig. S5. Strain numbers and numbers of patches analyzed were as follows. WT strains and data are the same as in Figs. 2 and 3. Arc35-GFP: WASp/las17Δacidic pan1Δacidic, YJC7104-6, N = 46, 44, 65. Cap1-GFP: WASp/las17Δacidic pan1Δacidic, YJC7027-9 N = 66, 66, 86. Abp1-GFP: WASp/las17Δacidic pan1Δacidic, YJC6961-3, N = 53, 75, 53. Cse4-GFP: YJC6725-6.
The differences between las17Δacidic and las17Δacidic pan1Δacidic cells were even greater when we examined CP and Abp1 (Fig. 6 C vs. Fig. 4 C). Peak levels of CP and Abp1 were higher in the double mutant (farther from normal) than in the single mutant (Fig. 6, C–F, Fig. 4, C–F).
In the assembly phase, the rate of increase for CP was higher, farther from WT, in the double mutant compared to the single mutant (WT 31.0 ± 1.9 molecules/s, las17Δacidic 33.8 ± 2.0 molecules/s, las17Δacidic pan1Δacidic 43.2 ± 2.4 molecules/s; Fig. S2 E, Fig. S5 E). For Abp1, the assembly rate was also increased in double-mutant cells compared to single-mutant cells (WT 129 ± 14 molecules/s, las17Δacidic 114 ± 14 molecules/s, las17Δacidic pan1Δacidic 158 ± 18 molecules/s; Fig. S2 I, Fig. S5 I).
In the disassembly phase, the rate of loss of CP was also higher, farther from WT, in the double mutant than in the single mutant (WT 34.3 ± 3.2 molecules/s, las17Δacidic 44.7 ± 5.0 molecules/s, las17Δacidic pan1Δacidic 51.5 ± 6.8 molecules/s; Fig. S2 F, Fig. S5 F). For Abp1, the results were similar (WT 152 ± 9 molecules/s, las17Δacidic 179 ± 14 molecules/s, las17Δacidic pan1Δacidic 222 ± 22 molecules/s; Fig. S2 J, Fig. S5 J).
The durations of assembly and disassembly were the same for CP in the single and double mutants (Fig. S2, G and H, Fig. S5, G and H). For Abp1, the duration of assembly was increased (WT 5.2 ± 0.3 s, las17Δacidic 7.4 ± 0.3 s, las17Δacidic pan1Δacidic 8.9 ± 0.2 s, Fig. S2 K, Fig. S5 K), but the duration of disassembly was not (Fig. S2 L, Fig. S5 L).
Overall, the results in this section illustrate that different molecule parameters can be observed in strains that have similar patch movement phenotypes. As above, there is no simple correlation between molecular composition and movement phenotype.
Type-I myosins and Pan1
Our previous study of actin patch motility did not include the myo3Δacidic myo5Δacidic pan1Δacidic mutant. Therefore, we studied the movement of Abp1-GFP patches in these cells, with MSD analysis as described (23), Fig. S6). The frequency of patch movement away from the cell cortex was similar to that of wt cells (Fig. S6 B). Patches in the mutant showed a slight decrease in the time they spent at the cortex before starting to move (Fig. S6 C). This phenotype is similar to the one observed for myo3Δacidic myo5Δacidic cells, but different from that of pan1Δacidic cells, which showed an increased time at the membrane (23). Therefore, in this respect, the effect of acidic regions mutations in type-I myosins is dominant to the effect of the analogous mutation in Pan1. In addition, myo3Δacidic myo5Δacidic pan1Δacidic cells displayed increased movement of patches, in the MSD analysis, especially for movements after the patch was >200 nm from the membrane (Fig. 6, A and D), which is similar to what was observed in myo3Δacidic myo5Δacidic cells (23).
Because the motility phenotypes were similar for myo3Δacidic myo5Δacidic pan1Δacidic cells and myo3Δacidic myo5Δacidic cells, we predicted that the behavior of Arp2/3 complex, CP, and Abp1 in myo3Δacidic myo5Δacidic pan1Δacidic cells would resemble their behavior in myo3Δacidic myo5Δacidic cells. However, this is not what was observed. Levels of Arp2/3 complex in myo3Δacidic myo5Δacidic pan1Δacidic were slightly decreased compared to WT levels (Fig. 7, A and B), not unchanged as observed in myo3Δacidic myo5Δacidic or pan1Δacidic cells. Furthermore, the rate of Arp2/3 accumulation in myo3Δacidic myo5Δacidic pan1Δacidic patches was significantly lower (WT 44.0 ± 3.2 molecules/s, mutant 25.8 ± 2.9 molecules/s, Fig. S7 A) and the duration of assembly was longer (WT 5.8 ± 0.3 s, mutant 7.1 ± 0.5 s, Fig. S7 C), again not what was observed in pan1Δacidic or myo3Δacidic myo5Δacidic cells. The rate of Arp2/3 disassembly from patches was also significantly decreased in myo3Δacidic myo5Δacidic pan1Δacidic cells (WT 56.9 ± 4.0 molecules/s, mutant 32.9 ± 3.6 molecules/s, Fig. S7 B), but the duration of the disassembly phase was unaffected (Fig. S7 D).
Figure 7.
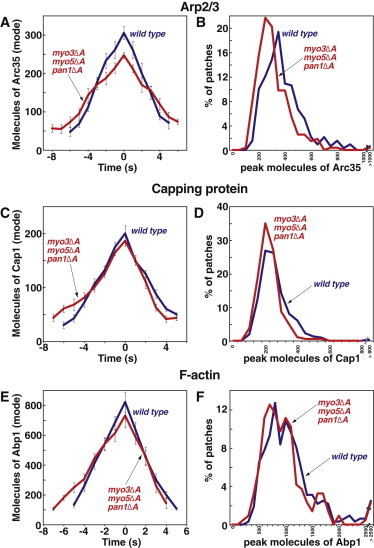
Assembly of Arp2/3 complex as GFP-Arc35 (A and B), CP as GFP-Cap1 (C and D) and F-actin as GFP-Abp1 (E and F) in WT (red) and myo3Δacidic myo5Δacidic pan1Δacidic (blue) cells. Panels as in Fig. 4. Supplemental data related to these strains are presented in Fig. S6 and Fig. S7. WT strains and data are the same as in Figs. 2 and 3. Arc35-GFP: myo3Δacidic myo5Δacidic pan1Δacidic, YJC7348-50, N = 93, 91, 93. Cap1-GFP: myo3Δacidic myo5Δacidic pan1Δacidic, YJC7344-6, N = 59, 61, 66. Abp1-GFP: myo3Δacidic myo5Δacidic pan1Δacidic, YJC7310-2, N = 68, 73, 75. Cse4-GFP: YJC6725-6.
The levels of CP in myo3Δacidic myo5Δacidic pan1Δacidic patches were essentially normal (Fig. 7, C and D), similar to the results for pan1Δacidic and myo3Δacidic myo5Δacidic cells. However, the myo3Δacidic myo5Δacidic pan1Δacidic cells displayed a significant decrease in the rate of CP assembly (WT 31.0 ± 1.9 molecules/s, mutant 19.0 ± 2.0 molecules/s, Fig. S6 E) and an increase in the duration of the assembly phase (WT 5.6 ± 0.3 s, mutant 6.7 ± 0.4 s, Fig. S6 G). These changes were not seen in the two other mutants. For CP, the rate and duration of disassembly were similar to wt (Fig. S6, F and H). The behavior of GFP-Abp1 patches in myo3Δacidic myo5Δacidic pan1Δacidic cells was affected in a manner similar to that of GFP-CP, with a decrease in the rate of assembly (WT 129 ± 14 molecule/s, mutant 96 ± 8 molecule/s, Fig. S6 I), and an increase in the duration of assembly (WT 5.2 ± 0.3 s, mutant 7.0 ± 0.2 s, Fig. S6 K). Abp1 disassembly was largely unaffected, except possibly having a shorter time of disassembly (Fig. S6, J and L).
Discussion
The formation of branched networks of actin filaments, at specific cellular sites, is essential for many cellular functions. A central component in this process is the Arp2/3 complex, which creates branch points and nucleates new filaments with free barbed ends. In biochemical studies, Arp2/3 complex alone has little activity; activator proteins are necessary to induce a conformational change that makes Arp2/3 active. In cells, various Arp2/3 regulators are known to be important for actin assembly and actin-based motility, but there have been very few investigations of the molecular mechanisms by which the regulators function.
Most research on the molecular mechanisms in cells has been in budding and fission yeast, where actin assembly induced by Arp2/3 complex is required for endocytosis and for the movement of endocytic vesicles away from the plasma membrane. One appealing hypothesis is that the regulation of actin assembly at these sites, termed cortical actin patches, occurs at the level of nucleation of filaments by Arp2/3. In a previous study comparing the functions of several regulators, we found that the individual regulators have both distinct and overlapping components with respect to actin filament assembly and actin patch movement (23). Here, we asked how loss of interaction between regulators and Arp2/3 complex influences the molecular composition of the filament network at actin patches. We compared the results for mutations of a number of regulator genes, alone and in combination, testing simple hypotheses for the molecular mechanism of regulator function. The most important conclusion from this study is that simple hypotheses regarding mechanism are not sufficient to explain experimental observations of motility.
In cells, Arp2/3 function might be regulated at three levels. First, a regulator might recruit Arp2/3 to an actin patch. Second, regulators might convert Arp2/3 to its active state, allowing it to nucleate new filaments. Of note, budding yeast Arp2/3 has significant activity on its own, so positive regulation of Arp2/3 activity may be less important in yeast than in other systems. Third, regulators might control the positions of actin filament branch points and their stability over time, thereby affecting the architecture of the filament network.
Failure to recruit Arp2/3 does not account for patch motility defects
The first hypothesis for why motility defects result from the lack of interaction between regulators and Arp2/3 is that not enough Arp2/3 is recruited to the patch. If this were the case, the defects in the motile behavior of actin patches would correlate with loss of some or all of Arp2/3. Our data do not support this prediction.
Most striking, between two mutants with modest decreases in the amount of Arp2/3 at the patch, one mutant displays a nearly complete loss of patch motility, whereas the other has essentially normal motility. The myo3Δacidic myo5Δacidic pan1Δacidic mutant had a ∼20% decrease in Arp2/3 (Fig. 7, Table S1), with essentially normal or increased patch motility (Fig. S7). The las17Δacidic myo5Δacidic mutant had a ∼30% decrease in Arp2/3, with very little to no patch motility (23). Furthermore, many individual patches in these two mutants had the same amount of Arp2/3, yet exhibited different motile behaviors. Thus, failure to recruit Arp2/3 cannot be the simple cause of the motility defects in Arp2/3 regulator mutants.
Failure to nucleate new filaments does not account for patch motility defects
A second hypothesis for why mutations in Arp2/3 regulators alter actin patch motility is that loss of Arp2/3 binding might decrease the activity of Arp2/3 to nucleate the growth of new filaments. Our results revealed little or no correlation between patch motility and either new filament barbed ends or the amounts of F-actin. The las17Δacidic myo3Δacidic myo5Δacidic mutant, with very little patch motility, had a decrease of ∼30% in the peak levels of capping protein, which reflects barbed ends. In fact, the ratio of CP/Arp2/3 in the patches of this mutant was nearly normal (Table S3), confirming that the nucleation activity of Arp2/3 was not affected. With respect to F-actin levels, only the myo3Δacidic myo5Δacidic mutant showed a decrease, (Fig. 5 F, Table S2) and this mutant had no defects in patch assembly or motility (23). In addition, the mutant with the severe motility defect, las17Δacidic myo3Δacidic myo5Δacidic, had normal amounts of F-actin.
Disruption of actin network architecture may account for patch motility defects
The ability of an actin network to generate force should depend on having the proper organization and architecture. The ratios of Arp2/3, CP, and F-actin (Abp1) to each other should provide insight into the architecture of the actin network. Here, we found that the mutant with the most severe defect in motility also had the largest change in the ratios of these three proteins. The las17Δacidic myo3Δacidic myo5Δacidic mutant had more F-actin per Arp2/3 (WT 2.7, mutant 3.8, Table S3) and more F-actin per CP (WT 4.1, mutant 5.2, Table S3). However, the ratio of CP/Arp2/3 was not changed (Table S3). One might speculate that the filament network in the mutant actin patches is less densely branched, with greater distance between branches. Such a network with a lower density of branches might be more flexible and less effective at supporting the forces that push an object.
Modeling the relationship between F-actin and Arp2/3 activators
Because the experimental data contradicted our initial expectations for the dependence of the F-actin content on the branching activity of the Arp2/3 regulators, we have developed a simple two-variable model to calculate this dependence. The two variables are [NPF], the protein count of a representative Arp2/3 regulator (i.e., nucleation-promoting factor) at an endocytic protein patch, and [F], the corresponding count of F-actin. The model makes the following assumptions:
-
1.
[NPF] grows cooperatively, in the sense that when it reaches a certain critical value [NPF]c (related to the size of a critical nucleus) it begins to increase; below the critical value [NPF] decreases. This assumption is motivated by the spontaneous appearance of the patches.
-
2.
[F] causes removal of [NPF] from the membrane. This assumption is motivated by studies in other cell types showing that [F] accelerates NPF dynamics at the cell membrane, and also reduces the total membrane-bound [NPF] (42,43).
-
3.
The growth rate of [F] is proportional to [NPF].
-
4.
The decay rate of [F] is independent of [NPF] and is determined by severing as suggested by (44).
The mathematical form of the model is chosen to be a simple one which embodies assumptions 1–4:
where kdet and ksev are decay-rate parameters; kbr is the actin filament branching rate per NPF, which might be reduced by acidic-domain mutations. Finally,
This function is positive, so that [NPF] grows, if [NPF] > [NPF]c. Because of the simplicity of the model, we do not identify the model parameters directly with biochemically measured quantities, but rather explore the range of possible behaviors of the model by varying the parameters.
In this model, the life cycle of a protein patch is as follows. First, a random fluctuation causes [NPF] to exceed [NPF]c. Then, as illustrated in Fig. 8 A, [NPF] grows, with [F] trailing. Finally, increasing [F] reduces [NPF], and subsequently [F] decays.
Figure 8.
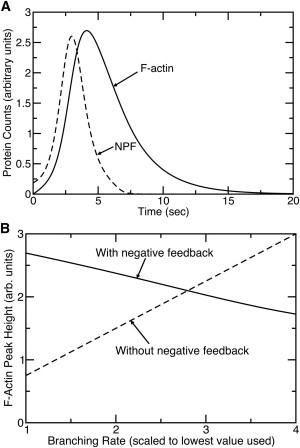
Mathematical modeling of relationships between F-actin and NPF levels. (A) Time courses of NPF (dashed line) and F-actin (solid line) accumulation in simple two-variable model of actin patch dynamics. (B) Dependence of F-actin peak height on branching rate in two-variable model. Dashed line gives results for a modified model in which negative feedback from F-actin onto NPFs has been removed.
Within this model, we have calculated (Fig. 8 B) the dependence of the F-actin peak height (the maximum value of [F]) on kbr. If there were no feedback of F-actin on NPF disassembly, this dependence would simply be a straight line, as indicated by the dashed line. However, the negative feedback of [F] on [NPF] weakens the dependence. As shown by the solid line in Fig. 8 B, it is even possible for the dependence to change sign, therefore increasing kbr causes a decrease in the peak value of [F].
Because the precise dependence of peak height versus kbr is strongly affected by the form of the function H([NPF]), we are not able to make a definite prediction for this dependence. However, the calculations have shown that strong departures from a linear dependence can result from established negative-feedback effects. This finding may be relevant to our unexpected results for the weak dependence of the F-actin peak height on acidic-domain mutations. The weakness of the dependence was surprising, and in the case of las17Δacidic, the dependence had the wrong sign. These effects may be partly due to the effects of negative feedback considered here.
Concluding Remarks
Understanding how the activities of regulators of Arp2/3 are coordinated with each other in a complex multistep process is critical to our understanding of how actin filament networks grow and generate force. Removing the acidic Arp2/3-binding regions of Arp2/3 regulators had deleterious effects on the assembly and movement of actin patches in previous work. Based on biochemical studies with these proteins, we hypothesized that the cellular defects were the result of poor recruitment of Arp2/3 to the patch or failure to activate Arp2/3 at the patch. We found no evidence that the motility phenotypes in the regulator mutants were the result of changes in recruitment or activation of Arp2/3. These findings are summarized in Fig. S9.
On the basis of our results, it is likely that the major role of these regulator proteins in vivo is something other than activation of Arp2/3. Of note, complete loss of any of these proteins results in much more severe phenotypes than mutations of the Arp2/3-binding region, in terms of cell growth and patch motility (23,24,45,46). Interestingly, in humans, most disease-causing WASp mutations decrease the level of the protein or alter the WIP-binding region (WH1) and not the Arp2/3-binding (acidic/DDW) region (47–49). One possible explanation is that the actin assembly system is sufficiently robust so that the changes in Arp2/3 recruitment or activation caused by mutations of these regulators are not sufficient to perturb the system. For example, in the mathematical model discussed previously, negative feedback can cause the F-actin peak height to be independent of the branching rate. In this case other factors, such as the feedback of F-actin onto Arp2/3 regulator activity seen in our modeling, may play a more significant role in influencing the assembly of the actin networks in patches.
Another, and not mutually exclusive, possible explanation is that Arp2/3 regulators are responsible for ensuring very specific subcellular localization and coordination of Arp2/3 activity to construct functional, force-generating actin structures. Our examination of the ratios of Arp2/3, CP, and F-actin provides some evidence that the fine architecture of patches in mutants may be affected by regulator mutants. However, current light microscopy assays may not be sufficient to reveal such fine changes in actin network architecture.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01 GM38542 to JAC and R01 GM86882 to A.E.C.
Supporting Material
References
- 1.Goley E.D., Welch M.D. The ARP2/3 complex: an actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006;7:713–726. doi: 10.1038/nrm2026. [DOI] [PubMed] [Google Scholar]
- 2.Mullins R.D., Heuser J.A., Pollard T.D. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollard T.D., Blanchoin L., Mullins R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 4.Pollard T.D., Borisy G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 5.Campellone K.G., Welch M.D. A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010;11:237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rottner K., Hänisch J., Campellone K.G. WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol. 2010;20:650–661. doi: 10.1016/j.tcb.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Derivery E., Sousa C., Gautreau A. The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev. Cell. 2009;17:712–723. doi: 10.1016/j.devcel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Campellone K.G., Webb N.J., Welch M.D. WHAMM is an Arp2/3 complex activator that binds microtubules and functions in ER to Golgi transport. Cell. 2008;134:148–161. doi: 10.1016/j.cell.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuchero J.B., Coutts A.S., Mullins R.D. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat. Cell Biol. 2009;11:451–459. doi: 10.1038/ncb1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snapper S.B., Takeshima F., Alt F.W. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat. Cell Biol. 2001;3:897–904. doi: 10.1038/ncb1001-897. [DOI] [PubMed] [Google Scholar]
- 11.Legg J.A., Bompard G., Machesky L.M. N-WASP involvement in dorsal ruffle formation in mouse embryonic fibroblasts. Mol. Biol. Cell. 2007;18:678–687. doi: 10.1091/mbc.E06-06-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benesch S., Polo S., Rottner K. N-WASP deficiency impairs EGF internalization and actin assembly at clathrin-coated pits. J. Cell Sci. 2005;118:3103–3115. doi: 10.1242/jcs.02444. [DOI] [PubMed] [Google Scholar]
- 13.Innocenti M., Gerboth S., Scita G. Abi1 regulates the activity of N-WASP and WAVE in distinct actin-based processes. Nat. Cell Biol. 2005;7:969–976. doi: 10.1038/ncb1304. [DOI] [PubMed] [Google Scholar]
- 14.Suetsugu S., Yamazaki D., Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev. Cell. 2003;5:595–609. doi: 10.1016/s1534-5807(03)00297-1. [DOI] [PubMed] [Google Scholar]
- 15.Yamazaki D., Oikawa T., Takenawa T. Rac-WAVE-mediated actin reorganization is required for organization and maintenance of cell-cell adhesion. J. Cell Sci. 2007;120:86–100. doi: 10.1242/jcs.03311. [DOI] [PubMed] [Google Scholar]
- 16.Yan C., Martinez-Quiles N., Alt F.W. WAVE2 deficiency reveals distinct roles in embryogenesis and Rac-mediated actin-based motility. EMBO J. 2003;22:3602–3612. doi: 10.1093/emboj/cdg350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamazaki D., Suetsugu S., Takenawa T. WAVE2 is required for directed cell migration and cardiovascular development. Nature. 2003;424:452–456. doi: 10.1038/nature01770. [DOI] [PubMed] [Google Scholar]
- 18.Bryce N.S., Clark E.S., Weaver A.M. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr. Biol. 2005;15:1276–1285. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 19.Helwani F.M., Kovacs E.M., Yap A.S. Cortactin is necessary for E-cadherin-mediated contact formation and actin reorganization. J. Cell Biol. 2004;164:899–910. doi: 10.1083/jcb.200309034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tehrani S., Faccio R., Cooper J.A. Cortactin has an essential and specific role in osteoclast actin assembly. Mol. Biol. Cell. 2006;17:2882–2895. doi: 10.1091/mbc.E06-03-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler B., Kastendieck D.H., Cooper J.A. Differently phosphorylated forms of the cortactin homolog HS1 mediate distinct functions in natural killer cells. Nat. Immunol. 2008;9:887–897. doi: 10.1038/ni.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez T.S., McCarney S.D., Burkhardt J.K. HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity. 2006;24:741–752. doi: 10.1016/j.immuni.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galletta B.J., Chuang D.Y., Cooper J.A. Distinct roles for Arp2/3 regulators in actin assembly and endocytosis. PLoS Biol. 2008;6:e1. doi: 10.1371/journal.pbio.0060001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y., Martin A.C., Drubin D.G. Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev. Cell. 2006;11:33–46. doi: 10.1016/j.devcel.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Schäfer G., Weber S., Onel S.F. The Wiskott-Aldrich syndrome protein (WASP) is essential for myoblast fusion in Drosophila. Dev. Biol. 2007;304:664–674. doi: 10.1016/j.ydbio.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Gildor B., Massarwa R., Schejter E.D. The SCAR and WASp nucleation-promoting factors act sequentially to mediate Drosophila myoblast fusion. EMBO Rep. 2009;10:1043–1050. doi: 10.1038/embor.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaksonen M., Toret C.P., Drubin D.G. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2006;7:404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- 28.Galletta B.J., Cooper J.A. Actin and endocytosis: mechanisms and phylogeny. Curr. Opin. Cell Biol. 2009;21:20–27. doi: 10.1016/j.ceb.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan F., Egile C., Li R. ARPC1/Arc40 mediates the interaction of the actin-related protein 2 and 3 complex with Wiskott-Aldrich syndrome protein family activators. J. Biol. Chem. 2004;279:54629–54636. doi: 10.1074/jbc.M402357200. [DOI] [PubMed] [Google Scholar]
- 30.Winter D., Podtelejnikov A.V., Li R. The complex containing actin-related proteins Arp2 and Arp3 is required for the motility and integrity of yeast actin patches. Curr. Biol. 1997;7:519–529. doi: 10.1016/s0960-9822(06)00223-5. [DOI] [PubMed] [Google Scholar]
- 31.Winter D.C., Choe E.Y., Li R. Genetic dissection of the budding yeast Arp2/3 complex: a comparison of the in vivo and structural roles of individual subunits. Proc. Natl. Acad. Sci. USA. 1999;96:7288–7293. doi: 10.1073/pnas.96.13.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brachmann C.B., Davies A., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Sheff M.A., Thorn K.S. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast. 2004;21:661–670. doi: 10.1002/yea.1130. [DOI] [PubMed] [Google Scholar]
- 34.Waddle J.A., Karpova T.S., Cooper J.A. Movement of cortical actin patches in yeast. J. Cell Biol. 1996;132:861–870. doi: 10.1083/jcb.132.5.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffman D.B., Pearson C.G., Salmon E.D. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell. 2001;12:1995–2009. doi: 10.1091/mbc.12.7.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joglekar A.P., Bouck D.C., Salmon E.D. Molecular architecture of a kinetochore-microtubule attachment site. Nat. Cell Biol. 2006;8:581–585. doi: 10.1038/ncb1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coffman V.C., Wu P., Wu J.Q. CENP-A exceeds microtubule attachment sites in centromere clusters of both budding and fission yeast. J. Cell Biol. 2011;195:563–572. doi: 10.1083/jcb.201106078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawrimore J., Bloom K.S., Salmon E.D. Point centromeres contain more than a single centromere-specific Cse4 (CENP-A) nucleosome. J. Cell Biol. 2011;195:573–582. doi: 10.1083/jcb.201106036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leake M.C., Chandler J.H., Armitage J.P. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 2006;443:355–358. doi: 10.1038/nature05135. [DOI] [PubMed] [Google Scholar]
- 40.Hedges S.B., Shah P. Comparison of mode estimation methods and application in molecular clock analysis. BMC Bioinformatics. 2003;4:31–41. doi: 10.1186/1471-2105-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaksonen M., Toret C.P., Drubin D.G. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell. 2005;123:305–320. doi: 10.1016/j.cell.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 42.Weiner O.D., Marganski W.A., Kirschner M.W. An actin-based wave generator organizes cell motility. PLoS Biol. 2007;5:e221. doi: 10.1371/journal.pbio.0050221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Millius A., Dandekar S.N., Weiner O.D. Neutrophils establish rapid and robust WAVE complex polarity in an actin-dependent fashion. Curr. Biol. 2009;19:253–259. doi: 10.1016/j.cub.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berro J., Sirotkin V., Pollard T.D. Mathematical modeling of endocytic actin patch kinetics in fission yeast: disassembly requires release of actin filament fragments. Mol. Biol. Cell. 2010;21:2905–2915. doi: 10.1091/mbc.E10-06-0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winter D., Lechler T., Li R. Activation of the yeast Arp2/3 complex by Bee1p, a WASP-family protein. Curr. Biol. 1999;9:501–504. doi: 10.1016/s0960-9822(99)80218-8. [DOI] [PubMed] [Google Scholar]
- 46.Tang H.Y., Cai M. The EH-domain-containing protein Pan1 is required for normal organization of the actin cytoskeleton in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996;16:4897–4914. doi: 10.1128/mcb.16.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Notarangelo L.D., Ochs H.D. Wiskott-Aldrich Syndrome: a model for defective actin reorganization, cell trafficking and synapse formation. Curr. Opin. Immunol. 2003;15:585–591. doi: 10.1016/s0952-7915(03)00112-2. [DOI] [PubMed] [Google Scholar]
- 48.Notarangelo L.D., Miao C.H., Ochs H.D. Wiskott-Aldrich syndrome. Curr. Opin. Hematol. 2008;15:30–36. doi: 10.1097/MOH.0b013e3282f30448. [DOI] [PubMed] [Google Scholar]
- 49.Thrasher A.J. New insights into the biology of Wiskott-Aldrich syndrome (WAS) Hematology Am. Soc. Hematol. Educ. Program. 2009;209:132–138. doi: 10.1182/asheducation-2009.1.132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.