

Abstract
Human fetal membranes express 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), which reduces biologically inert cortisone to active cortisol and may provide an extraadrenal source of cortisol mediating fetal development and parturition. The reductase activity of 11β-HSD1 depends on the availability of the cofactor reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) derived from the enzymatic activity of hexose-6-phosphodehydrogenase (H6PD). Based on the feed-forward induction of 11β-HSD1 by glucocorticoids in human fetal membranes, we hypothesize that glucocorticoids simultaneously induce H6PD in the fetal membranes. We found a parallel distribution of H6PD and 11β-HSD1 in the amnion, chorion, and decidua. In cultured human amnion fibroblasts, small interfering RNA-mediated knockdown of H6PD expression significantly attenuated the conversion of cortisone to cortisol. Cortisol (0.01–1 μm) induced H6PD expression in a concentration-dependent manner, which was attenuated by glucocorticoid receptor (GR) antagonist RU486. Cortisol induced the expression of p300, a histone acetyltransferase, whereas C646, an inhibitor of p300, attenuated the induction of H6PD by cortisol. Coimmunoprecipitation revealed GR and p300 in the same nuclear protein complex upon cortisol stimulation. Chromatin immunoprecipitation showed that cortisol increased the binding of p300 and GR to H6PD promoter and the acetylation of histone 3 lysine 9 on the promoters. In conclusion, the induction of H6PD by cortisol requires the participation of GR and p300 as well as the acetylation of H3K9 by p300. This may be a prerequisite for the parallel induction of reductase activity of 11β-HSD1 in human amnion fibroblasts in a feed-forward loop that may influence fetal development and the onset of parturition.
Glucocorticoids play key roles in fetal maturation in anticipation of extrauterine life and in several species appear to be mediators in the initiation of labor (1). Toward the end of human gestation, cortisol levels increase in both maternal and fetal circulations as well as in the amniotic fluid (2–4). Although the adrenal glands are normally regarded as a robust source of cortisol, the human fetal membranes may also provide an extraadrenal source of cortisol both for local actions in the membranes and systemically via amniotic fluid (5, 6). This unique role of fetal membranes in cortisol production may be of particular importance in human pregnancy before the onset of parturition because human fetal adrenal glands are characterized by a large fetal zone, which synthesizes dehydroepiandrosterone sulfate rather than cortisol during gestation. However, a modest increase of cortisol production from the definitive zone, which comprises a small proportion of the fetal adrenal glands, does occur (7).
This unique capability of the fetal membranes for production of cortisol relies on the expression of 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) (5, 8, 9), a microsomal enzyme that uses the abundant inert cortisone in the amniotic fluid as its substrate for the reductive regeneration of biologically active cortisol. Our previous studies have demonstrated that 11β-HSD1 is highly expressed in human amnion, chorion, and decidua (8, 9), affirming earlier work showing the membranes retained a high degree of reductive conversion of cortisone to cortisol (5, 10, 11). However, 11β-HSD1 is a bidirectional enzyme in vitro catalyzing the oxidation of 11β-hydroxyglucocorticoids as well as the reduction of 11-ketoglucocorticoids, whereas in vivo it normally reduces 11-ketoglucocorticoids to the active 11β-hydroxyderivates (12). This reductase activity of 11β-HSD1 depends on the availability of the cofactor reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), which derives from the enzymatic activity of hexose-6-phospho-dehydrogenase (H6PD) (13, 14).
H6PD, a microsomal counterpart of the cytosolic glucose-6-phosphate dehydrogenase, has emerged as a key NADPH generating enzyme of the endoplasmic reticulum (ER) lumen (13, 15). Although H6PD has a broad substrate range, under physiological conditions within the ER lumen, it uses glucose-6-phosphate and the oxidized form of nicotinamide adenine dinucleotide phosphate (NADP+) to generate NADPH (14). A functional collaboration between 11β-HSD1 and H6PD based on the mutual generation of pyridine nucleotide cofactors within the ER has been suggested (16). A direct physical interaction between 11β-HSD1 and H6PD allows the supply of NADPH generated by H6PD in close proximity to 11β-HSD1 for the efficient reduction of cortisone to cortisol and oxidation of NADPH back to the oxidized form of nicotinamide adenine dinucleotide phosphate despite a rather oxidative environment within the ER lumen (17, 18). The crucial role of H6PD in the reductase activity of 11β-HSD1 is well demonstrated by studies showing that overexpressing H6PD consistently increased 11β-HSD1 reductase activity (15, 16, 19), whereas decreasing H6PD expression with small interfering RNA (siRNA) attenuated 11β-HSD1 reductase activity (18–20). The findings that the intact microsomes prepared from H6PD knockout mice possessed only oxidase activity of 11β-HSD1 lend further convincing evidence for the pivotal role of H6PD in the reductase activity of 11β-HSD1 (21–23).
Of interest, cortisol, the product of the reductase activity of 11β-HSD1, induces the expression of 11β-HSD1 via the glucocorticoid receptor (GR) and CCAAT enhancer binding proteins in the fetal membranes (24–26). This may form a feed-forward loop for cortisol regeneration assuming that there is an adequate amount of NADPH. We hypothesize that in keeping with the up-regulation of 11β-HSD1 by cortisol, there might be concurrent induction of H6PD and 11β-HSD1 expression by cortisol in the fetal membranes, which would ensure 11β-HSD1 reductase activity to meet the increasing demand of cortisol toward the end of gestation.
Acetylation of histones is commonly associated with transcriptional activation of genes and is responsible both for a local open chromatin structure required for promoter activation (27) and for regulating the binding of transcription factors (28). The transcriptional coactivator p300 possesses intrinsic histone acetyl transferase activity (29, 30) and has been recognized as one of the key molecules involved in the interaction between transcription factors and the transcriptional machinery. It thus is an important element of transcription regulation networks (31). Studies have shown that p300 can be recruited by a number of transcription factors including GR and CCAAT enhancer binding proteins, which are involved in 11β-HSD1 gene regulation (31–35). In addition, glucocorticoids have been shown to induce the expression of p300 in other cell types (36, 37). We thus hypothesize that p300 could be up-regulated and involved in the regulation of H6PD transcription by cortisol in the fetal membranes. We studied these hypotheses in cultured primary human amnion fibroblasts.
Materials and Methods
Immunohistochemical staining of H6PD and 11β-HSD1 in human fetal membranes
Fetal membranes were collected at term from patients not in labor and delivered by elective cesarean section under a protocol approved by the Ethics Committee of the School of Life Sciences, Fudan University. Patients treated with steroids or other antiinflammatory agents or with clinical indication of inflammation were excluded from this study. To compare the distribution of H6PD and 11β-HSD1 in the fetal membranes, immunohistochemical staining for H6PD and 11β-HSD1 was carried out on paraffin embedded serial tissue sections (5 μm) of the whole membrane roll using the avidin-biotin-peroxidase method following a protocol provided by the manufacturer (Vector ABC; Vector Laboratories, Burlingame, CA). The H6PD antibody (Abgent, San Diego, CA), 11β-HSD1 antibody (Cayman Chemicals, Ann Arbor, MI) and vimentin antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:100 dilutions were applied, respectively, as the primary antibodies. The color reactions were developed using 3-amino-9-ethyl carbazole (red color). Slices were counterstained with Carazzi's hematoxylin. To test the specificity of immunocytochemical staining, slices were also stained with preimmune serum instead of the primary antibodies and 11β-HSD1 antibody preabsorbed with the 11β-HSD1 peptide.
Human amnion fibroblast cell culture
Human amnion fibroblasts were prepared from the fetal membranes collected as described above. Amnion was peeled from the chorion/decidua and digested with 0.125% trypsin (Sigma, St. Louis, MO) twice for 0.5 h each. The tissue was washed with PBS three times vigorously to remove the residual epithelial cells. The remaining amnion tissue was digested with 0.1% collagenase type I (Worthington, Lakewood, NJ) for 0.5 h. After centrifugation the fibroblasts were collected for culture in DMEM (Life Technologies, Inc., Grand Island, NY) containing 10% newborn calf serum (Life Technologies) and antibiotic-antimycotic (Life Technologies). The identity of fibroblasts has been previously verified, and more than 95% of the cells are fibroblasts (24).
siRNA-mediated knockdown of H6PD expression on the conversion of cortisone to cortisol in amnion fibroblasts
To determine the role of H6PD in the reductase activity of 11β-HSD1, cultured amnion fibroblasts were transfected with siRNA (5′-UUAUGGAGACAUGUCCCUGGAGCUC-3, 60 nm) against H6PD (GenePharma, Shanghai, China) or randomly scrambled negative siRNA using lipofectamine LTX in Opti-MEM (Invitrogen, Carlsbad, CA). After overnight transfection, the cells were cultured for another 48 h before incubation with cortisone (1 μm) in newborn calf serum-free medium for 12 h, a time point was chosen according to the preliminary time course study. Cortisol level in the collected medium was measured with enzyme immunoassay kit (R & D Systems, Minneapolis, MN). The effect of siRNA transfection on H6PD mRNA and protein levels were checked before incubation with cortisone using quantitative real-time PCR (qRT-PCR) and Western blotting analyses.
Treatment of human amnion fibroblasts and determination of H6PD and p300 mRNA and protein using qRT-PCR and Western blotting
On the third day of cell culture, the culture medium was changed to serum-free medium, and the cells were then treated with cortisol (0.01, 0.1, 1 μm) for 24 h to examine the expression of H6PD and p300. To study the role of GR in the induction of H6PD by cortisol, the cells were treated with cortisol (1 μm) in the presence or absence of the GR antagonist RU486 (1 μm). To study whether the induction of H6PD mRNA by cortisol requires the participation of de novo protein synthesis, the cells were treated with cortisol (1 μm) in the presence or absence of cycloheximide (CHX; 10 μm; Sigma), an inhibitor of protein synthesis. To study the effect of p300 inhibitor on cortisol's induction of H6PD, the cells were treated with cortisol (1 μm) in the presence or absence of C646 (10 μm) for 24 h. Total RNA was extracted from the cells using a RNeasy miniextraction kit (QIAGEN, Valencia, CA). Total cellular protein was extracted from the cells using radioimmunoprecipitation assay buffer. After reverse transcription of the RNA, the amounts of H6PD and p300 mRNA were measured using qRT-PCR. The reaction solution for qRT-PCR consisted of 1.0 μl diluted cDNA, 0.2 μm of each paired primers, and power SYBR Green PCR master mix (Toyobo, Osaka, Japan). The annealing temperature was set at 61 C. The absolute mRNA levels in each sample were calculated according to a standard curve set-up using serial dilutions of known amounts of specific templates against corresponding cycle threshold values. To control for sampling errors, qRT-PCR for the housekeeping gene β-actin was routinely performed on each sample. The ratio of the target genes over β-actin for each sample was obtained to normalize the expression of the target gene. The primer sequences used for qRT-PCR are illustrated in Table 1. H6PD and p300 protein levels were measured by Western blotting. Briefly, 25 μg total protein of each sample were electrophoresed in 4–20% sodium dodecyl sulfate-polyacrylamide gel and transferred to the nitrocellulose blot. After blocking, the blot was probed with 1:500 dilution of rabbit antibodies against H6PD (Abgent) and p300 (Santa Cruz Biotechnology) overnight. After incubation with antirabbit IgG antibody conjugated with horseradish peroxidase, the enhanced chemiluminescence detection system (Millipore, Billerica, CA) was used to detect the bands. To control sampling error, the same blots were probed with 1:5000 dilution of mouse antibody against β-actin (Sigma) for total cellular protein. The bands were visualized using a G Box iChemi chemiluminescence image capture system (Syngene, Cambridge, UK). The ratio of band intensities of target protein over β-actin was obtained as indication of the target protein levels.
Table 1.
Primer sequences used in qRT-PCR
| Genes | Primer sequence (5′–3′) | Accession no. | Product size (bp) |
|---|---|---|---|
| Primers for mRNA measurement | |||
| H6PD | Forward, CTGCAGCACGTCCGGATCCC | NM_004285.3 | 298 |
| Reverse, TGGCGCGGTTGATGAGAGGC | |||
| P300 | Forward, AGGCGTCGAGGACCTGGTGT | NM_001429.3 | 197 |
| Reverse, TGGCCATTGTAACCAGAAATGGGC3 | |||
| β-Actin | Forward, TCCTCCTGAGCGC AAGTACTCT | NM_001101.3 | 297 |
| Reverse, GCTCAGTAACAGTCCGCCTAGAA | |||
| Primers for ChIP assay | |||
| Name | Primer sequence | Location | Product size (bp) |
| PP1 | Forward, GACGCGGCAGGACAGCTCTA | −244, 146 | 390 |
| Reverse, GCCCGTCACTCCGACAAGTGC | |||
| PP2 | Forward, GCGTGTGCGCTCCGACTTCT | −445, −269 | 176 |
| Reverse, GGCCGAGGGACGTGGTTGTG | |||
| DP | Forward, TTCTCCTGAGATCCCGCTCA | −8993, −8825 | 169 |
| Reverse, TGCACGTGGTGTAAGCTAGG |
PP1, Proximal primer set 1; PP2, proximal primer set 2; DP, distal primer set.
Coimmunoprecipitation showing the presence of both GR and p300 in the same nuclear protein complex
On the third day of cell culture, cells were treated with cortisol (1 μm) for 12 h in the medium without serum. Nuclear protein was then extracted from the cells using nuclear extraction kit (Active Motif, Carlsbad, CA). Ten micrograms of nuclear protein was incubated with 2 μg rabbit antibody against human p300 (Santa Cruz Biotechnology) or preimmune rabbit IgG, respectively, for overnight. Protein A agarose beads were added and incubated for 1 h on ice to pull down the antibody-antigen complex. The antibody-antigen-agarose complex was washing adequately and was denatured in Western blotting loading buffer at 95 C for the subsequent analysis with Western blotting using 1:500 dilution of GR antibody (Santa Cruz Biotechnology). The bands were visualized as mentioned above.
Chromatin immunoprecipitation (ChIP) assay demonstrating the enrichment of GR, p300, and acetylated H3K9 (H3K9ac) to H6PD promoter in human amnion fibroblasts
On the third day of cell culture, cells were treated with cortisol (1 μm) for 12 h in the medium without serum. Upon termination of treatment, a ChIP assay was conducted using a kit from Upstate Biotechnology (Millipore). Briefly, the cells were fixed with 1% formaldehyde to cross-link the proteins bound to the chromatin DNA. After washing with PBS, the cells were incubated with glycine and then scraped off the dish in PBS containing protease inhibitor cocktail. After spinning down, the cells were resuspended with 1% sodium dodecyl sulfate lysis buffer supplemented with protease inhibitor. The chromatin DNA was sheared by sonication to produce DNA fragments of approximately 500-1000 bp. The same amounts of sheared DNA were used for subsequent immunoprecipitation with rabbit antibody against human p300 (Santa Cruz Biotechnology) or GR (Santa Cruz Biotechnology) or H3K9ac (Millipore) or an equal amount of preimmune rabbit IgG (Millipore). The immunoprecipitate was then incubated with protein A agarose-salmon sperm DNA (Millipore) and the antibody-protein-DNA-agarose complex was washed adequately and collected for subsequent reverse cross-linking by sodium chloride. The same amount of sheared DNA without antibody precipitation was also processed for reverse cross-linking and served as input control. The sheared DNA recovered from reverse cross-linking was extracted with a DNA extraction kit (QIAGEN) for further quantitative analysis with real-time PCR. The sequences and location of the primers for amplification of H6PD DNA with qRT-PCR are illustrated (see Fig. 6). In addition to the two primer sets spanning the proximal putative glucocorticoid response element (GRE) in the promoter, a further upstream primer set (∼−8.8 kb to the transcription start site) promoter region was also used in qRT-PCR as a control. The primer sequences are illustrated in Table 1. The amount of H6PD DNA precipitated was calculated from the threshold cycle number of the amplification curve of qRT-PCR. The percentage of antibody- or preimmune IgG-precipitated H6PD DNA to input H6PD DNA was used for statistical analysis. Real-time PCR products were also collected at a time point before saturation according to the amplification curve for demonstration of their abundance upon electrophoresis in 1.5% agarose gel.
Fig. 6.

A, Coimmunoprecipitation assay showing the detection of GR in the nuclear protein complex precipitated by p300 antibody upon stimulation of human amnion fibroblasts with cortisol (F; 1 μm). Preimmune IgG served as negative control. B, Diagram illustrating the positions of putative GREs and primer sets used for the ChIP assay. C and D, Top panels are the representative gel images showing the abundance of PCR products, and bottom panels are the mean data of qRT-PCR showing the enrichment of p300, GR to H6PD promoter upon cortisol (F; 1 μm) stimulation of human amnion fibroblasts. Preimmune IgG served as negative control (n = 3–4). *, P < 0.05 vs. control (CTR). TSS, Transcription start site; PP1, proximal primer set 1; PP2, proximal primer set 2; DP, distal primer set.
Statistical analysis
All data are reported as mean ± sem. A paired Student's t test or one-way ANOVA test followed by the Student-Newman-Keuls test was used to assess significant differences where appropriate using Statistical Package for the Social Sciences software version 12.0 (SPSS Inc., Chicago, IL). Significance was set at P < 0.05.
Results
Distribution and functional correlation of H6PD and 11β-HSD1 in human fetal membranes
Before immnohistochemical staining, we examined the specificity of the antibodies using protein extracted from human amnion fibroblasts with Western blotting. Our data showed that H6PD antibody was relatively specific revealing a strong band with an expected size of approximately 90 kDa (Fig. 1A). Although 11β-HSD1 antibody revealed multiple bands, the strongest band was observed at approximately 34 kDa (Fig. 1A), which was abolished by preabsorption with 11β-HSD1 peptide used for generation of the antibody (Fig. 1A). Staining of the serial sections demonstrated that the distribution of H6PD and 11β-HSD1 protein was strikingly similar in the fetal membranes. H6PD and 11β-HSD1 proteins were found in the decidual cells, chorion trophoblasts, and amnion epithelial and fibroblast cells (Fig. 1, D and E), suggesting that these two proteins are possibly functionally correlated in these cells. The staining with 11β-HSD1 antibody was largely abolished by peptide-preabsorption (Fig. 1B), defining the specificity of staining with 11β-HSD1 antibody. The functional correlation between H6PD and 11β-HSD1 was demonstrated in the study of siRNA-mediated knockdown of H6PD expression in amnion fibroblasts. Transfection of H6PD siRNA caused a 40–70% decrease of H6PD protein level, which led to a corresponding reduction of the conversion of cortisone to cortisol from 62 ± 13% in the scrambled siRNA group to 25 ± 7% in the H6PD siRNA group (Fig. 2).
Fig. 1.

Immunohistochemical staining of H6PD and 11β-HSD1 in human fetal membranes at term without labor. A, Western blots illustrating the specificity of the antibodies. B, Staining with 11β-HSD1 antibody preabsorbed by its peptide. C, Staining with preimmune serum. D, Staining with H6PD antibody. E, Staining with 11β-HSD1 antibody. F, Staining with vimentin antibody, a marker for mesoderm-derived cells. a, Amnion; c, chorion; d, decidua; m, mesenchymal layer of the amnion. Arrows indicate fibroblasts.
Fig. 2.
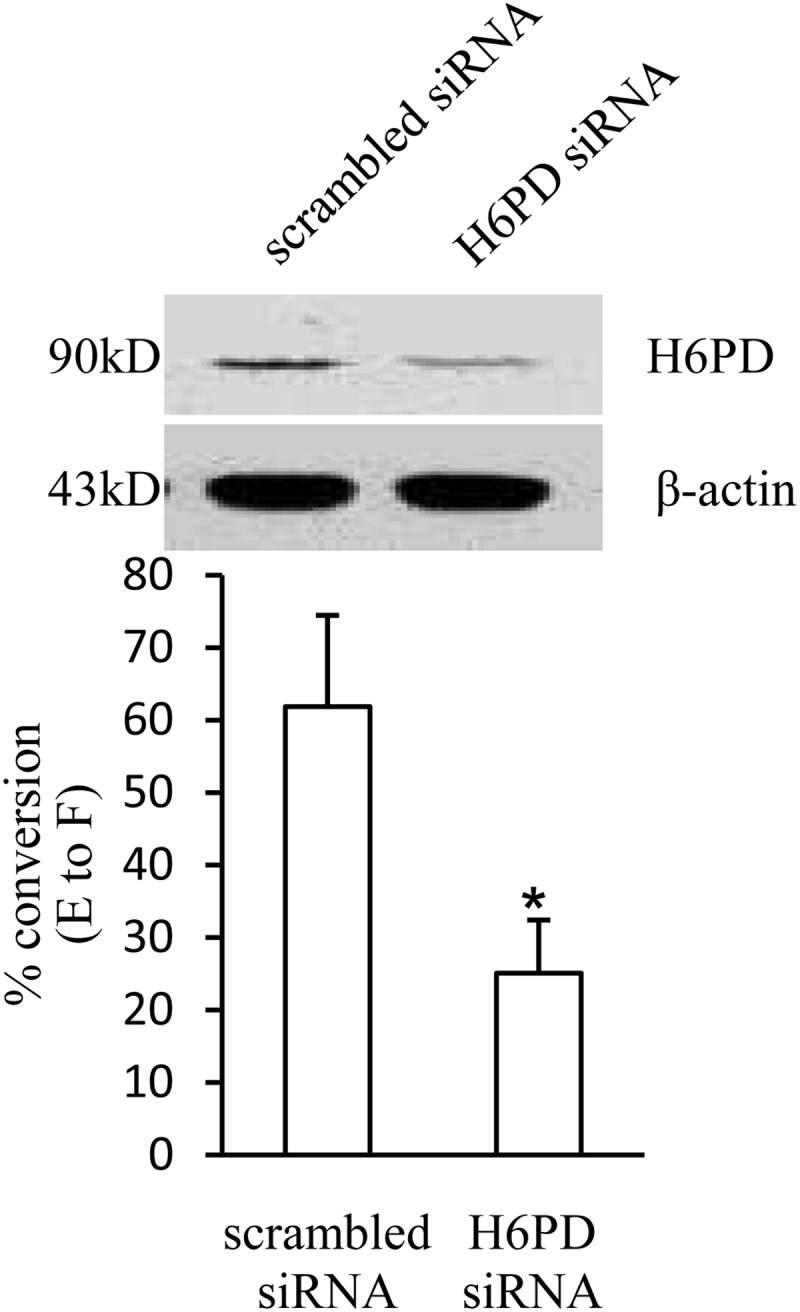
siRNA-mediated knockdown of H6PD expression significantly attenuated the conversion of cortisone (E) to cortisol (F) in human amnion fibroblasts. Top panel is the representative Western blot showing the reduced expression of H6PD with siRNA transfection. Bottom panel is the mean data of three separate experiments showing the percent conversion of cortisone to cortisol in H6PD siRNA and scrambled siRNA-transfected human amnion fibroblasts. *, P < 0.05.
Induction of H6PD mRNA and protein by cortisol
Cortisol (0.01, 0.1, and 1 μm) increased H6PD mRNA and protein levels in a concentration-dependent manner in human amnion fibroblasts (Fig. 3, A and D). The induction of H6PD mRNA and protein by cortisol (1 μm) could be significantly attenuated by the GR antagonist RU486 (1 μm) (Fig. 3, B and D), suggesting the involvement of GR in the induction of H6PD by cortisol. This GR-dependent induction of H6PD resembles the induction of 11β-HSD1 by cortisol as we reported earlier (25). Moreover, the fold induction of H6PD mRNA expression by cortisol (1 μm) was significantly reduced in the presence of CHX (10 μm), suggesting the requirement of de novo protein synthesis in the induction of H6PD mRNA expression by cortisol (Fig. 3C).
Fig. 3.

Induction of H6PD expression by cortisol in human amnion fibroblasts. A, Dose-dependent induction of H6PD mRNA by cortisol. B, Comparison of induction of H6PD by cortisol (1 μm) in the presence and absence of RU486 (1 μm). C, Comparison of induction of H6PD by cortisol (1 μm) in the presence and absence of protein synthesis inhibitor CHX (10 μm). D, Dose-dependent induction of H6PD protein by cortisol and the attenuation by RU486. The upper panel is the representative Western blot and the bottom panel is the mean data (n = 4–9). *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. vehicle control.
Induction of p300 mRNA and protein by cortisol
Cortisol (0.01, 0.1 and 1 μm) increased p300 mRNA and protein levels in a concentration-dependent manner in human amnion fibroblasts (Fig. 4, A and C). The induction of p300 mRNA and protein by cortisol (1 μm) could be significantly attenuated by the GR antagonist RU486 (1 μm) (Fig. 4, B and C). The induction of H6PD mRNA and protein by cortisol (1 μm) was significantly attenuated by cotreatment with p300 inhibitor C646 (10 μm) (Fig. 5, A and B).
Fig. 4.

Induction of p300 expression by cortisol in human amnion fibroblasts. A, Dose-dependent induction of p300 mRNA by cortisol. B, Comparison of induction of P300 by cortisol (1 μm) in the presence and absence of RU486 (1 μm). C, Dose-dependent induction of p300 protein by cortisol and the attenuation by RU486. The upper panel is the representative Western blot and the bottom panel is the mean data (n = 3–4). *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. vehicle control.
Fig. 5.

Attenuation of cortisol (F; 1 μm)-induced increases of H6PD mRNA (A) and protein (B) by p300 inhibitor C646 (10 μm) in human amnion fibroblasts (n = 3–4). *, P < 0.05, ***, P < 0.001 vs. control (CTR); #, P < 0.05, ##, P < 0.01 vs. F.
Enrichment of GR, p300, and H3K9ac to H6PD promoter by cortisol
Coimmunoprecipitation assay showed that GR protein could be identified in the nuclear protein complex precipitated by the p300 antibody after cortisol treatment (1 μm) for 12 h (Fig. 6A). Bioinformatic analysis revealed several putative GREs in the proximal promoter region of H6PD (within −600 bp). By designing primers spanning the two most proximal putative GREs (Fig. 6B), we found that the binding of p300 and GR to the H6PD promoter could be significantly increased on both sites but not on a distal site (−8.8 kb) by cortisol treatment (1 μm) for 12 h with a ChIP assay (Fig. 6, C and D). Furthermore, the ChIP assay also revealed the enrichment of H3K9ac to the same promoter region upon cortisol treatment (1 μm) for 12 h, which could be attenuated by cotreatment with C646 (Fig. 7).
Fig. 7.
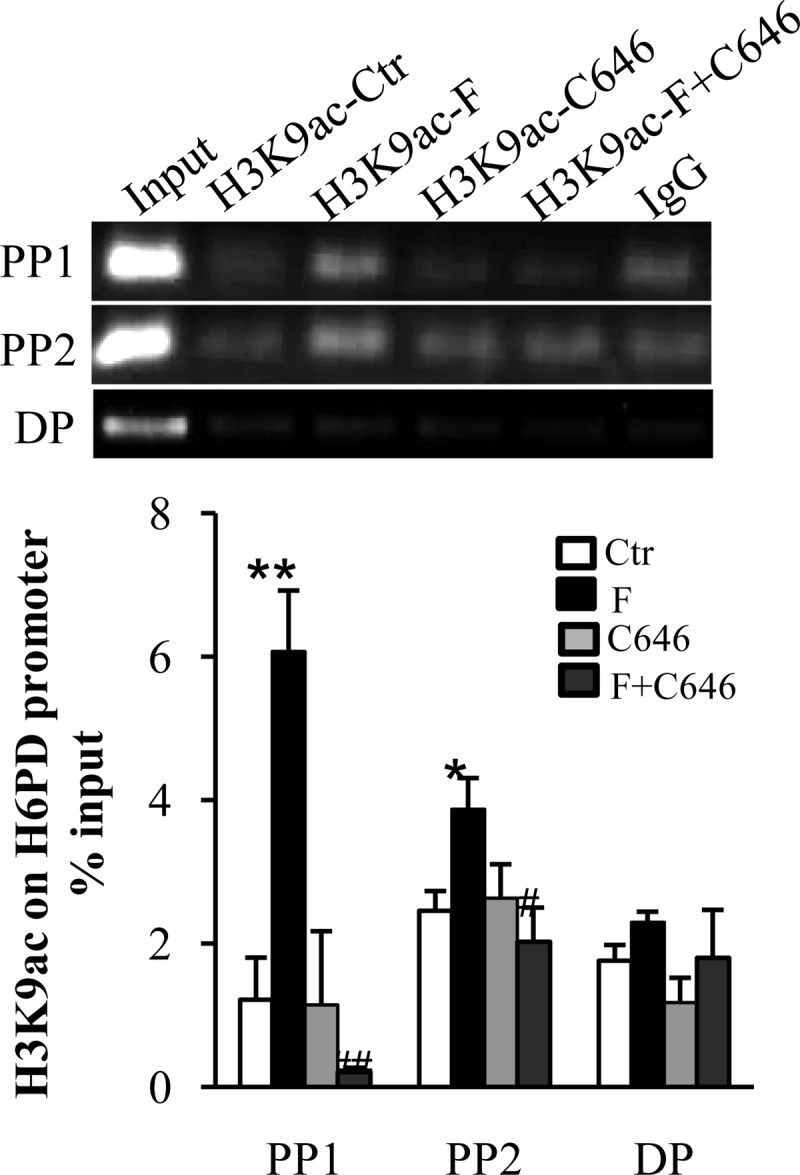
ChIP assay revealed the enrichment of H3K9ac on the proximal promoter region of H6PD upon cortisol (F; 1 μm) stimulation of human amnion fibroblasts, which was attenuated by cotreatment with C646 (10 μm), a P300 inhibitor. Preimmune IgG served as the negative control. Top panels are representative gel images showing the abundance of PCR products, and bottom panels are the mean data of qRT-PCR (n = 3). *, P < 0.05, **, P < 0.01 vs. control (CTR); #, P < 0.05, ##, P < 0.01 vs. F. PP1, Proximal primer set 1; PP2, proximal primer set 2; DP, distal primer set.
Discussion
Compelling evidence indicates a functional link between H6PD and 11β-HSD1 in terms of mutual provision of pyridine nucleotide cofactors within the ER (13, 38). The crucial role of H6PD in the reductase activity of 11β-HSD1 is best demonstrated by the reversal of the reductase to oxidase activity of 11β-HSD1 in H6PD knockout mice and by mutations in H6PD gene causing cortisone reductase deficiency (23, 38, 39). We demonstrated, in this study, that siRNA-mediated knockdown H6PD significantly reduced cortisol production upon incubation with cortisone in human amnion fibroblasts, supporting a similar finding in HT-1080 cells, a fibrosarcoma cell line (18). The reduced cortisol production seen upon cortisone incubation may reflect the net effect of decreased reductase activity and enhanced oxidase activity of 11β-HSD1 with the knockdown of H6PD expression. Therefore, concurrent increases of H6PD and 11β-HSD1 appear to be the prerequisite for the enhanced regeneration of cortisol and the consequent up-regulation of glucocorticoid action.
The importance of this concurrent induction is well illustrated in physiological as well as in pathological conditions in which enhanced reductase activity of 11β-HSD1 is necessary. In patients with type 2 diabetes mellitus, both H6PD and 11β-HSD1 expression are increased in adipose tissue (40), whereas in db/db mice, increased H6PD expression contributes to the enhanced amplification of glucocorticoid action through 11β-HSD1 in the liver (41). Moreover, in the offspring exposed to alcohol in utero, parallel and persistent increases in H6PD and 11β-HSD1 levels were observed in both liver and adipose tissues, which might account for the development of insulin-resistant diabetes in later life (42). Previously we demonstrated that glucocorticoids induced 11β-HSD1 mRNA expression as well as its redutase activity in cells prepared from human amnion and chorion (24, 26). Based on these findings, we proposed there might exist a feed-forward loop in cortisol regeneration in the fetal membranes toward the end of gestation (6). Glucocorticoids should induce H6PD expression concurrently with the reductase activity of 11β-HSD1 in the fetal membranes. Not surprisingly, we found a similar distribution of 11β-HSD1 and H6PD in the fetal membranes, suggesting a functional coupling of these two enzymes. Moreover, cortisol induced H6PD expression in a concentration-dependent manner via GR in human amnion fibroblasts, a situation resembling the induction of 11β-HSD1 expression by cortisol in the same cell type (24, 25). This induction of H6PD by cortisol would comprise an important link in the feed-forward cortisol regeneration in the fetal membranes in which the reductase activity of 11β-HSD1 toward the end of gestation appears to be crucial to the local proparturition effects of cortisol (43, 44).
Because human fetal membranes are in an ideal place to receive both maternal and fetal signals and transmit signals to uterine myometrium, there has been a specific focus on the role of membranes in the initiation and maintenance of parturition, particularly with regard to their synthesis of steroids and prostaglandins. Because intraamniotic fluid administration of glucocorticoids appeared to be more effective in provoking labor than systemic administration (45, 46), a possible role for cortisol produced locally in the intrauterine tissues in human parturition is suggested. Studies from our, as well as other, laboratories have demonstrated that glucocorticoids stimulate prostaglandin synthesis in the amnion fibroblasts and estradiol synthesis in the placenta (43, 47–50). Estradiol and prostaglandins (PGs), particularly PGE2 and PGF2α, play a crucial role in parturition (51). In addition to the role of cortisol in parturition, Murphy (5) believed that cortisol derived from the fetal membranes may diffuse to the fetal body via amniotic fluid and serve as a supplemental source of cortisol to the fetal adrenal glands for the maturation of fetal organs before the onset of labor. This notion is supported by the findings that the cortisol to cortisone ratio of amniotic fluid increases steadily with gestational age and is considerably higher than that of cord serum (4). Moreover, this ratio is significantly lower in the amniotic fluid of infants who develop respiratory distress syndrome (52), suggesting that this cortisol source might be important for fetal lung maturation. Amniotic cortisol absorption through the lung or open cranium was further supported by the observation that some anencephalic infants seemed to breathe at birth despite poor adrenal function (53).
Bioinformatic analysis of the H6PD promoter sequence reveals several putative GREs as well as CpG islands within −600 bp. The potential methylation of the CpG islands in the promoter could significantly affect the binding of regulatory transcription factors. We presented evidence in this study for the involvement of GR in the induction of H6PD expression by cortisol. In addition to the GR, another cortisol-inducible protein appears to be required for the induction of H6PD mRNA transcription because the induction of H6PD mRNA by cortisol could be significantly attenuated by the protein synthesis inhibitor CHX. We found that p300 was one of the inducible proteins in the induction of H6PD mRNA transcription by cortisol. P300 belongs to the p300-CREB (cAMP response element-binding protein) binding protein (CBP) family originally identified on the basis of their interaction with the transcription factor CREB (54). Both p300 and CBP interact with numerous transcription factors (35) and act to increase the expression of their target genes by relaxing the chromatin structure at the gene promoter through their intrinsic histone acetyltransferase activity and recruiting the basal transcriptional machinery including RNA polymerase II to the promoter (29, 30). GR is among numerous transcription factors that can recruit p300 in transcriptional activation (32, 34). We found that GR and p300 were in the same nuclear protein complex upon cortisol stimulation of human amnion fibroblasts and bound to the same promoter region, suggesting the interaction of GR and p300 in the induction of H6PD by cortisol. However, we are not clear whether this interaction of GR and p300 is direct or involves other bridging proteins.
The intrinsic histone acetyltransferase activity of p300 is well known. Because the increased acetylation of H3K9 bound with H6PD promoter by cortisol could be attenuated by p300 inhibitor C646, a reversible pyrazolone p300 inhibitor competing with acetyl-CoA for the p300 Lys-CoA binding pocket, we believe that p300 is involved in the acetylation of H3K9 induced by cortisol. However, p300 is also able to acetylate transcription factors and other lysine residues of histone 3 or the lysine residues of other histones (55, 56). Therefore, in addition to the acetylation of H3K9 by p300, there exists the possibility that p300 may also acetylate the transcription factors and other lysine residues of histone 3 or the lysine residues of other histones in the induction of H6PD by cortisol.
In conclusion, we have provided evidence in this study that cortisol induces H6PD expression in human amnion fibroblasts via promoting the binding of GR and p300 to H6PD promoter and subsequent acetylation of H3K9. This induction of H6PD by cortisol may be a prerequisite for the increased reductase activity of 11β-HSD1 regenerating bioactive cortisol in a feed-forward loop that may influence fetal development and onset of parturition.
Acknowledgments
This work was supported by the National Key Basic Research Program of China (Grant 2011CB944403), the Natural Science Foundation of China (Grant 30911120485), and National Institutes of Health Grant R01 HD 31514.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ChIP
- Chromatin immunoprecipitation
- CHX
- cycloheximide
- ER
- endoplasmic reticulum
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- H3K9ac
- acetylated H3K9
- H6PD
- hexose-6-phospho-dehydrogenase
- 11β-HSD1
- 11β-hydroxysteroid dehydrogenase 1
- NADPH
- reduced form of nicotinamide adenine dinucleotide phosphate
- PG
- prostaglandin
- qRT-PCR
- quantitative real-time PCR
- siRNA
- small interfering RNA.
References
- 1. Whittle WL, Patel FA, Alfaidy N, Holloway AC, Fraser M, Gyomorey S, Lye SJ, Gibb W, Challis JR. 2001. Glucocorticoid regulation of human and ovine parturition: the relationship between fetal hypothalamic-pituitary-adrenal axis activation and intrauterine prostaglandin production. Biol Reprod 64:1019–1032 [DOI] [PubMed] [Google Scholar]
- 2. Carr BR, Parker CR, Jr, Madden JD, MacDonald PC, Porter JC. 1981. Maternal plasma adrenocorticotropin and cortisol relationships throughout human pregnancy. Am J Obstet Gynecol 139:416–422 [DOI] [PubMed] [Google Scholar]
- 3. Murphy BE. 1982. Human fetal serum cortisol levels related to gestational age: evidence of a midgestational fall and a steep late gestational rise, independent of sex or mode of delivery. Am J Obstet Gynecol 144:276–282 [DOI] [PubMed] [Google Scholar]
- 4. Blankstein J, Fujieda K, Reyes FI, Faiman C, Winter JS. 1980. Cortisol, 11-desoxycortisol, and 21-desoxycortisol concentrations in amniotic fluid during normal pregnancy. Am J Obstet Gynecol 137:781–784 [DOI] [PubMed] [Google Scholar]
- 5. Murphy BE. 1977. Chorionic membrane as an extra-adrenal source of foetal cortisol in human amniotic fluid. Nature 266:179–181 [DOI] [PubMed] [Google Scholar]
- 6. Myatt L, Sun K. 2010. Role of fetal membranes in signaling of fetal maturation and parturition. Int J Dev Biol 54:545–553 [DOI] [PubMed] [Google Scholar]
- 7. Mesiano S, Jaffe RB. 1997. Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev 18:378–403 [DOI] [PubMed] [Google Scholar]
- 8. Sun K, Yang K, Challis JR. 1997. Differential expression of 11 beta-hydroxysteroid dehydrogenase types 1 and 2 in human placenta and fetal membranes. J Clin Endocrinol Metab 82:300–305 [DOI] [PubMed] [Google Scholar]
- 9. Sun K, Yang K, Challis JR. 1997. Differential regulation of 11β-hydroxysteroid dehydrogenase type 1 and 2 by nitric oxide in cultured human placental trophoblast and chorionic cell preparation. Endocrinology 138:4912–4920 [DOI] [PubMed] [Google Scholar]
- 10. Giannopoulos G, Jackson K, Tulchinsky D. 1982. Glucocorticoid metabolism in human placenta, decidua, myometrium and fetal membranes. J Steroid Biochem 17:371–374 [DOI] [PubMed] [Google Scholar]
- 11. Tanswell AK, Worthington D, Smith BT. 1977. Human amniotic membrane corticosteroid 11-oxidoreductase activity. J Clin Endocrinol Metab 45:721–725 [DOI] [PubMed] [Google Scholar]
- 12. Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 2004. 11β-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 25:831–866 [DOI] [PubMed] [Google Scholar]
- 13. Senesi S, Csala M, Marcolongo P, Fulceri R, Mandl J, Banhegyi G, Benedetti A. 2010. Hexose-6-phosphate dehydrogenase in the endoplasmic reticulum. Biol Chem 391:1–8 [DOI] [PubMed] [Google Scholar]
- 14. Senesi S, Legeza B, Balázs Z, Csala M, Marcolongo P, Kereszturi E, Szélenyi P, Egger C, Fulceri R, Mandl J, Giunti R, Odermatt A, Bánhegyi G, Benedetti A. 2010. Contribution of fructose-6-phosphate to glucocorticoid activation in the endoplasmic reticulum: possible implication in the metabolic syndrome. Endocrinology 151:4830–4839 [DOI] [PubMed] [Google Scholar]
- 15. Atanasov AG, Nashev LG, Schweizer RA, Frick C, Odermatt A. 2004. Hexose-6-phosphate dehydrogenase determines the reaction direction of 11β-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett 571:129–133 [DOI] [PubMed] [Google Scholar]
- 16. Czegle I, Piccirella S, Senesi S, Csala M, Mandl J, Bánhegyi G, Fulceri R, Benedetti A. 2006. Cooperativity between 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase is based on a common pyridine nucleotide pool in the lumen of the endoplasmic reticulum. Mol Cell Endocrinol 248:24–25 [DOI] [PubMed] [Google Scholar]
- 17. Atanasov AG, Nashev LG, Gelman L, Legeza B, Sack R, Portmann R, Odermatt A. 2008. Direct protein-protein interaction of 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the endoplasmic reticulum lumen. Biochim Biophys Acta 1783:1536–1543 [DOI] [PubMed] [Google Scholar]
- 18. Zhang YL, Zhong X, Gjoka Z, Li Y, Stochaj W, Stahl M, Kriz R, Tobin JF, Erbe D, Suri V. 2009. H6PDH interacts directly with 11β-HSD1: implications for determining the directionality of glucocorticoid catalysis. Arch Biochem Biophys 483:45–54 [DOI] [PubMed] [Google Scholar]
- 19. Bujalska IJ, Draper N, Michailidou Z, Tomlinson JW, White PC, Chapman KE, Walker EA, Stewart PM. 2005. Hexose-6-phosphate dehydrogenase confers oxo-reductase activity upon 11β-hydroxysteroid dehydrogenase type 1. J Mol Endocrinol 34:675–684 [DOI] [PubMed] [Google Scholar]
- 20. Száraz P, Bánhegyi G, Benedetti A. 2010. Altered redox state of luminal pyridine nucleotides facilitates the sensitivity towards oxidative injury and leads to endoplasmic reticulum stress dependent autophagy in HepG2 cells. Int J Biochem Cell Biol 42:157–166 [DOI] [PubMed] [Google Scholar]
- 21. Bujalska IJ, Hewitt KN, Hauton D, Lavery GG, Tomlinson JW, Walker EA, Stewart PM. 2008. Lack of hexose-6-phosphate dehydrogenase impairs lipid mobilization from mouse adipose tissue. Endocrinology 149:2584–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lavery GG, Hauton D, Hewitt KN, Brice SM, Sherlock M, Walker EA, Stewart PM. 2007. Hypoglycemia with enhanced hepatic glycogen synthesis in recombinant mice lacking hexose-6-phosphate dehydrogenase. Endocrinology 148:6100–6106 [DOI] [PubMed] [Google Scholar]
- 23. Lavery GG, Walker EA, Draper N, Jeyasuria P, Marcos J, Shackleton CH, Parker KL, White PC, Stewart PM. 2006. Hexose-6-phosphate dehydrogenase knock-out mice lack 11β-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J Biol Chem 281:6546–6551 [DOI] [PubMed] [Google Scholar]
- 24. Sun K, Myatt L. 2003. Enhancement of glucocorticoid-induced 11β-hydroxysteroid dehydrogenase type 1 expression by proinflammatory cytokines in cultured human amnion fibroblasts. Endocrinology 144:5568–5577 [DOI] [PubMed] [Google Scholar]
- 25. Yang Z, Guo C, Zhu P, Li W, Myatt L, Sun K. 2007. Role of glucocorticoid receptor and CCAAT/enhancer-binding protein α in the feed-forward induction of 11β-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts. J Endocrinol 195:241–253 [DOI] [PubMed] [Google Scholar]
- 26. Li W, Gao L, Wang Y, Duan T, Myatt L, Sun K. 2006. Enhancement of cortisol-induced 11β-hydroxysteroid dehydrogenase type 1 expression by interleukin 1β in cultured human chorionic trophoblast cells. Endocrinology 147:2490–2495 [DOI] [PubMed] [Google Scholar]
- 27. Sterner DE, Berger SL. 2000. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64:435–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Millar CB, Kurdistani SK, Grunstein M. 2004. Acetylation of yeast histone H4 lysine 16: a switch for protein interactions in heterochromatin and euchromatin. Cold Spring Harb Symp Quant Biol 69:193–200 [DOI] [PubMed] [Google Scholar]
- 29. Bannister AJ, Kouzarides T. 1996. The CBP co-activator is a histone acetyltransferase. Nature 384:641–643 [DOI] [PubMed] [Google Scholar]
- 30. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. 1996. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87:953–959 [DOI] [PubMed] [Google Scholar]
- 31. Schwartz C, Beck K, Mink S, Schmolke M, Budde B, Wenning D, Klempnauer KH. 2003. Recruitment of p300 by C/EBPβ triggers phosphorylation of p300 and modulates coactivator activity. EMBO J 22:882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kino T, Nordeen SK, Chrousos GP. 1999. Conditional modulation of glucocorticoid receptor activities by CREB-binding protein (CBP) and p300. J Steroid Biochem Mol Biol 70:15–25 [DOI] [PubMed] [Google Scholar]
- 33. Mink S, Haenig B, Klempnauer KH. 1997. Interaction and functional collaboration of p300 and C/EBPβ. Mol Cell Biol 17:6609–6617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Q, Su A, Chen J, Lefebvre YA, Haché RJ. 2002. Attenuation of glucocorticoid signaling through targeted degradation of p300 via the 26S proteasome pathway. Mol Endocrinol 16:2819–2827 [DOI] [PubMed] [Google Scholar]
- 35. Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A, Murray PJ, van Deursen JM, Brindle PK. 2006. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol 26:789–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alamdari N, Smith IJ, Aversa Z, Hasselgren PO. 2010. Sepsis and glucocorticoids upregulate p300 and downregulate HDAC6 expression and activity in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 299:R509–R520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang H, Menconi MJ, Wei W, Petkova V, Hasselgren PO. 2005. Dexamethasone upregulates the expression of the nuclear cofactor p300 and its interaction with C/EBPβ in cultured myotubes. J Cell Biochem 94:1058–1067 [DOI] [PubMed] [Google Scholar]
- 38. Zielinska AE, Walker EA, Stewart PM, Lavery GG. 2011. Biochemistry and physiology of hexose-6-phosphate knockout mice. Mol Cell Endocrinol 336:213–218 [DOI] [PubMed] [Google Scholar]
- 39. Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM, Arlt W, Lavery GG, Bedendo O, Ray DW, Laing I, Malunowicz E, White PC, Hewison M, Mason PJ, Connell JM, Shackleton CH, Stewart PM. 2003. Mutations in the genes encoding 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet 34:434–439 [DOI] [PubMed] [Google Scholar]
- 40. Uckaya G, Karadurmus N, Kutlu O, Corakci A, Kizildag S, Ural AU, Gul D, Kutlu M. 2008. Adipose tissue 11-β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase gene expressions are increased in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract 82(Suppl 2):S135–S140 [DOI] [PubMed] [Google Scholar]
- 41. Wang Y, Nakagawa Y, Liu L, Wang W, Ren X, Anghel A, Lutfy K, Friedman TC, Liu Y. 2011. Tissue-specific dysregulation of hexose-6-phosphate dehydrogenase and glucose-6-phosphate transporter production in db/db mice as a model of type 2 diabetes. Diabetologia 54:440–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nammi S, Dembele K, Nyomba BL. 2007. Increased 11-β-hydroxysteroid dehydrogenase type-1 and hexose-6-phosphate dehydrogenase in liver and adipose tissue of rat offspring exposed to alcohol in utero. Am J Physiol Regul Integr Comp Physiol 292:R1101–R1109 [DOI] [PubMed] [Google Scholar]
- 43. Casey ML, MacDonald PC, Mitchell MD. 1985. Despite a massive increase in cortisol secretion in women during parturition, there is an equally massive increase in prostaglandin synthesis. A paradox? J Clin Invest 75:1852–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun K, He P, Yang K. 2002. Intracrine induction of 11β-hydroxysteroid dehydrogenase type 1 expression by glucocorticoid potentiates prostaglandin production in the human chorionic trophoblast. Biol Reprod 67:1450–1455 [DOI] [PubMed] [Google Scholar]
- 45. Craft I, Brummer V, Horwell D, Morgan H. 1976. Betamethazone induction of labour. Proc R Soc Med 69:827–828 [PMC free article] [PubMed] [Google Scholar]
- 46. Nwosu UC, Wallach EE, Bolognese RJ. 1976. Initiation of labor by intraamniotic cortisol instillation in prolonged human pregnancy. Obstet Gynecol 47:137–142 [PubMed] [Google Scholar]
- 47. Guo C, Li J, Myatt L, Zhu X, Sun K. 2010. Induction of Gαs contributes to the paradoxical stimulation of cytosolic phospholipase A2α expression by cortisol in human amnion fibroblasts. Mol Endocrinol 24:1052–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sun K, Ma R, Cui X, Campos B, Webster R, Brockman D, Myatt L. 2003. Glucocorticoids induce cytosolic phospholipase A2 and prostaglandin H synthase type 2 but not microsomal prostaglandin E synthase (PGES) and cytosolic PGES expression in cultured primary human amnion cells. J Clin Endocrinol Metab 88:5564–5571 [DOI] [PubMed] [Google Scholar]
- 49. Zhu XO, Yang Z, Guo CM, Ni XT, Li JN, Ge YC, Myatt L, Sun K. 2009. Paradoxical stimulation of cyclooxygenase-2 expression by glucocorticoids via a cyclic AMP response element in human amnion fibroblasts. Mol Endocrinol 23:1839–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang W, Li J, Ge Y, Li W, Shu Q, Guan H, Yang K, Myatt L, Sun K. 2012. Cortisol induces aromatase expression in human placental syncytiotrophoblasts through the cAMP/Sp1 pathway. Endocrinology 153:2012–2022 [DOI] [PubMed] [Google Scholar]
- 51. Challis JR, Lye SJ, Gibb W. 1997. Prostaglandins and parturition. Ann NY Acad Sci 828:254–267 [DOI] [PubMed] [Google Scholar]
- 52. Smith BT, Worthington D, Maloney AH. 1977. Fetal lung maturation. III. The amniotic fluid cortisol/cortisone ratio in preterm human delivery and the risk of respiratory distress syndrome. Obstet Gynecol 49:527–531 [PubMed] [Google Scholar]
- 53. Burke RK, Lundy LE, Sisson TR, Jahed FM. 1973. Pulmonary surfactant. Is the fetal adrenal necessary? Obstet Gynecol 41:833–836 [PubMed] [Google Scholar]
- 54. Arany Z, Sellers WR, Livingston DM, Eckner R. 1994. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 77:799–800 [DOI] [PubMed] [Google Scholar]
- 55. Crump NT, Hazzalin CA, Bowers EM, Alani RM, Cole PA, Mahadevan LC. 2011. Dynamic acetylation of all lysine-4 trimethylated histone H3 is evolutionarily conserved and mediated by p300/CBP. Proc Natl Acad Sci USA 108:7814–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bannister AJ, Miska EA. 2000. Regulation of gene expression by transcription factor acetylation. Cell Mol Life Sci 57:1184–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]