

Abstract
Recent technological advancements in gene expression analysis have led to the discovery of a promising new group of prostate cancer (PCa) biomarkers that have the potential to influence diagnosis and the prediction of disease severity. The accumulation of deleterious changes in gene expression is a fundamental mechanism of prostate carcinogenesis. Aberrant gene expression can arise from changes in epigenetic regulation or mutation in the genome affecting either key regulatory elements or gene sequences themselves. At the epigenetic level, a myriad of abnormal histone modifications and changes in DNA methylation are found in PCa patients. In addition, many mutations in the genome have been associated with higher PCa risk. Finally, over- or underexpression of key genes involved in cell cycle regulation, apoptosis, cell adhesion and regulation of transcription has been observed. An interesting group of biomarkers are emerging from these studies which may prove more predictive than the standard prostate specific antigen (PSA) serum test. In this review, we discuss recent results in the field of gene expression analysis in PCa including the most promising biomarkers in the areas of epigenetics, genomics and the transcriptome, some of which are currently under investigation as clinical tests for early detection and better prognostic prediction of PCa.
Keywords: Prostate cancer, biomarker, epigenetics, methylation, acetylation, ncRNA, genomics, SNP, transcriptomics, miRNA, lncRNA
Introduction
The Knudson multiple-hit model hypothesizes that cancer results from the accumulation of multiple mutations in the genome [1], however recent work suggests that this may not be quite accurate. Marked changes in gene expression can occur without a single base change in the genetic code. More likely is that “hits” accumulate in the complex regulatory system which governs gene expression and the combined result of these changes is cellular transformation, or cancer. In recent decades, research has shown that the regulation of gene expression is far more complex than initially predicted.
Including the regulatory sequences encoded within the genome, there are a staggering number of controls for achieving proper protein expression. For example, simple modifications made to the nucleosome can change how often transcription occurs at a given locus. What was thought to be a simple structural scaffold used to package the genome into the nucleus is actually a sophisticated system which contributes to the regulation of gene expression. Alterations in chromatin structure influence how tightly or loosely the DNA is packaged, resulting in significant changes in gene transcription. This “new” field is called epigenetics. Within epigenetics, DNA and histone modifications including methylation and acetylation have proven to be critical regulators of gene expression which are undeniably altered in cancer cells. Similarly, what was thought to be “junk” DNA between genes in the human genome, in fact, encodes small RNA molecules that play a key role not only in the control of protein production but also in regulating the DNA-nucleosome complex. Some small non-protein coding transcripts can recruit chromatin remodeling enzymes and exert control over gene transcription, while others can effectively inhibit the translation of a protein. When aberrantly regulated, non-coding RNAs can have profound effects on transcription and translation-a fact that has been under intense investigation in PCa. The study of the effects of non-coding RNAs coupled with the global analysis of transcript levels is called transcriptomics. Finally, Knudson’s prediction that mutations in the genome can affect tumorigenesis is still relevant: single base changes and larger chromosomal rearrangements have a well-known role in cancer progression. Extensive studies on the role of gene fusion and single nucleotide polymorphisms (SNPs) in PCa are included in the field of genomics. Given the penetrance of PCa diagnosis within the population, the fields of epigenetics, genomics and transcriptomics have been extensively investigated and excellent progress has been made in determining which hits are most deleterious to the prostate epithelium.
Emerging PCa biomarkers are of increasing utility since the recent recommendation made by the United States Preventative Services Task Force (USPSTF) to discontinue the use of the PSA serum test as a diagnostic indicator of PCa. As many as 1 in 6 men are identified as having prostate cancer (PCa) in their lifetime, making it the most commonly diagnosed solid tumor aside from skin cancer in males [2]. The USPSTF’s recently released guide for PSA screening recommends against routine assay of PSA serum levels in men of all ages and races. According to the official statement published in the Annals of Internal Medicine in July 2012, PSA screening results in the treatment of clinically indolent cancers which can lead to significant morbidities. Importantly, the USPSTF did not advise against PSA levels as a marker for recurrence of PCa after treatment, indicating that it still serves as an appropriate biomarker for PCa advancement [3]. PSA has long been referred to as “the” PCa biomarker: It is expressed at low levels in normal prostate epithelium as part of the cell’s normal response to androgens, and in PCa, serum levels of PSA rise as androgen signaling increases. Unfortunately, despite its proven utility as a marker for recurrence of PCa after surgery or radiation, PSA as a diagnostic for primary PCa falls short. A better diagnostic marker is clearly needed to either strengthen the PSA test, or to replace it all together.
The perfect biomarker for primary and/or advanced PCa would have the following attributes: extremely high penetrance within the patient population, high specificity for PCa and detection via a non-invasive assay. Ideally, a biomarker would not be expressed in normal prostate epithelium and be detected at a level corresponding to disease severity in advancing PCa stages. An exemplary biomarker would also be very specific to PCa, to eliminate the occlusion of a diagnosis by the detection of other coincident cancers. Finally, serum, whole blood, urine or saliva tests are the most widely accepted by patients and therefore would be excellent platforms for an assay. In recent years, excellent progress has been made in identifying new markers for PCa at various stages. In this review, emerging biomarkers associated with the fields of epigenetics, genomics and transcriptomics will be discussed.
Epigenetics
The field of epigenetics has expanded exponentially in the last decade and has played a prominent role in the study of cancer genetics. Epigenetic gene regulation refers to non-coded heritable changes in gene expression and is comprised of DNA methylation, histone modifications and noncoding RNA-induced transcriptional changes. Aberrant epigenetic regulation can lead to global gene expression changes and genomic instability which can have clear implications in the development of cancer. Indeed, changes in the epigenome have long been associated with PCa risk and progression.
DNA methylation and PCa
Methylation of cytosine residues in the human genome contributes to transcriptional regulation and genomic stability [4-8]. A common phenomenon, 80-90% of coding CpG (5’- cytosine phosphate guanidine -3’) dinucleotides are methylated by DNA methyltransferases (DNMTs). DNMTs are responsible for the addition of a methyl group to the 5’ C position in cytosine, creating 5-methyl cytosine (5mC). The DNMT superfamily consists of those required for methylation of newly-synthesized DNA (e.g. DNMT3a, b) and those whose role is to maintain normal methylation throughout the genome (e.g. DNMT1). DNMTs also regulate methylation of CpG “islands,” which are made up of >65% CpG, and are found in half of human gene promoters [9]. Hypermethylation in promoter CpG islands is associated with gene silencing, either due to blockage of access of binding sites to transcription factors or changes in chromatin structure [10], while hypomethylation results in increased gene expression due to unchecked promoter binding. The delicate balance between hypermethylation and hypomethlyation of promoters is a key component of gene expression regulation in genomic imprinting and in X chromosome inactivation. For example, in the embryonic genome, some genes are silenced in either the paternal or maternally-inherited gene copy by hypermethylation, while others are hypomethylated to allow higher expression. These epigenetic changes are inherited and also involve histone marks such as methylation and acetylation [4]. As we age, global methylation changes are detected: Global genome-wide methylation of CpG residues is lost and unmethylated CpG islands in key tumor suppressor gene promoters become hypermethylated. Loss of pan-genome CpG methylation can result in chromosome aberrations including translocations and DNA strand breaks, while promoter-specific hypermethylation results in gene silencing [11]. Alterations in gene expression due to aberrant DNA methylation are well documented in prostate and many other cancers. Comprehensive lists of affected genes can be found in [4,6]. Hypermethylation and gene silencing have been documented for cell cycle regulators such as Anaphase Promoting Complex (APC) and Ras association domain-containing protein 1 (RASSF1a), detoxification enzymes (e.g., Glutathione S-Transferase Pi 1; GSTP1) and pro-apoptosis genes (e.g., certain caspase genes) [12-18]. In fact, a combined assay for GSTP1 and APC hypermethylation has shown great promise for detecting PCa in clinical samples with up to 100% certainty [12]. Promoter hypomethylation and subsequent upregulation of oncogenes such as WNT5A have been strongly linked with PCa diagnosis [19]. Moreover, nuclear hormone receptors such as estrogen receptor α and β, and AR (ERα, ERβ, respectively) are hypermethylated in PCa patients. Not surprisingly, given the androgen-dependent nature of most PCa’s, AR is only hypermethylated in 40% of clinical samples [20,21]. Classically, decreased detoxification enzyme expression leads to unchecked DNA damage and genomic instability, while increased growth factor signaling induces uncontrolled growth. Combined with the silencing of cell cycle regulators and the loss of proapoptosis gene expression, the situation is a “perfect storm” for the development of cancer. As PCa progresses, detection of hypermethylation in cell adhesion genes such as E-cadherin is observed, allowing a rapid transition from epithelial to mesenchymal morphology and behavior [22]. This growing body of evidence demonstrates that DNA methylation is a key epigenetic change contributing to the transformation of prostate epithelium and the subsequent invasion and metastasis phenotypes.
Experimental methods for detecting DNA methylation have been perfected over many decades [23-25]. Perhaps the biggest breakthrough in the field came from Hikoya Hiyatsu’s discovery that the treatment of DNA with high concentrations of bisulfite results in rapid deamination of cytosine and its conversion to uracil [26]. Although 5-methyl cytosine (5mC) can also undergo deamination to form thymine, the kinetics of this reaction are far slower-two orders of magnitude slower-which clearly favors the formation of uracil from unmethylated cytosine [26]. This discovery truly revolutionized the study of DNA methylation. For example, methylation-specific PCR or MS-PCR, is a widely-used technique which allows the amplification of either methylated or unmethylated template by designing primers to both the bisulfite-induced uracil-substituted form or the native methylated form. The amplified product can then be subject to a number of protocols to determine DNA sequence, restriction fragment length/fingerprinting or even taken a step further to quantitate the extent of methylation by realtime quantitative PCR (a technique called MethyLight [27]). There are significant advantages and drawbacks to each of these analytical methods including the amount of DNA required for each approach and the specificity of the target: Some are gene-specific and others more global.
Possibly most applicable to the field of biomarker discovery is the application of methylated DNA to microarray. The ability to compare a normal genome to the methylation state of a tumor genome is very powerful. Many methods of sample preparation for methylation detection by microarray have been developed throughout the years but two stand out as the most commonly used: Methylated DNA Immunoprecipitation (MeDIP) and bisulfite treatment followed by MS-PCR. MeDIP can employ a monoclonal antibody raised against 5mC, or a 5mC binding protein called Methyl CpG Binding Protein 2 (MeCP2). One disadvantage of this technique is that anti-5mC antibodies bind with higher efficiency to CpG-rich sequences, enriching the final pool for CpG-dense sequences. Despite this drawback, successful identification of hypermethylated CpG islands in promoter regions in cancer cells has been achieved. Elevating this technique to an ever faster and higher throughput level is the addition of Next Generation DNA sequencing. Bisulfite-treated, adapter-ligated and MS-PCR amplified products can be directly sequenced, bypassing the need for microarray analysis and the possibility of technical issues with hybridization and data analysis. The recent work of Yegnasubramanian, et al., truly combines all of these techniques into one elegant method by which the DNA methylome in normal and cancerous prostate culture cells are compared [16,17]. The comparison was made between normal fetal prostate epithelial cells (PrEC cells; Lonza, Inc.) and the classic PCa cell line, LNCaP. Using their streamlined and refined technique, microarray was used to compare methylated DNA from PrEC cells to LNCaP cells and futher to PCa patient samples. Hypermethylated regions in LNCaP cells (e.g. upstream of ADAMTS2 and GSTP1) were also hypermethylated in patients and vice versa, validating the technique. In addition, new areas of hyper- and hypomethylation were discovered [17].
The most useful PCa biomarkers discovered from DNA methylation research are clearly still being fully characterized since clinical tests for CpG methylation are not the standard of care for PCa patients. At this time, a combined test for hypermethylation of multiple gene promoters holds the most promise for accurate diagnosis and prediction of prognosis. Yegnasubramanian, et al., have demonstrated that a short list of genes including GSTP1, APC, PTGS2, MDR1, and RASSF1a are hypermethylated in PCa samples. Importantly, this subset of aberrantly methylated genes is positively correlated with disease stage and is unique to PCa as compared to other cancers. When investigated in combinations, predictive capability of this “test” reached 100% accuracy. Further, prostaglandin-endoperoxide synthase 2 (PTGS2) hypermethylation was correlated with increased risk of recurrence. The authors state that this assertion should be confirmed in a larger sample set [16]. In another preliminary study, a subset of the genes listed above were assayed in cell-free plasma samples from patients diagnosed with PCa [28]. Although the sample size was very small, hypermethylation at GSTP1 and APC were confirmed to be associated with increased disease severity. This study also established that cell-free DNA methylation assay is a good way to determine the presence of promoter hypermethylation in PCa samples, however, more work is required to ascertain whether all hypermethylated genes found in PCa tissues are translatable to a cell-free assay environment [28]. In summary, there are very promising future biomarkers in the DNA methylation field which, upon further research and validation, may become valuable diagnostic and prognostic aids.
Histone modifications in PCa
Chromatin remodeling has been shown to be an integral part of cancer progression (Figure 1) [29]. The delicate balance between active transcription and silencing of genes is critical for maintaining cellular homeostasis, and changes in chromatin structure can influence cellular transformation. Histone modifications including acetylation, methylation, phosphorylation and ubiquitination collectively make up the mechanisms affecting the complex structure of chromatin. We will focus on acetylation and methylation here. Each of these alterations in histone structure accomplish a similar task: blocking the normal electrostatic interaction between positively-charged lysine tails in the N-terminus of the histone and negatively-charged DNA phosphate groups. The tighter that the DNA is wrapped around histones due to these electrostatic interactions, the less accessible it is to transcription factors and other elements of the transcription machinery and the more a gene is effectively silenced. Conversely, the weaker the histone-DNA interaction, due to histone modification, the more transcriptional activity is seen. Acetylation of lysines in histone H2, H3 or H4 N-terminal tails is associated with a more exposed and active chromatin, called euchromatin. Acetyl groups are transferred to proteins from a donor molecule, acetyl-CoA, by Histone Acetyltransferases (HATs), and are removed by Histone Deacetylases (HDACs). The role of HATs and HDAC enzymes in PCa progression and therapy is complex and this topic is still under intense investigation.
Figure 1.

Epigenetic regulation of gene expression in PCa. This schematic illustrates how changes in the normal conformation of chromatin structure can impact transcription in PCa. In the left panel, low acetylation results in tightly wound DNA and transcription is rare. In the middle, methylation and acetylation of histones result in loose chromatin conformations and reduced ability of RNA polymerase (RNA Pol) to access the promoter region. Finally, the right panel illustrates the situation whereby histones are deacetylated by HDACs, promoter regions are hypermethylated by DNMTs and the chromatin conformation is closed. Transcription in this situation is very rare; the gene is effectively silenced. Orange “M” squares represent methyl groups; yellow “A” triangles represent acetyl groups.
HAT activity in PCa patients has both positive and negative effects on disease progression. HAT acetylation of loci containing negative cell cycle regulators like p21 (a cyclin-dependent kinase inhibitor) has been documented in PCa patients [30]. The loss of HAT activity results not only in a reduction in transcriptional activity in general, but also in inhibition of DNA repair due to the tightening of histone-DNA association [9,31]. Taken together, these observations represent a paradox in that increased HAT activity may be beneficial in regulating the cell cycle, but diminished HAT activity may also be advantageous since a buildup of errors in DNA replication could lead to cell death for rapidly dividing cancer cells. The latter has been exploited by the anti-cancer drug field. Treatment of rapidly dividing aggressive cancer cells with HAT inhibitors does indeed result in the accumulation of large double-strand breaks in the DNA which are lethal to the cell [32,33]. Consistent with the idea that HAT inhibition may be a beneficial anti-cancer therapy, increased HAT activity at oncogene loci has been detected in clinical samples. Specifically, a growing body of work has shown that the upregulation of androgen signaling in castrate men may be, in part, due to increased HAT activity [34,35]. Aberrant hyperacetylation at the AR gene locus leads to increased receptor expression and activation despite a lack of stimulation by androgens [36,37]. Therefore, HATs play competing roles in the progression of PCa, but they may represent a possible drug target as long as the anti-cancer effects outweigh the loss of important checkpoint genes which are regulated by HAT activity.
HDAC genes and their role in PCa progression have been widely studied with roles in silencing negative cell cycle regulators, cell adhesion molecules and anti-apoptosis genes. HDACs are separated into four groups or families based on structure: HDAC I, II, III and IV. Groups I and II are found in the nucleus where they modify chromatin structure, whereas groups III and IV are found throughout the cytoplasm, nucleus and mitochondria (reviewed in [9,38]). Increased HDAC expression, specifically HDAC1, is noted in a high percentage of PCas with the highest levels of expression found in hormone refractory disease [39-41]. HDAC activity and hypermethylation of promoter regions are intimately linked - both result in transcriptional silencing of tumor suppressor genes which is an integral first step in cancer development. In fact, p21, the cell cycle regulator highlighted above as a target of HAT activity, is silenced by HDACs in a high percentage of PCa patients, effectively removing negative cell cycle regulation [41,42]. Furthermore, HDAC1 specifically silences Phosphatase and Tensin homolog (PTEN) causing unchecked Phosphatidylinositol 3-kinase (PI3K) signaling and cell division [43,44]. Similarly, the E-cadherin locus has been shown to be heavily hypermethylated (at both the histone and promoter levels) and deacetylated [45]. This strong repression of cell adhesion molecules is thought to be a major driver of metastasis and invasion in PCa.
HDAC inhibitors (HDACi), have been proposed as powerful anti-cancer therapeutics due to the reliance of cancer cells on epigenetic modifications to silence tumor suppressor genes. HDACi molecules can be classified into five groups based on their chemical structure. HDAC1, the specific isoform which is upregulated in PCa patients, is potently inhibited by multiple classes of HDACi molecules [46-48]. Possibly the best known HDAC1 inhibitor is valproic acid (VPA). VPA belongs to the class of short-chain fatty acid inhibitors and also inhibits other HDACI family members as well as a portion of the HDACII family. VPA has a long history as an anti-epileptic and successful mood stabilizer. Like all known HDACs, VPA displays very low levels of toxicity in normal cells and the data from PCa cell lines like PC3 and LNCaP are convincing, with the most pronounced anti-proliferative effect on the hormone refractory PC3 line. Increased expression of E-cadherin, AR and p21 also indicate that VPA is successful in relieving HDAC-mediated transcriptional silencing in cancer cells in vitro [49-51]. PCa tumor volume in mouse xenografts is also significantly affected by VPA [49-51]. Although some studies seemed to indicate that VPA treatment may result in an increase in neuroendocrine differentiation - a fate which is linked with poor prognosis - recent work by Sidana, et al., showed that neuroendocrine markers are not upregulated in VPA-treated xenografts [52,53]. Although the molecular mechanism is still unknown, HDACi class I drugs such as VPA seem to block angiogenesis in tumors as assayed by repression of hypoxia inducible factor-α (HIF-1α) function [51]. The significance of decreased HIF1-α transcription factor activity is clear: without it, cancer cells lose a major mechanism of upregulating pro-angiogenic signaling such as vascular endothelial growth factor (VEGF). Truly, a PCa treatment which reverses gene silencing in tumor suppressors, anti-cell cycle regulators and anti-apoptosis genes while also blocking angiogenesis and simultaneously displaying low toxicity is a promising treatment indeed. Current clinical trials will determine whether these drugs are as effective as in in vitro models [54].
Similar to histone acetylation, methylation of lysines in histone proteins can result in a more open chromatin conformation. In contrast to acetylation, other lysine or arginine methylation events can cause tighter DNA-histone association and result in gene silencing [55]. Histone Methyltransferases (HMTs) regulate the addition of either one, two or three methyl groups to lysine residues using S-adenosyl methionine (SAM) as a donor and cofactor [56]. Although historically thought to be irreversible, Histone Demethylases (HDM) have been characterized which remove these modifications ([57] and reviewed in [58]). Histone methylation has not been studied as extensively as other histone modifications with respect to PCa, but some promising preliminary data are emerging.
Similar to other epigenetic marks, histone methylation has been associated with tumor suppressor and adhesion molecule gene silencing. Trimethylation of lysine 27 of histone H3 (H3K27) by the HMT Enhancer of Zeste Homologue 2 (EZH2) is a known silencing mark, and EZH2 is strongly upregulated in PCa [59]. EZH2 is a tempting marker for PCa diagnosis since its overexpression has been linked with the silencing of tumor suppressor genes and also to epithelial-to-mesenchymal transition via silencing of adhesion molecule expression [60]. As a biomarker, EZH2 lacks specificity since it is detected at higher levels than normal in melanoma, lymphoma and breast cancer. Subsequent studies have shown that the presence of EZH2 at a promoter results in recruitment of other Polycomb genes, heterochromatin proteins (HPs) and HDACs [61-63]. Clearly there is significant crosstalk between different types of histone modification enzymes. These data and others have led to the development and testing of anti-HMT drugs, however none are currently approved for PCa treatment [64]. Another common histone methylation event is the mono-, di-, or trimethylation of histone H4 lysine 20. Studies have indicated that trimethylation of H4K20 is associated with tumor suppressor gene silencing and PCa diagnosis, but further experiments did not significantly correlate this event with increased PCa severity which excludes it from the ideal biomarker category [65]. The study of HDAC and HMT inhibitors is complicated by the fact that epigenetic marks are important gene expression regulators.
Imprinting is established in the early embryo and allows for the silencing of one allele of a gene by epigenetic marks such as DNA and histone methylation/acetylation. Loss of imprinting can result in overexpression of an oncogene and can lead to cancer. For example, both alleles of Insulin Growth Factor-2 are expressed in PCa tissues, while in normal cells, this gene is imprinted and only the paternal allele is expressed [66,67]. In this case, overexpression can lead to unchecked cell proliferation. In a situation where HDAC and HMT inhibitors are being administered systemically, global imprinting changes can occur and a volatile or abnormal gene expression situation can be made worse.
In conclusion, two histone modifiers stand out as promising PCa biomarkers. The HAT p300 has not only been shown to be highly overexpressed in PCa, but also, expression levels of this protein are positively correlated with disease stage [68]. The HDM EZH2 was also shown to be upregulated with increasing severity in progressing PCa disease stages (discussed above). Perhaps the two together may make an excellent dual biomarker. Both molecules are at the center of intense investigation at this time and future work will elucidate whether these biomarkers translate to the clinic.
Long non-coding RNA regulation of gene expression
Expression, processing and function of non-coding RNA (ncRNA) is a relatively new field in PCa research [69,70]. The term ncRNA encompasses the well-studied functional RNAs like rRNA and tRNA, as well as microRNA (miRNA; previously known as small ncRNA), long ncRNA (lncRNA) and small interfering RNA (siRNA). For an excellent review of all of the subtypes within these groups of ncRNA, please see Gibb, et al., 2011 [71]. ncRNA is essential for cellular function: it makes up a portion of the ribosome, the spliceosome and carries amino acids to the growing protein chain. Although the field of ncRNAs encompasses over 30 different types of molecules [71,72], most relevant to the discussion of epigenetic PCa biomarkers are the lncRNAs that play a role in gene expression regulation. The human genome is reported to include a modest (compared to total genome size) ~20,000 protein coding genes, but astoundingly, there are an estimated ~23,000 lncRNAs in what used to unjustly be referred to as “junk” genomic DNA [73]. Functions for lncRNAs are extremely varied, poorly understood and may include: regulation of splicing, component of the nuclear scaffold, regulation of chromatin remodeling and regulation of transcription through imprinting (including DNA methylation [69,71]). Numerous lncRNAs have been shown to alter imprinting marks at oncogene and tumor suppressor gene loci. These changes in expression are proposed to be a major mechanism of tumorigenesis and cellular transformation [72,74,75].
lncRNAs have had a complicated history. First believed to be an artifact of “leaky” transcriptional regulation, they were dismissed as unimportant; however, lncRNAs in particular share many of the common traits of protein coding genes. They are spliced, polyadenylated, methylated, transcribed by RNA polymerase II and regulated by common transcription factors (reviewed in [69]). In contrast to mRNA, there is no open reading frame in lncRNAs and therefore no protein coding region. In fact, lncRNA transcripts commonly contain multiple stop codons in every frame. Further, unlike microRNA which are less than 200 bp long and will be discussed in detail in “Transcriptomics” in this review, lncRNAs are, on average, 800 bp. Genes encoding lncRNAs can be found in intergenic regions, introns, on the complimentary strand of a protein-coding gene or overlapping with another ncRNA-coding region (reviewed in [72]). lncRNAs have been reported to play a role in PCa turmorigenesis by multiple varying mechanisms.
Located in the well-known tumor suppressor region INK4b-ARF-INK4a, the lncRNA Antisense non-coding RNA in the INK4 locus (ANRIL) is detected at high levels or associated with polymorphisms in many disease states [76]. In normal prostate epithelium, expression of the three cell cycle regulators Ink4a, Ink4b and ARF is tightly controlled by epigenetic marks including histone methylation. In PCa, ANRIL has been shown to bind to and recruit epigenetic regulatory elements such as Polycomb repressive complex (PRC) proteins which results in a transcriptionally silenced chromatin conformation [77,78]. Although preliminary studies indicate that ANRIL overexpression is detected in PCa patient samples, ANRIL is not an ideal PCa biomarker due to its association with multiple other cancers [77]. In fact, ANRIL expression is detected in non-cancer related disease states such as atherosclerosis [79]. Although ANRIL does not qualify as a diagnostic biomarker, perhaps future studies will conclude that it is useful as a risk stratification marker.
Three lncRNAs have been shown to display prostate specificity and are actively under investigation as prostate biomarkers: Prostate cancer noncoding RNA-1 (PRNCR1), Prostate specific gene 1 (PSGEM1), and Prostate cancer antigen 3 (PCA3; also referred to as Differential Display 3 (DD3) in the literature) (for a review, see [71]). Probably the least characterized of this group is PRNCR1. This lncRNA was found while investigating 8q24, a region of the genome in which single nucleotide polymorphisms have been strongly associated with PCa [80,81]. The PRNCR1 gene encodes a ~13 Kb lncRNA which is expressed in all of the common PCa cell lines investigated and in 50% of the clinical PCa samples investigated. It should be noted that the small sample size and the racial bias (all samples were of Korean descent) make this an extremely preliminary study, however, the data are promising if the trend translates to other races. It has certainly been demonstrated previously that 8q24 is a region of great interest in PCa research (see “Single nucleotide polymorphisms section, this review; Figure 2). Many SNPs and even other ncRNAs have been associated with this locus in cancer patients [81-83].
Figure 2.
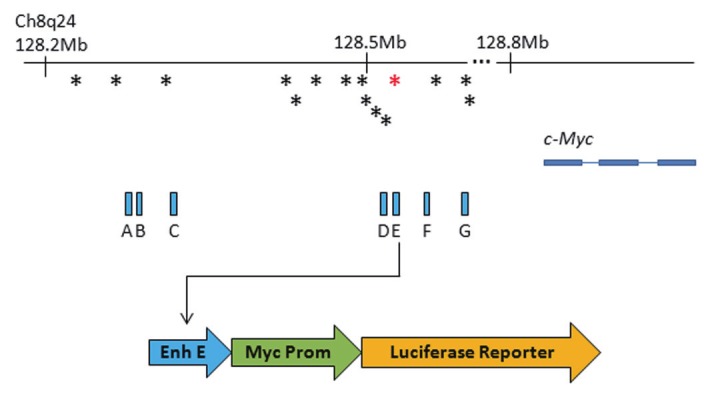
The 8q24 genomic locus. This locus spans over 1 Mb, but only a small relevant portion is shown. The c-Myc gene is the closest coding gene at 200 Kb from 8q24. This schematic illustrates not only the SNPs associated with 8q24 (asterisks), but also, in blue, the functional enhancer elements identified by Sotelo and colleagues [117]. In this study, each enhancer was subcloned into a vector containing a myc promoter and a luciferase reporter. In this way, the ability of each enhancer to drive reporter expression was assayed. The red asterisk represents the rs6983267 SNP. Alleles of this SNP have been associated with differential effects on c-Myc expression.
Perhaps better characterized is PCGEM1. Discovered in a differential display experiment comparing tumor and normal samples, PCGEM1 encodes a lncRNA which is extremely prostate-specific. In fact, among the 12 tissue types that were examined, only prostate expressed this transcript. Making it even more enticing, PCGEM1 expression was detected only in androgen-sensitive cell lines and clearly upregulated by the AR agonist, R1881 [84]. In patients, PCGEM1 is detected in 84% of PCa tumors with higher levels of expression found in African American PCa patients [84,85]. Further mechanistic studies indicated that PCGEM1 overexpression in LNCaP cells results in the inhibition of doxyrubicin-induced apoptosis [86]. Protection of cancer cells against apoptosis is a major hallmark of cancer progression. Since PCGEM1 is expressed at very low levels even in prostate, RT-PCR methods of detection have been less successful than fluorescent in situ hybridization (FISH). Certainly, FISH is a highly technical and expensive test for a diagnostic test. As newer technology emerges that allows for reliable detection of rare transcripts in cDNA populations, PCGEM1 has great potential as a PCa diagnostic biomarker - especially in the African American population.
By far the most well-known lncRNA in PCa is DD3/PCA3 (referred to as PCA3 hereafter). The first PCA3 urine test was FDA-approved in February 2012, 13 years after the discovery of PCA3 by Bussemakers and colleagues [87]. Again discovered using differential display comparing tumor and normal prostate samples, this lncRNA was shown to be expressed exclusively in the prostate and further, expressed only in AR-expressing cell lines [87]. Although correlation with disease stage and aggressiveness was not shown, PCA3 expression provides excellent distinction between tumor and normal tissue when used in combination with the standard PSA test [88-91]. For an excellent review of the clinical data relating to PCA3 expression in PCa patients, see Auprich, et al., 2010 [88]. Interestingly, PCA3 has been conclusively shown to be a better biomarker for PCa in biopsy samples than PSA; However the two together result in an even better predictor of PCa diagnosis [92]. The new urine test for PCA3 is an RT-PCR assay based on the premise that after digital rectal exam (DRE), prostate cells are shed into the urine. This platform has its limitations-variations in physician application of “pressure” during DRE and differing methods of urine collection and storage are inherent. Despite its use as a clinical biomarker, the mechanism and/or underlying reason for PCA3 upregulation in PCa is not well understood. In depth analysis of the gene indicates that multiple transcripts using different splicing patterns are likely, with the major isoform being ~2 Kb [93,94]. Recent work by Clark, et al., suggests that PCA3 may be evolutionarily linked to an overlapping protein-coding gene called BCH motif-containing molecule at the carboxyl terminal region 1 (BMCC-1). BMCC-1 and PCA3 were both shown to be upregulated in LNCaP cells treated with dihydrotestosterone (DHT), but clearly more studies are needed to determine whether they are true androgen-sensitive genes [93].
In summary, the biggest success in the search for PCa biomarkers in the lncRNA field is clearly the PCA3 story. With a urine test and FDA-approval, the future will decide whether the predictive ability of PCA3 remains in tact in very large sample sizes. The ncRNA field is extremely active as of late. New prostate specific lncRNAs are being documented regularly. Future work will determine whether they are as good, or better, than the PCA3 post-DRE urine test.
Genomic biomarkers in PCa
Single Nucleotide Polymorphisms (SNPs)
PCa is one of the most heritable cancers with up to 15% of cases linked to family history [95]. In fact, studies using genomic data from twins indicates that up to 42% of risk for developing PCa is linked to heritable components [96]. A great deal of research has been focused on discovering which genomic elements are responsible for this phenomenon and for PCa risk in general. The long-term viability of these studies has been variable: it is difficult to reproduce the data and difficult to determine what risk is significant. Despite these challenges, a large body of work has indicated that there are multiple complex genomic “hot spots” producing heritable sequence changes which are linked to disease risk [97-100].
Genome-wide association studies (GWAS) have been used to compare whole-genome single nucleotide polymorphism (SNP) arrays between populations of PCa patient genomic DNA’s (gDNA). First, a genomic region is identified as a hotspot by low resolution examination of hybridization differences between PCa patients and a control cohort on SNP array. Second, the locus is examined at high resolution by mapping and sequencing and each patient’s alleles are catalogued. In this way, alleles that are segregating within a population can be identified, and risk can be determined by correlating the SNP allele with the severity of disease in terms of clinico-histophathological variables [e.g. age, Gleason score (GS)]. Finally, each SNP is given a number and a statistical value representing the Population Attributable Risk (PAR). The result of this work is that one specific change in DNA sequence can be examined in thousands of patients to determine the probability of PCa development. Using this technique, many loci which link SNPs with increased PCa risk have been identified and are catalogued in Table 1. Most of the SNP-associated genes and genomic regions have not been studied in depth with respect to their role in PCa, however, some have been well-studied in other cancers or normal tissues. These SNPs are located within protein-coding genes, in intergenic regions and in loci without any known genes at all (reviewed in [97,98]).
Table 1.
Chromosomal regions in which SNP alleles are associated with PCa risk
| Chromosomal Region | Genes closest to SNP | Function and/or domains | p value (n) | REF |
|---|---|---|---|---|
| 2p15 | EHBP1 | Endocytic trafficking | 7.7x10-9 (38,747) | [252] |
| 2p21 | THADA | Unknown [253] | 1.6x10-8 (42,388) | [254] |
| 2q31 | ITGA6 | Signal transduction, adhesion | 8.7x10-23 (42,388) | [254] |
| 3p12 | None | N/A | 2.7x10-8 (11,338) | [255] |
| 3q21 | EEFSEC | Translation elongation; GTPase | 2.9x10-10 (61,388) | [256] |
| 4q22 | PDLIM5 | LIM-domain; protein-protein interaction | 1.3x10-11 (42,388) | [254] |
| 4q24 | FLJ20032 | hypothetical | 2.6x10-14 (42,388) | [254] |
| 6q25 | SLC22A3 | Cation transporter; removal of drugs/toxins | 5.5x10-10 (11,338) | [4] |
| 7p15 | JAZF1 | Zinc finger domain with nuclear localization | 2.14x10-6 (9,893) | [257] |
| 7q21 | LMTK2 | Endosomal membrane trafficking | 1.1x10-9 (11,338) | [255] |
| 8p21 | NKX3-1 | Homeodomain; tumor suppressor | 3.4x10-30 (38,747) | [252] |
| 8q24 | c-Myc | Transcription factor; oncogene | 1.1x10-12-6.4x10-18 | [101,106-112] |
| 10q11 | MSMB | Immunoglobulin binding factor | 8.7x10-29 (11,338)* | [255,257] |
| 10q26 | CTBP2 | Anti-apoptotic factor | 2.7x10-8 (9,893) | [257] |
| 11p15 | None | N/A | 2.7x10-33(42,388) | [254] |
| 11q13 | None | N/A | 6.7x10-12 (14,400) | [256-258] |
| 17q12 | HNF1B | Transcription factor | 5x10-20 (16,993) | [103,259] |
| 17q24 | None | N/A | 2.5x10-10 (17,837) | [9] |
| 19q13 | KLK2; KLK3 | Serine proteases | 1.6x10-11 (60,371) | [256] |
| 22q13.1 | TNRC6B | Gene silencing by miRNA, siRNA | 4.96x10-7 (11,955) | [260] |
| 22q13.2 | BIK | Induction of apoptosis | 5.9x10-29 (42,388) | [254] |
| Xp11.22 | NUDT10, NUDT11 | Trafficking, stress response, DNA repair, apoptosis | 3.95x10-13 (38,933) | [252] |
N/A = not applicable;
For situations where two groups discovered the locus, p value and n are given for the first published study.
The 17q12 locus contains the protein-coding gene, Hepatocyte Nuclear Factor 1B (HNF1B) or Transcription Factor 2 (TCF2). Two common SNP hotspots are actually located within this gene, making it of great interest to PCa research [101]. HNF1B has been studied extensively not only for its role in gene regulation in the liver including cholesterol, bile acid and lipoprotein metabolism, but also for its involvement in the progression of Maturity Onset Diabetes of the Young (MODY) [102,103]. This form of diabetes is similar to what we now call “Type 2” diabetes, but it differs in that it results from a simple mutation in one of the MODY genes, also known as the HNF1 genes. Within the general population of type 2 diabetics, a subset carry SNP rs757210 which is located in the HNF1B gene. Interestingly, the HNF1B risk allele (or SNP allele) for PCa was associated with protection against type 2 diabetes - and likewise, the risk allele for type 2 diabetes is associated with decreased risk for PCa [104,105]. The two alleles do not segregate purely randomly in the population since they are not found together in the same patient- a phenomenon referred to as linkage disequilibrium. The mechanism by which these SNPs contribute to both type 2 diabetes and PCa is not understood. HNF1B/TCF2/MODY5 contains a homeobox domain and is classified as a beta helix-loop-helix transcription factor. Known targets of this transcription factor are not obvious tumor suppressors, but the link to liver function is interesting from the viewpoint of type 2 diabetes progression. Clearly, in depth mechanistic studies are required to determine whether this gene has a role to play in the development of PCa, but the idea of a cancer-protective SNP allele is very interesting indeed.
Some SNP alleles are not associated with identifiable genes, as is the case for the 8q24 region. Studies indicate that there are up to seven distinct loci within 8q24 in which SNP alleles are correlated with up to a 50% increased risk of PCa [101,106-112]. 8q24 was first identified as an important region in familial PCa in 2006, and since then it has been linked with increased risk for other cancers such as colorectal, breast, bladder and ovarian cancer [113-115]. Interestingly, the region is over 1 Mb in length and yet it contains no canonical protein-coding genes. Recent studies have identified enhancer elements within 8q24 which are conserved in primates and canine species [116,117]. In an elegant study by Sotelo and colleagues, seven enhancers were identified which serve as functional transcriptional regulators in PCa cells in vitro [117]. When each enhancer was combined with the c-Myc promoter and a luciferase reporter in LNCaP, PC-3 and the CW620 colorectal cancer line, significant reporter expression was observed. The well-known proto-oncogene c-Myc is located 200 Kb upstream of 8q24 and has been proposed to be regulated by elements in this region in other cancers, however, this is the first study to link the enhancers in the region to gene expression in vitro specifically in PCa cells [117,118]. Further study by Ahmadeyeh, et al., showed that chromosomal looping leads to a direct interaction between PCa-associated SNP alleles and the c-Myc promoter [119]. Not surprisingly, given that misregulation of c-Myc is damaging to the cell, this region is heavily regulated at the epigenetic level by the Polycomb repressive complex (PRC; discussed in “Epigenetic regulation of PCa” [116]). The most important question at this time is whether or not these SNP alleles change c-Myc expression in vivo. Currently, there are conflicting reports on this issue. For rs1447295, for example, one study indicates that no difference in expression of c-Myc in the prostate exists between those carrying the normal and risk-associated allele, while other studies suggest the opposite [120-122]. Discrepancies are likely attributed to small sample size, contamination of normal tissue by small tumor islands (and vice versa) or the method by which c-Myc expression was assayed.
The racial disparity in PCa incidence, mortality and severity between African Americans (AAs) and Caucasians Americans (CAs) has been documented extensively [123]. It is clear from many large studies that AAs are at a higher risk for more severe PCa. Research on the various alleles of 8q24 SNPs may provide some insight into this discrepancy ([124], for example). A meta-analysis conducted by Troutman, et al., confirms that throughout the literature, a subset of risk-associated SNPs at 8q24 display differing patterns of risk association based on race [125]. For example, two alleles of SNP rs6983267, which is associated with enhancer E at 8q24, display differing phenotypes when overexpressed in the c-Myc promoter-reporter assay described above [117]. The T allele of SNP rs6983267 results in very strong reporter expression, while the G allele drives far weaker expression (although luciferase expression is detected). It is well known that low c-Myc expression is associated with increased cell proliferation while high c-Myc expression causes apoptosis and senescence, so it follows that the G allele is linked to higher PCa risk [117]. It is likely not a coincidence that rs6983267 falls in a consensus TCF transcription factor binding site and future studies on which TCF proteins bind at this site to regulate c-Myc will be very interesting. Remarkably, the G allele is found far more often in the AA population than in EWs and AWs (reviewed in [125]). It should be mentioned that sample size is very important for studies such as these since the association between 8q24/rs6983267 and AA PCa was not confirmed in studies using smaller sample sizes [126]. Unquestionably, these data are merely the first step in what could become an important biomarker in PCa racial disparity, but many further and more detailed studies in vivo are needed.
In conclusion, SNP analyses are an invaluable tool with which to identify new genomic biomarkers for PCa. Perhaps the best SNP marker that has been discovered so far for PCa are those included in the 8q24 locus. Over 100 studies have confirmed the linkage between SNP alleles at 8q24 and PCa risk, however, research has not elucidated the exact mechanism by which these SNPs impart this risk. A genetic test for 8q24 SNPs is likely in development at this time, although not yet published. One important discussion topic must be: Of what utility is this knowledge for a healthy individual? Similar to the Breast Cancer-1 and -2 (BRCA-1, 2) mutation which bestows greater risk for breast cancer in women, there are many possible outcomes for a patient who receives a positive test. Certainly, prophylactic prostatectomy is not something a person should enter into without careful consideration given the morbidities associated with the procedure. More research is needed to determine the long-term viability of 8q24 as a PCa risk predictor.
Gene fusion
Chromosomal aberrations resulting in gene fusions have a long history in cancer research. One of the most well-known is the Philadelphia Chromosome-so named because of its discovery at two of Philadelphia’s leading research institutes (Fox Chase Cancer Center and the University of Pennsylvania). In this situation, a reciprocal translocation between chromosomes 9 and 2 leads to the fusion of two genes which results in one fused transcript: the Breakpoint cluster region (BCR) gene and the V-abl Abelson murine leukemia viral oncogene homolog 1 (Abl1) gene. BCR-Abl has been extensively studied in the myelogenous and lymphoblastic leukemias (CML and CLL), but it is not often found outside of the leukemia family of cancers. Drugs directed against the BCR-Abl gene fusion product are extremely successful in reducing mortality from childhood CML (see [127-129] for reviews on BCR-Abl in leukemia).
Perhaps one of the most well-known PCa biomarkers is a gene fusion marker. The TMPRSS2-ETS gene fusion results from deletion or translocation on chromosome 21 which most commonly joins two independent genes: Transmembrane protease serine 2 (TMPRSS2) and a member of the v-ets erythroblastosis virus E26 oncogene family of transcription factors (ETS family). The discovery was initially made by reanalyzing published microarray data using the algorithm Cancer Outlier Profile Analysis (COPA). COPA was designed to identify genes that are extremely highly expressed in only a subset of cancer patients [130]. The first TMPRSS2 fusions that were identified involved the ETS-related gene (ERG) and the ETS variant 1 gene (ETV1). Subsequently, fusions between TMPRSS2 and ETS variant 4 and 5 (ETV4 and ETV5, respectively) were found [131,132]. TMPRSS2-ETS gene fusions can arise from either a deletion or a translocation which join the region between the 5’ Untranslated Region (UTR) of TMPRSS2 and protein-coding exons of an ETS family member (Figure 3). Normally, the ETS family members are not androgen-regulated genes and are expressed at modest levels in the prostate. Multiple fusions can be found within one patient and multiple types of TMPRSS2-ETS fusion (deletion, translocation with different breakpoints) can potentially be found in one prostate; however, multiple fusions are never found within one tumor focus [133,134]. Regardless of the etiology of the fusion (deletion or translocation and which exons are retained of the ETS family member), androgen-dependent overexpression of the ETS gene is observed [130].
Figure 3.

TMPRSS2-ETS gene fusion. Panel A illustrates the positions of TMPRSS2 and ERG in the human genome. The two genes are situated 2.8 Mb apart, with 36 predicted and coding genes in between. Deletion or translocation results in the fusion of the genes, as illustrated. Panel B displays the formation of the most common (30%) TE fusion product: fusion between TMPRSS2 exon 1 and ERG exons 4-11. Arrows indicate the start of translation in the normal mRNA. Since the canonical start is deleted in TE fusion of this type, an internal ATG is presumed to be the new translation initiation site. The table to the right shows the most commonly-found TMPRSS2-ETS family gene fusions [251].
TMPRSS2-ETS gene fusions have been studied in great detail over the last 7 years. The most common fusion, TMPRSS2-ERG, has been detected in anywhere between 40-70% of patients depending on the sample size, method of detection or racial bias amongst the sample [Please see [135-137] for excellent reviews of this literature]. Detection of TMPRSS2-ERG fusion (referred to hereafter as TE fusion) techniques range from standard Immunohistochemistry (IHC) to Fluorescence in situ Hybridization (FISH) or quantitative or standard RT-PCR (qRT-PCR or RT-PCR) using primers which capture one or more of the most common fusion transcripts. Within the family of TE aberrations, fusion can occur via deletion or translocation between TM exon 1 or 2 and ERG exons 2, 3, or 4. The resulting overexpression of ERG has been postulated to have significant impacts on prognosis for PCa patients; however, clinical data does not strongly support this theory. In fact, the literature is very controversial on this topic. Studies have indicated that TE fusion presence may help to predict relapse rate after radical prostatectomy to treat primary PCa. In a rather small cohort, Nam, et al., showed that TE fusion was present in almost 50% of PCa samples and of those, 47% experienced a rise in PSA post-surgery (indicating relapse) whereas only 7% of TE fusion-free patients relapsed [138]. Consistent with this, Guo, et al., showed that if the largest (index) focus of PCa present in a patient was TE fusion positive, then subsequent metastases were positive; a phenomenon which suggests that TE fusion status is correlated with metastasis [139]. Conversely, another small study by Saramaki, et al., indicated that the presence of TE fusion was a predictor of favorable prognosis and was not associated with a higher GS [140]. The complexity of this issue is highlighted by the presence of over 50 published studies on whether TE fusion status is a predictor of good or bad prognosis [135].
To make matters more complex, there appears to be a racial bias in the prevalence, type and etiology of TE fusions. Tissue microarray analysis by FISH indicates that TE fusion is less common in AA and Japanese Americans (JAs) than in CAs. Interestingly, the same study found that TE fusions in AA PCa’s arise more often from deletion than translocation [141]. It is not clear at this time whether or not the type of aberration which causes TE fusion is an important indicator of disease severity. A further study indicated that a SNP in the TMPRSS2 gene may predict future fusion events and downstream PCa diagnosis in CA, but this SNP was not associated with increased PCa risk in AA [142]. Taken together, these data imply that AAs have less TE fusions, and also that TE gene status is not concordant with risk for poor prognosis in AAs.
The impact of gross overexpression of ETS family members is an ongoing topic of research. ETS transcription factors are involved in the regulation of a wide range of cellular activities including cell migration, aging, apoptosis, angiogenesis and many others. Overexpression of ETS genes are known to cause cellular transformation (reviewed in [143-145]). In support of this, fibroblasts overexpressing ETS1 and ETS2 are highly tumorigenic in mice [146-148]. In the prostate, upregulation of certain ETS genes is linked to advanced metastatic disease [149]. Indeed, ERG is overexpressed in a staggeringly high number of PCa specimens, irrespective of the presence or absence of TE fusion [149].
As a biomarker, TE fusion is a useful diagnostic-but only for the 40-70% of patients that are positive for the fusion. Since TE fusion is never detected in normal, non-transformed cells, its presence is a clear indicator of PCa. In fact, Tomlins and colleagues recently conducted a study to determine the feasibility of using a urine test for the detection of TE fusion in men after DRE [150]. This work used urine voided after DRE from men presenting for prostate biopsy or radical prostatectomy to detect either TE fusion or PCA3 mRNA. As discussed previously in the ncRNA section of this review, PCA3 is an excellent diagnostic PCa biomarker with many of the same characteristics as TE fusion. The purpose of this study was to use both markers as a way to enhance early diagnosis of PCa to determine a need for invasive biopsy. The presence of TE fusion (and therefore overexpression of ERG) in urine was significantly associated with PCa diagnosis and severity, and the addition of PCA3 measurement gave the test more statistical power [150].
Given the high number of contradictory studies in the TMPRSS2-ETS fusion literature, definitive studies with large sample sizes and a standardized method of quantitation using all of the known fusion types are required. Until this study is completed, the utility of TE fusion as a biomarker is still under investigation. If the true frequency of TM-ETS fusions is as high as 70%, its utility as an initial diagnostic is clear. If it is a true indicator of poor prognosis or metastatic potential, even in a small subset of patients, it will be a useful tool in determining treatment paradigms for those that carry the aberration. Future follow up studies on these issues will be very important for the field of early diagnosis of PCa.
The genomics of PTEN and AR
Several critical tumor suppressors and oncogenes have been shown to be highly mutated in PCa. First, Phosphatase and tensin homolog (PTEN) is reported to be deleted, mutated or rearranged in 60-80% of PCas [151], although the literature is very conflicted on this topic. PTEN is also known as Mutated in multiple advanced cancers (MMAC; [152]) due to the prevalence of mutations in this gene in brain, breast, endometrium, kidney and prostate cancers [153]. Second, AR is the most mutated steroid receptor in PCa. Over 660 mutations have been catalogued in the AR gene to date, many of which are found in advanced PCa [154].
PTEN mutation and deletion
Investigation into the tumor suppressive activity of PTEN and its role in cell proliferation and migration has been an active topic of research for over a decade. PTEN can theoretically dephosphorylate proteins, but work by Myers, et al., showed that a mutation inactivating the protein phosphatase activity while retaining lipid phosphatase activity preserves the tumor suppressor function of the gene [155]. PTEN primarily dephosphorylates phosphatidylinositol 3,4,5-triphosphate (PIP3), an important regulator of cell proliferation and the product of active Phosphoinositide 3-kinase (PI3K). Indeed, stimulation of PIP3 signaling cascades mediated by PI3K results in increased proliferation, while activation of PTEN results in growth suppression (reviewed in [156]). Additionally, the presence of PTEN expression has been linked with cell cycle arrest, apoptosis and senescence [157-159]. In accordance with the classic characteristics of tumor suppressors, PTEN negatively regulates growth and is also commonly mutated in cancers. In fact, many types of mutations in a staggeringly high number of cancers have been associated with PTEN. Rearrangements and DNA base changes such as homozygous deletion, frame shift, nonsense, missense and splicing variants causing early termination are common in this gene [151]. Exactly how common mutations are in PTEN is an area of ongoing research. Germline (inherited) mutations in PTEN cause the problematic condition known as Cowden’s disease (or multiple hamartoma syndrome), in which patients develop many benign neoplasms called hamartomas throughout the body. Generally, these neoplasms cause no harm unless they grow in an organ where there is limited space like the spleen, kidney or hypothalamus. Later in life, however, once the natural aging process results in disruption of normal gene regulation and the accumulation of mutations, Cowden’s disease patients are at a far greater risk for developing cancer [160].
Despite the available contradictory literature, it is clear that regardless of the type of mutation, a correlation exists between PTEN mutation and PCa severity. The seminal work on PTEN gene loss in PCa was published by Gray and colleagues in 1995 and 1998 [161,162]. These studies established that the genomic region containing PTEN, 10q23.3, is deleted in 70% of PCas, and that loss of, or mutation in PTEN is responsible for the tumor suppressor activity of the region. Simultaneous with this work, another group determined that PTEN/MMAC was responsible for the tumor suppressor activity of 10q23.3 [152]. Since then, many groups have published analyses of the sequence of PTEN in PCa patients of various stages, grades and patient ethnicity. Vlietstra, et al., determined that 70% of PCa samples contained PTEN aberration [163]. However, Gray, et al., found that only 13% of tumors contained PTEN mutation [161]. Further, in a small study of PCa patients, PTEN mutations were found in 12% of cancers with a clear bias towards higher GS [164]. These discrepancies are explained by the study design in many cases. For example, Gray, et al., found 10q23.3 deletion at the level of either homozygous deletion, loss of heterozygosity or other, in up to 70% of patients using fluorescence-based allelotyping [165]. This technique will detect deletions (homozygous or heterozygous) in addition to DNA base changes. Later work by the same group employed direct sequencing to analyze mutations in the coding sequence [161]. Clearly, this method would not detect deletions (which would yield no sequence information and therefore be considered “inconclusive”) or mutations in the lengthy promoter sequence. Many publications are available which catalog the varying types of mutations in PTEN and correlate them with disease stage and metastasis potential, most of which fall victim to the same problem: Deletions are not detected by the chosen analysis method. Despite these pitfalls, one unifying theme is present: Mutation in PTEN is correlated with GS and metastatic potential. It is also formally possible that there is another tumor suppressor at 10q23.3 which contributes to the disparity between studies. Clearly, a definitive study is needed where a large number of patients including cohorts of low, medium and high GS are analyzed for PTEN coding mutations, promoter mutations and homozygous deletion. Until then, PTEN is not an ideal biomarker for initial PCa diagnosis given its low percentage of penetrance in the PCa population.
Genomic aberrations in AR
AR is highly mutated in advanced PCa, but rarely in localized primary tumors in CAs and europeans. In normal prostate epithelium, AR is absolutely required for cell proliferation and differentiation. A classic nuclear hormone receptor, AR binding to DHT causes translocation to the nucleus where it has been shown to partner with a multitude of cofactors before binding to DNA to activate gene transcription. Genomic amplification of the entire AR gene is a well-documented phenomenon that occurs in up to 30% of advanced PCa’s [166]. Somatic mutations are not common in localized primary tumors. In fact, less than 2% of primary PCa patients display AR mutation [167]. However, in AAs, up to 8% of primary PCa patients present with AR mutation [168]. The story is much different in advanced PCa. The relationship between AR signaling and PCa progression is clear: Blockade of the receptor or androgen depletion results in tumor shrinkage and cell death at early stages, but as the disease advances in stage, it becomes hormone refractory (HR) and deprivation is no longer able to arrest growth and metastasis. Simultaneous with the loss of androgen sensitivity, AR mutations are detected with much higher frequency. These mutations are most often gain-of-function changes, which allow the cell to bypass the scarcity of androgens during PCa therapy [154]. Base changes in the ligand binding domain are most commonly found, representing 49% of AR mutations in advanced PCa, while nuclear translocation domain mutations and DNA binding domain mutations represent 37% and 7% of the total, respectively. These data are publicly available at McGill University’s Androgen Receptor Mutations Database (http://androgendb.mcgill.ca/; [169]). Similar to the situation with the PTEN gene, studies detailing AR mutations in advanced PCa are commonplace. Unfortunately, this literature is also flawed by a lack of reproducibility and significant differences in analysis method and patient details such as disease stage, race, age, previous treatment, etc.
Significant controversy has surrounded the topic of trinucleotide repeat tract length in the AR gene. Within AR, two tracts of codon repeats exist, both of which are located in exon 1: polyglutamine is most proximal to the start site and poly-glycine is more distal (for a review on this phenomenon, see [170]). Given the importance of poly-glutamine repeats in neurodegenerative disease, there are far more studies published on this tract than on the poly-glycine tract. Racial disparity has been found in the number of CAG codon repeats, with CAs generally displaying more repeats than AAs [171]. In PCa, reduction in the number of glutamine codons in the polyglutamine (polyQ) tract has been correlated with increased PCa incidence and severity [172-174]. Shorter polyQ tracts were also associated with a likelihood of presenting with more advanced disease [172]. Further, some studies indicate that reduced polyQ tract length is associated with increased AR transactivation [175]. Conversely, longer polyQ regions render AR inefficient at binding coactivators [176]. It follows, then, that a causal relationship may exist between shorter polyQ tracts, and consequently more active AR, in AA patients and their propensity for more severe PCa. This theory is very attractive, as is polyQ as a biomarker for increased PCa severity. However, studies exist which do not find a significant correlation between codon repeat length and disease progression [177,178]. Moreover, the variations observed in polyQ tract length in PCa patient populations are not significantly different from those found in the general population [179]. Recently, studies with large cohorts of patient samples (normal and PCa) have disqualified any significance for polyQ tract length in PCa [180,181].
Specific inherited base changes in AR are linked with greater PCa risk in AAs. In many cases, germline mutation in AR results in androgen insensitivity syndrome (AIS). AIS is an inherited condition characterized by abnormal male genital development and a lack of secondary sex characteristic differentiation at puberty [170]. One particular base change, A1675T has been associated with early-onset PCa in AA families with a strong history of PCa. This change is a missense mutation, resulting in a T to S substitution in the DNA binding domain of AR [182]. Future studies will determine not only the mechanism by which this mutation leads to early-onset PCa, but also how penetrant this inherited mutation is in the AA population.
The discussed aberrations in the AR and PTEN genes are controversial due to the conflicting reports that can be found correlating each one with PCa risk and severity. It should be noted that the large GWAS that have been conducted on thousands of PCa patient samples compared with normal samples have failed to identify SNP hotspots in either AR or PTEN. Perhaps this is due to the fact that most PTEN aberrations are gene deletion, which wouldn’t be identified by SNP arrays. Given the high number of AR base changes that have been documented, the exclusion of the AR locus at X.q.11-12 in SNP array data is surprising, but may be explained by the experimental parameters.
Research into the genomics of PCa have not yielded the “perfect” biomarker as of yet. However, some very important markers of disease progression and severity have been discovered. Following the current trend towards personalized medicine, a diagnostic test for SNPs at 8q24, TE fusion, AR and PTEN mutation may be incredibly useful in determining treatment paradigms on an individual basis. The development of an inexpensive “panel” of tests for these markers would be very useful for this purpose.
Transcriptomics
The human transcriptome refers to the complete set of RNAs produced from the human genome at any given time. The products of gene transcription can be of use in cellular processes which are no longer limited to structural elements and protein translation. RNAs come in all shapes and sizes and their synthesis and destruction is extremely dynamic, unlike the relatively static genome. Some RNAs, such as microRNA (miRNA) and the previously discussed lncRNA are extremely influential at the level of protein or gene expression (respectively) but do not code for proteins. Even though most cells contain the same exact genome, transcriptome profiles can be completely different from cell-to-cell and account for the many varied shapes, sizes and functions of cells. Widespread and extensive gene expression changes are seen in cancer versus normal cells, a phenomenon which has been extensively investigated in cancer research. In PCa specifically, a growing number of studies have begun to elucidate the most common pathways that are up- and downregulated in PCa versus normal prostate epithelium and in advanced metastatic PCa compared to primary PCa.
RNA expression profiling by DNA microarray: challenges
One of the major challenges of transcriptome research in all fields has been the necessity for large quantities of RNA. Traditionally, most techniques required at least 5 μg of intact RNA to achieve robust expression analysis and profiles. In addition, retaining an undisturbed polyA tail was critical for cDNA synthesis since the universal primer used to complete the reaction binds specifically to this tail (oligo dT). Unfortunately, the use of an oligo dT primer for mRNA-to-cDNA library construction resulted in a clear 3’ bias in the resulting pool of cDNAs. The incorporation of random hexamers as the universal primer for cDNA synthesis has been a significant improvement on this technology, removing the issue of 3’ bias. Expanding on these improvements, whole-transcriptome amplification (WTA) techniques which boast a near zero bias and the need for miniscule amounts of input RNA are now common. RNA isolations from formalin fixed paraffin embedded (FFPE) tissues, flow cytometry isolates, laser microdissection outputs and the like, which generally yield a few nanograms of total RNA are no longer problematic. These new RNA amplification techniques are either based on PCR which creates exponential amplification of the template, or T7 polymerase-assisted linear amplification. Although both methods have substantial drawbacks and strengths, many studies have shown them to be an acceptable way to analyze small amounts of RNA from precious and even partially degraded clinical samples [183-187]. In the end, RNA handling and quality is perhaps most critical to achieving robust, reproducible results and data which are comparable across many patient samples.
Studies which interrogate patient samples isolated using differing methods and from varying sources can result in significantly divergent datasets. These differences are explained in most cases by modifications in sample preparation technique, RNA source and microarray platform. For most primary tumors, pathologist-verified tumor tissue will never approach 100% malignant cells with no contaminating normal stroma and epithelium. Using a technically difficult and time consuming approach, some groups have used laser microdissection (LMD) or laser capture microdissection (LCM) to remove unwanted cell types that may be associated with the cancerous glands. The major benefit of this approach is that there will be very little contamination from other cell types, however, this technique can introduce laser-induced tissue damage. Yet another sample preparation technique involves removal of all contaminating cell types by culturing explants of PCa tumors in epithelial growth medium for several weeks before extracting RNA from the surviving primary cell culture. Clearly there are benefits and drawbacks of this technique: ex vivo culture can result in global changes in gene expression within a short period of time. Many groups have attempted to preserve the in vivo nature of the tissue, choosing not to culture the tissue for any length of time and to simply use gross dissection to remove uninvolved areas before RNA and cDNA preparation. Samples treated in this way are often contaminated with normal prostate epithelium, stroma and endothelium. It is important to note that DNA microarray data must be interpreted in the context of the experiment at hand. For example, Chandran, et al., note that four common stroma-associated markers are listed as strongly downregulated in metastatic PCa samples [188]. Most likely, this phenomenon is attributed to contaminating stromal cells within the primary prostate or normal prostate comparison samples since no stromal cells are found in metastatic PCa tumors. Artifacts such as these must be taken into consideration when interpreting the data. Aside from the sample preparation technique, DNA microarray technology has become very diverse and complex with arrays containing anywhere from ten of thousands to hundreds of thousands of DNAs. The amount of redundancy at the level of repetitive oligos and at the level of multiple oligos per gene varies between manufacturers as does the cDNA labeling, detection and analysis package for each platform.
Using DNA microarray paired with advanced RNA amplification techniques, whole transcriptome analysis has been used to detect gene expression changes in prostate tumors as compared to paired normal samples. The general flow of such an experiment is as follows: RNA isolation from tumor and normal samples followed by RNA amplification and cDNA synthesis, hybridization to DNA microarray, data analysis and finally, validation of these data by northern blot, qRT-PCR. In some cases, the primary literature or follow up studies include protein analysis by either western blot or immunohistochemistry. A plethora of publications can be found that follow this paradigm and generate large datasets which are often publicly available [188-202] In addition, a sub-field of meta-analyses exists where these data are interrogated using new statistical techniques [194-196]. A review of this literature can be daunting since each study was conducted using different experimental parameters, samples, array platforms and statistical analyses. We have classified these data into three groups. First, there are a number of genes which were found to be up- or downregulated in PCa compared to normal samples in multiple (or even all) studies examined (Table 2). Second, several genes were highlighted in only one particular study-presumably due to the experimental parameters. These genes are not of lesser value, but are simply different from the more commonly identified genes (Table 3). Third, some genes were identified as robustly overexpressed particularly in metastatic castration-resistant PCa (MCRPCa) (Table 4). Examples in each of these three groups will be discussed in the text and tables hereafter.
Table 2.
Genes showing aberrant expression in primary and/or advanced PCa compared to normal prostate. The selected genes are not all-inclusive, rather, they represent genes that were found in multiple DNA microarray or meta-analysis studies
| Pathway or cellular function | Gene name | Gene symbol | Expression | REF |
|---|---|---|---|---|
| Cell cycle, cell proliferation | cellular-Myelocytomatosis viral oncogene | C-Myc | Up | [188,190,193,261] |
| Cyclin dependent kinase 1, 10 | CDK1, 10 | Up | ||
| DNA topoisomerase 2A | TOP2A | Up | ||
| Myb-related protein B | MYBL2 | Up | ||
| Androgen receptor | AR | Up | ||
| Estrogen receptor 1 | ESR1 | Up | ||
| Extracellular matrix, cell migration, cell adhesion | Tumor-associated calcium signal transducer 1 | TACSTD1 | Down | [188,191,192,197] |
| Cadherin-1 | CDH-1 | Down | ||
| Hyaluronan-mediated motility receptor | HMMR | Down | ||
| Metalloproteinase inhibitor 3 | TIMP3 | Up | ||
| Matric metalloprotease 9 | MMP9 | Up | ||
| Fatty acid, androgen, other metabolism | Alpha-methylacyl-CoA racemase | AMACR | Up | [195,200,201] |
| Fatty acid synthase | FASN | Up | ||
| Ornithine decarboxylase | ODC | Up | ||
| Signal transduction, transcription | Transcription factor E2F1 | E2F1 | Up | [190,194,196,198] |
| Type I inositol-3,4-bisphosphate 4-phosphatase | INPP4A | Up | ||
| Mitogen activated protein kinase | MAPK | Up | ||
| Histone-lysine N-methyltransferase | EZH2 | Up | ||
| c-Fos | FOS | Up | ||
| Forkhead box protein M1 | FOXM1 | Up | ||
| Ubiquitin carboxyl-terminal hydrolase 13 | USP-13 | Up | ||
| Transcription factor jun-B | JUNB | Up | ||
| Nuclear factor-κB | NFκB | Up | ||
| Signal transducers & activators of transcription 6 | STAT6 | Up | ||
| Homeobox protein NKX3.1 | NKX3.1 | Up | ||
| Function unknown | Hepsin | HPN | Up | [188,190,191,193,196,200] |
Table 3.
Potentially important genes upregulated in primary PCa which were reported once
| Gene name | Gene symbol | Function in PCa | REF |
|---|---|---|---|
| Serotonin receptor 2B | 5HTR2B | unknown | [192] |
| Histone 2A | H2AX | DSB repair | [189] |
| Macrophage inhibitory cytokine | MIC-1 | pro-survival | [200] |
| Mucin 1 | MUC-1 | Pro-migration/invasion | [191] |
| Platelet-derived growth factor receptor β | PDGFRβ | Pro-proliferation/migration | [199] |
| Proto-oncogene serine/threonine-protein kinase | PIM-1 | Proto-oncogene | [198] |
Table 4.
Selected genes with aberrant expression in metastatic castrate-resistant PCa versus primary PCa
| Gene name | Gene symbol | Expression | REF |
|---|---|---|---|
| Aldo-keto reductase family 1, member C3 | AKR1C3 | Up | [201] |
| Androgen receptor | AR | Up | [188,198,201] |
| Annexin A11 | ANXA11 | Down | [198] |
| Aurora A kinase | STK15 | Up | [190] |
| Autocrine motility factor receptor, isoform 2 | AMFR | Up | [197] |
| Eukaryotic translation initiation factor 1A | E1F1AX | Up | [198] |
| c-Fos | FOS | Down | [188] |
| Histone-lysine N-methyltransferase | EZH2 | Up | [201] |
| Hydroxy-delta-5-steroid dehydrogenase | HSD3B2 | Up | [201] |
| Transcription factor JunB | JUNB | Down | [188] |
| Krueppel-like factor 5 | KLF6 | Down | [201] |
| Matrix metalloprotease 9 | MMP9 | Up | [201] |
| Metastasis-associated 1 | MTA-1 | Up | [198,202] |
| Mucin 1 | MUC-1 | Up | [191] |
| Myb-related protein B | MYBL2 | Up | [190,191] |
| Steroid 5-alpha reductase 1 | SRD5A1 | Up | [201] |
| WNT5A | WNT5A | Up | [201] |
| WNT5B | WNT5B | Down | [198] |
Group 1: frequently reported genes in PCa transcriptomics research
A number of transcripts have been documented by multiple groups as aberrantly expressed in primary PCa. A portion of these gene products overlap with those found in comparison with MCRPCa samples. These genes are members of commonly studied cellular functions or signaling pathways and are listed in Table 2. Many have been studied (previously or since the pioneering microarray study) for their role in PCa in particular, or in other cancers. Genes such as c-Myc, AR, EZH2 and cadherins have been discussed in this review in other sections. It is no surprise that genes involved in promoting cell cycle progression and cell proliferation are upregulated in PCa. Likewise, extracellular matrix (ECM)-degrading proteins and those which play a prominent role in epithelial-to-mesenchymal transition (EMT) are obvious candidates for upregulation during PCa progression. Signal transduction proteins and transcription factors are clear mechanisms whereby large groups of genes can be altered simultaneously to effect cellular transformation and disease progression. Cellular energy production including fatty acid metabolism has been an active topic of PCa research for decades. The increased demand for energy in tumor cells is met by upregulation of many genes involved in fatty acid metabolism including the multi-enzyme protein, fatty acid synthase (FASN) [203]. In addition to the well-studied genes and pathways described, genes with an unknown link to PCa have been discovered by this method. For instance, almost every DNA microarray study and meta-analysis on prostate samples to date has uncovered overexpression of the hepsin (HPN) gene.
HPN was detected in transcriptome expression analysis comparing primary PCa with normal prostate or BPH samples in a remarkable number of studies to date [188,190,191,193,196,200]. Located at 19q11-13.2, many SNPs have been associated with HPN, however, the data do not clearly indicate whether or not specific SNP alleles lead to greater PCa risk or severity. Racial bias may contribute to the contradiction noted amongst these studies. For example, a study within the Korean population suggested that SNP allele rs1688030 was not associated with greater PCa risk, while another study indicated that rs1688030 did impart greater risk amongst Americans of European descent [204,205]. Similarly conflicting, Pal, et al., and Kim, et al., found increased risk associated with rs2305745 and rs2305747 in Europeans and Koreans, but Holt, et al., did not associate rs2305747 with increased risk in CAs or AAs [206,207]. It seems that a definitive consensus on whether or not SNPs in HPN confer a greater risk of PCa development and progression has yet to be determined, but it is clear that HPN overexpression is specifically associated with PCa.
HPN expression is strongly associated with PCa at various stages during the natural history of PCa-from the pre-cancerous level of highgrade prostatic intraepithelial neoplasms (HGPIN) to the moderate-grade of localized PCa (reviewed in [208]). First documented in the prostate by Dhanasekaran, et al., HPN overexpression is detected at the RNA and protein levels from low to moderate-grade PCa, not at all in normal prostate, and at very low levels in MCRPCa [198]. These data have largely been confirmed by further studies, however some contradictory data exist which suggest continued overexpression of HPN in MCRPCa [209,210]. Additional work has characterized HPN expression in prostate and other cancers with similar results; HPN upregulation as compared to normal tissues was found in ovarian, breast and kidney cancer [211-213].
The function of HPN in PCa remains unclear. The gene encodes a transmembrane serine protease with high homology to the pancreatic serine protease, trypsin [214]. HPN has been proposed to play a critical role in differentiation, since inhibition of expression and function with antisense oligonucleotides and antibodies, respectively, resulted in decreased proliferation and a loss of characteristic cell morphology in hepatoblastoma cells [215]. Other cell-surface proteases clearly play a role in cell migration by literally clearing a path through ECM. Transgenic mouse overexpression of HPN in the prostate indicates that too much HPN activity in the epithelium results in weakening of the adhesion between stroma and epithelium [216]. It is clear that HPN’s role in cell morphology and growth is much more complex, although a detailed mechanism for how this protein contributes to PCa progression is still unknown.
A number of intriguing studies have investigated HPN’s potential as a PCa biomarker and imaging agent. First, Kelly, et al., employed phage display technology to identify peptides that bind with high affinity to HPN in vivo. Using iterative rounds of phage display, they have characterized a number of labeled peptides which bind to LNCaP xenografts, and they are currently looking to expand these studies to the clinic with the long-term goal of improving live imaging of the prostate [217]. Further, a study examining prostate biopsies for HPN protein expression by IHC found that HPN expression was clearly associated with a cancer diagnosis. Of the 90 samples that were queried, 100% of the PCa samples were positive for HPN while only 11% of BPH samples were positive. Interestingly, the 11% of BPH samples showing HPN expression contained regions of HGPIN [218]. These and other data highly suggest that HPN is a promising PCa biomarker.
Group 2: potentially important genes which are reported once
Of the genes that were highlighted in only one PCa transcriptomics study to date, many have been characterized further in follow up studies, or even studied previously in a different context. For example, the mucin genes have a long history in cancer research. Mucin proteins line epithelial surfaces and their overexpression has been documented in lung, breast, ovarian, pancreatic, colon and now prostate cancers (for a review, see [219]). Similarly, metastasis-associated protein-1 (MTA-1) has been widely studied for its role in PCa. MTA-1 expression is highest in MCRPCa [220]. Recent work has suggested that MTA-1 overexpression leads to more invasive and migratory behaviors in cultured cells, as well as increased expression of VEGF, a fact which may suggest that MTA-1 plays a role in vascularization of the progressing tumor [221].
The histone 2A gene, H2AX, was identified by Nanni and colleagues in their ex vivo culture and DNA microarray study. In this work, one of the H2AX was highly underexpressed in recurrent but localized PCa cultures when compared to normal prostate epithelial cultures [189]. H2AX encodes one of the H2A histone family proteins which make up about 10% of H2A histones in the nucleosome of human fibroblasts (reviewed in [222]). Interestingly, H2AX is phosphorylated in situations where DNA double-strand breaks (DSB) occur, and this modification is thought to open the chromatin to allow for the repair machinery to access the DNA damage. Downregulation of this key damage repair sensor could be indicative of a more global phenomenon whereby DNA damage genes are transcriptionally silenced in advanced PCa-a concept which has been documented by other groups [223,224]. In addition, Nanni, et al., showed that phospho-H2AX, also known as γ-H2AX, is detected at high levels in primary PCa while displaying little-to-no detection in advanced PCas. A paradox exists in relation to DSB: Many groups have documented that they are cancer-causing and still others note that they are an effective anti-cancer treatment since DSB accumulation triggers apoptosis. For this treatment paradigm to be effective, however, the DSB sensing pathway must be paralyzed, a situation which seems to occur naturally as PCa advances. An intriguing study suggests that, in fact, the DSB sensory pathway may be an excellent target for drug therapy since the pathway may already be crippled in advanced PCa. Combination drug therapy against the ataxia telangiectasia mutated (ATM) kinase which is responsible for phosphorylation of H2AX may increase double strand breaks and render cells hypersensitive to DSB and apoptosis during standard chemotherapy [225]. The DSB detection and repair pathway is a well-known player in tumorigenesis in other cancers such as breast and non-Hodgkin lymphoma [226,227]. Although more studies are necessary to confirm and extend the findings presented here, if γ-H2AX expression continues to distinguish PCas with poor prognosis, it may become an important biomarker and treatment target in PCa as well.
Group 3: differentially expressed genes in primary versus metastatic PCa
DNA microarray has also been used to compare the transcriptome of MCRPCa with primary PCa (Table 4). A number of key observations have been made using this approach. First, metastatic PCas display heterogeneous transcriptome expression profiles. For example, even within the same patient, some metastatic foci express high levels of PSA while others express less. Importantly, this indicates that AR transactivation remains intact at some level. Indeed, AR RNA levels vary considerably, but absolute levels compared to primary PCa are high. Second, organ-specific transcriptome profiles were not found. This suggests that the metastatic site does not influence gene expression in the distant tumor [188]. Third, key pathways previously implicated in metastasis and disease progression were also identified by this method; a fact which validates the procedure. Such pathways or cellular functions include cell adhesion, transcription, cell cycle and metabolism. Similar to primary PCa samples, the HPN gene maintains stable high levels of expression in metastatic samples in many studies, while c-FOS and transcription factor JUNB (JUNB) are detected at low levels [188,228]. The significance of c-FOS and JUNB reduction is clear: Both are implicated in EMT [229,230]. Genes such as WNT5A, EZH2, MAPK pathway members, AR and various androgen metabolism genes appear on most lists of those overexpressed at high levels in metastatic PCa [188,190,201,228].
Despite the heterogeneity between metastatic samples, AR itself and androgen metabolism genes are one of the few gene clusters that display almost global upregulation as shown by Stanbrough, et al., [201]. Specifically, aldo-keto reductase family 1, member C3 (AKR1C3) may play an interesting role in metastatic PCa. AKR1C3 normally catalyzes the conversion of androstenedione, an adrenal androgen, to testosterone; an event which has long been postulated to be the source of residual androgens in PCa patients undergoing androgen deprivation therapy (ADT) [231-233]. AKR1C3 is not the only enzyme for which this holds true: Steroid 5-alpha reductase 1 (SRD5A1), an enzyme responsible for converting T to DHT is similarly overexpressed in metastatic PCa. Moreover, hydroxy-delta-5-steroid dehydrogenase/3 beta-and steroid delta-isomerase 2 (HSD3B2) which catalyzes the conversion of Dehydroepiandrosterone (DHEA) to androstenedione, a critical step in producing DHT from adrenal androgens, is also overexpressed at high levels [201]. Taken together, these data suggest a mechanism for how MCRPCa cells can continue to upregulate genes that require AR transactivation. The most obvious argument against this model is that the combination of ADT and anti-AR drugs are palliative and not curative. Undoubtedly, more research is needed into both the role that androgen metabolism enzymes play in the progression of MCRPCa and into the development of more effective drugs that block AR signaling.
In conclusion, transcriptome profiling using DNA microarray is a field plagued by inconsistencies and technical difficulties. However, many important discoveries have been either confirmed or made de novo using this technology. HPN is by far the most notable primary PCa marker which has great potential as a drug target and diagnostic. As PCa advances to MCRPCa, HPN biomarker utility is still questionable, as is its role in PCa progression. Future research on this gene is of great interest to the field.
microRNAs and PCa
As part of the transcriptome, miRNA has gained rapid acceptance as an important epigenetic regulator of protein expression with a clear role in cancer development and progression [234-237]. For a detailed review of miRNA biogenesis including processing and mature miRNA function, see [238]. miRNAs are small, non-coding RNA molecules which bind to complimentary sequences in the 3’ untranslated region (3’-UTR) of mRNA. These small RNAs do not work alone: they are part of a large complex called the RNA-induced silencing complex (RISC). Binding of complimentary sequences suppresses translation simply by blocking access to the ribosome, or marks the target mRNA for degradation through cleavage of the message by the RNase III family member Dicer - both of which effectively silence the gene. Genes encoding miRNAs are found within introns of other genes, within repetitive genomic elements or within transposable element sequences. miRNA transcripts, called miRs, are structurally similar to mRNAs, with a polyA tail and 5’ cap like protein-coding RNAs. Biogenesis of the mature miRNA is as follows (Figure 4): First, the primary miRNA (pri-miRNA) is transcribed which can be several kb in length and is characterized by a classic “hairpin” or “stemloop” structure. The pri-miR is cleaved by the RNase III, Drosha, to form the pre-miRNA. The pre-miR represents a long (60-70 bp) intermediate form which is transported to the cytoplasm by the Exportin-5 complex where it is cleaved a final time by Dicer, creating the shorter (on average, 22bp) mature form [239,240]. Finally, RISC proteins such as Argonaute (Ago) aid in the uncoiling of the mature miR duplex, creating the active RISC complex. The technique of using labeled miRNA to probe oligonucleotide arrays has been very informative in determining the extent of post-tanscriptional regulation by this mechanism: recent estimates predict that up to 1/3 of human mRNAs are modulated by miRNAs. Since each miRNA can bind and affect multiple mRNAs, the magnitude of the list of targets of this form of epigenetic gene regulation is growing rapidly.
Figure 4.

miRNA processing. This schematic illustrates the biogenesis of a mature RNA-induced silencing complex (RISC). First, the pri-miR is transcribed and clipped by Drosha. After translocation to the cytoplasm, the pre-miR is again processed by Dicer before unwinding by Argonaute (Ago) and binding to other complex members to form the RISC.
In general, aberrant miRNA expression has been found in PCa, however, these studies have not been fully validated. Questions such as whether improper miRNA expression is a cause or consequence of cancer remain unanswered. The literature on miRNA levels in PCa patients is clouded by inconsistencies which are most likely due to both the immature technology used to capture and quantitate miRNAs in bulk tissue, and the presence of normal cells in so-called “tumor” samples. The contamination of the sample with normal epithelium creates a significant background when assaying for the relatively low abundance miRNA expression profile. Approaches such as laser microdissection, flow cytometry and the like may prove useful in removing this significant element of inconsistency from the literature and these studies. For an excellent summary of the literature on miRNA expression in PCa samples, see [234,235,241]. Watahiki, et al., have used a novel approach to characterize miRNA levels in PCa samples: pathologist-validated tumor samples are transplanted to the sub-renal capsule in immune compromised mice and the resulting xenografts are used in Next Generation sequencing experiments to quantitate the levels of common miRNAs in each sample. In this way, the heterogeneity of prostate tumor tissues is avoided since only truly transformed tumor samples grow in the environment of the renal capsule [242].
By focusing on specific miRNAs instead of the entire collection, progress has been made in determining their role in the regulation of protein expression in PCa. A compelling story is emerging from studies on miR-34a and its regulation of tumor cell growth and migration via modulation of p53 and c-myc [243,244]. It has been demonstrated that miR-34a expression is down-modulated in PCa patients. In agreement with this, miR-34a overexpression in PC3 cells results in suppressed cell migration, colony formation in soft agar and reduced c-Myc, c-Met and E2F1 proteins [244]. Further, miR-34a is a potent inhibitor of AR protein expression in cell lines, making it a potential therapeutic target. Indeed, restoration of miR-34a in PCa cell lines leads to AR suppression and prevention of AR nuclear translocation [243]. miR-34a repression in PCa cell lines is reversed by administration of Bio Response- 3, 3′-diindolylmethane (BR-DIM), a phytochemical found in cruciferous vegetables such as broccoli, cabbage and kale [245]. A diet rich in these vegetables has long been associated with decreased cancer risk and evidence is mounting that BR-DIM administration can reduce the risk of cancer recurrence after radical prostatectomy (for a review of the effects of BR-DIM on cancer, see [246]). Although the mechanism is unknown, BR-DIM appears to stimulation demethylation at the promoter of the MiR-34a gene. Reduced methylation results in reestablishment of miR-34a levels and subsequently, AR protein levels are abolished. Consistent with this, treatment of LNCaP cells in vitro with BR-DIM results in downregulation of AR and PSA as well as causing increased apoptosis and reduced tumor size in LNCaP xenografts in vivo [245]. In this way, BR-DIM acts as an antiandrogen therapeutic, but is not an AR antagonist nor does it affect DHT levels. Phase I dose escalation studies are complete and later-Phase trials are ongoing to establish this compound as a standard PCa therapy [247].
miRNA research is admittedly in its infancy. As discussed above, the literature is complicated by inconsistencies and studies of specific miRNA genes are preliminary in most cases. Despite these obvious issues, there is great potential in miRNAs as biomarkers. Secretion of miRNAs through a ceramide-dependent pathway has been documented. Furthermore, secreted miRs that are packaged into exosomes remain functional, implying that they can be an important mechanism of communication between tumor and surrounding normal epithelium or stroma [248]. Given these data, secreted miRs are an attractive biomarker, provided that specificity for PCa and clear tumor/normal delineation can be demonstrated for each proposed miR biomarker. Future studies should be aimed at fulfilling these goals.
Conclusion
A number of molecules or modifications involved in gene expression regulation have emerged as potential biomarkers for primary and/or advanced PCa. From the field of epigenetics research, GSTP1, EZH2, p300 and PCA3 are of interest for further study. In addition to the classic genomic PCa markers PTEN and AR, the 8q24 locus is also an important area to pursue. Finally, HPN expression in PCa is an area of great importance. Although these new markers all have their own strengths and weaknesses, many of which were discussed in this review, none fit every characteristic of the “ideal” biomarker. For example, EZH2 seems an excellent diagnostic tool, but it lacks specificity since it is also a marker for advanced breast cancer [249]. HPN overexpression in primary PCa certainly possesses the necessary convincing preliminary data, but its expression in advanced PCa is controversial. PSA, on the other hand, is extremely prostate-specific in its expression, but is not reliable as a primary PCa marker. Therefore, a combined assay for EZH2, HPN and PSA may provide more specificity for PCa and better predictive capability at both early and late stages of PCa. So far, a study investigating these three markers together is not available.
Combined biomarker testing is not a new idea given the current migration of clinical oncology towards more personalized medicine. In fact, a panel of markers (some of which were discussed in this review) selected by Talesa and colleagues were included in a PCR-based combined assay. Urine samples were collected from both PCa patients and normal males after prostate massage/DRE and the extracted RNA was subjected to qRT-PCR for PSA, PCA3, prostate specific membrane antigen (PSMA) and HPN. Interestingly, this group found that PSA in combination with PSMA was the best predictor of PCa diagnosis and severity, followed by PCA3 alone. HPN was not a good marker in urine RNA isolates [250]. Although there are several issues associated with this study (the method of sample collection, the assay platform chosen, for example), it is an excellent illustration of the power of combining biomarkers into one possibly more predictive diagnostic. More studies of this nature are being published on a regular basis. Unquestionably, the field of PCa biomarkers is ever-expanding and the next several years of research will be very interesting.
Acknowledgements
This work was supported by R01MD005824, R21CA149137, R21CA120625, and R21CA143589 grants (to S.K.) and by Roswell Park Cancer Institute and National Cancer Institute (NCI) grant #P30 CA016056. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Cancer Institute.
References
- 1.Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Society AC. In: Cancer Facts & Figures. Society AC, editor. Atlanta, GA: 2012. [Google Scholar]
- 3.Moyer VA. Screening for Prostate Cancer: U. S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2012;157:120–134. doi: 10.7326/0003-4819-157-2-201207170-00459. [DOI] [PubMed] [Google Scholar]
- 4.Cooper CS, Foster CS. Concepts of epigenetics in prostate cancer development. Br J Cancer. 2009;100:240–245. doi: 10.1038/sj.bjc.6604771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoque MO. DNA methylation changes in prostate cancer: current developments and future clinical implementation. Expert Rev Mol Diagn. 2009;9:243–257. doi: 10.1586/erm.09.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majumdar S, Buckles E, Estrada J, Koochekpour S. Aberrant DNA methylation and prostate cancer. Curr Genomics. 2011;12:486–505. doi: 10.2174/138920211797904061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koochekpour S. DNA methylome and the complexity of discovering prostate cancer biomarkers. Asian J Androl. 2011;13:661–662. doi: 10.1038/aja.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koochekpour S. Genetic and epigenetic changes in human prostate cancer. Iran Red Crescent Med J. 2011;13:80–98. [PMC free article] [PubMed] [Google Scholar]
- 9.Perry AS, Watson RW, Lawler M, Hollywood D. The epigenome as a therapeutic target in prostate cancer. Nat Rev Urol. 2010;7:668–680. doi: 10.1038/nrurol.2010.185. [DOI] [PubMed] [Google Scholar]
- 10.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 11.Decottignies A, d’Adda di Fagagna F. Epigenetic alterations associated with cellular senescence: a barrier against tumorigenesis or a red carpet for cancer? Semin Cancer Biol. 2011;21:360–366. doi: 10.1016/j.semcancer.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Jeronimo C, Henrique R, Hoque MO, Mambo E, Ribeiro FR, Varzim G, Oliveira J, Teixeira MR, Lopes C, Sidransky D. A quantitative promoter methylation profile of prostate cancer. Clin Cancer Res. 2004;10:8472–8478. doi: 10.1158/1078-0432.CCR-04-0894. [DOI] [PubMed] [Google Scholar]
- 13.Jeronimo C, Usadel H, Henrique R, Silva C, Oliveira J, Lopes C, Sidransky D. Quantitative GSTP1 hypermethylation in bodily fluids of patients with prostate cancer. Urology. 2002;60:1131–1135. doi: 10.1016/s0090-4295(02)01949-0. [DOI] [PubMed] [Google Scholar]
- 14.Jeronimo C, Varzim G, Henrique R, Oliveira J, Bento MJ, Silva C, Lopes C, Sidransky D. I105V polymorphism and promoter methylation of the GSTP1 gene in prostate adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2002;11:445–450. [PubMed] [Google Scholar]
- 15.Enokida H, Shiina H, Urakami S, Igawa M, Ogishima T, Li LC, Kawahara M, Nakagawa M, Kane CJ, Carroll PR, Dahiya R. Multigene methylation analysis for detection and staging of prostate cancer. Clin Cancer Res. 2005;11:6582–6588. doi: 10.1158/1078-0432.CCR-05-0658. [DOI] [PubMed] [Google Scholar]
- 16.Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975–1986. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 17.Yegnasubramanian S, Wu Z, Haffner MC, Esopi D, Aryee MJ, Badrinath R, He TL, Morgan JD, Carvalho B, Zheng Q, De Marzo AM, Irizarry RA, Nelson WG. Chromosome-wide mapping of DNA methylation patterns in normal and malignant prostate cells reveals pervasive methylation of gene-associated and conserved intergenic sequences. BMC Genomics. 2011;12:313. doi: 10.1186/1471-2164-12-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, Hamilton SR, Rashid A. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod Pathol. 2005;18:1632–1640. doi: 10.1038/modpathol.3800490. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Williamson M, Bott S, Brookman-Amissah N, Freeman A, Nariculam J, Hubank MJ, Ahmed A, Masters JR. Hypomethylation of WNT5A, CRIP1 and S100P in prostate cancer. Oncogene. 2007;26:6560–6565. doi: 10.1038/sj.onc.1210472. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki M, Tanaka Y, Perinchery G, Dharia A, Kotcherguina I, Fujimoto S, Dahiya R. Methylation and inactivation of estrogen, progesterone, and androgen receptors in prostate cancer. J Natl Cancer Inst. 2002;94:384–390. doi: 10.1093/jnci/94.5.384. [DOI] [PubMed] [Google Scholar]
- 21.Reibenwein J, Pils D, Horak P, Tomicek B, Goldner G, Worel N, Elandt K, Krainer M. Promoter hypermethylation of GSTP1, AR, and 14-3-3sigma in serum of prostate cancer patients and its clinical relevance. Prostate. 2007;67:427–432. doi: 10.1002/pros.20533. [DOI] [PubMed] [Google Scholar]
- 22.Ke XS, Qu Y, Cheng Y, Li WC, Rotter V, Oyan AM, Kalland KH. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics. 2010;11:669. doi: 10.1186/1471-2164-11-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darst RP, Pardo CE, Ai L, Brown KD, Kladde MP. Bisulfite sequencing of DNA. Curr Protoc Mol Biol Chapter. 2010;Chapter 7:Unit 7.9.1–17. doi: 10.1002/0471142727.mb0709s91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fouse SD, Nagarajan RO, Costello JF. Genome-scale DNA methylation analysis. Epigenomics. 2010;2:105–117. doi: 10.2217/epi.09.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulewska A, Niklinska W, Kozlowski M, Minarowski L, Naumnik W, Niklinski J, Dabrowska K, Chyczewski L. Detection of DNA methylation in eucaryotic cells. Folia Histochem Cytobiol. 2007;45:315–324. [PubMed] [Google Scholar]
- 26.Hayatsu H. Discovery of bisulfite-mediated cytosine conversion to uracil, the key reaction for DNA methylation analysis--a personal account. Proc Jpn Acad Ser B Phys Biol Sci. 2008;84:321–330. doi: 10.2183/pjab/84.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 28.Delgado-Cruzata L, Hruby GW, Gonzalez K, McKiernan J, Benson MC, Santella RM, Shen J. DNA methylation changes correlate with Gleason score and tumor stage in prostate cancer. DNA Cell Biol. 2012;31:187–192. doi: 10.1089/dna.2011.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21:502–517. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 32.Chen CC, Tyler J. Selective sensitization of cancer cells to DNA damage by a HAT inhibitor. Cell Cycle. 2009;8:2867. [PubMed] [Google Scholar]
- 33.Bandyopadhyay K, Baneres JL, Martin A, Blonski C, Parello J, Gjerset RA. Spermidinyl-CoA-based HAT inhibitors block DNA repair and provide cancer-specific chemo- and radiosensitization. Cell Cycle. 2009;8:2779–2788. doi: 10.4161/cc.8.17.9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popov VM, Wang C, Shirley LA, Rosenberg A, Li S, Nevalainen M, Fu M, Pestell RG. The functional significance of nuclear receptor acetylation. Steroids. 2007;72:221–230. doi: 10.1016/j.steroids.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, Sawyers CL. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009;69:958–966. doi: 10.1158/0008-5472.CAN-08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, Parson W, Meyers DJ, Cole PA, Culig Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644–1655. doi: 10.1158/1535-7163.MCT-11-0182. [DOI] [PubMed] [Google Scholar]
- 37.Gong J, Zhu J, Goodman OB Jr, Pestell RG, Schlegel PN, Nanus DM, Shen R. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene. 2006;25:2011–2021. doi: 10.1038/sj.onc.1209231. [DOI] [PubMed] [Google Scholar]
- 38.Li LC, Carroll PR, Dahiya R. Epigenetic changes in prostate cancer: implication for diagnosis and treatment. J Natl Cancer Inst. 2005;97:103–115. doi: 10.1093/jnci/dji010. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Zou X, Berger AD, Twiss C, Peng Y, Li Y, Chiu J, Guo H, Satagopan J, Wilton A, Gerald W, Basch R, Wang Z, Osman I, Lee P. Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J Transl Res. 2009;1:62–71. [PMC free article] [PubMed] [Google Scholar]
- 40.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 41.Patra SK, Patra A, Dahiya R. Histone deacetylase and DNA methyltransferase in human prostate cancer. Biochem Biophys Res Commun. 2001;287:705–713. doi: 10.1006/bbrc.2001.5639. [DOI] [PubMed] [Google Scholar]
- 42.Roy S, Tenniswood M. Site-specific acetylation of p53 directs selective transcription complex assembly. J Biol Chem. 2007;282:4765–4771. doi: 10.1074/jbc.M609588200. [DOI] [PubMed] [Google Scholar]
- 43.Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, Majid S, Igawa M, Dahiya R. Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer. 2008;123:552–560. doi: 10.1002/ijc.23590. [DOI] [PubMed] [Google Scholar]
- 44.Rennie PS, Nelson CC. Epigenetic mechanisms for progression of prostate cancer. Cancer Metastasis Rev. 1998;17:401–409. doi: 10.1023/a:1006121219097. [DOI] [PubMed] [Google Scholar]
- 45.Kim NH, Kim SN, Kim YK. Involvement of HDAC1 in E-cadherin expression in prostate cancer cells; its implication for cell motility and invasion. Biochem Biophys Res Commun. 2011;404:915–921. doi: 10.1016/j.bbrc.2010.12.081. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Sang N. Histone deacetylase inhibitors: the epigenetic therapeutics that repress hypoxia-inducible factors. J Biomed Biotechnol. 2011;2011:197946. doi: 10.1155/2011/197946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller CP, Singh MM, Rivera-Del Valle N, Manton CA, Chandra J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J Biomed Biotechnol. 2011;2011:514261. doi: 10.1155/2011/514261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong ST, Stein MN. HDAC inhibitor research: still in its infancy. Oncology (Williston Park) 2010;24:188–189, 192. [PubMed] [Google Scholar]
- 49.Kortenhorst MS, Isharwal S, van Diest PJ, Chowdhury WH, Marlow C, Carducci MA, Rodriguez R, Veltri RW. Valproic acid causes dose- and time-dependent changes in nuclear structure in prostate cancer cells in vitro and in vivo. Mol Cancer Ther. 2009;8:802–808. doi: 10.1158/1535-7163.MCT-08-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wedel SA, Sparatore A, Soldato PD, Al-Batran SE, Atmaca A, Juengel E, Hudak L, Jonas D, Blaheta RA. New histone deacetylase inhibitors as potential therapeutic tools for advanced prostate carcinoma. J Cell Mol Med. 2008;12:2457–2466. doi: 10.1111/j.1582-4934.2008.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao D, Xia Q, Lv J, Zhang H. Chronic administration of valproic acid inhibits PC3 cell growth by suppressing tumor angiogenesis in vivo. Int J Urol. 2007;14:838–845. doi: 10.1111/j.1442-2042.2007.01823.x. [DOI] [PubMed] [Google Scholar]
- 52.Sidana A, Wang M, Chowdhury WH, Toubaji A, Shabbeer S, Netto G, Carducci M, Lupold SE, Rodriguez R. Does valproic acid induce neuroendocrine differentiation in prostate cancer? J Biomed Biotechnol. 2011;2011:607480. doi: 10.1155/2011/607480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Angelucci A, Muzi P, Cristiano L, Millimaggi D, Cimini A, Dolo V, Miano R, Vicentini C, Ceru MP, Bologna M. Neuroendocrine transdifferentiation induced by VPA is mediated by PPARgamma activation and confers resistance to antiblastic therapy in prostate carcinoma. Prostate. 2008;68:588–598. doi: 10.1002/pros.20708. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez R. Sidney kimmel Comprehensive Cancer Center at Johns Hopkins, Valproic acid in treating patients with progressive, non-metastatic prostate cancer. National Cancer Institute NCT00670046. 2009 [Google Scholar]
- 55.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 56.Sawan C, Herceg Z. Histone modifications and cancer. Adv Genet. 2010;70:57–85. doi: 10.1016/B978-0-12-380866-0.60003-4. [DOI] [PubMed] [Google Scholar]
- 57.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 58.Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 59.Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem. 2005;280:22437–22444. doi: 10.1074/jbc.M501379200. [DOI] [PubMed] [Google Scholar]
- 60.Kong D, Heath E, Chen W, Cher ML, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O, Hwang C, Gupta N, Chitale D, Sakr WA, Menon M, Sarkar FH. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS One. 2012;7:e33729. doi: 10.1371/journal.pone.0033729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17:2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 62.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 63.Sellers WR, Loda M. The EZH2 polycomb transcriptional repressor--a marker or mover of metastatic prostate cancer? Cancer Cell. 2002;2:349–350. doi: 10.1016/s1535-6108(02)00187-3. [DOI] [PubMed] [Google Scholar]
- 64.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Behbahani TE, Kahl P, von der Gathen J, Heukamp LC, Baumann C, Gutgemann I, Walter B, Hofstadter F, Bastian PJ, von Ruecker A, Muller SC, Rogenhofer S, Ellinger J. Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urol. 2012;12:5. doi: 10.1186/1471-2490-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobosy JR, Roberts JL, Fu VX, Jarrard DF. The expanding role of epigenetics in the development, diagnosis and treatment of prostate cancer and benign prostatic hyperplasia. J Urol. 2007;177:822–831. doi: 10.1016/j.juro.2006.10.063. [DOI] [PubMed] [Google Scholar]
- 67.Jarrard DF, Bussemakers MJ, Bova GS, Isaacs WB. Regional loss of imprinting of the insulin-like growth factor II gene occurs in human prostate tissues. Clin Cancer Res. 1995;1:1471–1478. [PubMed] [Google Scholar]
- 68.Isharwal S, Miller MC, Marlow C, Makarov DV, Partin AW, Veltri RW. p300 (histone acetyltransferase) biomarker predicts prostate cancer biochemical recurrence and correlates with changes in epithelia nuclear size and shape. Prostate. 2008;68:1097–1104. doi: 10.1002/pros.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prensner JR, Chinnaiyan AM. The Emergence of lncRNAs in Cancer Biology. Cancer Discov. 2011;1:391–407. doi: 10.1158/2159-8290.CD-11-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Romanuik TL, Ueda T, Le N, Haile S, Yong TM, Thomson T, Vessella RL, Sadar MD. Novel biomarkers for prostate cancer including non-coding transcripts. Am J Pathol. 2009;175:2264–2276. doi: 10.2353/ajpath.2009.080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gibb EA, Vucic EA, Enfield KS, Stewart GL, Lonergan KM, Kennett JY, Becker-Santos DD, MacAulay CE, Lam S, Brown CJ, Lam WL. Human cancer long non-coding RNA transcriptomes. PLoS One. 2011;6:e25915. doi: 10.1371/journal.pone.0025915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, Chalk AM, Chiu KP, Choudhary V, Christoffels A, Clutterbuck DR, Crowe ML, Dalla E, Dalrymple BP, de Bono B, Della Gatta G, di Bernardo D, Down T, Engstrom P, Fagiolini M, Faulkner G, Fletcher CF, Fukushima T, Furuno M, Futaki S, Gariboldi M, Georgii-Hemming P, Gingeras TR, Gojobori T, Green RE, Gustincich S, Harbers M, Hayashi Y, Hensch TK, Hirokawa N, Hill D, Huminiecki L, Iacono M, Ikeo K, Iwama A, Ishikawa T, Jakt M, Kanapin A, Katoh M, Kawasawa Y, Kelso J, Kitamura H, Kitano H, Kollias G, Krishnan SP, Kruger A, Kummerfeld SK, Kurochkin IV, Lareau LF, Lazarevic D, Lipovich L, Liu J, Liuni S, McWilliam S, Madan Babu M, Madera M, Marchionni L, Matsuda H, Matsuzawa S, Miki H, Mignone F, Miyake S, Morris K, Mottagui-Tabar S, Mulder N, Nakano N, Nakauchi H, Ng P, Nilsson R, Nishiguchi S, Nishikawa S, Nori F, Ohara O, Okazaki Y, Orlando V, Pang KC, Pavan WJ, Pavesi G, Pesole G, Petrovsky N, Piazza S, Reed J, Reid JF, Ring BZ, Ringwald M, Rost B, Ruan Y, Salzberg SL, Sandelin A, Schneider C, Schonbach C, Sekiguchi K, Semple CA, Seno S, Sessa L, Sheng Y, Shibata Y, Shimada H, Shimada K, Silva D, Sinclair B, Sperling S, Stupka E, Sugiura K, Sultana R, Takenaka Y, Taki K, Tammoja K, Tan SL, Tang S, Taylor MS, Tegner J, Teichmann SA, Ueda HR, van Nimwegen E, Verardo R, Wei CL, Yagi K, Yamanishi H, Zabarovsky E, Zhu S, Zimmer A, Hide W, Bult C, Grimmond SM, Teasdale RD, Liu ET, Brusic V, Quackenbush J, Wahlestedt C, Mattick JS, Hume DA, Kai C, Sasaki D, Tomaru Y, Fukuda S, Kanamori-Katayama M, Suzuki M, Aoki J, Arakawa T, Iida J, Imamura K, Itoh M, Kato T, Kawaji H, Kawagashira N, Kawashima T, Kojima M, Kondo S, Konno H, Nakano K, Ninomiya N, Nishio T, Okada M, Plessy C, Shibata K, Shiraki T, Suzuki S, Tagami M, Waki K, Watahiki A, Okamura-Oho Y, Suzuki H, Kawai J, Hayashizaki Y. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 74.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mallardo M, Poltronieri P, D’Urso OF. Non-protein coding RNA biomarkers and differential expression in cancers: a review. J Exp Clin Cancer Res. 2008;27:19. doi: 10.1186/1756-9966-27-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Popov N, Gil J. Epigenetic regulation of the INK4b-ARF-INK4a locus: in sickness and in health. Epigenetics. 2010;5:685–690. doi: 10.4161/epi.5.8.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956–1962. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, Ibrahim JG, Thomas NE, Sharpless NE. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One. 2009;4:e5027. doi: 10.1371/journal.pone.0005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chung S, Nakagawa H, Uemura M, Piao L, Ashikawa K, Hosono N, Takata R, Akamatsu S, Kawaguchi T, Morizono T, Tsunoda T, Daigo Y, Matsuda K, Kamatani N, Nakamura Y, Kubo M. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2011;102:245–252. doi: 10.1111/j.1349-7006.2010.01737.x. [DOI] [PubMed] [Google Scholar]
- 81.Joung JY, Park S, Yoon H, Lee SJ, Park WS, Seo HK, Chung J, Kim SY, Hong SH, Lee YS, Kim J, Lee KH. Association of common variations of 8q24 with the risk of prostate cancer in Koreans and a review of the Asian population. BJU Int. 2012;12:5. doi: 10.1111/j.1464-410X.2012.11211.x. [DOI] [PubMed] [Google Scholar]
- 82.He H, Nagy R, Liyanarachchi S, Jiao H, Li W, Suster S, Kere J, de la Chapelle A. A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Res. 2009;69:625–631. doi: 10.1158/0008-5472.CAN-08-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jin G, Sun J, Isaacs SD, Wiley KE, Kim ST, Chu LW, Zhang Z, Zhao H, Zheng SL, Isaacs WB, Xu J. Human polymorphisms at long non-coding RNAs (lncRNAs) and association with prostate cancer risk. Carcinogenesis. 2011;32:1655–1659. doi: 10.1093/carcin/bgr187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Srikantan V, Zou Z, Petrovics G, Xu L, Augustus M, Davis L, Livezey JR, Connell T, Sesterhenn IA, Yoshino K, Buzard GS, Mostofi FK, McLeod DG, Moul JW, Srivastava S. PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc Natl Acad Sci U S A. 2000;97:12216–12221. doi: 10.1073/pnas.97.22.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Petrovics G, Zhang W, Makarem M, Street JP, Connelly R, Sun L, Sesterhenn IA, Srikantan V, Moul JW, Srivastava S. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene. 2004;23:605–611. doi: 10.1038/sj.onc.1207069. [DOI] [PubMed] [Google Scholar]
- 86.Fu X, Ravindranath L, Tran N, Petrovics G, Srivastava S. Regulation of apoptosis by a prostate-specific and prostate cancer-associated noncoding gene, PCGEM1. DNA Cell Biol. 2006;25:135–141. doi: 10.1089/dna.2006.25.135. [DOI] [PubMed] [Google Scholar]
- 87.Bussemakers MJ, van Bokhoven A, Verhaegh GW, Smit FP, Karthaus HF, Schalken JA, Debruyne FM, Ru N, Isaacs WB. DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999;59:5975–5979. [PubMed] [Google Scholar]
- 88.Auprich M, Haese A, Walz J, Pummer K, de la Taille A, Graefen M, de Reijke T, Fisch M, Kil P, Gontero P, Irani J, Chun FK. External validation of urinary PCA3-based nomograms to individually predict prostate biopsy outcome. Eur Urol. 2010;58:727–732. doi: 10.1016/j.eururo.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 89.de Kok JB, Verhaegh GW, Roelofs RW, Hessels D, Kiemeney LA, Aalders TW, Swinkels DW, Schalken JA. DD3(PCA3), a very sensitive and specific marker to detect prostate tumors. Cancer Res. 2002;62:2695–2698. [PubMed] [Google Scholar]
- 90.Popa I, Fradet Y, Beaudry G, Hovington H, Tetu B. Identification of PCA3 (DD3) in prostatic carcinoma by in situ hybridization. Mod Pathol. 2007;20:1121–1127. doi: 10.1038/modpathol.3800963. [DOI] [PubMed] [Google Scholar]
- 91.Sokoll LJ, Ellis W, Lange P, Noteboom J, Elliott DJ, Deras IL, Blase A, Koo S, Sarno M, Rittenhouse H, Groskopf J, Vessella RL. A multicenter evaluation of the PCA3 molecular urine test: pre-analytical effects, analytical performance, and diagnostic accuracy. Clin Chim Acta. 2008;389:1–6. doi: 10.1016/j.cca.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 92.Wang R, Chinnaiyan AM, Dunn RL, Wojno KJ, Wei JT. Rational approach to implementation of prostate cancer antigen 3 into clinical care. Cancer. 2009;115:3879–3886. doi: 10.1002/cncr.24447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clarke RA, Zhao Z, Guo AY, Roper K, Teng L, Fang ZM, Samaratunga H, Lavin MF, Gardiner RA. New genomic structure for prostate cancer specific gene PCA3 within BMCC1: implications for prostate cancer detection and progression. PLoS One. 2009;4:e4995. doi: 10.1371/journal.pone.0004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schalken JA, Hessels D, Verhaegh G. New targets for therapy in prostate cancer: differential display code 3 (DD3(PCA3)), a highly prostate cancer-specific gene. Urology. 2003;62:34–43. doi: 10.1016/s0090-4295(03)00759-3. [DOI] [PubMed] [Google Scholar]
- 95.MacInnis RJ, Antoniou AC, Eeles RA, Severi G, Guy M, McGuffog L, Hall AL, O’Brien LT, Wilkinson RA, Dearnaley DP, Ardern-Jones AT, Horwich A, Khoo VS, Parker CC, Huddart RA, Mc-Credie MR, Smith C, Southey MC, Staples MP, English DR, Hopper JL, Giles GG, Easton DF. Prostate cancer segregation analyses using 4390 families from UK and Australian population-based studies. Genet Epidemiol. 2010;34:42–50. doi: 10.1002/gepi.20433. [DOI] [PubMed] [Google Scholar]
- 96.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 97.Wiklund F. Prostate cancer genomics: can we distinguish between indolent and fatal disease using genetic markers? Genome Med. 2010;2:45. doi: 10.1186/gm166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Witte JS. Prostate cancer genomics: towards a new understanding. Nat Rev Genet. 2009;10:77–82. doi: 10.1038/nrg2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nakagawa H, Akamatsu S, Takata R, Takahashi A, Kubo M, Nakamura Y. Prostate cancer genomics, biology, and risk assessment through genome-wide association studies. Cancer Sci. 2012;103:607–613. doi: 10.1111/j.1349-7006.2011.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, Habegger L, Ambrogio L, Fennell T, Parkin M, Saksena G, Voet D, Ramos AH, Pugh TJ, Wilkinson J, Fisher S, Winckler W, Mahan S, Ardlie K, Baldwin J, Simons JW, Kitabayashi N, MacDonald TY, Kantoff PW, Chin L, Gabriel SB, Gerstein MB, Golub TR, Meyerson M, Tewari A, Lander ES, Getz G, Rubin MA, Garraway LA. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Xu J, Blondal T, Kostic J, Sun J, Ghosh S, Stacey SN, Mouy M, Saemundsdottir J, Backman VM, Kristjansson K, Tres A, Partin AW, Albers-Akkers MT, Godino-Ivan Marcos J, Walsh PC, Swinkels DW, Navarrete S, Isaacs SD, Aben KK, Graif T, Cashy J, Ruiz-Echarri M, Wiley KE, Suarez BK, Witjes JA, Frigge M, Ober C, Jonsson E, Einarsson GV, Mayordomo JI, Kiemeney LA, Isaacs WB, Catalona WJ, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 102.Armendariz AD, Krauss RM. Hepatic nuclear factor 1-alpha: inflammation, genetics, and atherosclerosis. Curr Opin Lipidol. 2009;20:106–111. doi: 10.1097/mol.0b013e3283295ee9. [DOI] [PubMed] [Google Scholar]
- 103.Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson JT, Thorleifsson G, Manolescu A, Rafnar T, Gudbjartsson D, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Blondal T, Stacey SN, Helgason A, Gunnarsdottir S, Olafsdottir A, Kristinsson KT, Birgisdottir B, Ghosh S, Thorlacius S, Magnusdottir D, Stefansdottir G, Kristjansson K, Bagger Y, Wilensky RL, Reilly MP, Morris AD, Kimber CH, Adeyemo A, Chen Y, Zhou J, So WY, Tong PC, Ng MC, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Tres A, Fuertes F, Ruiz-Echarri M, Asin L, Saez B, van Boven E, Klaver S, Swinkels DW, Aben KK, Graif T, Cashy J, Suarez BK, van Vierssen Trip O, Frigge ML, Ober C, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Palmer CN, Rotimi C, Chan JC, Pedersen O, Sigurdsson G, Benediktsson R, Jonsson E, Einarsson GV, Mayordomo JI, Catalona WJ, Kiemeney LA, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 104.Kang J, Chen MH, Zhang Y, Moran BJ, Dosoretz DE, Katin MJ, Braccioforte MH, Salenius SA, D’Amico AV. Type of diabetes mellitus and the odds of Gleason score 8 to 10 prostate cancer. Int J Radiat Oncol Biol Phys. 2012;82:e463–467. doi: 10.1016/j.ijrobp.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 105.Moses KA, Utuama OA, Goodman M, Issa MM. The association of diabetes and positive prostate biopsy in a US veteran population. Prostate Cancer Prostatic Dis. 2012;15:70–74. doi: 10.1038/pcan.2011.40. [DOI] [PubMed] [Google Scholar]
- 106.Cheng I, Plummer SJ, Jorgenson E, Liu X, Rybicki BA, Casey G, Witte JS. 8q24 and prostate cancer: association with advanced disease and meta-analysis. Eur J Hum Genet. 2008;16:496–505. doi: 10.1038/sj.ejhg.5201959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schumacher FR, Feigelson HS, Cox DG, Haiman CA, Albanes D, Buring J, Calle EE, Chanock SJ, Colditz GA, Diver WR, Dunning AM, Freedman ML, Gaziano JM, Giovannucci E, Hankinson SE, Hayes RB, Henderson BE, Hoover RN, Kaaks R, Key T, Kolonel LN, Kraft P, Le Marchand L, Ma J, Pike MC, Riboli E, Stampfer MJ, Stram DO, Thomas G, Thun MJ, Travis R, Virtamo J, Andriole G, Gelmann E, Willett WC, Hunter DJ. A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res. 2007;67:2951–2956. doi: 10.1158/0008-5472.CAN-06-3591. [DOI] [PubMed] [Google Scholar]
- 108.Severi G, Hayes VM, Padilla EJ, English DR, Southey MC, Sutherland RL, Hopper JL, Giles GG. The common variant rs1447295 on chromosome 8q24 and prostate cancer risk: results from an Australian population-based case-control study. Cancer Epidemiol Biomarkers Prev. 2007;16:610–612. doi: 10.1158/1055-9965.EPI-06-0872. [DOI] [PubMed] [Google Scholar]
- 109.Suuriniemi M, Agalliu I, Schaid DJ, Johanneson B, McDonnell SK, Iwasaki L, Stanford JL, Ostrander EA. Confirmation of a positive association between prostate cancer risk and a locus at chromosome 8q24. Cancer Epidemiol Biomarkers Prev. 2007;16:809–814. doi: 10.1158/1055-9965.EPI-06-1049. [DOI] [PubMed] [Google Scholar]
- 110.Wang L, McDonnell SK, Slusser JP, Hebbring SJ, Cunningham JM, Jacobsen SJ, Cerhan JR, Blute ML, Schaid DJ, Thibodeau SN. Two common chromosome 8q24 variants are associated with increased risk for prostate cancer. Cancer Res. 2007;67:2944–2950. doi: 10.1158/0008-5472.CAN-06-3186. [DOI] [PubMed] [Google Scholar]
- 111.Witte JS. Multiple prostate cancer risk variants on 8q24. Nat Genet. 2007;39:579–580. doi: 10.1038/ng0507-579. [DOI] [PubMed] [Google Scholar]
- 112.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 113.Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE, Cox DR, Ponder BA. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier JB, Houlston R. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–988. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 115.Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, Gudmundsson J, Jakobsdottir M, Bergthorsson JT, Sigurdsson A, Blondal T, Witjes JA, Vermeulen SH, Hulsbergen-van de Kaa CA, Swinkels DW, Ploeg M, Cornel EB, Vergunst H, Thorgeirsson TE, Gudbjartsson D, Gudjonsson SA, Thorleifsson G, Kristinsson KT, Mouy M, Snorradottir S, Placidi D, Campagna M, Arici C, Koppova K, Gurzau E, Rudnai P, Kellen E, Polidoro S, Guarrera S, Sacerdote C, Sanchez M, Saez B, Valdivia G, Ryk C, de Verdier P, Lindblom A, Golka K, Bishop DT, Knowles MA, Nikulasson S, Petursdottir V, Jonsson E, Geirsson G, Kristjansson B, Mayordomo JI, Steineck G, Porru S, Buntinx F, Zeegers MP, Fletcher T, Kumar R, Matullo G, Vineis P, Kiltie AE, Gulcher JR, Thorsteinsdottir U, Kong A, Rafnar T, Stefansson K. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet. 2008;40:1307–1312. doi: 10.1038/ng.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, Stephens RM, Harris TJ, Munroe DJ, Wu X. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010;107:3001–3005. doi: 10.1073/pnas.0906067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grisanzio C, Freedman ML. Chromosome 8q24-Associated Cancers and MYC. Genes Cancer. 2010;1:555–559. doi: 10.1177/1947601910381380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M, Caswell JL, Beckwith CA, Hills A, Macconaill L, Coetzee GA, Regan MM, Freedman ML. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A. 2010;107:9742–9746. doi: 10.1073/pnas.0910668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sole X, Hernandez P, de Heredia ML, Armengol L, Rodriguez-Santiago B, Gomez L, Maxwell CA, Aguilo F, Condom E, Abril J, Perez-Jurado L, Estivill X, Nunes V, Capella G, Gruber SB, Moreno V, Pujana MA. Genetic and genomic analysis modeling of germline c-MYC overexpression and cancer susceptibility. BMC Genomics. 2008;9:12. doi: 10.1186/1471-2164-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, Yao K, Kehoe SM, Lenz HJ, Haiman CA, Yan C, Henderson BE, Frenkel B, Barretina J, Bass A, Tabernero J, Baselga J, Regan MM, Manak JR, Shivdasani R, Coetzee GA, Freedman ML. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pomerantz MM, Beckwith CA, Regan MM, Wyman SK, Petrovics G, Chen Y, Hawksworth DJ, Schumacher FR, Mucci L, Penney KL, Stampfer MJ, Chan JA, Ardlie KG, Fritz BR, Parkin RK, Lin DW, Dyke M, Herman P, Lee S, Oh WK, Kantoff PW, Tewari M, McLeod DG, Srivastava S, Freedman ML. Evaluation of the 8q24 prostate cancer risk locus and MYC expression. Cancer Res. 2009;69:5568–5574. doi: 10.1158/0008-5472.CAN-09-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carter BS, Bova GS, Beaty TH, Steinberg GD, Childs B, Isaacs WB, Walsh PC. Hereditary prostate cancer: epidemiologic and clinical features. J Urol. 1993;150:797–802. doi: 10.1016/s0022-5347(17)35617-3. [DOI] [PubMed] [Google Scholar]
- 124.Freedman ML, Haiman CA, Patterson N, Mc-Donald GJ, Tandon A, Waliszewska A, Penney K, Steen RG, Ardlie K, John EM, Oakley-Girvan I, Whittemore AS, Cooney KA, Ingles SA, Altshuler D, Henderson BE, Reich D. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci U S A. 2006;103:14068–14073. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Troutman SM, Sissung TM, Cropp CD, Venzon DJ, Spencer SD, Adesunloye BA, Huang X, Karzai FH, Price DK, Figg WD. Racial disparities in the association between variants on 8q24 and prostate cancer: a systematic review and meta-analysis. Oncologist. 2012;17:312–320. doi: 10.1634/theoncologist.2011-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Robbins C, Torres JB, Hooker S, Bonilla C, Hernandez W, Candreva A, Ahaghotu C, Kittles R, Carpten J. Confirmation study of prostate cancer risk variants at 8q24 in African Americans identifies a novel risk locus. Genome Res. 2007;17:1717–1722. doi: 10.1101/gr.6782707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cilloni D, Saglio G. Molecular pathways: BCR-ABL. Clin Cancer Res. 2012;18:930–937. doi: 10.1158/1078-0432.CCR-10-1613. [DOI] [PubMed] [Google Scholar]
- 128.Liu-Dumlao T, Kantarjian H, Thomas DA, O’Brien S, Ravandi F. Philadelphia-Positive Acute Lymphoblastic Leukemia: Current Treatment Options. Curr Oncol Rep. 2012;14:387. doi: 10.1007/s11912-012-0247-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yeung DT, Hughes TP. Therapeutic targeting of BCR-ABL: prognostic markers of response and resistance mechanism in chronic myeloid leukaemia. Crit Rev Oncog. 2012;17:17–30. doi: 10.1615/critrevoncog.v17.i1.30. [DOI] [PubMed] [Google Scholar]
- 130.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 131.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 132.Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, Prensner JR, Cao X, Singla N, Montie JE, Varambally S, Mehra R, Chinnaiyan AM. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- 133.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, Setlur S, Tchinda J, Tomlins SA, Hofer MD, Pienta KG, Kuefer R, Vessella R, Sun XW, Meyerson M, Lee C, Sellers WR, Chinnaiyan AM, Rubin MA. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 134.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 135.Shah RB, Chinnaiyan AM. The discovery of common recurrent transmembrane protease serine 2 (TMPRSS2)-erythroblastosis virus E26 transforming sequence (ETS) gene fusions in prostate cancer: significance and clinical implications. Adv Anat Pathol. 2009;16:145–153. doi: 10.1097/PAP.0b013e3181a12da7. [DOI] [PubMed] [Google Scholar]
- 136.Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, Schalken JA. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol. 2009;56:275–286. doi: 10.1016/j.eururo.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 137.Morris DS, Tomlins SA, Montie JE, Chinnaiyan AM. The discovery and application of gene fusions in prostate cancer. BJU Int. 2008;102:276–282. doi: 10.1111/j.1464-410X.2008.07665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nam RK, Sugar L, Yang W, Srivastava S, Klotz LH, Yang LY, Stanimirovic A, Encioiu E, Neill M, Loblaw DA, Trachtenberg J, Narod SA, Seth A. Expression of the TMPRSS2:ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br J Cancer. 2007;97:1690–1695. doi: 10.1038/sj.bjc.6604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guo CC, Dancer JY, Wang Y, Aparicio A, Navone NM, Troncoso P, Czerniak BA. TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum Pathol. 2011;42:11–17. doi: 10.1016/j.humpath.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saramaki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, Visakorpi T. TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res. 2008;14:3395–3400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- 141.Magi-Galluzzi C, Tsusuki T, Elson P, Simmerman K, LaFargue C, Esgueva R, Klein E, Rubin MA, Zhou M. TMPRSS2-ERG gene fusion prevalence and class are significantly different in prostate cancer of Caucasian, African-American and Japanese patients. Prostate. 2011;71:489–497. doi: 10.1002/pros.21265. [DOI] [PubMed] [Google Scholar]
- 142.FitzGerald LM, Agalliu I, Johnson K, Miller MA, Kwon EM, Hurtado-Coll A, Fazli L, Rajput AB, Gleave ME, Cox ME, Ostrander EA, Stanford JL, Huntsman DG. Association of TMPRSS2-ERG gene fusion with clinical characteristics and outcomes: results from a population-based study of prostate cancer. BMC Cancer. 2008;8:230. doi: 10.1186/1471-2407-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. Eur J Cancer. 2005;41:2462–2478. doi: 10.1016/j.ejca.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 144.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/s0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- 145.Hsu T, Trojanowska M, Watson DK. Ets proteins in biological control and cancer. J Cell Biochem. 2004;91:896–903. doi: 10.1002/jcb.20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Seth A, Watson DK, Blair DG, Papas TS. cets-2 protooncogene has mitogenic and oncogenic activity. Proc Natl Acad Sci U S A. 1989;86:7833–7837. doi: 10.1073/pnas.86.20.7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Seth A, Papas TS. The c-ets-1 proto-oncogene has oncogenic activity and is positively autoregulated. Oncogene. 1990;5:1761–1767. [PubMed] [Google Scholar]
- 148.Hart AH, Corrick CM, Tymms MJ, Hertzog PJ, Kola I. Human ERG is a proto-oncogene with mitogenic and transforming activity. Oncogene. 1995;10:1423–1430. [PubMed] [Google Scholar]
- 149.Oettgen P, Finger E, Sun Z, Akbarali Y, Thamrongsak U, Boltax J, Grall F, Dube A, Weiss A, Brown L, Quinn G, Kas K, Endress G, Kunsch C, Libermann TA. PDEF, a novel prostate epithelium-specific ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J Biol Chem. 2000;275:1216–1225. doi: 10.1074/jbc.275.2.1216. [DOI] [PubMed] [Google Scholar]
- 150.Tomlins SA, Aubin SM, Siddiqui J, Lonigro RJ, Sefton-Miller L, Miick S, Williamsen S, Hodge P, Meinke J, Blase A, Penabella Y, Day JR, Varambally R, Han B, Wood D, Wang L, Sanda MG, Rubin MA, Rhodes DR, Hollenbeck B, Sakamoto K, Silberstein JL, Fradet Y, Amberson JB, Meyers S, Palanisamy N, Rittenhouse H, Wei JT, Groskopf J, Chinnaiyan AM. Urine TMPRSS2:ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA. Sci Transl Med. 2011;3:94ra72. doi: 10.1126/scitranslmed.3001970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ali I, Schriml L, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91:1922–1932. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 152.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 153.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, Mc-Combie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 154.Koochekpour S. Androgen receptor signaling and mutations in prostate cancer. Asian J Androl. 2010;12:639–657. doi: 10.1038/aja.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol. 2004;22:2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 157.Persad S, Attwell S, Gray V, Delcommenne M, Troussard A, Sanghera J, Dedhar S. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc Natl Acad Sci U S A. 2000;97:3207–3212. doi: 10.1073/pnas.060579697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Davies MA, Koul D, Dhesi H, Berman R, Mc-Donnell TJ, McConkey D, Yung WK, Steck PA. Regulation of Akt/PKB activity, cellular growth, and apoptosis in prostate carcinoma cells by MMAC/PTEN. Cancer Res. 1999;59:2551–2556. [PubMed] [Google Scholar]
- 159.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gray IC, Stewart LM, Phillips SM, Hamilton JA, Gray NE, Watson GJ, Spurr NK, Snary D. Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br J Cancer. 1998;78:1296–1300. doi: 10.1038/bjc.1998.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gray IC, Phillips SM, Lee SJ, Neoptolemos JP, Weissenbach J, Spurr NK. Loss of the chromosomal region 10q23-25 in prostate cancer. Cancer Res. 1995;55:4800–4803. [PubMed] [Google Scholar]
- 163.Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998;58:2720–2723. [PubMed] [Google Scholar]
- 164.Pourmand G, Ziaee AA, Abedi AR, Mehrsai A, Alavi HA, Ahmadi A, Saadati HR. Role of PTEN gene in progression of prostate cancer. Urol J. 2007;4:95–100. [PubMed] [Google Scholar]
- 165.Gray IC, Nobile C, Muresu R, Ford S, Spurr NK. A 2.4-megabase physical map spanning the CYP2C gene cluster on chromosome 10q24. Genomics. 1995;28:328–332. doi: 10.1006/geno.1995.1149. [DOI] [PubMed] [Google Scholar]
- 166.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 167.Gottlieb B, Beitel LK, Wu J, Elhaji YA, Trifiro M. Nuclear receptors and disease: androgen receptor. Essays Biochem. 2004;40:121–136. doi: 10.1042/bse0400121. [DOI] [PubMed] [Google Scholar]
- 168.Koochekpour S, Hu S, Subramani D, Majumdar S, Liu Z, Velasco-Gonzalez C, Buckles E, Wood B, Christian Winters J. Somatic and genomic instability of androgen receptor in African Americans with prostate cancer. J Urol. 2011;185 [Google Scholar]
- 169.Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012;33:887–894. doi: 10.1002/humu.22046. [DOI] [PubMed] [Google Scholar]
- 170.Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A. Androgen insensitivity syndrome: clinical features and molecular defects. Hormones (Athens) 2008;7:217–229. doi: 10.14310/horm.2002.1201. [DOI] [PubMed] [Google Scholar]
- 171.Edwards A, Hammond HA, Jin L, Caskey CT, Chakraborty R. Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics. 1992;12:241–253. doi: 10.1016/0888-7543(92)90371-x. [DOI] [PubMed] [Google Scholar]
- 172.Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Dahl D, Brufsky A, Talcott J, Hennekens CH, Kantoff PW. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci U S A. 1997;94:3320–3323. doi: 10.1073/pnas.94.7.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hsing AW, Gao YT, Wu G, Wang X, Deng J, Chen YL, Sesterhenn IA, Mostofi FK, Benichou J, Chang C. Polymorphic CAG and GGN repeat lengths in the androgen receptor gene and prostate cancer risk: a population-based case-control study in China. Cancer Res. 2000;60:5111–5116. [PubMed] [Google Scholar]
- 174.Stanford JL, Just JJ, Gibbs M, Wicklund KG, Neal CL, Blumenstein BA, Ostrander EA. Polymorphic repeats in the androgen receptor gene: molecular markers of prostate cancer risk. Cancer Res. 1997;57:1194–1198. [PubMed] [Google Scholar]
- 175.Beilin J, Ball EM, Favaloro JM, Zajac JD. Effect of the androgen receptor CAG repeat polymorphism on transcriptional activity: specificity in prostate and non-prostate cell lines. J Mol Endocrinol. 2000;25:85–96. doi: 10.1677/jme.0.0250085. [DOI] [PubMed] [Google Scholar]
- 176.Irvine RA, Ma H, Yu MC, Ross RK, Stallcup MR, Coetzee GA. Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Hum Mol Genet. 2000;9:267–274. doi: 10.1093/hmg/9.2.267. [DOI] [PubMed] [Google Scholar]
- 177.Bratt O, Borg A, Kristoffersson U, Lundgren R, Zhang QX, Olsson H. CAG repeat length in the androgen receptor gene is related to age at diagnosis of prostate cancer and response to endocrine therapy, but not to prostate cancer risk. Br J Cancer. 1999;81:672–676. doi: 10.1038/sj.bjc.6690746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Robins DM. Androgen receptor gene polymorphisms and alterations in prostate cancer: of humanized mice and men. Mol Cell Endocrinol. 2012;352:26–33. doi: 10.1016/j.mce.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wallen MJ, Linja M, Kaartinen K, Schleutker J, Visakorpi T. Androgen receptor gene mutations in hormone-refractory prostate cancer. J Pathol. 1999;189:559–563. doi: 10.1002/(SICI)1096-9896(199912)189:4<559::AID-PATH471>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 180.Price DK, Chau CH, Till C, Goodman PJ, Baum CE, Ockers SB, English BC, Minasian L, Parnes HL, Hsing AW, Reichardt JK, Hoque A, Tangen CM, Kristal AR, Thompson IM, Figg WD. Androgen receptor CAG repeat length and association with prostate cancer risk: results from the prostate cancer prevention trial. J Urol. 2010;184:2297–2302. doi: 10.1016/j.juro.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lindstrom S, Ma J, Altshuler D, Giovannucci E, Riboli E, Albanes D, Allen NE, Berndt SI, Boeing H, Bueno-de-Mesquita HB, Chanock SJ, Dunning AM, Feigelson HS, Gaziano JM, Haiman CA, Hayes RB, Henderson BE, Hunter DJ, Kaaks R, Kolonel LN, Le Marchand L, Martinez C, Overvad K, Siddiq A, Stampfer M, Stattin P, Stram DO, Thun MJ, Trichopoulos D, Tumino R, Virtamo J, Weinstein SJ, Yeager M, Kraft P, Freedman ML. A large study of androgen receptor germline variants and their relation to sex hormone levels and prostate cancer risk. Results from the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. J Clin Endocrinol Metab. 2010;95:E121–127. doi: 10.1210/jc.2009-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hu SY, Liu T, Liu ZZ, Ledet E, Velasco-Gonzalez C, Mandal DM, Koochekpour S. Identification of a novel germline missense mutation of the androgen receptor in African American men with familial prostate cancer. Asian J Androl. 2010;12:336–343. doi: 10.1038/aja.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tomlins SA, Mehra R, Rhodes DR, Shah RB, Rubin MA, Bruening E, Makarov V, Chinnaiyan AM. Whole transcriptome amplification for gene expression profiling and development of molecular archives. Neoplasia. 2006;8:153–162. doi: 10.1593/neo.05754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang E. RNA amplification for successful gene profiling analysis. J Transl Med. 2005;3:28. doi: 10.1186/1479-5876-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Diboun I, Wernisch L, Orengo CA, Koltzenburg M. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics. 2006;7:252. doi: 10.1186/1471-2164-7-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Analysis of gene expression in single live neurons. Proc Natl Acad Sci U S A. 1992;89:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li Y, Li T, Liu S, Qiu M, Han Z, Jiang Z, Li R, Ying K, Xie Y, Mao Y. Systematic comparison of the fidelity of aRNA, mRNA and T-RNA on gene expression profiling using cDNA microarray. J Biotechnol. 2004;107:19–28. doi: 10.1016/j.jbiotec.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 188.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nanni S, Priolo C, Grasselli A, D’Eletto M, Merola R, Moretti F, Gallucci M, De Carli P, Sentinelli S, Cianciulli AM, Mottolese M, Carlini P, Arcelli D, Helmer-Citterich M, Gaetano C, Loda M, Pontecorvi A, Bacchetti S, Sacchi A, Farsetti A. Epithelial-restricted gene profile of primary cultures from human prostate tumors: a molecular approach to predict clinical behavior of prostate cancer. Mol Cancer Res. 2006;4:79–92. doi: 10.1158/1541-7786.MCR-05-0098. [DOI] [PubMed] [Google Scholar]
- 190.LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, Gerald WL. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 191.apointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, Ekman P, DeMarzo AM, Tibshirani R, Botstein D, Brown PO, Brooks JD, Pollack JR. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, Catalona WJ, Watson MA, Milbrandt J. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 2001;61:5692–5696. [PubMed] [Google Scholar]
- 193.Luo J, Duggan DJ, Chen Y, Sauvageot J, Ewing CM, Bittner ML, Trent JM, Isaacs WB. Human prostate cancer and benign prostatic hyperplasia: molecular dissection by gene expression profiling. Cancer Res. 2001;61:4683–4688. [PubMed] [Google Scholar]
- 194.Ning QY, Wu JZ, Zang N, Liang J, Hu YL, Mo ZN. Key pathways involved in prostate cancer based on gene set enrichment analysis and meta analysis. Genet Mol Res. 2011;10:3856–3887. doi: 10.4238/2011.December.14.10. [DOI] [PubMed] [Google Scholar]
- 195.Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62:4427–4433. [PubMed] [Google Scholar]
- 196.Xu L, Tan AC, Naiman DQ, Geman D, Winslow RL. Robust prostate cancer marker genes emerge from direct integration of inter-study microarray data. Bioinformatics. 2005;21:3905–3911. doi: 10.1093/bioinformatics/bti647. [DOI] [PubMed] [Google Scholar]
- 197.Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, Stephens RM, Caporaso NE, Loffredo CA, Ambs S. Tumor immuno-biological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 198.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 199.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP, Lander ES, Loda M, Kantoff PW, Golub TR, Sellers WR. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–209. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 200.Welsh JB, Sapinoso LM, Su AI, Kern SG, Wang-Rodriguez J, Moskaluk CA, Frierson HF Jr, Hampton GM. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- 201.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 202.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 203.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006;9:230–234. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 204.Kim HJ, Han JH, Chang IH, Kim W, Myung SC. Variants in the HEPSIN gene are associated with susceptibility to prostate cancer. Prostate Cancer Prostatic Dis. 2012 doi: 10.1038/pcan.2012.17. [DOI] [PubMed] [Google Scholar]
- 205.Burmester JK, Suarez BK, Lin JH, Jin CH, Miller RD, Zhang KQ, Salzman SA, Reding DJ, Catalona WJ. Analysis of candidate genes for prostate cancer. Hum Hered. 2004;57:172–178. doi: 10.1159/000081443. [DOI] [PubMed] [Google Scholar]
- 206.Pal P, Xi H, Kaushal R, Sun G, Jin CH, Jin L, Suarez BK, Catalona WJ, Deka R. Variants in the HEPSIN gene are associated with prostate cancer in men of European origin. Hum Genet. 2006;120:187–192. doi: 10.1007/s00439-006-0204-3. [DOI] [PubMed] [Google Scholar]
- 207.Holt SK, Kwon EM, Lin DW, Ostrander EA, Stanford JL. Association of hepsin gene variants with prostate cancer risk and prognosis. Prostate. 2010;70:1012–1019. doi: 10.1002/pros.21135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sardana G, Dowell B, Diamandis EP. Emerging biomarkers for the diagnosis and prognosis of prostate cancer. Clin Chem. 2008;54:1951–1960. doi: 10.1373/clinchem.2008.110668. [DOI] [PubMed] [Google Scholar]
- 209.Chen Z, Fan Z, McNeal JE, Nolley R, Caldwell MC, Mahadevappa M, Zhang Z, Warrington JA, Stamey TA. Hepsin and maspin are inversely expressed in laser capture microdis-sectioned prostate cancer. J Urol. 2003;169:1316–1319. doi: 10.1097/01.ju.0000050648.40164.0d. [DOI] [PubMed] [Google Scholar]
- 210.Stamey TA, Warrington JA, Caldwell MC, Chen Z, Fan Z, Mahadevappa M, McNeal JE, Nolley R, Zhang Z. Molecular genetic profiling of Gleason grade 4/5 prostate cancers compared to benign prostatic hyperplasia. J Urol. 2001;166:2171–2177. [PubMed] [Google Scholar]
- 211.Stephan C, Yousef GM, Scorilas A, Jung K, Jung M, Kristiansen G, Hauptmann S, Kishi T, Nakamura T, Loening SA, Diamandis EP. Hepsin is highly over expressed in and a new candidate for a prognostic indicator in prostate cancer. J Urol. 2004;171:187–191. doi: 10.1097/01.ju.0000101622.74236.94. [DOI] [PubMed] [Google Scholar]
- 212.Tanimoto H, Yan Y, Clarke J, Korourian S, Shigemasa K, Parmley TH, Parham GP, O’Brien TJ. Hepsin, a cell surface serine protease identified in hepatoma cells, is overexpressed in ovarian cancer. Cancer Res. 1997;57:2884–2887. [PubMed] [Google Scholar]
- 213.Roemer A, Schwettmann L, Jung M, Stephan C, Roigas J, Kristiansen G, Loening SA, Lichtinghagen R, Jung K. The membrane proteases adams and hepsin are differentially expressed in renal cell carcinoma.Are they potential tumor markers? . J Urol. 2004;172:2162–2166. doi: 10.1097/01.ju.0000144602.01322.49. [DOI] [PubMed] [Google Scholar]
- 214.Leytus SP, Loeb KR, Hagen FS, Kurachi K, Davie EW. A novel trypsin-like serine protease (hepsin) with a putative transmembrane domain expressed by human liver and hepatoma cells. Biochemistry. 1988;27:1067–1074. doi: 10.1021/bi00403a032. [DOI] [PubMed] [Google Scholar]
- 215.Torres-Rosado A, O’Shea KS, Tsuji A, Chou SH, Kurachi K. Hepsin, a putative cell-surface serine protease, is required for mammalian cell growth. Proc Natl Acad Sci U S A. 1993;90:7181–7185. doi: 10.1073/pnas.90.15.7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Klezovitch O, Chevillet J, Mirosevich J, Roberts RL, Matusik RJ, Vasioukhin V. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell. 2004;6:185–195. doi: 10.1016/j.ccr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 217.Kelly KA, Setlur SR, Ross R, Anbazhagan R, Waterman P, Rubin MA, Weissleder R. Detection of early prostate cancer using a hepsin-targeted imaging agent. Cancer Res. 2008;68:2286–2291. doi: 10.1158/0008-5472.CAN-07-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Goel MM, Agrawal D, Natu SM, Goel A. Hepsin immunohistochemical expression in prostate cancer in relation to Gleason’s grade and serum prostate specific antigen. Indian J Pathol Microbiol. 2011;54:476–481. doi: 10.4103/0377-4929.85078. [DOI] [PubMed] [Google Scholar]
- 219.Gendler SJ. MUC1, the renaissance molecule. J Mammary Gland Biol Neoplasia. 2001;6:339–353. doi: 10.1023/a:1011379725811. [DOI] [PubMed] [Google Scholar]
- 220.Hofer MD, Kuefer R, Varambally S, Li H, Ma J, Shapiro GI, Gschwend JE, Hautmann RE, Sanda MG, Giehl K, Menke A, Chinnaiyan AM, Rubin MA. The role of metastasis-associated protein 1 in prostate cancer progression. Cancer Res. 2004;64:825–829. doi: 10.1158/0008-5472.can-03-2755. [DOI] [PubMed] [Google Scholar]
- 221.Kai L, Wang J, Ivanovic M, Chung YT, Laskin WB, Schulze-Hoepfner F, Mirochnik Y, Satcher RL Jr, Levenson AS. Targeting prostate cancer angiogenesis through metastasis-associated protein 1 (MTA1) Prostate. 2011;71:268–280. doi: 10.1002/pros.21240. [DOI] [PubMed] [Google Scholar]
- 222.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 224.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 225.Shaheen FS, Znojek P, Fisher A, Webster M, Plummer R, Gaughan L, Smith GC, Leung HY, Curtin NJ, Robson CN. Targeting the DNA double strand break repair machinery in prostate cancer. PLoS One. 2011;6:e20311. doi: 10.1371/journal.pone.0020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Srivastava N, Gochhait S, Gupta P, Bamezai RN. Copy number alterations of the H2AFX gene in sporadic breast cancer patients. Cancer Genet Cytogenet. 2008;180:121–128. doi: 10.1016/j.cancergencyto.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 227.Prabhu LV, Kini H, Nayak SR, Pai MM, K N. Ilio-femoral aneurysm masquerading as an inguinal abscess. Rom J Morphol Embryol. 2007;48:161–163. [PubMed] [Google Scholar]
- 228.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, Kim R, Rubin MA, Pienta KJ. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 229.Hulboy DL, Matrisian LM, Crawford HC. Loss of JunB activity enhances stromelysin 1 expression in a model of the epithelial-to-mesenchymal transition of mouse skin tumors. Mol Cell Biol. 2001;21:5478–5487. doi: 10.1128/MCB.21.16.5478-5487.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hay ED, Zuk A. Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis. 1995;26:678–690. doi: 10.1016/0272-6386(95)90610-x. [DOI] [PubMed] [Google Scholar]
- 231.Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3 alpha-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3 alpha/17 beta-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 232.El-Alfy M, Luu-The V, Huang XF, Berger L, Labrie F, Pelletier G. Localization of type 5 17beta-hydroxysteroid dehydrogenase, 3beta-hydroxysteroid dehydrogenase, and androgen receptor in the human prostate by in situ hybridization and immunocytochemistry. Endocrinology. 1999;140:1481–1491. doi: 10.1210/endo.140.3.6585. [DOI] [PubMed] [Google Scholar]
- 233.Dufort I, Rheault P, Huang XF, Soucy P, Luu-The V. Characteristics of a highly labile human type 5 17beta-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:568–574. doi: 10.1210/endo.140.2.6531. [DOI] [PubMed] [Google Scholar]
- 234.Coppola V, De Maria R, Bonci D. MicroRNAs and prostate cancer. Endocr Relat Cancer. 2010;17:F1–17. doi: 10.1677/ERC-09-0172. [DOI] [PubMed] [Google Scholar]
- 235.Gandellini P, Folini M, Zaffaroni N. Towards the definition of prostate cancer-related microRNAs: where are we now? Trends Mol Med. 2009;15:381–390. doi: 10.1016/j.molmed.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 236.Heneghan HM, Miller N, Kerin MJ. MiRNAs as biomarkers and therapeutic targets in cancer. Curr Opin Pharmacol. 2010;10:543–550. doi: 10.1016/j.coph.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 237.Sevli S, Uzumcu A, Solak M, Ittmann M, Ozen M. The function of microRNAs, small but potent molecules, in human prostate cancer. Prostate Cancer Prostatic Dis. 2010;13:208–217. doi: 10.1038/pcan.2010.21. [DOI] [PubMed] [Google Scholar]
- 238.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 239.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 240.Piriyapongsa J, Marino-Ramirez L, Jordan IK. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176:1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gandellini P, Folini M, Zaffaroni N. Emerging role of microRNAs in prostate cancer: implications for personalized medicine. Discov Med. 2010;9:212–218. [PubMed] [Google Scholar]
- 242.Watahiki A, Wang Y, Morris J, Dennis K, O’Dwyer HM, Gleave M, Gout PW. MicroRNAs associated with metastatic prostate cancer. PLoS One. 2011;6:e24950. doi: 10.1371/journal.pone.0024950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ostling P, Leivonen SK, Aakula A, Kohonen P, Makela R, Hagman Z, Edsjo A, Kangaspeska S, Edgren H, Nicorici D, Bjartell A, Ceder Y, Perala M, Kallioniemi O. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011;71:1956–1967. doi: 10.1158/0008-5472.CAN-10-2421. [DOI] [PubMed] [Google Scholar]
- 244.Yamamura S, Saini S, Majid S, Hirata H, Ueno K, Deng G, Dahiya R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS One. 2012;7:e29722. doi: 10.1371/journal.pone.0029722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kong D, Heath E, Chen W, Cher M, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O, Hwang C, Gupta N, Chitale D, Sakr WA, Menon M, Sarkar FH. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-DIM treatment. Am J Transl Res. 2012;4:14–23. [PMC free article] [PubMed] [Google Scholar]
- 246.Banerjee S, Kong D, Wang Z, Bao B, Hillman GG, Sarkar FH. Attenuation of multi-targeted proliferation-linked signaling by 3,3’-diindolylmethane (DIM): from bench to clinic. Mutat Res. 2011;728:47–66. doi: 10.1016/j.mrrev.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Heath EI, Heilbrun LK, Li J, Vaishampayan U, Harper F, Pemberton P, Sarkar FH. A phase I dose-escalation study of oral BR-DIM (BioResponse 3,3’- Diindolylmethane) in castrate-resistant, non-metastatic prostate cancer. Am J Transl Res. 2010;2:402–411. [PMC free article] [PubMed] [Google Scholar]
- 248.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442–17452. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Talesa VN, Antognelli C, Del Buono C, Stracci F, Serva MR, Cottini E, Mearini E. Diagnostic potential in prostate cancer of a panel of urinary molecular tumor markers. Cancer Biomark. 2009;5:241–251. doi: 10.3233/CBM-2009-0109. [DOI] [PubMed] [Google Scholar]
- 251.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Manolescu A, Gudbjartsson D, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Blondal T, Jakobsdottir M, Stacey SN, Kostic J, Kristinsson KT, Birgisdottir B, Ghosh S, Magnusdottir DN, Thorlacius S, Thorleifsson G, Zheng SL, Sun J, Chang BL, Elmore JB, Breyer JP, McReynolds KM, Bradley KM, Yaspan BL, Wiklund F, Stattin P, Lindstrom S, Adami HO, McDonnell SK, Schaid DJ, Cunningham JM, Wang L, Cerhan JR, St Sauver JL, Isaacs SD, Wiley KE, Partin AW, Walsh PC, Polo S, Ruiz-Echarri M, Navarrete S, Fuertes F, Saez B, Godino J, Weijerman PC, Swinkels DW, Aben KK, Witjes JA, Suarez BK, Helfand BT, Frigge ML, Kristjansson K, Ober C, Jonsson E, Einarsson GV, Xu J, Gronberg H, Smith JR, Thibodeau SN, Isaacs WB, Catalona WJ, Mayordomo JI, Kiemeney LA, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40:281–283. doi: 10.1038/ng.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Drieschner N, Kerschling S, Soller JT, Rippe V, Belge G, Bullerdiek J, Nimzyk R. A domain of the thyroid adenoma associated gene (THADA) conserved in vertebrates becomes destroyed by chromosomal rearrangements observed in thyroid adenomas. Gene. 2007;403:110–117. doi: 10.1016/j.gene.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 254.Eeles RA, Kote-Jarai Z, Al Olama AA, Giles GG, Guy M, Severi G, Muir K, Hopper JL, Henderson BE, Haiman CA, Schleutker J, Hamdy FC, Neal DE, Donovan JL, Stanford JL, Ostrander EA, Ingles SA, John EM, Thibodeau SN, Schaid D, Park JY, Spurdle A, Clements J, Dickinson JL, Maier C, Vogel W, Dork T, Rebbeck TR, Cooney KA, Cannon-Albright L, Chappuis PO, Hutter P, Zeegers M, Kaneva R, Zhang HW, Lu YJ, Foulkes WD, English DR, Leongamornlert DA, Tymrakiewicz M, Morrison J, Ardern-Jones AT, Hall AL, O’Brien LT, Wilkinson RA, Saunders EJ, Page EC, Sawyer EJ, Edwards SM, Dearnaley DP, Horwich A, Huddart RA, Khoo VS, Parker CC, Van As N, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Cooper CS, Southey MC, Lophatananon A, Liu JF, Kolonel LN, Le Marchand L, Wahlfors T, Tammela TL, Auvinen A, Lewis SJ, Cox A, FitzGerald LM, Koopmeiners JS, Karyadi DM, Kwon EM, Stern MC, Corral R, Joshi AD, Shahabi A, McDonnell SK, Sellers TA, Pow-Sang J, Chambers S, Aitken J, Gardiner RA, Batra J, Kedda MA, Lose F, Polanowski A, Patterson B, Serth J, Meyer A, Luedeke M, Stefflova K, Ray AM, Lange EM, Farnham J, Khan H, Slavov C, Mitkova A, Cao G, Easton DF. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009;41:1116–1121. doi: 10.1038/ng.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, Donovan JL, Hamdy FC, Dearnaley DP, Muir KR, Smith C, Bagnato M, Ardern-Jones AT, Hall AL, O’Brien LT, Gehr-Swain BN, Wilkinson RA, Cox A, Lewis S, Brown PM, Jhavar SG, Tymrakiewicz M, Lophatananon A, Bryant SL, Horwich A, Huddart RA, Khoo VS, Parker CC, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Fisher C, Jamieson C, Cooper CS, English DR, Hopper JL, Neal DE, Easton DF. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 256.Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, Gylfason A, Agnarsson BA, Benediktsdottir KR, Magnusdottir DN, Orlygsdottir G, Jakobsdottir M, Stacey SN, Sigurdsson A, Wahlfors T, Tammela T, Breyer JP, McReynolds KM, Bradley KM, Saez B, Godino J, Navarrete S, Fuertes F, Murillo L, Polo E, Aben KK, van Oort IM, Suarez BK, Helfand BT, Kan D, Zanon C, Frigge ML, Kristjansson K, Gulcher JR, Einarsson GV, Jonsson E, Catalona WJ, Mayordomo JI, Kiemeney LA, Smith JR, Schleutker J, Barkardottir RB, Kong A, Thorsteinsdottir U, Rafnar T, Stefansson K. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009;41:1122–1126. doi: 10.1038/ng.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 258.Zheng SL, Stevens VL, Wiklund F, Isaacs SD, Sun J, Smith S, Pruett K, Wiley KE, Kim ST, Zhu Y, Zhang Z, Hsu FC, Turner AR, Johansson JE, Liu W, Kim JW, Chang BL, Duggan D, Carpten J, Rodriguez C, Isaacs W, Gronberg H, Xu J. Two independent prostate cancer risk-associated Loci at 11q13. Cancer Epidemiol Biomarkers Prev. 2009;18:1815–1820. doi: 10.1158/1055-9965.EPI-08-0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sun J, Zheng SL, Wiklund F, Isaacs SD, Purcell LD, Gao Z, Hsu FC, Kim ST, Liu W, Zhu Y, Stattin P, Adami HO, Wiley KE, Dimitrov L, Li T, Turner AR, Adams TS, Adolfsson J, Johansson JE, Lowey J, Trock BJ, Partin AW, Walsh PC, Trent JM, Duggan D, Carpten J, Chang BL, Gronberg H, Isaacs WB, Xu J. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat Genet. 2008;40:1153–1155. doi: 10.1038/ng.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sun J, Zheng SL, Wiklund F, Isaacs SD, Li G, Wiley KE, Kim ST, Zhu Y, Zhang Z, Hsu FC, Turner AR, Stattin P, Liu W, Kim JW, Duggan D, Carpten J, Isaacs W, Gronberg H, Xu J, Chang BL. Sequence variants at 22q13 are associated with prostate cancer risk. Cancer Res. 2009;69:10–15. doi: 10.1158/0008-5472.CAN-08-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lapointe J, Li C, Giacomini CP, Salari K, Huang S, Wang P, Ferrari M, Hernandez-Boussard T, Brooks JD, Pollack JR. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. 2007;67:8504–8510. doi: 10.1158/0008-5472.CAN-07-0673. [DOI] [PubMed] [Google Scholar]