

Abstract
A forward genetic screen in the ascidian Ciona intestinalis identified a mutant line (frimousse) with a profound disruption in neural plate development. In embryos with the frimousse mutation, the anteriormost neural plate cells, which are products of an FGF induction at the blastula and gastrula stages, initially express neural plate-specific genes but fail to maintain the induced state and ultimately default to epidermis. The genetic lesion in the frimousse mutant lies within a connexin gene (cx-11) that is transiently expressed in the developing neural plate in a temporal window corresponding to the period of a-lineage neural induction. Using a genetically encoded calcium indicator we observed multiple calcium transients throughout the developing neural plate in wild-type embryos, but not in mutant embryos. A series of treatments at the gastrula and neurula stages that block the calcium transients, including gap junction inhibition and calcium depletion, were also found to disrupt the development of the anterior neural plate in a similar way to the frimousse mutation. The requirement for cx-11 for anterior neural fate points to a crucial role for intercellular communication via gap junctions, probably through mediation of Ca2+ transients, in Ciona intestinalis neural induction.
Keywords: Ciona, Calcium transients, Connexin, Neural induction
INTRODUCTION
The ascidian larval CNS is a comparatively simple organ. It is composed of only ∼330 cells in Ciona intestinalis and is organized along the rostral/caudal axis into several morphologically distinct regions: the sensory vesicle, neck, visceral ganglion and caudal nerve cord (Meinertzhagen et al., 2004). The close evolutionary relationship of ascidians to their vertebrate cousins (Delsuc et al., 2006) can be seen in the morphogenesis and anatomy of their respective CNSs. Although a one-to-one correspondence of the anatomical regions of the ascidian CNS to those of vertebrates may not be exact, reflecting their extensive divergence, and is the subject of conflicting interpretation (Dufour et al., 2006), gene expression and anatomical data have equated the sensory vesicle with the vertebrate forebrain, the neck region with the vertebrate midbrain-hindbrain boundary, the visceral ganglion with the hindbrain, and the caudal nerve cord with the vertebrate spinal cord (Meinertzhagen et al., 2004; Imai and Meinertzhagen, 2007).
Unlike in vertebrates, the ascidian CNS develops according to a fixed, and well-described, cell lineage (Nishida, 1987; Cole and Meinertzhagen, 2004). The three primary lineages that contribute to the ascidian CNS trace back to the 8-cell stage. The A-lineage, which gives rise to the posterior sensory vesicle, neck, visceral ganglion and ventral nerve cord, is so-called because it originates from the A4.1 pair of blastomeres. In a similar fashion, the a-lineage descends from the a4.2 blastomeres and gives rise to the anterior sensory vesicle, as well as to two non-neural derivatives of the neural plate, the adhesive palps, which are found at the anterior pole of the larva, and the oral siphon primordium, which is found immediately anterior to the sensory vesicle (Nishida, 1987; Veeman et al., 2010). The final lineage to contribute to the ascidian CNS, the b-lineage, originates from the b4.2 blastomeres and contributes to the dorsal nerve cord.
The early specification and development of the ascidian CNS have been most extensively studied in the distantly related species Ciona intestinalis and Halocynthia roretzi (Bertrand et al., 2003; Miya and Nishida, 2003; Meinertzhagen et al., 2004; Wada et al., 2004; Imai et al., 2006; Lemaire et al., 2008). Blastomere isolation experiments in H. roretzi have shown that neural specification occurs in isolated A4.1 blastomeres, but not in a4.2 or b4.2 blastomeres, suggesting that the A-lineage arises cell-autonomously, whereas the a- and b-lineages require induction (Nishida, 1991). For the a-lineage, induction by FGF signaling starting in the early cleavage stages has been shown to be crucial in both C. intestinalis and H. roretzi (Kim and Nishida, 2001; Bertrand et al., 2003; Miya and Nishida, 2003). Induction can be observed as early as the 32- to 64-cell stage by the expression of the transcription factors otx and dmrt1 in the neural precursor cells (Bertrand et al., 2003; Tresser et al., 2010). The source of the a-lineage inducer has been identified in C. intestinalis as the vegetally localized FGF9/16/20-producing A4.1 descendants (Bertrand et al., 2003).
The induction of the a-lineage in ascidians is hypothesized to be evolutionarily conserved with vertebrate anterior neural induction (Meinertzhagen et al., 2004). Although both require FGF signaling (Launay et al., 1996; Sasai et al., 1996; Bertrand et al., 2003), BMP inhibitors do not play a role in the ascidian process (Darras and Nishida, 2001). We have previously described a spontaneous mutant line, frimousse (frm), in C. intestinalis in which the development of the a-lineage is profoundly disrupted (Deschet and Smith, 2004). Homozygous frm embryos lack palps, the oral siphon precursor and the anterior sensory vesicle, as seen by the absence of pigment cells and Arrestin staining (Fig. 1). Lineage-tracing and expression studies demonstrated that the a-lineage neural plate derivatives in frm/frm embryos become misspecified as epidermis after having initially expressed, and subsequently lost, markers of neural specification. The conclusion was that the gene disrupted by the frm mutation plays a role in maintaining neural plate identity in the a-lineage (Deschet and Smith, 2004). Additionally, the a-lineage cells misfated from the sensory vesicle in frm/frm embryos remained on the surface of the embryo as a thickened epidermis (Fig. 1B, arrow), rather than neurulating, giving an open rostral neural tube phenotype, whereas the rostral A-lineage components of the CNS, including the posterior sensory vesicle and visceral ganglion, appeared to be intact in frm/frm embryos (Fig. 1C,D, CRALBP staining). We report here that the causative mutation in the frm line lies within a connexin gene that is transiently expressed in the neural plate.
Fig. 1.

Characterization of the frimousse mutation. (A,B) Wild-type (A) and frm/frm (B) late tailbud stage Ciona intestinalis embryos. (C,D) Wild-type (C) and frm/frm (D) larvae immunostained for CRALBP (pan-CNS) and Arrestin (sensory vesicle). VG, visceral ganglion; SV, sensory vesicle. (E) Mapping of the frm mutation on chromosome 2q. Sequence traces (top) show linkage by SNP mapping to position 2.67 Mb. Finer mapping in the region 1.0 to 2.5 Mb with INDEL markers is shown (bottom). Single recombinant frm/frm tadpoles are indicated (F9, B4, G12, H5, F10). Circles indicate where only the linked INDEL allele was observed, and X indicates where a recombination resulted in both INDEL alleles being present. (F) The C. intestinalis connexin-like 11 (cx-11) gene, which in frm embryos contains a 4 bp deletion at the 5′ splice site of the single intron. (G) Comparison of the cx-11 5′ splice sites in wild type (wt) and in the frm allele with the consensus sequence (Wang and Burge, 2008). Variation from consensus are indicated in red. (H) RT-PCR for cx-11 using intron-spanning primers. Splicing is greatly reduced in cDNA from frm/frm embryos. RT–, controls without reverse transcriptase. (I) Injection of morpholinos (MOs) to cx-11 phenocopies the frm phenotype. (J) RT-PCR for three anterior neural plate-expressed genes (HO9, brain; six3/6, oral siphon primordium; SP-8, palp) and actin (loading control) in embryos injected with control MO or cx-11 MOs and in homozygous frm embryos (frm/frm). No temp., no template control.
MATERIALS AND METHODS
SNP and INDEL mapping
The frm mutation was mapped using SNP markers, as described previously (Veeman et al., 2011). The first round of linkage analysis used primers designed at two loci equidistant along each of the 14 chromosome arms of C. intestinalis, with the exception of chromosome arms (4p, 5p and 6p) containing rDNA. Finer resolution mapping was then achieved using either pooled genomic DNA or genomic DNA from single larvae with region-specific primers. Recombination was quantified either by measuring peak heights from sequencing traces for SNPs (Veeman et al., 2011) or by gel electrophoresis on a 3.5% agarose gel using a 50:50 mixture of standard molecular grade agarose and MetaPhor agarose (Cambrex Bio Science, Rockland, ME, USA)
PCR products spaced within the linked region were analyzed for the presence of INDELs differing between wild-type and frm/frm embryos. Single tadpoles were sorted and placed in individual wells of a 96-well plate. Genomic DNA was extracted as described (Veeman et al., 2011) and then PCR amplified for the INDEL-containing region. PCR products were then analyzed by gel electrophoresis as above.
RT-PCR
Heterozygous frm adults were spawned to generate pools of 150 phenotypically wild-type (wt/wt and wt/frm) and phenotypically mutant (frm/frm) larvae. RNA isolation was performed using Trizol (Invitrogen) followed by phenol/chloroform extraction and ethanol precipitation. cDNA was prepared using the Advantage RT for PCR Kit (Clontech). Primer sequences for RT-PCR of cx-11 were 5′-CGATACTAAAAACCCCAAGC-3′ and 5′-GGCATCTTCGTCACTTTCTT-3′.
Whole-mount embryo staining
Immunostaining for Arrestin and cellular retinaldehyde-binding protein (CRALBP) (Tsuda et al., 2003; Tresser et al., 2010), Bodipy-FL phallacidin staining and embryo imaging (Veeman et al., 2010) and fluorescent in situ hybridizations (Christiaen et al., 2009b) were performed as described previously. For cx-11, 1211 bp digoxigenin-labeled sense and antisense probes were prepared from the full-length cDNA. Colorimetric in situ staining was performed on embryos as previously described (Christiaen et al., 2009b), except that after tyramide amplification and washes the embryos were incubated with a 1:1000 dilution of anti-fluorescein AP-conjugated antibody (Perkin-Elmer) in PBST (PBS with 0.1% Tween 20) for 1 hour at room temperature then at 4°C overnight. Embryos were then washed five times in PBST and stained with NBT/BCIP.
Gene knockdown/inhibition
Cx-11 was knocked down using a 1:1 mixture of translation-blocking and splice-blocking morpholinos (MOs): cx-11 translational block, 5′-ATATGTTCCACATCGTGTAATATGT-3′; cx-11 splice block, 5′-TCTCGCTGTGGAAGAAAGATAAGTA-3′. MO injections were performed as described previously (Yamada et al., 2003). Control embryos received the MO 5′-CCTCTTACCTCAGTTACAATTTATA-3′.
RT-PCR with cDNA from MO-injected embryos was performed with primers: HO9, 5′-ATGTAAACCACTCCCCGTGA-3′ and 5′-GCACTTGAAATGCAAACAGG-3′; six3/6, 5′-GCTCTGTCTCGTTGCCTTCT-3′ and 5′-CAATTGTTGCGTTTCACCAG-3′; SP-8, 5′-CAATTGTTGCGTTTCACCAG-3′ and 5′-ACGTAGGGTGACGAACTTGG-3′; and actin, 5′-GTGCTTTCATGGTACGCTTCT-3′ and 5′-CGGCGATTCCAGGGAACATAG-3′.
Embryo treatments
β-glycyrrhetinic acid treatment
β-glycyrrhetinic acid was dissolved in DMSO to make a 1000× stock solution, and then added to dechorionated embryos at a final concentration of 100 μM. Control embryos received DMSO only. After treatment (2-3 hours at 18°C, depending on protocol) the embryos were transferred through several Petri dishes of sea water to wash out the drugs. Treated embryos were cultured at 13°C or 15°C to hatching stage, at which point they were fixed and assessed for developmental defects.
Low Ca2+ treatment
Fertilized, dechorionated embryos were grown in filtered natural sea water with 2.5 μg/ml each streptomycin and kanamycin until early gastrula stage at 18°C. Embryos were transferred into artificial sea water (ASW) with either 11 mM (regular) or 0.5 mM (low) CaCl2. Embryos were grown until initial tailbud stage (∼3.5 hours), at which time the 0.5 mM Ca2+-treated embryos were transferred through three washes with 11 mM Ca2+ ASW. Embryos were then grown to late tailbud stage to assess phenotype.
Transgenic constructs
etr1p::GCaMP5 was constructed using the Gateway system (Invitrogen). GCaMP5 (gift of J. Akerboom) (Akerboom et al., 2012) was amplified with primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCAGAAAAAATGGGTTCTCATCATCATCA-3′ and 5′-GGGGACACTTTGTACAAGAAAGCTGGGTTTATCACTTCGCTGTCATCATTTGTAC-3′. This was then recombined into pDONR 221 (Invitrogen) and the resultant entry vector (L1/GCaMP-5/L2) was recombined using LR Clonase (Invitrogen) with L3/etr1p/L5 into destination vector pSP1.72BSSPE-R3-R5-RFA (Roure et al., 2007). Fertilized, dechorionated embryos were then electroporated (Christiaen et al., 2009a) with 50 μg etr1p::GCaMP-5 and imaged for 2.5-4 minutes on a Leica DMI6000B inverted microscope with a Hamamatsu ImageEM C9100-13 electron multiplier charge-coupled device (EM-CCD) camera at 31 frames per second. Calcium transient movies were created using QuickTime Pro (Apple). To calculate relative fluorescence, cells in digital images were manually outlined and quantified using ImageJ (Abramoff et al., 2004).
For Cx-11p::Cx-11-H2B::GFP, primers 5′-TTTCGCAGCGGTATGTGCTTCTGTGG-3′ and 5′-CGGAGCACTCGGAAACAGAATATGTG-3′ were used to PCR amplify a 3.75 kb genomic fragment containing the two cx-11 exons and intervening intron, and 2.1 kb of upstream sequence from the predicted start ATG. The PCR product was cloned into the PCR8/GW/TOPO TA vector and recombined with RFA-H2B::GFP to generate a C-terminal nuclear-localized GFP fusion protein (Roure et al., 2007).
etr1p::OtxHD:enR was created using the Gateway system by first amplifying OtxHD:enR from pSix3:OtxHD:enR (Haeussler et al., 2010) using primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCAGAAAAAATGGTATACAGTTCGTCTAG-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTAATTCTATACGTTCAGGTCCT-3′ with att B1/att B2-kozac feet attached. The resulting PCR product was then BP cloned into pDONR 221 to create an L1/OtxEnR/L2 entry vector. This entry vector was then recombined using LR Clonase with L3/etr1p/L5 into the destination vector pSP1.72BSSPE-R3-R5-RFA (Roure et al., 2007); the construct was electroporated as above.
RESULTS
The frimousse mutation disrupts a connexin gene
We mapped the frm mutation using pooled genomic samples from frm/frm and wild-type larvae with a set of single-nucleotide polymorphism (SNP) markers distributed along the C. intestinalis chromosomes (Veeman et al., 2011). An initial round of mapping linked the mutant locus to the 8.06 Mb chromosome arm 2q (Fig. 1E). Finer analysis of SNP markers in pooled samples showed strongest linkage between 0.5 Mb and 3.5 Mb on 2q (supplementary material Fig. S1A). Higher resolution mapping required analysis of genomic DNA from single frm/frm larvae for recombination in this region. Several hundred frm/frm larvae were first screened using an INDEL marker for recombination in this genomic region. Linkage analyses with five selected recombinant larvae using a set of nine INDEL markers in the region 1.20 to 2.50 Mb are shown in Fig. 1E. This final linkage analysis was able to narrow down the linked region to a genomic segment of ∼0.41 Mb (Fig. 1E, yellow box).
A combination of quantitative RT-PCR and sequencing was then used to narrow down the ∼40 predicted genes in this region. Our previous mutation-mapping projects in Ciona have shown that transcript downregulation is a good predictor of mutant genes (Jiang et al., 2005; Veeman et al., 2008; Chiba et al., 2009; Tresser et al., 2010). One transcript in particular within the region showed strong downregulation: a predicted connexin gene annotated as connexin like-11 (cx-11) (supplementary material Fig. S1B). C. intestinalis cx-11 is a predicted two-exon gene with a short intron (Fig. 1F). Comparison of the nucleotide sequence of the cx-11 genes from frm/frm and wild-type genomic DNAs identified a 4 bp deletion in the frm/frm gene at the boundary between the first exon and the intron (Fig. 1F). Although the deletion did not result in the most highly conserved nucleotides at the splice junction being altered, several bases downstream from the junction were altered from the consensus (Fig. 1G). An analysis was performed on the wild-type and mutated splice sites using SplicePort, which calculates the probability of splice junctions (Dogan et al., 2007). When the wild-type cx-11 genomic sequence was analyzed, the strongest predicted candidate sequences for splice donor and acceptor sites were those indicated in Fig. 1F. Both sites scored a value of over +1.4, which the algorithm gives as the lowest false-positive rate (FPR) and highest probability. However, when the analysis was run to include the 4 bp deletion in the frm/frm sequence, the splice donor site, which had previously scored over +1.4, now scored –0.22, significantly decreasing probability and increasing the FPR.
In order to directly assess whether these changes would disrupt cx-11 splicing, PCR primers spanning the intron were used to amplify cDNA from frm/frm and wild-type embryos. Although the cx-11 transcript was downregulated in frm/frm embryos (supplementary material Fig. S1B), we were able to detect transcript by increasing the number of PCR cycles (Fig. 1H). The amount of properly spliced cx-11 transcript was dramatically reduced in cDNA samples from frm/frm embryos in comparison to cDNA from wild-type embryos, indicating that the 4 bp deletion detected in the mutant cx-11 allele was sufficient to greatly reduce splicing at this site. Unspliced transcript contains a premature stop codon following exon 1.
The strong genetic linkage, downregulation and splicing defect in the mutant allele of cx-11 made it a strong candidate for the mutation causing the frm phenotype. Final support was obtained when it was observed that injection of mixed splice-blocking and translation-blocking cx-11 morpholino antisense oligonucleotides (cx-11 MOs) into wild-type embryos phenocopied the frm phenotype in 16 of 18 cx-11 MO-injected embryos scored. The cx-11 MO-injected embryos showed reduced or absent palps, pigment cells and Arrestin staining (Fig. 1I). RT-PCR was performed on pooled cDNA from six cx-11 MO-injected and six control MO-injected embryos. The cx-11 MO-injected embryos showed knockdown of the brain marker HO9, the oral siphon primordium marker six3/6 and the palp marker SP-8 comparable to that in frm/frm embryos (Fig. 1J).
cx-11 is transiently expressed in neural precursor cells
The temporal expression of cx-11 was examined by RT-PCR in staged cDNA samples (Fig. 2A) [see Hotta et al. (Hotta et al., 2007) for C. intestinalis staging]. A low level of maternal transcript was found in eggs and appears to persist through early cleavage stages. Much stronger expression of cx-11 was observed starting at the 32-cell stage and persisted through neurula stages. At the neurula stage, transcript levels were already declining, and by early tailbud stage the transcript level was greatly reduced. Thus, elevation of cx-11 expression is observed in a very narrow temporal window corresponding to the period of a-lineage neural induction in C. intestinalis.
Fig. 2.

Cx-11 expression during C. intestinalis development. (A) RT-PCR for cx-11 and actin (control) at various stages from egg to late tailbud. l. neurula, late neurula; e. tailbud, early tailbud; l. tailbud, late tailbud. (B-H) Cx-11 expression in embryos at the 32-cell (B,C), 112-cell (D-F) and neurula (G,H) stages as detected by fluorescent in situ hybridization. In the diagrams, cx-11-expressing cells are colored: orange, A-lineage; red, a-lineage; dark blue, secondary muscle cells; light blue, primary muscle cells. (I) Expression of a cx-11/nuclear-GFP fusion protein (green) driven by a 2.1 kb genomic region 5′ of the predicted cx-11 translation start. Embryos were also stained with phallacidin (red) to outline cells. Image shows a three-dimensional reconstruction from a laser-scanning confocal microscope.
The spatial expression of cx-11 was examined by fluorescent whole-mount in situ hybridization in embryos at the peak stages of cx-11 expression (32-cell to neurula). At all stages examined, specific hybridization was observed in both the neural and muscle precursor cells. At the 32-cell stage (Fig. 2B,C), cx-11 expression was strongest in the B6.2 and B6.4 primary muscle precursor cells, but weak expression was observed in the A6.2 and A6.4 cells, which are the precursors of both the CNS A-lineage and primary notochord cells (Minokawa et al., 2001). At the next round of division of the A6.2 and A6.4 cells, the notochord and CNS fates segregate (44-cell stage). We were unable to detect expression of cx-11 at this stage in the a-lineage, although we could not rule out a very low level of expression. No specific hybridization was observed with a control sense probe (supplementary material Fig. S2). At the initial gastrula (112-cell) stage, expression of cx-11 was observed in the CNS a-lineage, as well as in the eight A-lineage neural precursor cells, including the A8.16 cells, which at this stage are fated to give rise to both lateral caudal nerve cord and secondary muscle cells (Fig. 2D-F) (Hudson and Yasuo, 2008). Expression was also observed in the primary muscle lineage (Fig. 2F, light blue cells) and in the b8.17 cells (dark blue), which give rise to both the b-lineage of the CNS and to secondary muscle cells. Expression of cx-11 was already declining by the neural plate stage (Fig. 2A), making the hybridization signals difficult to detect, although faint cx-11 expression could be detected in the neural plate in both the A- and a-lineages (Fig. 2G,H), with the strongest hybridization seen in the presumptive posterior muscles.
Cx-11 expression was also investigated using a transiently expressed transgenic construct (Cx-11p::Cx-11-H2B::GFP) that encodes a nuclear-localized GFP fused at the end of the second cx-11 exon driven by 2.1 kb of 5′ upstream genomic sequence. Because of the perdurance of GFP we were able to detect cx-11/nuclear-GFP fusion-expressing cells at the tailbud stage (Fig. 2I). Consistent with the in situ results, we observed GFP-labeled nuclei throughout the CNS, as well as the atrial siphon primordium, palps and posterior muscle.
Gap junction inhibition mimics the frm phenotype
The genetic, expression and MO knockdown data all point to a mutation in cx-11 as being the cause of the frm phenotype. To further investigate a possible role for gap junctions in the development of the C. intestinalis CNS, embryos were treated with β-glycyrrhetinic acid (βGA), a widely used gap junction channel blocker with broad specificity (Juszczak and Swiergiel, 2009). In a preliminary experiment to determine the efficacy and reversibility of gap junction blockage by βGA, swimming C. intestinalis larvae were soaked in 100 μM βGA. Within 5 minutes of βGA addition, larvae stopped swimming, presumably due to inhibition of the spread of excitation by gap junctions between muscle cells (Bone, 1992; Horie et al., 2010). Transferring the βGA-treated larvae back to plain sea water gradually restored normal swimming over a period of 15-30 minutes.
The ease with which βGA could be added to, and washed out from, the embryos allowed us to precisely determine if, and when, there was a requirement for gap junction communication in C. intestinalis CNS development. In these experiments, C. intestinalis embryos were treated with βGA for either 2 or 3 hours starting at various time points, the earliest being mid-gastrulation (Fig. 3A). The embryos were scored for regional morphological markers including palps, pigment cells, and neural tube closure, and were immunostained for both CRALBP and Arrestin (Tsuda et al., 2003) (see Fig. 1C,D). Addition of βGA at the 32-cell stage resulted in grossly abnormal embryos that failed to gastrulate and could not be scored. The defects from βGA treatments starting at the gastrula stage were much more discrete. Two-hour βGA treatments starting at mid-gastrula, late gastrula or mid-neurula stages (treatments 1, 2 and 3; Fig. 3A) were all effective at preventing the appearance of the palps as assessed at late tailbud stage (palps were identified on only 27%, 16% and 15% of embryos, respectively), whereas the majority of embryos treated for 2 hours at either late neurula or tailbud stages (treatments 4 and 5; Fig. 3A) had palps (58% and 87%, respectively). Three-hour βGA treatments starting either at mid- or late gastrula were even more effective at blocking the development of the palps. The induction of the sensory vesicle was assessed by Arrestin staining. Two-hour βGA treatment resulted in 48% of the embryos having greatly reduced Arrestin staining and 17% completely lacked Arrestin staining (Fig. 3A,B). Three-hour βGA treatment increased these percentages to 53% and 42%, respectively. Consistent with the timing of induction of pigment cells (Nishida, 1991), we observed that later (from late gastrula) and longer (3-hour) βGA treatment was required to block pigment cell induction. Of these embryos, 47% had weak pigment expression and 21% had no detectable pigment. Finally, the 3-hour βGA treatment was effective in generating an open brain phenotype at percentages similar to those seen in frm embryos.
Fig. 3.
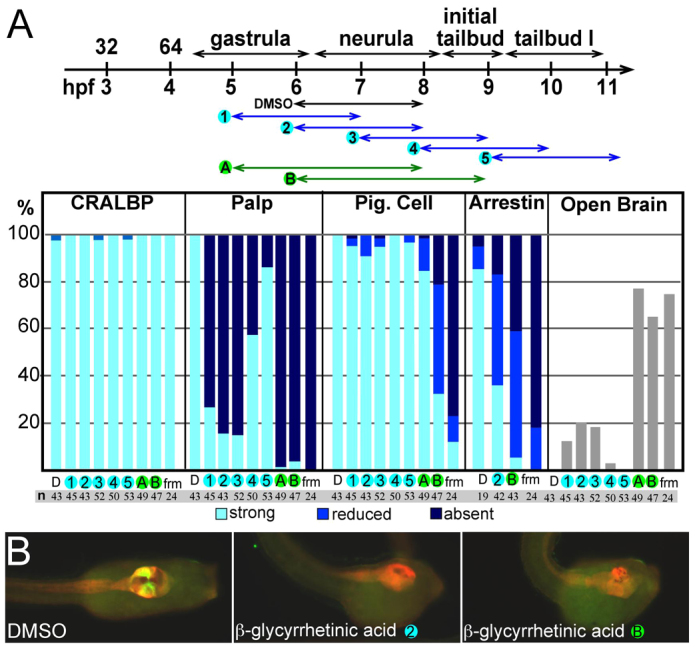
β-glycyrrhetinic acid disrupts CNS development. (A) β-glycyrrhetinic acid (βGA) disrupts CNS development. At the top is a timecourse of embryo treatments with βGA (or DMSO as control). Blue arrows indicate 2-hour treatments and green arrows indicate 3-hour treatments. The treated embryos (and frm/frm embryos) were scored for both morphological defects (disruption of palps and open brains) and molecular markers [pigment (pig.) cells (melanin) and immunostaining for CRALBP and Arrestin]. The column labels at the bottom of the bar chart correspond to numbers/letters in the timecourse and indicate number of embryos examined (n). hpf, hours post-fertilization; D, DMSO control; frm, frm/frm embryo. (B) Representative control (DMSO) and βGA-treated embryos from two different treatment courses. Embryos are immunostained for CRALBP (red) and Arrestin (green/yellow).
In summary, βGA establishes a crucial role for gap junctions in the development of anterior neural plate derivatives in C. intestinalis. Moreover, it identifies different sensitivity periods for palp, pigment cell and sensory vesicle induction. The defects caused by βGA treatment appeared to be confined to the a-lineage; the A-lineage was largely normal, as seen by the persistence of CRALBP staining in the posterior sensory vesicle and visceral ganglion.
Calcium transients in the Ciona neural plate
Having identified a cx-11 mutation as the cause of the frm phenotype, we investigated possible mechanisms of action for gap junctions in C. intestinalis CNS development. Studies in Xenopus have shown an essential role for Ca2+ transients in neural induction (Leclerc et al., 2000; Leclerc et al., 2006; Moreau et al., 2008), and, although gap junction communication has not been shown to be involved in Ca2+ transients in the Xenopus neural plate, in other contexts gap junctions are essential for Ca2+ transients (Orellana et al., 2012). To investigate the presence of Ca2+ transients in the C. intestinalis neural plate, we transiently expressed a transgene construct containing the fluorescent Ca2+ reporter protein GCaMP5 (Akerboom et al., 2012) using the cis-regulatory region from the pan-neural gene etr1 (etr1p::GCaMP5) (Yagi and Makabe, 2001; Tresser et al., 2010). The transient expression method in Ciona, which involves electroporating hundreds of embryos at a time, gives variable levels of expression, and Ca2+ transients could typically be seen in half or fewer of the electroporated embryos. Additionally, we could not image embryos for Ca2+ transients longer than ∼5 minutes without severely damaging the embryos. Expression of the transgene was detectable as early as the late gastrula stage, when sporadic Ca2+ transients were seen in the neural plate (Fig. 4A; supplementary material Movie 1). By mid-neurula stage the Ca2+ transients were much more abundant and moved between clusters of adjacent cells throughout the a- and A-lineages of the neural plate (Fig. 4B; supplementary material Movie 2). The etr1 promoter also drives expression in a subset of muscle cells (Veeman et al., 2010) and in supplementary material Movie 2 a muscle transient can also be seen. Fig. 4C shows the relative fluorescence intensity in seven cells in one embryo [one cell being a non-transgenic negative control (cell 1)] over a 90-second interval. All of the measured cells showed one or two Ca2+ transients during this period, each of ∼10 seconds in duration (with the exception of the negative control cell).
Fig. 4.

Ca2+ transients in the neural plate. (A) Ca2+ transients (arrow) in a late gastrula stage embryo expressing the fluorescent Ca2+ indicator GCaMP5 in the neural plate. Selected frames covering 417 seconds of a time-lapse movie are shown. (B) Ca2+ transients at mid-neurula stage. Colored arrows indicate a wave of Ca2+ transients moving between adjacent cells (cells 2-7; cell 1 is a non-transgenic control). (C) Relative fluorescence intensities of the cells indicated in B plotted versus time.
In order to assess whether the frm mutation disrupts Ca2+ transients in the neural plate, one-cell stage embryos from heterozygous frm parents were electroporated with etr1p::GCaMP5. The GCaMP5 reporter was expressed at much lower levels in the a-lineage of the homozygous frm mutants (frm/frm) because in the mutant these cells are transfated to epidermis (Fig. 5A), thus restricting our analysis to the A-lineage. The lower level of GCaMP5 expression in the a-lineage did, however, allow us to easily identify the frm/frm mutants, which constitute only 25% of the embryos. Embryos from several parents used in independent electroporations were imaged at the neurula stage. The percentage of electroporated embryos showing Ca2+ transients in the A-lineage during ∼3-minute imaging sessions is presented in Fig. 5B. Ca2+ transients were observed in 32% (n=34) of phenotypically wild-type embryos (which included heterozygous frm embryos). By contrast, only one frm/frm embryo out of 15 recorded (7%) was observed to have a single weak Ca2+ transient [frm(pos), Fig. 5A].
Fig. 5.

frm mutation disrupts Ca2+ transients. (A) Time-lapse recordings of Ca2+ transients in wild-type (wt) and homozygous frm embryos expressing GCaMP5 in the neural plate. The first column shows neurula stage embryos at the beginning of the time-lapse. frm(neg) is an example of an embryo showing no Ca2+ transients, whereas the single frm embryo showing a weak Ca2+ transient is labeled frm(pos). The second column shows a temporal projection of images collected for 2.8 minutes. Ca2+ transients are indicated by arrowheads. The final column shows the same embryos at the tailbud stage. Homozygous frm embryos are confirmed by reduced expression of GCaMP5 in the anterior CNS. (B) Percentage of wild-type and homozygous frm embryos showing Ca2+ transients. (C) Addition of βGA eliminates Ca2+ transients. Relative intensities of three cells before and after addition of βGA (at dashed line). The dashed line itself corresponds to a ∼3-minute interval in which βGA was added and mixed.
We observed that addition of the gap junction channel blocker βGA could phenocopy the frm mutation (Fig. 3). We further observed that addition of 100 μM βGA to neurula stage embryos expressing etr1p::GCaMP5 eliminated Ca2+ transients (Fig. 5C). Fig. 5C shows the relative fluorescence intensities in three cells before and ∼5 minutes after the addition of βGA. There was a complete absence of Ca2+ transients throughout the neural plate after the addition of βGA (supplementary material Movie 3).
As an additional test for the function of the Ca2+ transients, embryos were partially depleted of Ca2+ by incubation in low Ca2+ (0.5 mM versus 11 mM Ca2+ for control embryos) artificial sea water (ASW) starting at late gastrulation. The low Ca2+ ASW completely eliminated Ca2+ transients, whereas re-addition of Ca2+ to 11 mM quickly resulted in prolonged Ca2+ transients (Fig. 6A). To phenotypically assess the effects of Ca2+ depletion on development, embryos were exposed to low Ca2+ ASW for a 3-hour period starting at late gastrula stage and extending through the initial tailbud stage, and then returned to natural sea water and cultured to the late tailbud stage. Embryos were assessed for the presence of pigment cells, palps and neural tube closure, and were immunostained for CRALBP and Arrestin. Defects in the low Ca2+-treated embryos appeared to be highly specific (e.g. there were no observable defects in gastrulation, tailbud elongation or caudal neural tube closure). The most conspicuous defects were the absence of palps and pigment cells in the majority of the low Ca2+-treated embryos (Fig. 6D). Additionally, approximately half (45%) of the low Ca2+-treated embryos had open rostral neural tubes. Immunostaining showed that the majority of the low Ca2+-treated embryos had no, or greatly reduced, Arrestin staining, whereas CRALBP staining remained at, or near, control levels in all embryos (Fig. 6B,C). In summary, the defects arising from the low Ca2+ treatment starting at the late gastrula stage appeared to be primarily in the a-lineage of the CNS, whereas caudal portions of the CNS, as assessed by CRALBP staining, appeared largely intact.
Fig. 6.
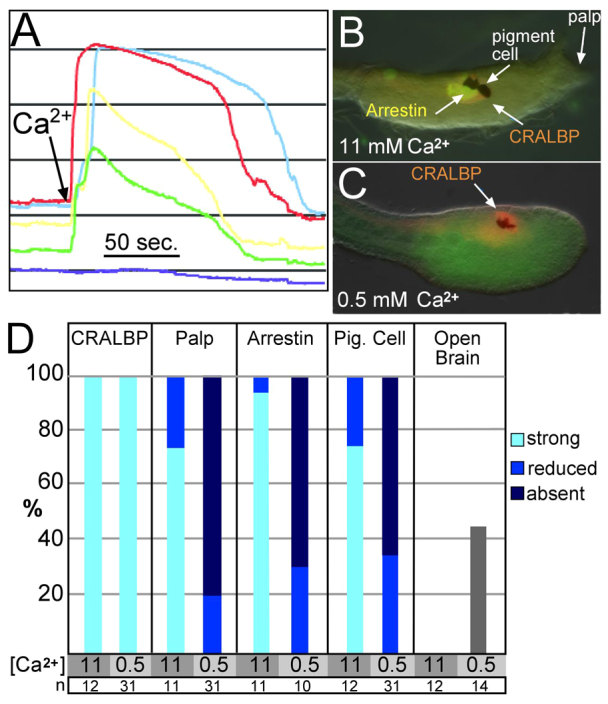
Ca2+ depletion disrupts calcium transients. (A) No Ca2+ transients were observed in the neural plate of embryos incubated in low Ca2+ artificial sea water (ASW). Addition of Ca2+ to 11 mM (arrow) causes an immediate wave of fluorescence in the cells. The dark blue line indicates a non-expressing cell. (B,C) Representative embryos incubated in ASW containing either 11 mM or 0.5 mM Ca2+ and immunostained for the indicated markers. (D) Incubation of embryos in low Ca2+ ASW (0.5 mM Ca2+) for 3 hours (gastrula though initial tailbud stage) resulted in an absence of palp development, caused an open brain phenotype, and greatly reduced the presence of pigment and Arrestin-expressing cells in the sensory vesicle. Control embryos were incubated in normal ASW (11 mM Ca2+).
DISCUSSION
The frm mutation results in severe disruption of the development of the anterior (a-lineage) neural plate derivatives in C. intestinalis (Deschet and Smith, 2004). In the present study we have shown that a genetic lesion in a connexin gene (cx-11) is responsible for the frm phenotype. Connexin proteins are best known for their role in forming gap junctions, which join the cytoplasm of adjacent cells, although connexins also form hemichannels, which can function as channels to the extracellular fluid (Bruzzone and Dermietzel, 2006; Mese et al., 2007). The mapping studies presented here show a tight genetic linkage to a cx-11 allele that carries a splice-disrupting mutation that results in the incorporation of a premature stop codon. Additionally, the frm mutation is closely phenocopied by injection of translation-blocking and splice-blocking MOs and by the addition of a broad specificity gap junction inhibitor during restricted temporal windows of CNS development.
It is difficult to assign an exact vertebrate ortholog to the C. intestinalis cx-11 gene. Ascidians, such as C. intestinalis, are tunicates, which together with the vertebrates and cephalochordates comprise the chordates. Connexin genes are found only in vertebrates and tunicates and in no other living group; even the cephalochordates lack connexin genes (Shestopalov and Panchin, 2008). Seventeen connexin-like genes have been annotated for the C. intestinalis genome (Okamura et al., 2005), although our analysis of these sequences suggests that the correct number is fifteen. Phylogenetic analyses of chordate connexin genes usually result in the tunicate genes clustering together, reflecting their extensive divergence from the vertebrate connexin genes (Abascal and Zardoya, 2012). One interpretation of this result is that the last common ancestor of the vertebrates and tunicates had only a single connexin gene that was then independently expanded in each group. However, the extreme sequence divergence of the tunicate connexin genes may result in clustering due to long-branch attraction. Attempts to correct for long-branch attraction suggest that there might have been at least two connexin genes in the last common ancestor (Cruciani and Mikalsen, 2007).
The temporal expression of cx-11 in the neural precursor cells correlates precisely with the period of a-lineage induction. Expression above the low baseline level is first detected at the 32-cell stage, persists through gastrulation, and then declines at neurula stage. Moreover, there appears to be a progressive specification of a-lineage fates. Previous work with isolated ascidian blastomeres showed that the palps are induced first, followed by the sensory vesicle, and finally the pigment cells, reflecting a progressive specification of cell fates between the 32-cell and gastrula stages (Nishida, 1991; Wada et al., 1999). This same progression of specification is apparent in the results from staged βGA treatment, with early treatments most profoundly disrupting palps, whereas sustained treatments are required to significantly reduce pigment cells. Previous work in C. intestinalis using voltage clamping and dye transfer has described gap junction coupling between cells as early as the 2-cell stage, with a noticeable increase in coupling starting at the 16- to 32-cell stages (Serras et al., 1988; Dale et al., 1991). Moreover, blastomere isolation and cleavage arrest experiments in H. roretzi have demonstrated gap junction coupling between a-lineage and A-lineage cells, which declines during the late neurula to tailbud stages in neurally committed but not epidermally committed a-lineage cells (Saitoe et al., 1996).
We hypothesize that the role of cx-11 in C. intestinalis anterior neural plate development is through mediation of Ca2+ transients (Fig. 7). We have shown that Ca2+ transients can be detected in the C. intestinalis neural plate as early as late gastrula, and that in homozygous frm embryos Ca2+ transients are essentially eliminated. Moreover, addition of a gap junction inhibitor to late gastrula and neurula stage embryos eliminates Ca2+ transients while at the same time mimicking the frm phenotype. Additionally, Ca2+ depletion starting at the late gastrula stage eliminated Ca2+ transients and caused defects similar to those seen in frm/frm embryos. In Xenopus, Ca2+ transients have been shown to be an essential feature of neural induction (Moreau et al., 1994; Leclerc et al., 2000; Leclerc et al., 2006; Moreau et al., 2008). Taken together, our data point to a conserved role for Ca2+ transients in C. intestinalis neural induction.
Fig. 7.
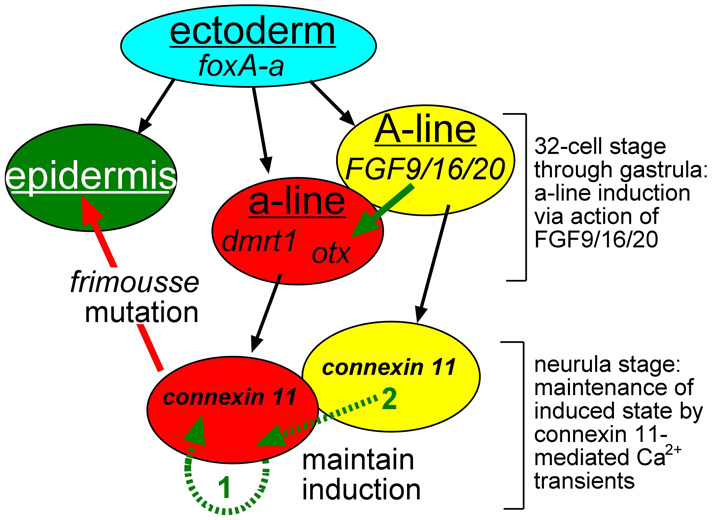
Model for cx-11 function in anterior neural plate induction in C. intestinalis. The a-lineage is induced by FGF9/16/20 from the neighboring A-lineage. At the neurula stage, cx-11 is expressed throughout the A- and a-lineages, and thus may contribute to signal propagation (most likely Ca2+ transients) between the A- and a-lineages (green arrow 2) or within the a-lineage (green arrow 1). a-line, a-lineage.
Connexins are known to mediate Ca2+ waves in mammalian cells (Toyofuku et al., 1998; Lacar et al., 2011; Orellana et al., 2012), although a role in vertebrate neural plate induction has not been demonstrated. Gap junctions appear to function via several mechanisms in Ca2+ wave propagation. In one mechanism, gap junctions act intercellularly to propagate Ca2+ waves by allowing the spread of Ca2+ releasing factors, including Ca2+ itself (Leybaert et al., 1998; Isakson et al., 2006; Lacar et al., 2011). A second mechanism has been described in the developing mammalian CNS and involves extracellular propagation via ATP released from hemichannels acting via P2 receptors to trigger Ca2+ waves (Dale, 2008; Orellana et al., 2012). This second mechanism is unlikely to be present in C. intestinalis because ascidians appear to lack purinergic receptors (Okamura et al., 2005). Further research will be needed to uncover the source of the Ca2+ of the transients (intracellular or extracellular) and to identify other essential gene products, such as Ca2+ channels.
Further investigation will also be needed to place the requirement for cx-11 and Ca2+ transients into the context of the well-characterized pathway of a-lineage CNS induction in ascidians (Bertrand et al., 2003; Miya and Nishida, 2003; Meinertzhagen et al., 2004; Imai et al., 2006). Early responses to induction in the a-lineage include the expression of the transcription factors otx and dmrt1 (Haeussler et al., 2010; Tresser et al., 2010). In the frm mutation, otx expression is maintained in the a-lineage, whereas the expression of downstream genes such as six3/6 (Haeussler et al., 2010) is lost following an initial brief expression (Deschet and Smith, 2004). We speculate that cx-11 is required to maintain a-lineage induction through the mediation of Ca2+ transients (Fig. 7). Because cx-11 expression and Ca2+ transients are observed widely in the neural plate, cx-11 could function to mediate Ca2+ transients either within the induced a-lineage (Fig. 7, green arrow 1), between the A-lineage and the a-lineage (Fig. 7, green arrow 2), or both. Because GCaMP5 is not expressed in the a-lineage of the frm mutant, preventing us from assessing the presence or absence of Ca2+ transients, it remains a possibility that Ca2+ transients persist in these cells, although βGA treatment was able to eliminate Ca2+ transients throughout the neural plate. Results from Xenopus demonstrating that Ca2+ transients are required in neurally induced ectodermal explants (animal caps) indicate that, at least in this species, Ca2+ signaling within the induced tissue is sufficient (Leclerc et al., 2006). It has been proposed that whereas the A-lineage of the ascidian CNS develops autonomously, a-lineage induction shows similarity to vertebrate neural induction and might reflect a conserved mechanism that predates the split of the vertebrate and tunicate subphyla (Bertrand et al., 2003; Meinertzhagen et al., 2004). Likewise, the requirement for Ca2+ transients in Xenopus neural induction suggests that this mechanism was also present in the common ancestor of the tunicates and vertebrates.
Supplementary Material
Acknowledgments
We thank Takehiro Kusakabe for anti-Arrestin and anti-CRALBP antibodies.
Footnotes
Funding
This work was supported by the National Institutes of Health [R01 HD038701 to W.C.S.]; G.J.K. was supported by the Gangneung-Wonju National University Visiting Professors Programs (2011). Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.084681/-/DC1
These authors contributed equally to this work
References
- Abascal F., Zardoya R. (2012). Evolutionary analyses of gap junction protein families. Biochim. Biophys. Acta (in press). [DOI] [PubMed] [Google Scholar]
- Abramoff M. D., Magalhaes P. J., Ram S. J. (2004). Image processing with ImageJ. Biophotonics International 11, 36–42 [Google Scholar]
- Akerboom J., Chen T. W., Wardill T. J., Tian L., Marvin J. S., Mutlu S., Calderón N. C., Esposti F., Borghuis B. G., Sun X. R., et al. (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 32, 13819–13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand V., Hudson C., Caillol D., Popovici C., Lemaire P. (2003). Neural tissue in ascidian embryos is induced by FGF9/16/20, acting via a combination of maternal GATA and Ets transcription factors. Cell 115, 615–627 [DOI] [PubMed] [Google Scholar]
- Bone Q. (1992). On the locomotion of ascidian tadpole larvae. J. Mar. Biol. Assoc.UK 72, 161–186 [Google Scholar]
- Bruzzone R., Dermietzel R. (2006). Structure and function of gap junctions in the developing brain. Cell Tissue Res. 326, 239–248 [DOI] [PubMed] [Google Scholar]
- Chiba S., Jiang D., Satoh N., Smith W. C. (2009). Brachyury null mutant-induced defects in juvenile ascidian endodermal organs. Development 136, 35–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiaen L., Wagner E., Shi W., Levine M. (2009a). Electroporation of transgenic DNAs in the sea squirt Ciona. Cold Spring Harb. Protoc. 2009, pdb prot5345 [DOI] [PubMed] [Google Scholar]
- Christiaen L., Wagner E., Shi W., Levine M. (2009b). Whole-mount in situ hybridization on sea squirt (Ciona intestinalis) embryos. Cold Spring Harb. Protoc. 2009, pdb prot5348 [DOI] [PubMed] [Google Scholar]
- Cole A. G., Meinertzhagen I. A. (2004). The central nervous system of the ascidian larva: mitotic history of cells forming the neural tube in late embryonic Ciona intestinalis. Dev. Biol. 271, 239–262 [DOI] [PubMed] [Google Scholar]
- Cruciani V., Mikalsen S. O. (2007). Evolutionary selection pressure and family relationships among connexin genes. Biol. Chem. 388, 253–264 [DOI] [PubMed] [Google Scholar]
- Dale N. (2008). Dynamic ATP signalling and neural development. J. Physiol. 586, 2429–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale B., Santella L., Tosti E. (1991). Gap-junctional permeability in early and cleavage-arrested ascidian embryos. Development 112, 153–160 [DOI] [PubMed] [Google Scholar]
- Darras S., Nishida H. (2001). The BMP/CHORDIN antagonism controls sensory pigment cell specification and differentiation in the ascidian embryo. Dev. Biol. 236, 271–288 [DOI] [PubMed] [Google Scholar]
- Delsuc F., Brinkmann H., Chourrout D., Philippe H. (2006). Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439, 965–968 [DOI] [PubMed] [Google Scholar]
- Deschet K., Smith W. C. (2004). Frimousse – a spontaneous ascidian mutant with anterior ectodermal fate transformation. Curr. Biol. 14, R408–R410 [DOI] [PubMed] [Google Scholar]
- Dogan R. I., Getoor L., Wilbur W. J., Mount S. M. (2007). SplicePort – an interactive splice-site analysis tool. Nucleic Acids Res. 35, W285–W291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour H. D., Chettouh Z., Deyts C., de Rosa R., Goridis C., Joly J. S., Brunet J. F. (2006). Precraniate origin of cranial motoneurons. Proc. Natl. Acad. Sci. USA 103, 8727–8732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M., Jaszczyszyn Y., Christiaen L., Joly J. S. (2010). A cis-regulatory signature for chordate anterior neuroectodermal genes. PLoS Genet. 6, e1000912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T., Nakagawa M., Sasakura Y., Kusakabe T. G., Tsuda M. (2010). Simple motor system of the ascidian larva: neuronal complex comprising putative cholinergic and GABAergic/glycinergic neurons. Zoolog. Sci. 27, 181–190 [DOI] [PubMed] [Google Scholar]
- Hotta K., Mitsuhara K., Takahashi H., Inaba K., Oka K., Gojobori T., Ikeo K. (2007). A web-based interactive developmental table for the ascidian Ciona intestinalis, including 3D real-image embryo reconstructions: I. From fertilized egg to hatching larva. Dev. Dyn. 236,1790–1805 [DOI] [PubMed] [Google Scholar]
- Hudson C., Yasuo H. (2008). Similarity and diversity in mechanisms of muscle fate induction between ascidian species. Biol. Cell 100, 265–277 [DOI] [PubMed] [Google Scholar]
- Imai J. H., Meinertzhagen I. A. (2007). Neurons of the ascidian larval nervous system in Ciona intestinalis: I. Central nervous system. J. Comp. Neurol. 501, 316–334 [DOI] [PubMed] [Google Scholar]
- Imai K. S., Levine M., Satoh N., Satou Y. (2006). Regulatory blueprint for a chordate embryo. Science 312, 1183–1187 [DOI] [PubMed] [Google Scholar]
- Isakson B. E., Olsen C. E., Boitano S. (2006). Laminin-332 alters connexin profile, dye coupling and intercellular Ca2+ waves in ciliated tracheal epithelial cells. Respir. Res. 7, 105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D., Munro E. M., Smith W. C. (2005). Ascidian prickle regulates both mediolateral and anterior-posterior cell polarity of notochord cells. Curr. Biol. 15, 79–85 [DOI] [PubMed] [Google Scholar]
- Juszczak G. R., Swiergiel A. H. (2009). Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 181–198 [DOI] [PubMed] [Google Scholar]
- Kim G. J., Nishida H. (2001). Role of the FGF and MEK signaling pathway in the ascidian embryo. Dev. Growth Differ. 43, 521–533 [DOI] [PubMed] [Google Scholar]
- Lacar B., Young S. Z., Platel J. C., Bordey A. (2011). Gap junction-mediated calcium waves define communication networks among murine postnatal neural progenitor cells. Eur. J. Neurosci. 34, 1895–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay C., Fromentoux V., Shi D. L., Boucaut J. C. (1996). A truncated FGF receptor blocks neural induction by endogenous Xenopus inducers. Development 122, 869–880 [DOI] [PubMed] [Google Scholar]
- Leclerc C., Webb S. E., Daguzan C., Moreau M., Miller A. L. (2000). Imaging patterns of calcium transients during neural induction in Xenopus laevis embryos. J. Cell Sci. 113, 3519–3529 [DOI] [PubMed] [Google Scholar]
- Leclerc C., Néant I., Webb S. E., Miller A. L., Moreau M. (2006). Calcium transients and calcium signalling during early neurogenesis in the amphibian embryo Xenopus laevis. Biochim. Biophys. Acta 1763, 1184–1191 [DOI] [PubMed] [Google Scholar]
- Lemaire P., Smith W. C., Nishida H. (2008). Ascidians and the plasticity of the chordate developmental program. Curr. Biol 18, R620–R631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leybaert L., Paemeleire K., Strahonja A., Sanderson M. J. (1998). Inositol-trisphosphate-dependent intercellular calcium signaling in and between astrocytes and endothelial cells. Glia 24, 398–407 [PubMed] [Google Scholar]
- Meinertzhagen I. A., Lemaire P., Okamura Y. (2004). The neurobiology of the ascidian tadpole larva: recent developments in an ancient chordate. Annu. Rev. Neurosci. 27, 453–485 [DOI] [PubMed] [Google Scholar]
- Mese G., Richard G., White T. W. (2007). Gap junctions: basic structure and function. J. Invest. Dermatol. 127, 2516–2524 [DOI] [PubMed] [Google Scholar]
- Minokawa T., Yagi K., Makabe K. W., Nishida H. (2001). Binary specification of nerve cord and notochord cell fates in ascidian embryos. Development 128, 2007–2017 [DOI] [PubMed] [Google Scholar]
- Miya T., Nishida H. (2003). An Ets transcription factor, HrEts, is target of FGF signaling and involved in induction of notochord, mesenchyme, and brain in ascidian embryos. Dev. Biol. 261, 25–38 [DOI] [PubMed] [Google Scholar]
- Moreau M., Leclerc C., Gualandris-Parisot L., Duprat A. M. (1994). Increased internal Ca2+ mediates neural induction in the amphibian embryo. Proc. Natl. Acad. Sci. USA 91, 12639–12643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau M., Néant I., Webb S. E., Miller A. L., Leclerc C. (2008). Calcium signalling during neural induction in Xenopus laevis embryos. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 1371–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida H. (1987). Cell lineage analysis in ascidian embryos by intracellular injection of a tracer enzyme. III. Up to the tissue restricted stage. Dev. Biol. 121, 526–541 [DOI] [PubMed] [Google Scholar]
- Nishida H. (1991). Induction of brain and sensory pigment cells in the ascidian embryo analyzed by experiments with isolated blastomeres. Development 112, 389–395 [Google Scholar]
- Okamura Y., Nishino A., Murata Y., Nakajo K., Iwasaki H., Ohtsuka Y., Tanaka-Kunishima M., Takahashi N., Hara Y., Yoshida T., et al. (2005). Comprehensive analysis of the ascidian genome reveals novel insights into the molecular evolution of ion channel genes. Physiol. Genomics 22, 269–282 [DOI] [PubMed] [Google Scholar]
- Orellana J. A., Sánchez H. A., Schalper K. A., Figueroa V., Sáez J. C. (2012). Regulation of intercellular calcium signaling through calcium interactions with connexin-based channels. Adv. Exp. Med. Biol. 740, 777–794 [DOI] [PubMed] [Google Scholar]
- Roure A., Rothbächer U., Robin F., Kalmar E., Ferone G., Lamy C., Missero C., Mueller F., Lemaire P. (2007). A multicassette Gateway vector set for high throughput and comparative analyses in Ciona and vertebrate embryos. PLoS ONE 2, e916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoe M., Inazawa T., Takahashi K. (1996). Neuronal expression in cleavage-arrested ascidian blastomeres requires gap junctional uncoupling from neighbouring cells. J. Physiol. 491, 825–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai Y., Lu B., Piccolo S., De Robertis E. M. (1996). Endoderm induction by the organizer-secreted factors chordin and noggin in Xenopus animal caps. EMBO J. 15, 4547–4555 [PMC free article] [PubMed] [Google Scholar]
- Serras F., Baud C., Moreau M., Guerrier P., Van den Biggelaar J. A. M. (1988). Intercellular communication in the early embryo of the ascidian Ciona intestinalis. Development 102, 55–63 [Google Scholar]
- Shestopalov V. I., Panchin Y. (2008). Pannexins and gap junction protein diversity. Cell. Mol. Life Sci. 65, 376–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T., Yabuki M., Otsu K., Kuzuya T., Hori M., Tada M. (1998). Intercellular calcium signaling via gap junction in connexin-43-transfected cells. J. Biol. Chem. 273, 1519–1528 [DOI] [PubMed] [Google Scholar]
- Tresser J., Chiba S., Veeman M., El-Nachef D., Newman-Smith E., Horie T., Tsuda M., Smith W. C. (2010). doublesex/mab3 related-1 (dmrt1) is essential for development of anterior neural plate derivatives in Ciona. Development 137, 2197–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M., Kusakabe T., Iwamoto H., Horie T., Nakashima Y., Nakagawa M., Okunou K. (2003). Origin of the vertebrate visual cycle: II. Visual cycle proteins are localized in whole brain including photoreceptor cells of a primitive chordate. Vision Res. 43, 3045–3053 [DOI] [PubMed] [Google Scholar]
- Veeman M. T., Nakatani Y., Hendrickson C., Ericson V., Lin C., Smith W. C. (2008). Chongmague reveals an essential role for laminin-mediated boundary formation in chordate convergence and extension movements. Development 135, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeman M. T., Newman-Smith E., El-Nachef D., Smith W. C. (2010). The ascidian mouth opening is derived from the anterior neuropore: reassessing the mouth/neural tube relationship in chordate evolution. Dev. Biol. 344, 138–149 [DOI] [PubMed] [Google Scholar]
- Veeman M. T., Chiba S., Smith W. C. (2011). Ciona genetics. Methods Mol. Biol. 770, 401–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada S., Katsuyama Y., Saiga H. (1999). Anteroposterior patterning of the epidermis by inductive influences from the vegetal hemisphere cells in the ascidian embryo. Development 126, 4955–4963 [DOI] [PubMed] [Google Scholar]
- Wada S., Sudou N., Saiga H. (2004). Roles of Hroth, the ascidian otx gene, in the differentiation of the brain (sensory vesicle) and anterior trunk epidermis in the larval development of Halocynthia roretzi. Mech. Dev. 121, 463–474 [DOI] [PubMed] [Google Scholar]
- Wang Z., Burge C. B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi K., Makabe K. W. (2001). Isolation of an early neural maker gene abundantly expressed in the nervous system of the ascidian, Halocynthia roretzi. Dev. Genes Evol. 211, 49–53 [DOI] [PubMed] [Google Scholar]
- Yamada L., Shoguchi E., Wada S., Kobayashi K., Mochizuki Y., Satou Y., Satoh N. (2003). Morpholino-based gene knockdown screen of novel genes with developmental function in Ciona intestinalis. Development 130, 6485–6495 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.