

Abstract
Coiled-coil formation of four different oligopeptides was characterized in solution, on hydrogels, and on membranes by employing circular dichroism spectroscopy, surface plasmon resonance spectroscopy, attenuated total reflection infrared spectroscopy, and ellipsometry. Peptide sequences rich in either glutamic acid (E: E3Cys, i-E3Cys) or lysine (K: K3Cys, i-K3Cys) were used to represent minimal mimics of eukaryotic SNARE motifs. Half of the peptides were synthesized in reverse sequence, so that parallel and antiparallel heptad coiled-coil structures were formed. Either E-peptides or K-peptides were attached covalently to phospholipid anchors via maleimide chemistry, and served as receptors for the recognition of the corresponding binding partners added to solution. Attenuated total reflection infrared spectroscopy of single bilayers confirmed the formation of coiled-coil complexes at the membrane interface. Coiled-coil formation in solution, as compared with association at the membrane surface, displays considerably larger binding constants that are largely attributed to loss of translational entropy at the interface. Finally, the fusogenicity of the various coiled-coil motifs was explored, and the results provide clear evidence that hemifusion followed by full fusion requires a parallel orientation of α-helices, whereas antiparallel oriented coiled-coil motifs display only docking.
Introduction
Membrane fusion plays a pivotal role in processes that require transport of molecules that would otherwise not be capable of crossing the lipid bilayer (1). The initial adhesion between the opposing membranes of eukaryotic cells is predominantly achieved by a coiled-coil interaction involving two or more amino acid strands that form a rope-like superhelical structure (2). Coiled-coil-forming peptides and proteins display a heptad repeat (a-b-c-d-e-f-g) in which apolar amino acids occupy a and d positions, and charged amino acids are positioned at positions e and g, resulting in an amphiphilic helix (3). By estimating the free energy per heptad repeat from the overall gain in free energy for a soluble N-ethylmaleimide-sensitive-factor attachment protein receptor (SNARE), Li et al. (4) found that ∼5–6 kBT of energy are released per heptad repeat during coiled-coil formation. This gain in free energy upon formation of coiled-coil strands is predominantly due to the packing of the hydrophobic a- and d-residues facing against each other. The geometry and aggregation state (i.e., the number of strands that participate in bundle formation) are governed mainly by the amino acid sequence (5). In nature, coiled-coil interactions are able to overcome the initial energy barrier of membrane fusion. One of the most intricate fusion processes is the calcium-stimulated exocytosis of synaptic vesicles to release neurotransmitters in the synaptic cleft involving a variety of proteins (i.e., SNAREs) assembling into a parallel oriented ternary coiled-coil bundle (6,7). An eight-heptad repeat segment is responsible for the highly stable coiled-coil motif that facilitates fusion, because it releases enough free energy to overcome the intrinsic energy barrier originating from dehydration of the bilayer. However, the energetics of fusion remains controversial, with different results obtained with continuum-mechanics models and molecular-dynamics simulations seeking quantitative thermodynamic data (8). Recently, Smith and Weisshaar (9) suggested that docking rather than fusion is the rate-limiting step in SNARE-driven fusion assays, putting the focus on the initial contact of two bilayers that may require many collisions until a docked pair of two vesicles forms. Li and co-workers (4) determined the energetics and dynamics of SNARE protein folding upon coiled-coil formation to be 35 kBT, a folding energy that is close to the proposed energy needed for membrane fusion (≈50 kBT) (10,11). Enhancement of the overall rate may only be achieved by more efficient docking. These findings call for additional methods to study the recognition kinetics of coiled-coil peptides in a native environment.
Robson Marsden and co-workers (12) recently reported on a reduced model for membrane fusion based on a parallel coiled-coil system with a recognition domain encompassing three heptad repeats. The authors found that this minimal system mimics all aspects of native membrane fusion comprising docking followed by lipid and content mixing. Similar results were found by Meyenberg et al. (13) in fusion experiments with a more complex model using the same recognition peptide sequences but attached to native transmembrane domains derived from SNARE proteins. The peptide sequence used was first described by Litowski and Hodges (14) and was designed to form short but specifically interacting heterodimeric coiled-coils with a considerable binding strength (∼10−7 M) and great stability. Those sequences were named E and K due to their prevailing content of the charged residues glutamic acid (E) and lysine (K).
Here, we investigate the thermodynamics and kinetics of coiled-coil formation between the peptides with either a E- or K-sequence (E3Cys and K3Cys) taking place in solution and at lipid bilayers. By inverting the primary sequence of the recognition domain (i-E3Cys and i-K3Cys), we were able to compare coiled-coil peptides with different superhelical macrodipoles and a predominantly parallel or antiparallel orientation (Fig. 1). Peptides were covalently attached via a cysteine linker to maleimide-functionalized lipids incorporated into lipid bilayers (15) to investigate fusogenic activity. Although Monera et al. (16) showed that in the case of similar electrostatic interactions, antiparallel coiled-coils are more stable, Lygina et al. (17) proved that a parallel orientation of peptide hybrids leads to a higher fusogenicity. We found that all peptidic dimers display approximately identical binding affinities, but coiled-coil formation in the context of membranes generates less free energy compared with complexation in solution due to loss of translational degrees of freedom. Additionally, a closer proximity of membranes is achieved through the formation of parallel coiled-coils. Only parallel coiled-coil formation eventually results in fusion, and antiparallel coiled-coils exhibit only docking events.
Figure 1.
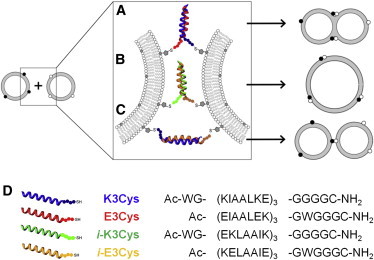
Schematic drawing of the envisioned peptide-mediated membrane-membrane interaction through coiled-coil formation. Parallel coiled-coil formation of peptides E3Cys and K3Cys (A) and inverted peptides i-E3Cys and i-K3Cys (B) are thought to be capable of inducing lipid mixing similarly to hemifusion or fusion, whereas for antiparallel coiled-coil formation (C) only docking events are expected. (D) Schematics, abbreviations, and primary sequences of the peptides used in this work. The N-terminus is acetylated, and the C-terminus is amidated.
Materials and Methods
For details regarding the materials and methods used in this work, see the Supporting Material.
Results and Discussion
The thermodynamics of parallel- and antiparallel-aligned coiled-coil formation composed of E- and K-peptides was scrutinized in solution, on hydrogels, and at membrane surfaces. We employed a variety of methods, including ellipsometry, circular dichroism (CD) spectroscopy, surface plasmon resonance (SPR) spectroscopy, and attenuated total reflection infrared (ATR-IR) spectroscopy, to quantify the binding affinity of E- and K-peptide sequences in the context of lipid bilayers compared with their association in solution. In addition, we investigated the capability of coiled-coil dimers to induce lipid mixing or eventually content mixing of two vesicle populations.
Peptide-peptide interaction in solution and within a hydrogel
Before examining the intricate interfacial chemistry that takes place at phospholipid membranes, we investigated the coiled-coil formation of E- and K-peptides in solution by means of CD spectroscopy (18–20). We found that K-peptides show a substantial α-helical content, whereas E-peptides adopt a predominantly random coil structure before dimerization (see Table 1). After the formation of parallel- and antiparallel-aligned heterodimeric coiled-coil structures, the α-helix content increases considerably (>70%), whereas the β-sheet content vanishes. The ratio of α-helix to random-coil content of the coiled-coil dimers reflects the ratio of amino acids that form the heptad repeat (21 aa) to amino acids that act merely as an anchor group (6–7 aa). In Fig. 2, A and B, the CD spectra show two characteristic minima at 208 nm and 220 nm, which are indicative of an α-helix. Previous studies showed that the intensities of the two above-mentioned minima are virtually equal for coiled-coil motifs, whereas for single-stranded α-helices the ratio of [θ]220nm/[θ]208nm is ∼0.86 (21). Our calculated ratios for the heterodimeric pairs are all ∼1 (see Table 1, last row), which confirms the formation of coiled-coil structures.
Table 1.
Secondary structure of peptides before and after formation of coiled-coil dimers, obtained from CD measurements in solution (cpeptide = 0.1 mM; in phosphate buffer (50 mM Na2HPO4, pH 6.8) PB 6.8)
| E3Cys | K3Cys | i-E3Cys | i-K3Cys | E3Cys + K3Cys | i-E3Cys + i-K3Cys | i-E3Cys + K3Cys | E3Cys + i-K3Cys | |
|---|---|---|---|---|---|---|---|---|
| α-helix/ % | 31 | 46 | 31 | 44 | 79 | 78 | 73 | 79 |
| β-sheet/ % | 12 | 23 | 12 | 23 | 0 | 0 | 2 | 0 |
| random/ % | 57 | 31 | 57 | 33 | 21 | 21 | 24 | 21 |
| [θ]220/[θ]208 | 0.69 | 0.87 | 0.71 | 0.75 | 1.00 | 0.98 | 0.94 | 1.03 |
Fits were obtained with DichroWeb online analysis software (18,19).
Figure 2.
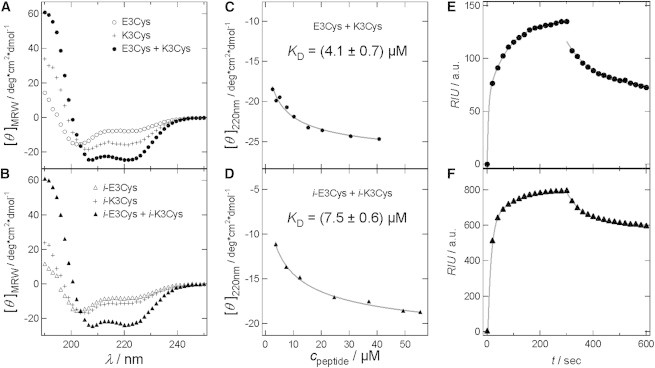
(A–D) CD spectra and analysis to determine the dissociation constant of coiled-coil dimers. (A) E3Cys (open circles), K3Cys (crosses), and a mixture of both (solid circles) in PB 6.8. (B) i-E3Cys (open triangles), i-K3Cys (crosses), and the corresponding mixture (solid triangles). (C and D) Concentration dependence of [θ]MRW at 220 nm of heterodimeric coiled-coil to determine KD for E3Cys and K3Cys (C), and for i-E3Cys and i-K3Cys (D; fits of Eq. 1 are shown as gray lines (22)). (E and F) Association (t = 0–300 s) and dissociation (t = 300–600 s) of E-peptides coupled to immobilized K-peptides on a hydrogel monitored by SPR spectroscopy. Association of peptides was fitted by a double exponential function to obtain kon, whereas dissociation of the peptide assembly was fitted by a monoexponential function (koff; see Supporting Material; gray lines). (E) E3Cys was added to immobilized K3Cys at a concentration of 15 μM (solid circles). KD = (0.5 ± 0.3) μM. (F) i-E3Cys was added to immobilized i-K3Cys at a concentration of 15 μM (solid triangles). KD = (2.3 ± 1.8) μM.
The intensity of the ellipticity at 220 nm can be used to determine the dissociation constant of coiled-coil complexes by dilution experiments (Fig. 2, C and D). Here, successively reducing the peptide concentration leads to a dissociation of coiled-coil assemblies, which results in a corresponding decrease of the α-helix content (22). The change in ellipticity at 220 nm [θ]220nm can be described by the following equation:
| (1) |
where c is the peptide concentration. In addition to the dissociation constant KD as the fit parameter, the ellipticity for a monomeric unfolded peptide ([θ]mon) and the maximum ellipticity for a complete coiled-coil structure ([θ]cc) need to be specified. Here, we determined the minima at 220 nm for an unfolded peptide from the CD spectra of single peptides before dimerization, and used this value as [θ]mon in the fit function. We determined [θ]cc by fitting Eq. 1 to the data. Of note, the resulting dissociation constants are intrinsically inaccurate due to the low signal/noise ratio for the lowest peptide concentrations. The dissociation constants determined for inverted and noninverted peptides, both of which lead to parallel aligned dimeric coiled-coil structures, are in the lower micromolar regime. The KD for a coiled-coil formed of i-K3Cys and i-E3Cys (KD = (7.5 ± 0.6) μM) is nearly double the KD found for K3Cys and E3Cys (KD = (4.1 ± 0.7) μM). The values determined for an antiparallel packing (and thus dimerization of K3Cys and i-E3Cys or i-K3Cys and E3Cys, respectively) are found to be generally smaller but very similar for both coiled-coil heterodimers (KD(K3Cys/i-E3Cys) = (2.9 ± 0.8) μM; and KD(i-K3Cys/E3Cys) = (2.5 ± 0.6) μM; see Fig. S1). Literature values for KD of the noninverted coiled-coil complexes using similar sequences were reported to be between 10−7 and 10−8 M employing CD spectroscopy, isothermal titration calorimetry measurements (23), and guanidine hydrochloride denaturation studies (14). The KD-values are smaller by one order of magnitude, which we attribute to the inherent inaccuracy of the method as well as a slightly different peptide sequence used in our study.
Additionally, due to the limited signal/noise ratio of CD spectroscopy at low peptide concentrations, we carried out SPR spectroscopy, which in addition to thermodynamic values from isotherm data allowed us to measure the adsorption and desorption kinetics (24). Coiled-coil formation can be best described by a double exponential time dependence (τa, τb), where the smaller time constant τa provides kon, which describes the prevailing interaction. Dissociation of the dimers could be fitted with a monoexponential function, providing the off rate, koff. Day et al. (25) showed that KD-values determined from measurements in solution and by SPR are well comparable. Instead of tethering the sample to a plain surface, the peptide is attached to a wide-meshed hydrogel. Therefore, in SPR experiments the sample retains most of its rotational entropic properties and its diffusional freedom (26); hence, in principle, the measurement can be treated like a solution-based method.
The resulting KD-values computed from kinetic data (see Fig. 2, E and F; Fig. S2, and Table S1) at various peptide concentrations are smaller but still in the same regime as the dissociation constants determined by CD spectroscopy. SPR reveals KD-values in the low micromolar range, with a significantly lower dissociation constant for the coiled-coil formed with K3Cys tethered to the hydrogel. Both the parallel coiled-coil formation with E3Cys and the antiparallel coiled-coil formation with i-E3Cys reveal a KD of (0.5 ± 0.3) μM. In comparison, coiled-coil formation with tethered i-K3Cys shows higher KD-values. Here, the antiparallel heterodimerization with E3Cys gives a KD of (1.2 ± 1.0) μM, whereas the dissociation constant for the parallel heterodimer formed with i-E3Cys was found to be (2.3 ± 1.8) μM.
Because all dissociation constants from the various peptide combinations, determined with two independent methods, are approximately in the same regime, we can conclude that coiled-coil formation is rather independent of helix orientation. This is remarkable, because Monera et al. (16) reported that antiparallel coiled-coil structures are more stable when similar electrostatic interactions are given. However, the authors used cysteine-bridged peptides for their denaturation studies, i.e., the two helices were covalently coupled to each other, imposing constraints we do not need. In our case, the electrostatic interactions at the e and g positions are always between lysine and glutamic acid residues; therefore, we attribute the difference between their findings and ours to differences in entropy changes upon assembly/disassembly due to constraining disulfide bridges, which covalently couple the two coiled-coil-forming peptides.
Coiled-coil formation at the membrane interface
The following experiments describe coiled-coil formation in the context of solid supported lipid bilayers that mimic the native situation more closely. In these experiments we sought to monitor the coiled-coil formation of peptides added in solution to preformed lipopeptides (LPs) embedded in a lipid bilayer deposited on a solid support. Therefore, we first needed to ensure successful coiled-coil formation on lipid bilayers using IR spectroscopy, and in a second step we used time-resolved ellipsometry to investigate the binding affinity of a peptide in solution binding to a membrane-anchored LP.
ATR-IR spectroscopy was used to confirm and quantify successful formation of coiled-coil structures at the membrane interface. Covalent coupling to solid supported membranes (SSM) deposited on the ATR-IR crystal was monitored before addition of the binding partner. An advantage of this method is that lipids and peptides both show strong absorption bands that do not overlap. Lipids have a characteristic band pattern in the regime of 2800–3000 cm−1 originating from the different stretching vibrations of the fatty acid alkyl chains, whereas the most prominent absorption band of peptides and proteins is the amide I band at ∼1650 cm−1 (27). In the course of the experiment, a bilayer consisting of dioleoylphosphatidylcholine (DOPC) with 10 mol% of receptor lipid 1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4(p-maleimidomethyl)cyclohexane-carboxamide (MCCDOPE) was prepared on the ATR crystal. Subsequently, K-peptides were coupled covalently to the surface using maleimide chemistry covering a large portion of the surface. Finally, coiled-coil structures were formed on the bilayer by addition of the corresponding peptide (Fig. 3, A and D). The successful coupling and heterodimerization of the peptides is visible in the spectra due to the increasing intensity of the amide I band, which could be monitored in a time-dependent fashion due to the use of a flow cell (Fig. 3, B and E). The LP coupling reaction and the coiled-coil formation can be detected by a steep increase of amide I intensity. Rinsing the surface with buffer results in partial dissolution of the complexes. Control experiments with peptides added to neat membranes lacking MCCDOPE show a transient increase in amide I absorption for K-peptides, whereas E-peptides added to neat PC-bilayers do not show a nonspecific interaction. However, K-peptides are removed by further addition of E-peptides that presumably compete with the bilayer (Fig. S5).
Figure 3.
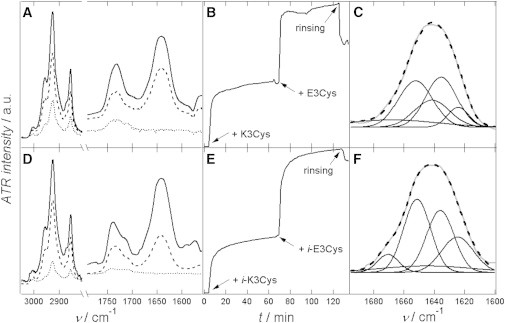
(A and D) ATR-IR spectra of plain SSMs (⋯··) consisting of DOPC/MCCDOPE 90:10 in D2O with 50 mM NaCl spread on an Si-covered ZnSe crystal. Lipid bands and the amide I region are shown. LPs were formed with K3Cys (A) or i-K3Cys (D) covalently attached to MCCDOPE (- - -). Spectra obtained after coiled-coil formation with E3Cys (A) or i-E3Cys (D) are also shown (—). (B and E) Time course of LP coupling reaction followed by coiled-coil formation shown in A and D, respectively. The intensity of the amide I band is plotted versus time. The time course starts after bilayer formation (0 min). Addition of peptides and rinsing is indicated by arrows. Panel C shows the deconvoluted amide I band for the coiled-coil build of LP-K3Cys + E3Cys. In panel F deconvoluted amide I band of LP-i-K3Cys + i-E3Cys is shown. (C and F) Single Gaussian fits are shown in black, and the amide I band of the spectra is in gray. Black scattered lines represent the sum of all Gaussian fits.
The amide I band is mainly generated by exciting C=O stretching vibrations in the protein backbone, stabilizing the secondary structure. Therefore, this region is sensitive to conformational changes such as coiled-coil formation. Heimburg et al. (28) showed that dimeric coiled-coil structures display at least three separable bands in the amide I region. This separation can be achieved by deconvoluting the FTIR spectra and reassembling the amide I band by multiple Gaussian fits (Fig. 3, C and F). As a consequence, we can safely assume that coiled-coil formation also occurs at the membrane interface, an important prerequisite for further thermodynamic and fusion studies.
In addition to ATR-IR spectroscopy, we also monitored in situ coupling of peptides to an SSM through maleimide chemistry by time-resolved ellipsometry as previously described (15). The principal angle del determined by this method is proportional to the layer thicknesses; hence, absolute height changes resulting from peptide coupling and coiled-coil formation on a spread SSM (Fig. S3) can be computed (29,30). We found that the in situ coupling reaction of E-peptides to MCCDOPE is very slow in contrast to LP formation of K-peptides. However, subsequent coiled-coil formation with corresponding binding partners followed similar kinetics independently of the LPs used. We attribute this finding to the negative charge of E-peptides, which results in an electrostatic repulsion of the membrane and thus slows down the adsorption and subsequent binding. Therefore, we used predominantly K-LPs (hereafter denoted as LP-K) as the membrane-based receptor. Because the binding of K-peptides to SSM is linearly related to the MCCDOPE content in the membrane (Fig. S5), we rule out the possibility that nonspecifically bound K-peptides participate in our binding assay.
Adsorption isotherms were measured to determine the affinity between E- and K-peptides on lipid bilayers (31). Coiled-coil forming E-peptides were added in increasing concentrations to a membrane containing 3 mol % LP-K, resulting in a concomitant increase in apparent thickness corresponding to an increase in peptide coverage on the bilayer. The adsorption isotherms originating from parallel coiled-coil formation, such as E3Cys added to LP-K3Cys, and i-E3Cys added to LP-i-K3Cys, are shown in Fig. 4 A. Assembly into antiparallel coiled-coil structures is shown in Fig. 4 B. The maximum layer thickness Δhmax and the dissociation constant KD can be determined by fitting the data with a Langmuir adsorption isotherm (Eq. 2) or a Bragg-Williams isotherm (Eq. 3):
| (2) |
| (3) |
In contrast to the Langmuir isotherm, the regression with a Bragg-Williams model reflects the sigmoidal shape of one of the experimental data, indicative of a slightly cooperative binding. Here, the cooperativity parameter χ is introduced, which is in the range of 0–2 for weak cooperativity (30). In our experiments, only the antiparallel interaction of LP-K3Cys with i-E3Cys shows a Bragg-Williams behavior with a marginal cooperativity of χ = 1.2. The other three analyzed coiled-coil formations can be fitted with high accuracy using the Langmuir equation corresponding to χ = 1.
Figure 4.
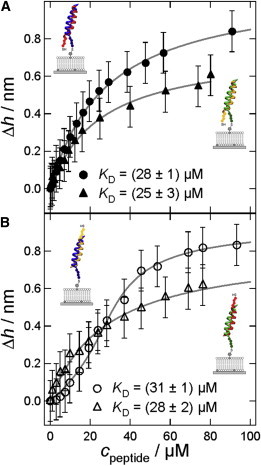
Adsorption isotherms obtained from ellipsometry represent coiled-coil formation of E-peptides binding to K-LPs. Bilayers were formed from DOPC/MCCDOPE 97:3, and K-peptides were then coupled covalently to the surface. The isotherm was measured by subsequently increasing the added E-peptide concentration. (A) Parallel coiled-coil formation. Solid circles: LP-K3Cys + E3Cys; solid triangles: LP-i-K3Cys + i-E3Cys; data were fitted according to Eq. 2 (fits: gray line). (B) Antiparallel coiled-coil formation. Open circles: LP-K3Cys + i-E3Cys; open triangles: LP-i-K3Cys + E3Cys. Data were fitted according to Langmuir for LP-i-K3Cys + E3Cys, and a Bragg-Williams isotherm (Eq. 3) was used for LP-K3Cys + i-E3Cys (fits: gray line).
In summary, we found no significant difference in dissociation constant between antiparallel and parallel coiled-coil formations at the membrane interface (see Table 2). All isotherms obtained from lipid bilayers display a similar affinity between E- and K-peptides, with KD-values ranging between 25 μM and 31 μM. Compared with measurements of peptide dimerization in solution (CD spectroscopy) or within a hydrogel (SPR spectroscopy), the KD-values found for coiled-coil dissociation at the membrane interface are increased by one order of magnitude. Of note, the covalent immobilization of one peptide to a 3D hydrogel does not impair the release of free assembly energy associated with coiled-coil formation. We attribute this decrease in apparent affinity at the membrane interface to loss of translational entropy, which inevitably occurs due to a restriction in mobility of the surface-bound peptides. We consider the binding of peptides from solution to their corresponding LP counterparts embedded in a membrane as an adsorption process in which at least one degree of translational freedom is lost. Generally, loss of entropy upon adsorption on a membrane surface is due to the conversion of free translational and rotational degrees of freedom into bound motions, i.e., soft vibrations of only a few kBT. We found that upon formation of coiled-coil structures on the surface, the lateral movement was significantly reduced compared with the mobility of lipids. Along these lines, diffusion measurements suggest that coiled-coil LPs are less laterally mobile in the bilayer matrix (Fig. 5). The average lateral diffusion constant D is reduced almost by a factor of 10 in comparison with the lateral mobility of phospholipids in a plain SSM (DPOPC = (5.5 ± 2.5) μm2/s; DLP-K3Cys+E3Cys = (0.8 ± 0.1) μm2/s; and DLP-i-K3Cys+i-E3Cys = (0.5 ± 0.1) μm2/s). Also, the mobile fraction is reduced from ∼74% in a plain POPC SSM to ∼43–45% when coiled-coil complexes are present. Therefore, we assume that entropy loss contributes to the free energy in an appreciable way. By following the argument of Ben-Tal et al. (32), we may be able to put this argument in the form of numbers. The authors estimated a loss of entropy that corresponds to a free energy increase of −TΔS° of ∼1.5 kBT per degree of translational freedom from the adsorbing molecule. In general, despite the quantitative estimations from theory, we can safely assume that our reduction in binding enthalpies compared with association of peptides in solution arises from a loss of entropy upon binding to membrane-based LPs. This correction adds up to an additional maximum of free-energy loss of ≈−4.5 kBT for coiled-coil formation on a membrane surface, leading to essentially identical affinities at the membrane surface and in solution (see Table 2).
Table 2.
Dissociation constants KD and corresponding free enthalpies ΔG° for coiled-coil formation determined by CD spectroscopy and SPR, and at the membrane interface by ellipsometry (Langmuir)
| Method | K3Cys + E3Cys |
i-K3Cys + i-E3Cys |
K3Cys + i-E3Cys |
i-K3Cys + E3Cys |
||||
|---|---|---|---|---|---|---|---|---|
| KD/μM | ΔG°/kBT | KD/μM | ΔG°/kBT | KD/μM | ΔG°/kBT | KD/μM | ΔG°/kBT | |
| CD | 4.1 ± 0.7 | −12.4 | 7.5 ± 0.6 | −11.8 | 2.9 ± 0.8 | −12.8 | 2.5 ± 0.6 | −12.9 |
| SPR | 0.5 ± 0.3 | −14.5 | 2.3 ± 1.8 | −13.0 | 0.5 ± 0.3 | −14.5 | 1.2 ± 1.0 | −13.6 |
| Langmuir | 28 ± 1 | −10.5 | 25 ± 3 | −10.6 | 31 ± 1∗ | −10.4 | 28 ± 2 | −10.5 |
Ellipsometry data were fitted with the Bragg-Williams isotherm.
Figure 5.
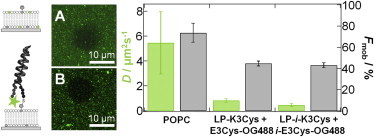
Lateral mobility of peptides measured by fluorescence recovery after photobleaching. Left: Schematic drawings of the labeling techniques. Plain SSM was labeled with boron-dipyrromethene (BODIPY, top) and the coiled-coil structure was labeled with Oregon Green 488 maleimide (OG488). (A and B) Fluorescence micrograph of (A) plain POPC/MCCDOPE/BODIPY 89:10:1 and (B) POPC/MCCDOPE 90:10 functionalized with LP-K3Cys and E3Cys (OG488 labeled; scale bars: 10 μm). Images were collected shortly after the bleaching pulse. The graph on the right side shows diffusion coefficients (green) and corresponding mobile fractions (gray) of a plain SSM consisting of POPC/MCCDOPE/BODIPY 89:10:1 (POPC), and SSM consisting of POPC/MCCDOPE 90:10 functionalized with LP-K3Cys bound to E3Cys (OG488 labeled; LP-K3Cys + E3Cys-OG488), and functionalized with LP-i-K3Cys bound to i-E3Cys (OG488 labeled; LP-i-K3Cys + i-E3Cys-OG488).
Finally, we note that otherwise coiled-coil formation on SSM is independent of the environment. Furthermore, the thermodynamics of peptide association is also independent of helix orientation regardless of whether parallel or antiparallel coiled-coil structures are formed. Also, for inverted peptides with the opposite direction of helical dipole moment, no substantial changes in binding strength could be found. All values are in the same regime, with slightly higher dissociation constants for antiparallel coiled-coil formation. Interestingly, it has been proposed that ∼16 kBT are required for hemifusion (8,10). As a consequence, a single dimer of this size is not sufficient to induce fusion, mainly due to the missing 3–4 kBT of free energy spent to reduce entropy at the membrane interface. Therefore, the necessary amount of free energy can only be recruited by forming a larger number of coiled-coil complexes or by using longer helices with more heptad repeats.
Fusogenicity of parallel and antiparallel coiled-coil complexes
Fusion efficiency with regard to both lipid mixing and content mixing was explored as a function of peptide assembly. We were mainly interested in determining whether parallel coiled-coil structures lead to higher fusion rates compared with antiparallel assemblies according to the zipper model, which predicts a shorter distance between the two opposing lipid bilayers if the peptides form a parallel coiled-coil bundle (Fig. 1). Apart from the alignment, we also addressed the question of whether reversal of the sequence changes the fusion efficiencies. Because all peptide combinations show virtually identical binding constants, differences in fusogenic activity can be solely attributed to differences in molecular orientation.
We quantified lipid mixing and content mixing by carrying out dequenching fluorescence assays (13,33). For this purpose, we prepared two liposome populations: one containing a fluorescent dye in self-quenching concentration, and one devoid of fluorophore. We used 10 mol % Texas Red DHPE in the membrane shell for lipid mixing, and 20 mM sulforhodamine B (SRB), a water-soluble dye enriched in the liposome lumen, for content mixing. The fluorescently labeled liposome populations were decorated with K-peptides, and the second, unlabeled vesicle population was functionalized with E-peptides. After the two vesicle populations were mixed, we detected lipid mixing or content mixing by increasing fluorescence intensity due to dilution of the corresponding fluorescence dye (Fig. 6).
Figure 6.
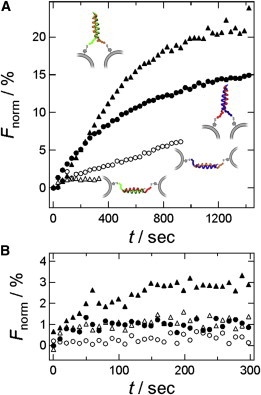
Lipid-mixing and content-mixing experiments with small unilamellar vesicles (SUVs) decorated with E- and K-peptides (100% refers to 1:1 vesicle mixing). (A) Texas Red self-quenching assay for lipid mixing. Labeled SUVs were functionalized with K-peptides and mixed with unlabeled E-peptide bearing SUVs (start of mixing: t = 0). Parallel peptide packing facilitates lipid mixing (solid circles: K3Cys + E3Cys; solid triangles: i-K3Cys + i-E3Cys) compared with antiparallel coiled-coil formation (open circles: K3Cys + i-E3Cys, open triangles: i-K3Cys + E3Cys). (B) Content mixing monitored with an SRB self-quenching assay. K-peptide-functionalized SUVs were filled with SRB (20 mM) and mixed with buffer-filled SUVs displaying E-peptides. At time t = 0, Ca2+ ions were added (cfinal = 8 mM). Same legend as used in panel A.
We define 100% fusion as a one-to-one mixture of vesicles, i.e., one fluorescently labeled vesicle interacts with exactly one unlabeled liposome, resulting in a calculable dilution of fluorophore concentration (see Supporting Material). Because the quenching mechanism for lipids covalently coupled to a fluorescent dye, such as Texas Red DHPE, depends on the membrane composition (34), we measured the concentration dependence of fluorescence for our lipid system using the Stern-Volmer equation (Fig. S4). A concentration-dependent Stern-Volmer constant for SRB was previously published (35). From these data, we determined a value for 100% fusion, which was used for normalization.
From Fig. 6 A, it is evident that parallel coiled-coil formation leads to a substantial lipid mixing, which is visible by the increasing fluorescence intensity of the Texas Red dye, whereas antiparallel dimerization shows slower or negligible lipid mixing of the two vesicle populations. This result also proves a distinct peptide specificity for the vesicle-vesicle interaction. The observed fusogenicity cannot be due to charge effects, because both K-peptides and both E-peptides carry the same charges. Therefore, we attribute the different docking and fusion efficiencies of the four peptide pairs to purely structural effects. A higher fusogenicity of parallel coiled-coil pairs was expected, because in this case the two membranes are forced into close contact with each other, whereas in the antiparallel alignment the peptides instead create a spacer that holds the two membranes apart. In the content-mixing experiments (Fig. 6 B), only the parallel interaction mediated by i-K3Cys and i-E3Cys showed a small but detectable increase in Fnorm, and the antiparallel coiled-coil assembly was virtually indistinguishable from control measurements in the absence of fusion peptides. Furthermore, it is important to note that content mixing was only detectable after the addition of Ca2+ ions, which was not needed for lipid-mixing experiments. We attribute this to the bridging effect of Ca2+ binding to PC, PE, and nonreacted MCCDOPE carrying a negative charge. Similar observations were made by Höök et al. (36) for otherwise zwitterionic lipids. However, full fusion is generally low compared with reconstituted SNAREs (37).
The only peptide assembly that showed a positive result in both fluorescence fusion assays was the coiled-coil consisting of i-K3Cys and i-E3Cys. Concerning the dissociation constant, this particular dimer showed the weakest binding in solution, but on an SSM its KD was the lowest. However, it is unlikely that this small difference in free energy explains the fusion activity of the peptides. Instead, we attribute the difference in content mixing to the dipole orientation affecting the peptide arrangement in the contact zone. Other important factors comprise higher-order assemblies as observed for peptides on the membrane surface. Preliminary fluorescence (Fig. 5) and atomic force microscopy images (G. Pähler, B. Lorenz, and A. Janshoff, unpublished) suggest that fusion peptides are organized in small domains whose sizes seem to be dependent on the peptide sequence.
The slow but consistent lipid mixing observed for the antiparallel coiled-coil consisting of K3Cys and i-E3Cys is also notable. This could be rationalized by the binding cooperativity found in ellipsometry studies for this particular dimer. Here, we also assume that this cooperativity mirrors the lateral rearrangement of LPs required to accommodate lipid mixing. Further investigations are planned to quantify this effect.
In summary, we found efficient lipid mixing for parallel coiled-coil heterodimerization as opposed to the corresponding antiparallel assembly. Content mixing, however, required addition of Ca2+ and was only observed for a single parallel combination of the coiled-coil dimers formed between the two opposing membranes. The sole mixing of both vesicle populations before addition of Ca2+ (Fig. 6 B, t = 0) led to no increase in fluorescence intensity (data not shown).
Conclusions
The impact of peptide sequence on coiled-coil formation in solution and in the context of lipid bilayers was addressed with respect to docking thermodynamics and fusion efficiency. We found that neither antiparallel/parallel packing nor inversion of the helical dipole moment had a significant influence on the thermodynamics of coiled-coil dimerization. The dissociation constants of all peptide dimers were in the same regime. Free-enthalpy changes were significantly reduced when one peptide was coupled to a lipid bilayer compared with coiled-coil formation in solution. The difference of 3–4 kBT between coiled-coil formation in solution and at the membrane interface was largely attributed to a loss of translational degrees of freedom upon binding to the membrane, and corroborated by measurements of lateral diffusion suggesting that coiled-coil dimers organize in rather immobile clusters. The lateral diffusion constant of coiled-coil structures on the surface decreased by a factor of 10, whereas the completely immobile fraction increased by 30%.
The fusion assays reveal that a parallel coiled-coil formation is needed for significant lipid mixing. We attribute this finding to the difference in proximity needed to overcome the hydration barrier. The zipper-like arrangement of the two peptides ensures a closer vicinity of the two opposing membranes, thereby facilitating hemifusion. Simonsson et al. (36) found a sixfold increased number of fusion events for a zipper-like orientation of two complementary DNA strands compared with DNA strands that form antiparallel double helices. In our case, however, full fusion of the two leaflets was rarely observed, and an appreciable efficiency was observed only for a single sequence. There are two possible explanations for this observation. First, the reduced lateral mobility prevents accumulation of coiled-coil dimers in the contact zone of the two vesicles, thereby limiting fusion efficiency. Second, thus far we have only employed lipids to anchor the recognition elements. Meyenberg et al. (13) found that peptidic transmembrane anchors may boost fusion due to the finite stiffness of the helix and more severe perturbation of the membrane.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 937).
Supporting Material
References
- 1.Jahn R., Lang T., Südhof T.C. Membrane fusion. Cell. 2003;112:519–533. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 2.Tamm L.K., Crane J., Kiessling V. Membrane fusion: a structural perspective on the interplay of lipids and proteins. Curr. Opin. Struct. Biol. 2003;13:453–466. doi: 10.1016/s0959-440x(03)00107-6. [DOI] [PubMed] [Google Scholar]
- 3.Robson Marsden H., Kros A. Self-assembly of coiled coils in synthetic biology: inspiration and progress. Angew. Chem. Int. Ed. Engl. 2010;49:2988–3005. doi: 10.1002/anie.200904943. [DOI] [PubMed] [Google Scholar]
- 4.Li F., Pincet F., Tareste D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat. Struct. Mol. Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 5.Steinmetz M.O., Jelesarov I., Kammerer R.A. Molecular basis of coiled-coil formation. Proc. Natl. Acad. Sci. USA. 2007;104:7062–7067. doi: 10.1073/pnas.0700321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jahn R., Scheller R.H. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 7.Südhof T.C., Rothman J.E. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chernomordik L.V., Kozlov M.M. Current Topics in Membranes. Academic Press; New York: 2011. Membrane fusion; pp. i–iii. [Google Scholar]
- 9.Smith E.A., Weisshaar J.C. Docking, not fusion, as the rate-limiting step in a SNARE-driven vesicle fusion assay. Biophys. J. 2011;100:2141–2150. doi: 10.1016/j.bpj.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markin V.S., Albanesi J.P. Membrane fusion: stalk model revisited. Biophys. J. 2002;82:693–712. doi: 10.1016/S0006-3495(02)75432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen F.S., Melikyan G.B. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol. 2004;199:1–14. doi: 10.1007/s00232-004-0669-8. [DOI] [PubMed] [Google Scholar]
- 12.Robson Marsden H., Nina A., Kros A. A reduced SNARE model for membrane fusion . Angew. Chem. Int. Ed. Engl. 2009;48:2330–2333. doi: 10.1002/anie.200804493. [DOI] [PubMed] [Google Scholar]
- 13.Meyenberg K., Lygina A.S., Diederichsen U. SNARE derived peptide mimic inducing membrane fusion. Chem. Commun. (Camb.) 2011;47:9405–9407. doi: 10.1039/c1cc12879e. [DOI] [PubMed] [Google Scholar]
- 14.Litowski J.R., Hodges R.S. Designing heterodimeric two-stranded α-helical coiled-coils. Effects of hydrophobicity and α-helical propensity on protein folding, stability, and specificity. J. Biol. Chem. 2002;277:37272–37279. doi: 10.1074/jbc.M204257200. [DOI] [PubMed] [Google Scholar]
- 15.Schuy S., Treutlein B., Janshoff A. In situ synthesis of lipopeptides as versatile receptors for the specific binding of nanoparticles and liposomes to solid-supported membranes. Small. 2008;4:970–981. doi: 10.1002/smll.200701006. [DOI] [PubMed] [Google Scholar]
- 16.Monera O.D., Kay C.M., Hodges R.S. Electrostatic interactions control the parallel and antiparallel orientation of α-helical chains in two-stranded α-helical coiled-coils. Biochemistry. 1994;33:3862–3871. doi: 10.1021/bi00179a010. [DOI] [PubMed] [Google Scholar]
- 17.Lygina A.S., Meyenberg K., Diederichsen U. Transmembrane domain peptide/peptide nucleic acid hybrid as a model of a SNARE protein in vesicle fusion. Angew. Chem. Int. Ed. Engl. 2011;50:8597–8601. doi: 10.1002/anie.201101951. [DOI] [PubMed] [Google Scholar]
- 18.Whitmore L., Wallace B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32(Web Server issue) doi: 10.1093/nar/gkh371. W668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 20.Pace C.N., Vajdos F., Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou N.E., Kay C.M., Hodges R.S. Synthetic model proteins: the relative contribution of leucine residues at the nonequivalent positions of the 3-4 hydrophobic repeat to the stability of the two-stranded α-helical coiled-coil. Biochemistry. 1992;31:5739–5746. doi: 10.1021/bi00140a008. [DOI] [PubMed] [Google Scholar]
- 22.Chao H., Houston M.E.J., Jr., Hodges R.S. Kinetic study on the formation of a de novo designed heterodimeric coiled-coil: use of surface plasmon resonance to monitor the association and dissociation of polypeptide chains. Biochemistry. 1996;35:12175–12185. doi: 10.1021/bi9530604. [DOI] [PubMed] [Google Scholar]
- 23.Apostolovic B., Klok H.-A. pH-sensitivity of the E3/K3 heterodimeric coiled coil. Biomacromolecules. 2008;9:3173–3180. doi: 10.1021/bm800746e. [DOI] [PubMed] [Google Scholar]
- 24.Myszka D.G. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr. Opin. Biotechnol. 1997;8:50–57. doi: 10.1016/s0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- 25.Day Y.S.N., Baird C.L., Myszka D.G. Direct comparison of binding equilibrium, thermodynamic, and rate constants determined by surface- and solution-based biophysical methods. Protein Sci. 2002;11:1017–1025. doi: 10.1110/ps.4330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlsson R., Roos H.k., Persson B. Kinetic and concentration analysis using BIA technology. Methods. 1994;6:99–110. [Google Scholar]
- 27.Goormaghtigh E., Raussens V., Ruysschaert J.-M. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta. 1999;1422:105–185. doi: 10.1016/s0304-4157(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 28.Heimburg T., Schünemann J., Geisler N. FTIR-spectroscopy of multistranded coiled coil proteins. Biochemistry. 1999;38:12727–12734. doi: 10.1021/bi983079h. [DOI] [PubMed] [Google Scholar]
- 29.Faiss S., Schuy S., Janshoff A. Phase transition of individually addressable microstructured membranes visualized by imaging ellipsometry. J. Phys. Chem. B. 2007;111:13979–13986. doi: 10.1021/jp073671t. [DOI] [PubMed] [Google Scholar]
- 30.Schuy S., Schäfer E., Janshoff A. Coiled-coil lipopeptides mimicking the prehairpin intermediate of glycoprotein gp41. Angew. Chem. Int. Ed. Engl. 2009;121:765–768. doi: 10.1002/anie.200803080. [DOI] [PubMed] [Google Scholar]
- 31.Hinderliter A., May S. Cooperative adsorption of proteins onto lipid membranes. J. Phys. Condens. Matter. 2006;18:S1257–S1270. doi: 10.1088/0953-8984/18/28/S09. [DOI] [PubMed] [Google Scholar]
- 32.Ben-Tal N., Honig B., Ben-Shaul A. Association entropy in adsorption processes. Biophys. J. 2000;79:1180–1187. doi: 10.1016/S0006-3495(00)76372-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lorenz B., Keller R., Janshoff A. Colloidal probe microscopy of membrane-membrane interactions: from ligand-receptor recognition to fusion events. Biophys. Chem. 2010;150:54–63. doi: 10.1016/j.bpc.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 34.MacDonald R.I. Characteristics of self-quenching of the fluorescence of lipid-conjugated rhodamine in membranes. J. Biol. Chem. 1990;265:13533–13539. [PubMed] [Google Scholar]
- 35.Plant A.L. Mechanism of concentration quenching of a xanthene dye encapsulated in phospholipid vesicles. Photochem. Photobiol. 1986;44:453–459. doi: 10.1111/j.1751-1097.1986.tb04692.x. [DOI] [PubMed] [Google Scholar]
- 36.Simonsson L., Jönsson P., Höök F. Site-specific DNA-controlled fusion of single lipid vesicles to supported lipid bilayers. ChemPhysChem. 2010;11:1011–1017. doi: 10.1002/cphc.200901010. [DOI] [PubMed] [Google Scholar]
- 37.Parlati F., Weber T., Rothman J.E. Rapid and efficient fusion of phospholipid vesicles by the α-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc. Natl. Acad. Sci. USA. 1999;96:12565–12570. doi: 10.1073/pnas.96.22.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castorph S., Schwarz Henriques S., Salditt T. Synaptic vesicles studied by dynamic light scattering. Eur Phys J E Soft Matter. 2011;34:1–11. doi: 10.1140/epje/i2011-11063-2. [DOI] [PubMed] [Google Scholar]
- 39.Andrushchenko V.V., Vogel H.J., Prenner E.J. Optimization of the hydrochloric acid concentration used for trifluoroacetate removal from synthetic peptides. J. Pept. Sci. 2007;13:37–43. doi: 10.1002/psc.793. [DOI] [PubMed] [Google Scholar]
- 40.Axelrod D., Koppel D.E., Webb W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soumpasis D.M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 1983;41:95–97. doi: 10.1016/S0006-3495(83)84410-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leonenko Z.V., Finot E., Cramb D.T. Investigation of temperature-induced phase transitions in DOPC and DPPC phospholipid bilayers using temperature-controlled scanning force microscopy. Biophys. J. 2004;86:3783–3793. doi: 10.1529/biophysj.103.036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.