

Abstract
Each segment along arterial vessels adapts to different circumstances, including blood pressure and sympathetic innervation. PKC and Rho-associated kinase (ROCK) Ca2+-sensitizing pathways leading to myosin phosphatase inhibition are critically involved in α1-adrenoceptor-mediated vascular smooth muscle contraction in distinctive time-dependent manners. We tested whether the amplitude and time course of each pathway varies dynamically between arterial segments. Using pharmacological approaches, we determined the time-dependent roles of Ca2+ release, Ca2+ influx, PKC and ROCK in α1-agonist-induced contraction and phosphorylation of key proteins in denuded rat small mesenteric artery, midsized caudal artery and thoracic aorta. SR Ca2+ release and voltage-dependent Ca2+ influx were essential for the initial rising and late sustained phases, respectively, of phenylephrine-induced contraction, regardless of arterial size. In small mesenteric arteries, α1A-subtype-specific antagonists and inhibitors of PKC, but not ROCK, markedly reduced the initial and late phases of contraction in a non-additive manner and suppressed phosphorylation of myosin light chain (MLC) and CPI-17, but not myosin targeting subunit of myosin light chain phosphatase (MYPT1). In aorta, an α1D-specific antagonist reduced both the initial and late phases of contraction with a significant decrease in MLC but not CPI-17 or MYPT1 phosphorylation. ROCK inhibitors, but not PKC inhibitors, suppressed the sustained phase of contraction with a decrease in MLC and MYPT1 phosphorylation in the aorta. The effect of ROCK inhibitors was additive with the α1D-antagonist. The results for midsized arteries were intermediate. Thus, the PKC–CPI-17 Ca2+-sensitizing pathway, which is dependent on PKC subtype and a Ca2+-handling mechanism, and is downstream of α1A receptors, plays a major role in α1-agonist-induced contraction of small resistance arteries in the splanchnic vascular beds. The effect of PKC and ROCK increases and decreases, respectively, with decreasing arterial size.
Key points
Each segment along arterial vessels adapts to different circumstances, such as high blood pressure in the central and low pressure in the peripheral arteries and high sympathetic innervation in the peripheral and low innervation in the central arteries.
We tested, using pharmacological tools, whether the amplitude and time course of each signalling pathway varies dynamically between arterial segments in rat.
In small mesenteric arteries, α1-agonist produced a contraction and myosin light chain phosphorylation through sequential activation of α1A-subtype receptors, Ca2+, PKC and protein kinase C-potentiated protein phosphatase inhibitor protein 17 kDa (CPI-17).
In large aorta, α1-agonist-induced contraction and phosphorylation were produced through activation of α1D receptors followed by a Ca2+ increase and constitutively active Rho-kinase in an independent manner. The results for midsized arteries were intermediate.
Our findings provide insights into the development of new therapeutic agents controlling the size-dependent vasoconstriction.
Introduction
Smooth muscle contraction is primarily regulated by reversible 20 kDa myosin light chain (MLC) phosphorylation, the extent of which is determined by the balance between MLC kinase (MLCK) and MLC phosphatase (MLCP) activity. Contractile agonists increase both [Ca2+]i, which upregulates Ca2+–calmodulin-dependent MLCK (Kamm & Stull, 2001), and contractile Ca2+ sensitivity (Ca2+ sensitization) through G protein-mediated downregulation of MLCP (Somlyo & Somlyo, 1994) and these increases are dually regulated in fully differentiated smooth muscle (Dimopoulos et al. 2007). [Ca2+]i increases following sarcoplasmic reticulum (SR) Ca2+ release and Ca2+ influx through voltage-dependent Ca2+ channels while Ca2+ sensitization is mediated by PKC and Rho-associated kinase (ROCK). Nobe & Paul (2001) analysed in porcine coronary artery the temporal relationship between [Ca2+]i and amplitude of contraction in response to the thromboxane A2 analogue U46619 and found that the initial rising phase of contraction was associated with Ca2+ release and PKC-mediated Ca2+ sensitization. In the sustained phase of contraction, where the force level is much higher than that of the initial phase, Ca2+ influx and ROCK-mediated Ca2+ sensitization are dominant. Similarly, in rabbit femoral artery smooth muscle, an α1-agonist rapidly increased [Ca2+]i and resulted in MLC phosphorylation through the classical Gq–PLCβ–IP3–SR-Ca2+–calmodulin–MLCK pathway (Dimopoulos et al. 2007). Simultaneously, the smooth muscle-specific myosin phosphatase inhibitor protein CPI-17 (Eto, 2009) is phosphorylated at Thr38 to significant levels within seconds through the Gq–PLCβ–(SR-Ca2++DAG)–PKC pathway, which leads to rapid MLCP inhibition. In fact, inhibition of either Ca2+ release from the SR or PKC potently inhibited the rapid phosphorylation of both CPI-17 and MLC as well as the initial rising phase of contraction, but the slow development of contraction remained. These results demonstrate that CPI-17-mediated rapid MLCP inhibition together with MLCK activation synergistically triggers immediate MLC phosphorylation and contraction. After transient Ca2+ release from the SR, Ca2+ influx through voltage-dependent Ltype Ca2+ channels maintains a tonic level of cytoplasmic Ca2+, which in turn activates MLCK (Somlyo & Somlyo, 1994; Isotani et al. 2004). In parallel, agonist-induced stimulation of the Gα12/13 G protein and partial Ca2+ influx activate the small G protein RhoA, which then activates ROCK (Matsui et al. 1996; Ishizaki et al. 1996; Gohla et al. 2000; Somlyo & Somlyo, 2003; Sakurada et al. 2003). Activated ROCK phosphorylates the myosin targeting subunit of MLCP, MYPT1, at Thr853 and Thr696 (in human 133 kDa sequence; Kitazawa et al. 2003; Niiro et al. 2003; Wilson et al. 2005), resulting in MLCP inhibition. RhoA/ROCK-mediated MLCP inhibition, in addition to the partial activation of MLCK via Ca2+ influx, may therefore contribute to MLC phosphorylation in the tonic phase of contraction so that the order of the pathway is G12/13–RhoA–ROCK–MYPT1. Thus, the biphasic inhibition of MLCP through the sequential activation of PKC followed by ROCK in co-operation with the biphasic activation of MLCK by Ca2+ release and Ca2+ influx, accounts for the rapid increase and subsequent maintenance of MLC phosphorylation in femoral artery (Dimopoulos et al. 2007). The role of the Ca2+-independent Gα12/13 G protein pathway in α1-agonist-induced contraction, however, is questionable because all α1-adrenoceptor subtypes in smooth muscle are thought to be linked only to the Gq G protein (Wirth et al. 2008; Docherty, 2010). Thus, the specific coupling of G protein subtype to downstream signalling may determine the response of smooth muscle contraction to agonist stimuli, although the entire pathway remains unclear.
Arteries are blood vessels that carry oxygenated blood under high pressure away from the heart through large conduit vessels like the aorta, then through midsized muscular arteries, small peripheral resistance arteries and arterioles to reach the peripheral tissue capillaries throughout the body. Each segment along arterial vessels adapts to specific conditions including blood pressure, flow speed and nerve innervation, suggesting that different signal transduction mechanisms may support different functions at various locations. The expression and function of α1-adrenoceptor subtypes in arterial smooth muscle varies according to location, with the α1A-adrenoceptor subtype being substantially more expressed in peripheral arteries than in central conduit arteries of mice (Rokosh & Simpson, 2002) while the α1D and α1B subtypes have ubiquitous distribution with much higher mRNA content for the 1D- compared with the 1B-subtype (Methven et al. 2009a,b; see Docherty, 2010 for review). However, how the combination of α1-adrenoceptor subtype and the signals mediating Ca2+-sensitizing kinases influences arterial smooth muscle responsiveness is not fully understood. Mueed et al. (2004) used kinase inhibitors to show that steady-state α1-agonist-induced contraction in rat caudal artery is predominantly mediated by ROCK rather than PKC, while both kinases were equally involved in vascular contraction of the aorta. By contrast, Budzyn et al. (2006) found that steady-state contractile responses of rat small mesenteric artery to α1-agonists were almost exclusively mediated by PKC rather than ROCK, while the contraction of the aorta and large mesenteric artery are regulated by both kinases to varying degrees. Thus, we hypothesized that the kinases coupled with G proteins were specific at each vascular locus, and the time-dependent change in kinase activity determined the complex time course of agonist-induced contraction. We therefore examined time-dependent PKC and ROCK signalling by measuring the time courses of contraction, [Ca2+]i, MLC (at Ser19), CPI-17 (at Thr38) and MYPT1 (at Thr853) phosphorylation, and the effects of kinase inhibitors and channel blockers in intact rat small mesenteric artery. We also compared these results to those for the larger caudal artery and thoracic aorta. Our study revealed that PKC and ROCK, as well as Ca2+ release and Ca2+ influx, involve α1-agonist-induced contraction in arteries in size- and time-dependent manners.
Methods
External solution compositions
The external solutions for intact smooth muscle rings were prepared as described previously (Kitazawa et al. 2009). Normal external solution for intact smooth muscle rings was 150 mm NaCl, 4 mm KCl, 2 mm calcium methanesulphonate, 2 mm magnesium methanesulphonate, 5.6 mm glucose and 5 mm Hepes. Potassium methanesulphonate (124 mm) was substituted for NaCl in the depolarizing external solution with all other components used at the same concentration. Both solutions were adjusted to pH 7.4 with Tris.
Tissue preparation, force measurement and quick freezing
All animal procedures were approved by the Animal Care and Use Committee of the Boston Biomedical Research Institute. Sprague–Dawley rats (250–350 g) of either sex were killed with CO2 gas inhalation. After thoracotomy, the thoracic aorta, caudal, mesenteric, intrarenal and ovarian arteries were isolated. Following dissection of fat and soft connective tissue and removal of endothelial layers, each arterial segment with a diameter specified in the Results section was cut into rings of 0.75 or 1.0 mm in length. Two fine tungsten rod tips were inserted into the lumens of the arterial rings. One rod was connected to a force transducer (AM801, SensoNor, Horten, Norway) and the other to a micromanipulator to adjust the muscle length, in which the arterial rings produced a maximum force. For force measurements, each 0.75mm-long ring was mounted in a well on a Bubble chamber plate to allow for rapid solution changes as described previously (Dimopoulos et al. 2007). The solution temperature was maintained at 35°C throughout the experiments. Each arterial ring except the aorta was repeatedly stimulated for 3 min with 124 mm K+ solution at 15 min intervals until the peak contraction no longer increased. For aortas, arterial rings were stimulated for 5 min with high K+ solution at 20 min intervals. The rings were then alternately stimulated with high K+ and 10 μm phenylephrine (PE) until the PE-induced contraction no longer increased. Treatment with high K+ between the PE-induced contractions was required to maintain constant SR Ca2+ loading along with a reproducible time course and amplitude of PE-induced contractions. Arterial ring endothelial layer denudation was confirmed when they displayed no relaxation in response to 10 μm acetylcholine during PE-induced contraction. PE concentrations greater than 1 μm produced a large contraction with a latency time between PE stimulation and onset of contraction that was estimated using the method of Horiuti et al. (1989). To deplete SR Ca2+ stores, arterial rings were incubated in normal external solution containing 1 μm ryanodine (BioMol, Plymouth Meeting, PA, USA) and 20 mm caffeine for 15 min and washed with the same solution without caffeine for another 15 min whereupon caffeine no longer evoked a transient contraction (Dimopoulos et al. 2007). To block L-type voltage-dependent Ca2+ influx, rings were incubated in normal external solution containing 1 μm nicardipine for 10 min and stimulated with PE in the presence of the drug. After treatment, high K+ did not evoke significant contraction in arteries of varying sizes. The time course and amplitude of PE-induced contraction and the effects of inhibitors were not significantly different between male and female animals.
To accommodate the number of small mesenteric arterial rings required for phosphorylation measurements, several 1.0mm-long rings were perpendicularly held together with U-shaped tungsten clips, each end of which was connected with a monofilament silk ring to either the force transducer or micromanipulator extension rod (Supplemental Fig. S1). The arterial ring strings were quick-frozen by plunging into liquid nitrogen-cooled propane (−150°C) at rest and 10 s, 30 s and 3 min after PE stimulation.
Cytoplasmic Ca2+ measurements
Methods for measuring intracellular Ca2+ were as described in detail previously (Dimopoulos et al. 2007). Briefly, conditioned arterial strips were incubated in an external solution containing 10 μm fura-2 AM, 0.5% DMSO and 0.01% Pluronic F127 (Molecular Probes) for 2–3 h at 37°C. After loading, strips were washed in fresh external solution. Measurements of fura-2 fluorescence (by alternating excitations at 340 nm and 380 nm) ratio signal were carried out with the Muscle Research System (SI GmbH, Heidelberg, Germany; Gűth & Wojciechowski, 1986). The fluorescence signal for each excitation light and the ratio signal (F340/F380) were digitized using PowerLab/8SP (ADInstruments, Colorado Springs, CO, USA) and displayed on a computer. The F340/F380 ratio was simply used as a relative [Ca2+]i signal (Konishi et al. 1988) and the resting value was represented as one. The solutions in the tube cuvette were maintained at 35°C. Since the solution exchange with a peristaltic pump in the Muscle Research System was much slower than that of the Bubble chamber system used for force measurements, the time course of Ca2+ signals could not be compared with that of force and MLC phosphorylation obtained in the Bubble chamber system (Kitazawa et al. 2009).
Western blotting
Western blotting experiments were carried out as previously described (Dimopoulos et al. 2007). Briefly, frozen rings were transferred to frozen acetone containing 10% trichloroacetic acid, incubated at –80°C overnight and then gradually warmed to room temperature. The acid-fixed rings were washed with acetone and dried under vacuum. The dried strips were homogenized in Laemmli sample buffer. To examine CPI-17 and MYPT1 phosphorylation levels in the same sample, western blotting experiments were always carried out in duplicate. Equal amounts of each extract were loaded onto 8–15% gradient polyacrylamide gels with a stacking gel. Separated proteins were transferred to the same (0.22 μm in pore size) nitrocellulose membranes in 10% methanol–bicarbonate transfer buffer (pH 9.9; Dunn, 1986) for 1.5 h in a wet-transfer tank (TE22, Hoefer) at 15°C. Thereafter, the membranes were blocked in a Tris-buffered saline solution containing 0.05% Tween-20 and 5% non-fat milk and then incubated with a primary antibody followed by an alkaline phosphatase-conjugated secondary antibody. The immunoblots were developed with an alkaline phosphatase substrate solution (Promega) to visualize immunoreactive proteins. The bands of alkaline phosphatase products were digitized with a colour scanner and analysed with image processing software (Signal Analytics Co., Vienna, VA, USA) that permitted the subtraction of background obtained from regions adjacent to the focused proteins. We compared the ratios of phosphorylated to total amounts of CPI-17, MLC and MYPT1 in paired sets of western blots.
To estimate the stoichiometric amounts of total and phosphorylated CPI-17, SDS extracts of small mesenteric artery and aorta stimulated with PE for 30 s were probed along with various concentrations of phosphorylated recombinant CPI-17 (Dimopoulos et al. 2007). The protein content of the typical mammalian cell was assumed to be 18% of total cell weight and the molecular weight of CPI-17 is 17,000 kDa.
Two-dimensional (2-D) isoelectric focusing–SDS-polyacrylamide gel electrophoresis
The 2-D isoelectric focusing–SDS-polyacrylamide gel electrophoresis was used to determine the stoichiometric amounts of MLC phosphorylation in arteries as described previously (Kitazawa et al. 1991). Briefly, quick-frozen, acid-fixed and dried samples were homogenized in glycerol sample buffer (containing 1% SDS, 10% glycerol, 20 mm dithiothreitol and 100 μg ml−1 bovine serum albumin). Each supernatant of the homogenates was applied to an isoelectric focusing polyacrylamide tube gel with 5% pH ampholytes 4.5/5.4 (Pharmalyte, Pharmacia Biotechnology Inc.), and run overnight. Then, an appropriate portion of gel was grafted onto the top of a SDS–polyacrylamide slab gel (15%) and the second dimension was run. Protein transfer was carried out from polyacrylamide gels to nitrocellulose membranes. The membranes were extensively washed overnight and stained with colloidal gold (BioRad) (Kitazawa et al. 1991). The colloidal gold-stained blots of MLC were digitized and analysed as described above.
It is known that, during the 2-D gel electrophoresis, unphosphorylated non-muscle MLC and doubly phosphorylated smooth muscle MLC co-migrate (Kitazawa et al. 1991). We estimated the amount of non-muscle MLC in MA and aorta assuming that total intensity of the spot at the same position as doubly phosphorylated MLC in 2-D gels was from unphosphorylated non-muscle MLC when resting arterial tissues were treated with 1 μm staurosporine in the Ca2+-free, 2 mm EGTA-containing solution for 1 h at 37°C. We revealed that the average unphosphorylated non-muscle MLC was 4 ± 1 and 21 ± 3% (n= 8) of the total MLC in MA and aorta, respectively. These average values were respectively subtracted from the doubly phosphorylated MLC spots in the 2-D gels for quantification of muscle MLC phosphorylation. The percentage of MLC phosphorylation was calculated by dividing {P1 + (P2 – UN)}/{U + P1 + (P2 – UN)} × 100, where U was the amount of unphosphorylated, P1 monophosphorylated and P2 diphosphorylated muscle MLC, and UN unphosphorylated non-muscle MLC. If UN was more than P2, the component (P2 – UN) was assumed to be zero.
Drugs, chemical reagents and antibodies
Y27632 was a gift from Yoshitomi Pharmaceutical (Mitsubishi Pharma). PDBu, GF109203X, Gö-6976, calphostin C and H-1152 were from BioMol (Enzo Life Sciences). Phenylephrine, acetylcholine and nicardipine were from Sigma–Aldrich. Ryanodine and GSK-429286 were from Tocris Bioscience. The following primary antibodies and dilutions were used in this study: anti-CPI-17 IgY (Kitazawa et al. 2000; 1:5000); anti-pCPI17 IgY (from Dr M. Eto of Thomas Jefferson University; 1:1000), polyclonal anti-PKCα (Sigma; 1:1000); polyclonal anti-PKCβ1/2 (BD Biosciences; 1:200); polyclonal anti-PKCδ (Santa Cruz; 1:200); polyclonal anti-PKCɛ (Sigma; 1:500); polyclonal anti-PP1Cδ (from Dr Eto; 1:5000); polyclonal anti-ROCK1 (Sigma; 1:2000); polyclonal anti-ROCK2 (Santa Cruz Biotechnology; 1:1000); monoclonal anti-RhoA (Santa Cruz Biotechnology; 1:1000); polyclonal anti-MYPT1 (BabCO; 1:5000); polyclonal pMYPT1 Thr696 (Kitazawa et al. 2003; 1:2000); polyclonal anti-pMYPT1 Thr853 (Upstate; 1:5000); monoclonal anti-MLCIgM (Sigma; 1:1000); polyclonal anti-pMLC Ser19 (Cell Signaling; 1:1000); polyclonal anti-pan-actin(Sigma; 1:1000); monoclonal anti-smooth muscle-specific α-actin (Sigma;1:5000); monoclonal anti-β-actin (Sigma; 1:5000). Secondary antibody against chicken IgY was from Promega (1:5000). Anti-mouse and anti-rabbit IgG secondary antibodies (1:5000) were from Chemicon. Anti-mouse IgM secondary antibody was from Sigma (1:5000).
Statistics
Results are expressed as the mean ± SEM of n experiments. Statistical significance was evaluated using ANOVA among all groups and post hoc two-tailed t test between two groups, P < 0.05 being considered significant.
Results
Effect of PKC and ROCK inhibitors on the time course of α1-agonist-induced contraction in rat arteries of varying sizes
We first examined the time course and amplitude of contraction in response to a maximum phenylephrine (PE) concentration (30 μm) and the effect of pre-treatment with the PKC inhibitor GF109203X (3 μm), the ROCK inhibitor Y27632 (10 μm), and a combination of the two. Denuded rat arteries of three different sizes were first used: small mesenteric resistance artery (Fig. 1A; arterial segments from the second- and third-order vessels of mesenteric arterial beds; 203 ± 6 μm inner diameter (i.d.) and 285 ± 5 μm outer diameter (o.d.) measured under Ca2+-free conditions), midsized caudal artery (Fig. 1B; arterial segments from the middle portion of tails; 254 ± 9 μm i.d.; 454 ± 10 μm o.d.) and large thoracic aorta (Fig. 1C; 1503 ± 28 μm i.d.; 1854 ± 52 μm o.d.). The peak of PE-induced contraction was 122 ± 2% (n= 21) of 124 mm K+-induced contraction in mesenteric artery, 127 ± 4% (n= 15) in caudal artery, and 140 ± 10% (n= 14) in aorta. Inhibitor efficacy varied with artery size. In small mesenteric artery (Fig. 1A), GF109203X markedly (80–90%) inhibited both the initial rising and late sustained phases of PE-induced contraction with a significant delay in onset whereas Y27632 had no effect on the initial rising phase of contraction and partially inhibited (20–30%) the sustained phase of contraction. Increasing the Y27632 concentration to 30 μm had no additional effect on the initial phase of contraction (not shown), whereas a combination of both GF109203X and Y27632 diminished PE-induced contraction, suggesting a dominant role of Ca2+ sensitization signalling in mesentery artery contraction (Fig. 1A). To confirm such differential effects of the two inhibitors, we examined the response of arteries from other tissues. Small intrarenal arteries (n= 4; 180±6 μm i.d.; 271 ± 6 μm o.d.) and ovarian arteries (n= 4; 158 ± 10 μm i.d.; 238 ± 8 μm o.d.) essentially showed similar inhibitor responses: GF109203X strongly but Y27632 only partially inhibited PE-induced contraction (Fig. 2A and B). In contrast, both inhibitors almost equally reduced PE-induced contraction of midsized caudal artery (Fig. 1B) and superior mesenteric artery from the proximal part of first-order vessels of mesenteric arterial beds (Fig. 2C; n= 4; 633 ± 34 μm i.d.; 894 ± 32 μm o.d.) in both the initial and sustained phases. In large conduit aorta, GF109203X only partially (20–30%) and Y27632 almost completely abolished the sustained phase, but neither compound induced a clear delay in the initial rising phase in aorta (Fig. 1C). A mixture of GF109203X and Y27632 totally abolished the sustained phase of contraction in all three artery sizes. In both caudal artery and aorta, the initial transient contraction was considerably more resistant to the two inhibitors than the sustained phase (Fig. 1B and C). Figure 3 shows the correlation between artery diameter and kinase inhibitor response, with PE-induced contraction more effectively inhibited by GF109203X in smaller arteries. Together, these results suggest that the efficacy of PKC and ROCK inhibitors on α1-agonist-induced contraction is dependent on tissue size but not localization. In all cases, the inhibitory effects of the two inhibitors on PE-induced contraction were additive.
Figure 1. PE-induced contraction.
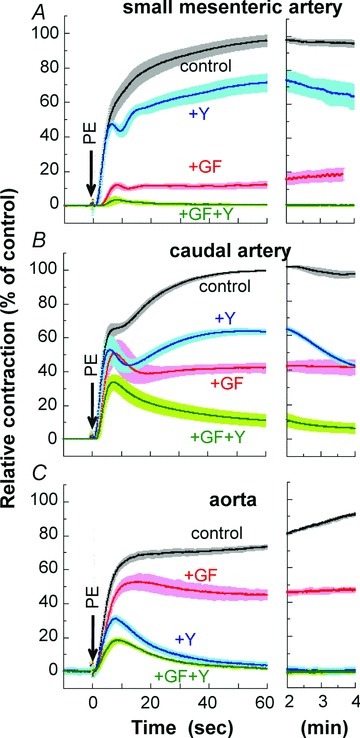
Time course of 30 μm PE-induced contraction with ± 1 SEM in the absence and presence of 3 μm GF109203X, 10 μm Y27632 or a combination of the two inhibitors in rat small mesenteric arteries (A, n= 6–10), midsized caudal arteries (B, n= 4–6) and large aorta (C, n= 4–6). GF, GF109203X; Y, Y27632.
Figure 2. Effect of GF109203X and Y27632 on PE-induced contraction.
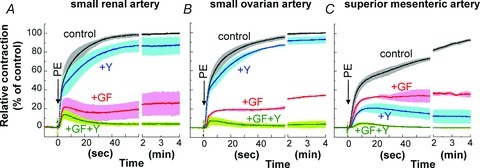
Effect of 3 μm GF109203X, 10 μm Y27632 and a combination of the two inhibitors on PE-induced contraction in small intrarenal (A) and ovarian arteries (B) and midsized primary mesenteric arteries (C). n= 3–6.
Figure 3. Correlation of artery inner diameter (ID) with the ratio (FY-27632/FGF-109203X) between contractions in the presence of 10 μm Y27632 and 3 μm GF109203X.
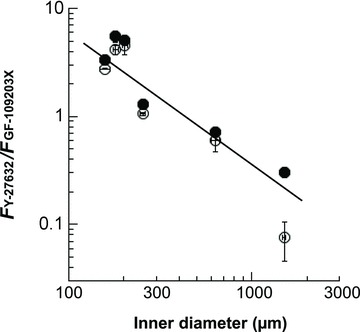
Filled and open circles represent the values 20 s and 3 min, respectively, after PE stimulation. A ratio of one indicates that the two drugs had an equal inhibition efficacy, while ratios larger than one indicates that the efficacy of the GF-compound was higher than that of the Y-compound, and ratios less than one represent a GF efficacy that is less than Y. Data points (filled circles) were fitted with a line of FY-27632/FGF-109203X= 1543 × ID−1.212 (R= 0.9935). The results for outer diameter were similar. n= 4–8.
Role of PKC isoforms in PE-induced contraction of mesentery artery
We compared the effects of three classes of PKC inhibitors (GF109203X, calphostin C and Gö6976) and PKC downregulation on PE-induced contraction of small mesentery arteries. Low concentrations of GF109203X suppressed the initial rising phase more strongly than the late sustained phase of contraction (Fig. 4A). Calphostin C has a high inhibitory potency that is similar to GF109203X, but its inhibitory mechanism involves binding to the regulatory domain of both conventional and novel PKC isoforms (Kobayashi et al. 1989; Bruns et al. 1991), indicating that this microbial compound has an inhibitory spectrum distinct from GF109203X, which antagonizes ATP binding. Calphostin C at 1 μm inhibited both the initial rising and sustained phases of contraction (Fig. 4B), which is similar to the effect of 3 μm GF109203X (Fig. 4A) in small mesenteric artery. Small intrarenal and ovarian arteries showed essentially similar responses to calphostin C (not shown). Gö-6976 selectively inhibits the kinase domain of conventional rather than novel isoform PKCs (Martiny-Baron et al. 1993), and its inhibitory spectrum differs from that of GF109203X (Bain et al. 2007). Similar to GF109203X and calphostin C, Gö-6976 inhibited the initial rising phase of contraction but only partially inhibited the sustained phase of contraction (Fig. 4C). The three inhibitors had similar inhibitory patterns during the initial rising phase of contraction (Fig. 4, inset panels). Together, these results suggest that Ca2+-dependent and Ca2+-independent PKCs play a significant role in the initial rising and sustained phases, respectively, of PE-induced contraction. Sensitivity to GF-109023X for 30 μm PE-induced contraction was similar between small mesenteric artery and aorta, whereas the extent of inhibition was largely different (Supplemental Fig. S2A).
Figure 4. Effect of GF109203X (A), calphostin C (B) and Gö-6976 (C) during the time course of 30 μm PE-induced contraction in small mesenteric arteries.
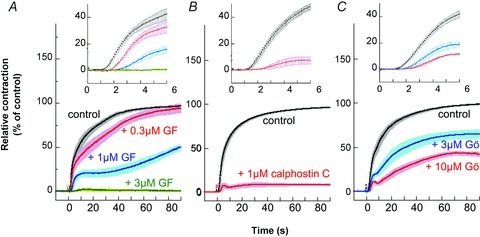
Each ring was incubated in external solution containing a PKC inhibitor for 15 min before PE stimulation. For calphostin C, arterial rings were illuminated by a spotlight from 5 min before and during PE stimulation (Bruns et al. 1991). GF, GF109203X; Gö, Gö-6976. n= 4–6.
Ohanian et al. (1996) reported that among the five PKC isoforms expressed in rat mesenteric artery, downregulation of PKCα and δ by prolonged incubation with phorbol 12,13-dibutyrate (PDBu) caused a parallel loss of PDBu-induced contraction, but did not affect the maximum contractile response to noradrenaline. However, we found a significant decrease in the sensitivity of steady-state PE-induced contraction after 24 h pretreatment with 1 μm active 4β-PDBu, but not for the inactive 4α-PDBu (Fig. 5C). In addition, 4β-PDBu pretreatment caused a larger suppression in the initial rising phase than in the sustained phase of contraction, and the suppression was more profound at lower PE concentrations (Fig. 5D–F). In contrast, PDBu-induced contraction was completely abolished (Fig. 5A; Ohanian et al. 1996). There was a significant decrease in PKCα and δ isoform expression levels to 14 ± 2% and 54 ± 2% (n= 4) of the control, respectively, whereas the expression of PKCβ1/2 or ɛ isoforms was not changed (Fig. 5B). Levels of CPI-17, the major PKC downstream target in differentiated smooth muscle, were also not significantly decreased. This result is similar to that of Ca2+-dependent PKC inhibition (Fig. 4), suggesting that PKCα downregulation plays a significant role in the initial rising phase of PE-induced contraction after prolonged treatment of small mesenteric artery with 4β-PDBu.
Figure 5. Effect of PKC downregulation on contraction and protein expression in small mesenteric arteries.
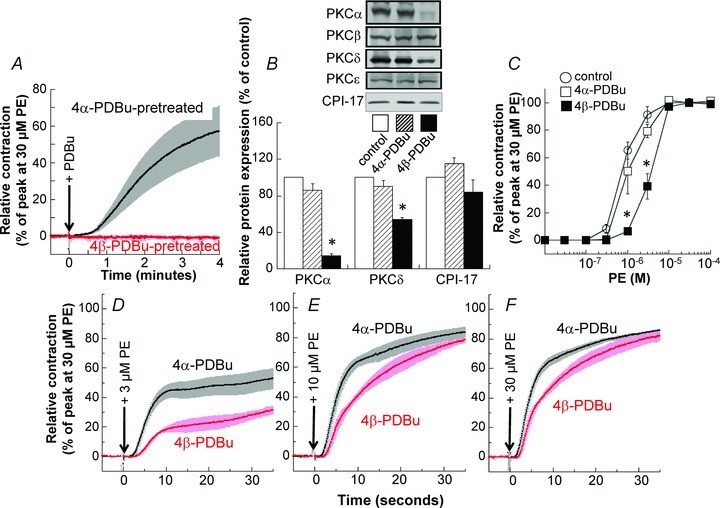
A, effect of pretreatment with active 4β-PDBu for 24 h on 1 μm (4β-)PDBu-induced contraction compared with that of inactive 4α-PDBu pretreatment. The time course of contraction in 4α-PDBu-pretreated arterial rings was comparable with that of control (0.1% DMSO pretreatment for 24 h, not shown). B, effect of 4β-PDBu pretreatment on relative protein expression compared with that of control and 4α-PDBu pretreatment. C, effect of 4β-PDBu pretreatment on steady-state PE concentration–contraction relationship. D, E and F, effect of 4β-PDBu pretreatment on the time course of 3, 10 and 30 μm PE-induced contraction, respectively. Asterisks represent a significant difference between the control and 4α-PDBu treatment. n= 4.
Role of ROCK in PE-induced aorta contraction
Y27632 has widely been used as a ROCK inhibitor, but it also eqi-potently inhibits several members of the AGC subfamily of protein kinases in vitro (Bain et al. 2007). To investigate whether or not Y27632 produces the potent inhibition of PE-induced contraction in arterial smooth muscle mainly through inhibition of ROCK, two other ROCK inhibitors, H-1152 and GSK-429286, were used to compare with Y27632 effects in aorta and mesenteric artery. The ROCK inhibitor H-1152 has a 10-fold higher potency compared with Y27632 and some specificity differences with respect to other protein kinases (Nichols et al. 2009). As shown in Fig. 6, H-1152 had the same inhibitory effect on the time course of PE-induced contraction in aorta as Y27632, albeit with nearly ten times higher potency (Fig. 6A vs. B; also see Supplemental Fig. S2B vs. C). GSK-429286 has an inhibitory potency to ROCK similar to that of H-1152, and shows no inhibitory effect on LRRK2, which is effectively inhibited by either Y27632 or H-1152 (Nichols et al. 2009). GSK-429286 similarly inhibited the sustained phase of PE contraction (Fig. 6C; Supplemental Fig. S2D). These results suggest that the Y-, H- and GSK-compounds suppress the sustained phase of PE contraction all by specifically inhibiting ROCK in rat aorta smooth muscle. Similar sensitivity was also observed for the three ROCK inhibitors in mesenteric artery, although they had much smaller effects compared with those seen for aorta (Supplemental Fig. S2).
Figure 6. Effect of various ROCK inhibitors on PE-induced contraction in rat aorta.
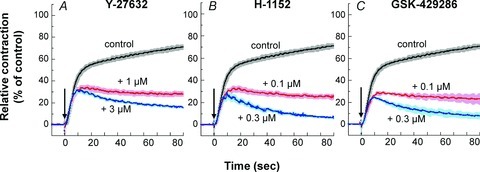
PE (30 μm) was added at time zero (arrow). Each aorta ring was incubated in external solution containing a ROCK inhibitor for 15 min before PE stimulation. n= 4.
Effects of inhibitors on Ca2+ rise
In rabbit femoral artery, both GF109203X at 3 μm and Y27632 at 10 μm significantly but only partially decreased the rate of initial rise of Ca2+ in response to PE but did not reduce the sustained level of Ca2+ (Dimopoulos et al. 2007). In both rat small mesenteric artery and aorta, the rate of initial rise of Ca2+ was not significantly reduced in the presence of either GF109203X or Y27632 (Fig. 7). The sustained level of Ca2+ in small mesenteric artery was significantly but partially decreased by the presence of GF109203X but not Y27632 (Fig. 7A) whereas in aorta the sustained Ca2+ level was slightly but significantly decreased by the presence of Y27632 but not GF109203X (Fig. 7B). However, another potent ROCK inhibitor GSK-429286 at 1 μm had no significant effect on Ca2+ level in either the initial rising or sustained phase of PE-induced contraction in aorta (Supplemental Fig. S3).
Figure 7.
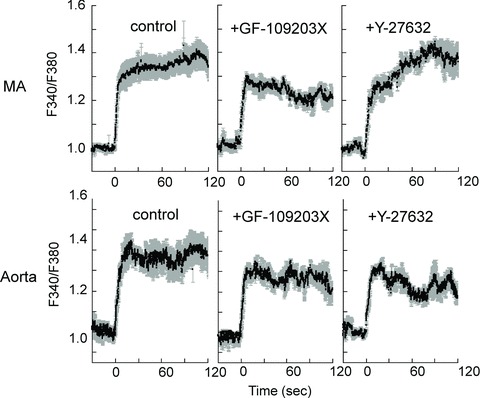
Effect of 3 μm GF109203X and 10 μm Y27632 on time course of Ca2+ signal during PE-induced contraction in small mesenteric artery (MA; A) and aorta (B). A, n= 3–9. B, n= 3.
Effects of inhibiting Ca2+ release and blocking Ca2+ influx
As previously shown in rabbit femoral artery (Dimopoulos et al. 2007), depletion of intracellular Ca2+ stores by ryanodine treatment (Ashida et al. 1988; see Methods for protocol) diminished the initial rapid Ca2+ rise in response to PE but the sustained phase of Ca2+ was slowly developed in small mesenteric artery (Fig. 8A). Treatment with the voltage-dependent Ca2+ channel blocker nicardipine (Nakayama et al. 1985) strongly inhibited the sustained but not initial rapid phase of Ca2+ rise (Fig. 8B). A mixture of ryanodine and nicardipine totally abolished an increase in Ca2+ in response to PE (Fig. 8B) as seen in rabbit femoral artery (Dimopoulos et al. 2007).
Figure 8. Effect of treatment with 1 μm ryanodine (rye, A) and 1 μm nicardipine (nic, B) on time course of Ca2+ rise in response to 30 μm PE in small mesenteric artery.
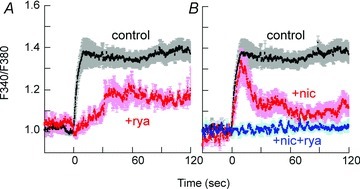
n= 3–4.
Figure 9 illustrates the effects of ryanodine and nicardipine on the time course of PE-induced contraction in small mesenteric artery (A), midsized caudal artery (B) and large aorta (C). Ryanodine pretreatment largely delayed the onset of contraction in all rat arteries of varying sizes as seen in rabbit femoral artery (Dimopoulos et al. 2007; see Supplemental Fig. S4 for more details). The late sustained phase of contraction in the presence of ryanodine was restored to a level similar to control in small mesenteric artery but to a significantly lower level than control in caudal artery and aorta (Fig. 9B and C). Treatment with nicardipine primarily inhibited the sustained phase of PE-induced contraction in all three rat artery sizes although the amplitude and time course of nicardipine-induced inhibition varied with artery size. The significant inhibition induced by nicardipine pretreatment occurred a few seconds after PE stimulation in small mesenteric artery, 10 s in caudal artery and later than 20 s in aorta, suggesting that significant Ca2+ influx occurs immediately after PE stimulation in small mesenteric artery compared with the long delay seen for caudal artery and aorta. The late sustained phase of contraction in small mesenteric artery was markedly diminished by nicardipine but was maintained at substantial levels for at least several minutes in caudal artery and aorta (Fig. 9). In aorta, an initial transient component of contraction that remained in the presence of Y27632 (Fig. 1C) was almost completely abolished by a combination of Y27632 and ryanodine treatment (Fig. 9C), whereas nicardipine with Y27632 had no inhibitory effect on transient contraction (not shown). When Ca2+ entry was totally blocked by the removal of extracellular Ca2+ and addition of 2 mm EGTA, PE produced a large transient contraction without the sustained phase in all arteries of varying sizes (Supplemental Fig. S5).
Figure 9. Effect of 1 μm ryanodine, 1 μm nicardipine, and a combination of the two blockers on 30 μm PE-induced contraction in mesenteric (A; n= 3–6), caudal (B; n= 4–6) and aortic arteries (C; n= 4).
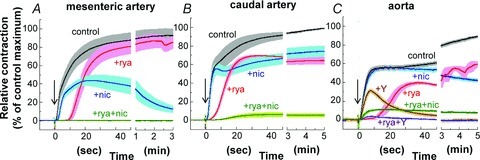
After ryanodine treatment (see Methods), 20 mm caffeine no longer evoked a contraction. After nicardipine treatment (see Methods), high K+-induced contraction was no longer elicited. +rya and +nic represents, respectively, the contraction of ryanodine- and nicardipine-treated arteries with ± 1 SEM (see Methods). +rya+nic represents a contraction of arteries treated with first ryanodine and then nicardipine in the presence of ryanodine. In aorta (C), +Y and +rya+Y represent, respectively, a contraction of aorta treated with 10 μm Y27632 and a combination of ryanodine and Y27632.
Blocking both SR Ca2+ release with ryanodine and voltage-dependent Ca2+ influx with nicardipine almost totally inhibited PE-induced increases in Ca2+ (Fig. 8B) and the initial rising phase of PE-induced contraction in all rat arteries of varying sizes (Fig. 9). The steady-state peak of PE-induced contraction remaining in the presence of the two blockers was 0 ± 0% in mesenteric artery, 6 ± 2% in caudal artery and 8 ± 1% in aorta (n= 4), suggesting that some tissue type-dependent Ca2+ sensitization is present in intact rat artery. Under the same conditions as for PE in the presence of both blockers, 10 μm serotonin and 0.3 μm ET-1 evoked, respectively, 3 ± 0 and 35 ± 3% (n= 4) of PE-induced contraction in small mesenteric artery, indicating an agonist type-dependent Ca2+ sensitization (Kitazawa et al. 2000). A combination of ryanodine treatment and the extracellular Ca2+-free conditions almost totally abolished either initial or sustained phase of PE contraction even in aorta (Supplemental Fig. S5).
Effect of α1A-specific antagonist and inhibition of PKC and ROCK
We investigated the effect of α1-adrenoceptor subtype-specific antagonists on PE-induced contraction in small mesenteric, caudal and aortic arteries. The α1A-specific antagonist RS100329 has a pKi of 9.6 for α1A with 100-fold higher potency compared with those of α1B and α1D adrenoceptors (Williams et al. 1999) and markedly shifted the steady-state concentration–response relationship of PE-induced contraction of small mesenteric artery to the left (Supplemental Fig. S6). RS-100329 at 1 nm almost completely suppressed the initial rising phase of PE-induced contraction for at least 60 s in small mesenteric artery (Fig. 10A), intermediately in caudal artery (B) and only partly in aorta (C). RS-100329 also delayed the onset of contraction in small mesenteric and caudal arteries but not aorta. GF109203X even at 3 μm had no additional inhibitory effect on PE-induced contraction in the presence of RS-100329 at least for the initial 60 s in mesenteric and caudal arteries whereas the late sustained phase of contraction was more potently suppressed in the presence of a mixture of RS-100329 and GF109203X compared with RS-100329 alone (Fig. 10A and B). A combination of RS-100329, GF109203X and 10 μm Y27632 almost completely abolished PE-induced contraction in all three types of arteries except for an initial small transient contraction in aorta (Fig. 10C).
Figure 10. Effect of 1 nm RS-100329 in combination with 3 μm GF109203X or GF109203X + 10 μm Y27632 on the time course of PE-induced contraction in small mesenteric arteries (A), caudal arteries (B) and aorta (C).
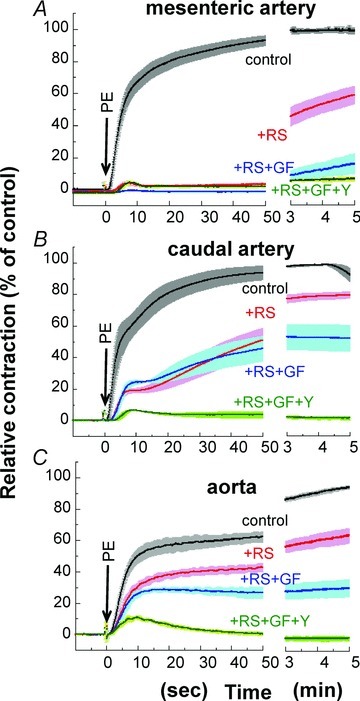
RS, RS-100329; GF, GF109203X; Y, Y27632. n= 4.
The α1A-specific agonist A-61603 (Knepper et al. 1995) at 30 nm generated a large contraction equivalent to that of 30 μm PE in small mesenteric artery (Supplemental Fig. S7). GF109203X at 3 μm markedly reduced both the initial rising and late sustained phases of A-61603-induced contraction to 7 ± 4% (n= 3) of control, whereas neither the initial nor late phase of contraction was significantly inhibited by the presence of 1 μm GSK-429286.
Effect of α1D-specific antagonist and inhibition of PKC and ROCK
BMY7378 is an α1D-specific antagonist, which has about 100-fold potency towards α1D compared with α1A and α1B (Yamamoto & Koike, 2001), although at high concentrations the compound can have antagonistic action against a wide range of receptors, e.g. 5-HT1, H1 and D2 (Goetz et al. 1995). BMY-7378 at 0.1 μm had no significant effect on the time course of PE-induced contraction in small mesenteric artery (Fig. 11A) whereas contraction in aorta was almost abolished at the same concentration except for a small contraction during the sustained phase (Fig. 11C). A 10fold increase in BMY7278 to 1 μm significantly inhibited the initial rising and sustained phases of contraction in mesenteric and caudal arteries (Fig. 11A and B). High BMY-7378 concentrations (1–3 μm) also delayed the onset of 10 μm 5-HT- and histamine-induced contractions with reduced plateau levels (not shown), suggesting that 1 μm BMY7278-induced inhibition of PE-induced contraction in mesenteric and caudal arteries is due not only to blocking of the α1D receptor but also to non-specific inhibition of agonist-induced contraction. The ROCK inhibitor GSK-429286 further reduced the sustained phase of contraction in the presence of even high concentrations (1 μm) of BMY7278 in mesenteric and caudal arteries (Fig. 11A and B) and in the presence of 0.1 μm BMY7278 in aorta (Fig. 11C). Addition of 3 μm GF109203X also markedly suppressed the sustained phase of PE-induced contraction in the presence of 1 μm BMY7278 in mesenteric and caudal arteries whereas the small contraction in the sustained phase remaining in the presence of 0.1 μm BMY7278 in aorta was resistant to GF109203X (Fig. 11C).
Figure 11. Effect of BMY-7378 on 30 μm PE-induced contraction in small mesenteric arteries (A), caudal arteries (B) and aorta (C).
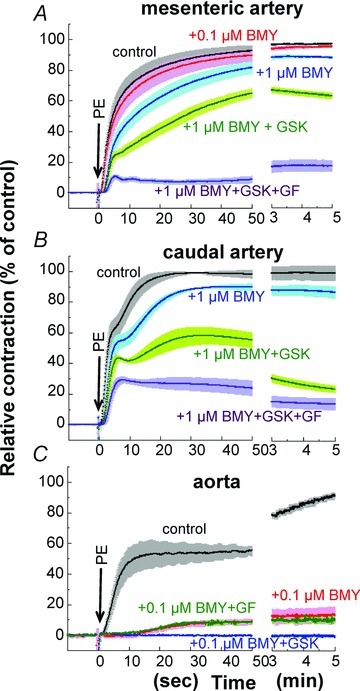
GSK, 1 μm GSK-429286; GF, 3 μm GF109203X. n= 4–6.
Recently, Ca2+-independent phospholipase A2 (iPLA2) was proposed to be involved in the sustained phase of agonist- and KCl-induced vascular contraction, suggesting that the free arachidonic acid produced by iPLA2 regulates RhoA-independent ROCK activity and contractile Ca2+ sensitivity of vascular smooth muscle (Guo et al. 2003; Ratz et al. 2009). The iPLA2 inhibitor bromoenol lactone (BEL) at 10 μm decreased the sustained phase of PE-induced contraction to 63 ± 7% (n= 6) of the control with no significant delay in the initial rapid phase of contraction in caudal artery (not shown). Addition of 1 μm GSK-429286 to 10 μm BEL-containing solution further reduced the contraction to 36 ± 12% (n= 3) of the control. This result suggests that the inhibitory effects of ROCK and iPLA2 inhibitors are rather additive and, thus, ROCK is not downstream of BEL-sensitive iPLA2 during α1-agonist-induced contraction.
Expression of proteins linked to the contractile signalling pathway in rat mesenteric, caudal and aortic arteries
To investigate the molecular mechanism responsible for PE-induced contraction in arterial smooth muscle, we examined expression levels of several regulatory/contractile proteins in small mesenteric artery compared with those of aorta and caudal artery. Total smooth muscle-specific α-actin content in small mesenteric and caudal artery was slightly but significantly higher (115 ± 3% and 110 ± 4%, respectively; n= 5) than that of aorta when total protein contents were matched among the three tissues. When the expression level of α-actin was matched using immunoblotting with smooth muscle-specific α-actin antibody to equalize the contractile region of cells (α-actin bands in Fig. 12A), the average expression levels of β-actin and total (pan) actin in small mesenteric artery were maintained at levels similar to that of aorta and caudal artery, suggesting no change in actin isoform content in arteries of different sizes. PKCα, protein phosphatase type 1C δ-isoform (PP1Cδ) and ROCK1 and 2 were also comparable among the three artery types (Fig. 12A). MYPT1, CPI-17 and MLC expression was significantly higher in small mesenteric artery than in aorta, whereas RhoA was significantly lower in the former (Fig. 12B). These protein expression measurements were performed in endothelium-intact arteries. However, since the number of intimal cells was 8% of the total cell number in small rabbit mesenteric artery (Huh et al. 2011), the involvement of endothelial cells in the measured expression level of regulatory/contractile proteins appears to be low in small mesenteric artery and negligible in large aorta.
Figure 12. Expression of various contractile and regulatory proteins in rat small mesenteric artery (MA), caudal artery (CA) and aorta.
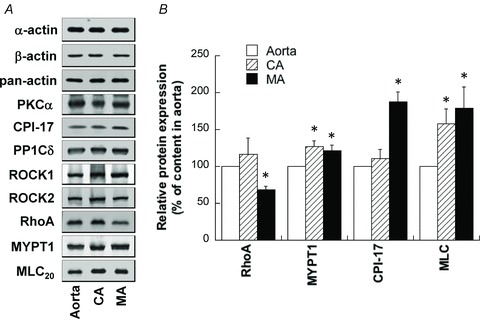
Protein contents in extracts were adjusted by equalizing smooth muscle-specific α-actin content between different arteries. A, representative Western blot images of various proteins in rat aorta, CA and MA. B, average expression levels of proteins, the contents of which were significantly different between arteries (P < 0.05). n= 6–30.
MLC, CPI-17 and MYPT1 phosphorylation and effect of RS-100329, GF109203X and Y27632 during PE-induced contraction in small mesenteric artery
Figure 13 illustrates the time courses of phosphorylation of MLC Ser19 (B), CPI-17 Thr38 (C) and MYPT1 Thr853 (D) at rest and after PE stimulation compared with contraction (A) in small mesenteric artery. The increases in MLC and CPI-17 phosphorylation reached their respective maximum within 10 s, which peaked before contraction plateaued (Fig. 13B and C). MLC phosphorylation was maintained at a high level until 3 min, whereas CPI-17 phosphorylation decreased by about 30% at 3 min. MYPT1 phosphorylation at Thr853 was already 50 ± 6% at rest and did not significantly increase 10 s after PE stimulation (Fig. 13D) whereas the contraction already increased to about 70% of maximum at the same time point. Thr853 phosphorylation was significantly increased at 30 s and 3 min compared with that at rest. The resting MYPT1 Thr696 phosphorylation was already 80 ± 8% of the control and was not significantly enhanced at 10 s (not shown).
Figure 13. Time course of phosphorylation of MLC (B), CPI-17 (C) and MYPT1 (D) compared with that of contraction (A) in response to 30 μm PE in small rat mesenteric artery.
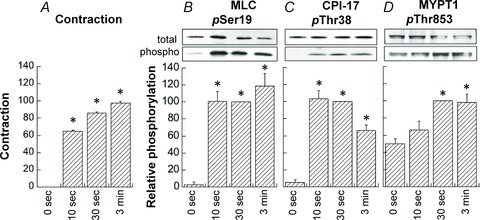
Average values of phosphorylation were normalized with a respective value at 30 s after PE stimulation. Contractions were normalized with a maximum at 3 min. *Significant difference (P < 0.05) from the resting value at 0 s. n= 4–8.
The α1A-specific antagonist RS-100329 potently reduced PE-induced contraction (Fig. 14A), MLC phosphorylation (B) and CPI-17 phosphorylation (C) to less than 10% of their respective controls at 30 s after PE stimulation in small mesenteric artery. However, MYPT1 phosphorylation at either Thr853 (Fig. 14D) or Thr696 (not shown) was not significantly decreased by the presence of RS-100329. The PKC inhibitor GF109203X, like RS-100329, markedly inhibited contraction (Fig. 14A), as well as MLC (B) and CPI-17 phosphorylation (C). GF109203X did not significantly reduce MYPT1 phosphorylation at either Thr853 (D) or Thr696 (not shown). The ROCK inhibitor Y27632 did not significantly inhibit phosphorylation of CPI-17 while MYPT1 phosphorylation at both Thr853 (Fig. 14D) and Thr696 were significantly but partially inhibited in response to Y27632, corresponding to a small inhibition of MLC phosphorylation and contraction (Fig. 14A and B).
Figure 14. Effects of 1 nm RS-100329, 3 μm GF109203X and 10 μm Y27632 on phosphorylation of MLC at Ser19 (B), CPI-17 at Thr38 (C), MYPT1 at Thr853 (D) and contraction (A) at 30 s after PE stimulation.
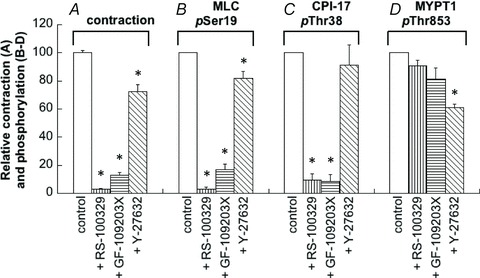
The antagonist and inhibitors were applied at least 10 min before and were continuously present during PE stimulation. Phosphorylation levels in the presence of the chemicals were normalized with a control value in the absence of chemicals. Levels of contraction in the presence of inhibitors were normalized with control 30 s after PE stimulation. *Significant difference from control level of phosphorylation. n= 4–9.
Phosphorylation of MLC, CPI-17 and MYPT1 and effect of BMY-7378, GF109203X and Y27632 during PE-induced contraction in aorta
In aorta, both MLC and CPI-17 were rapidly phosphorylated within 10 s to a value not significantly different from the value at 30 s after PE stimulation (Supplemental Fig. S8), which is similar to the results for mesenteric artery (Fig. 13). At 3 min, phosphorylation of MLC but not CPI-17 decreased to about 60% of the control at 30 s. MYPT1 phosphorylation at ROCK-specific Thr853 (Murányi et al. 2005) was already high (76 ± 7%) at rest and only slightly increased with time after PE stimulation (Supplemental Fig. S8D), suggesting an existence of constitutively active ROCK(s) at rest (Clelland et al. 2011).
In aorta, the α1D-antagonist BMY-7378 at 0.3 μm potently inhibited PE-induced contraction (Fig. 15A) and MLC phosphorylation (B), but had neither significant effect on phosphorylation of CPI-17 (C) nor MYPT1 (D). The presence of 10 μm Y27632 potently reduced contraction and phosphorylation of MLC, and significantly but partially decreased CPI-17 phosphorylation. In addition, Y27632 potently inhibited MYPT1 phosphorylation at both rest and 30 s after PE stimulation to 21 ± 3% (n= 4; not shown) and 23 ± 3%, respectively, of control in aorta compared with partial inhibition to 61 ± 3% in small mesenteric artery (Fig. 14D). GF-109023X had no significant effect on phosphorylation of MLC and CPI-17 in aorta (Fig. 15) in contrast to the marked reduction seen in small mesenteric artery (Fig. 14). Although GF109203X induced a partial but significant reduction of contraction in aorta without significant decrease in MLC phosphorylation at the same time point (Fig. 15), further detailed studies are needed to determine whether the MLC phosphorylation-independent mechanism is involved in the contractile reduction when PKC is inhibited.
Figure 15. Effect of 0.3 μm α1D-antagonist (BMY-7378), 3 μm GF109203X and 10 μm Y27632 on contraction (A), on phosphorylation of MLC (B), CPI-17 (C) and MYPT1 (D) at 30 s after PE stimulation in aorta.
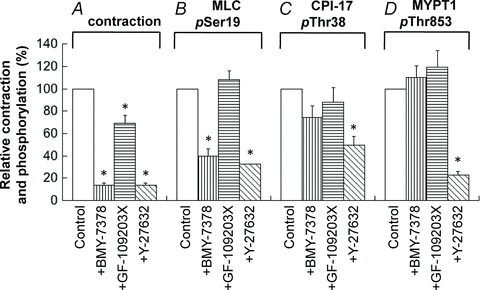
n= 3–8.
Quantitative amounts of phosphorylated MLC and CPI-17 in small mesenteric artery and aorta
To determine the physiological significance of increased MLC phosphorylation levels in response to PE in addition to relative changes in the phosphorylation level, isoelectric focusing–SDS polyacrylamide (2-D) gel electrophoresis was performed to separate amounts of mono- and di-phosphorylated from unphosphorylated MLC (see Methods). In both arterial tissues, MLC phosphorylation was augmented to a level of physiological significance at 30 s after PE-stimulation compared with that at rest (Table 1). The levels of PE-induced MLC phosphorylation as well as relative contraction in small mesenteric artery at 30 s were significantly larger than those of aorta.
Table 1.
Quantitative measurements of MLC20 phosphorylation in rat mesenteric artery and aorta
| Phosphorylated MLC (mol of Pi/mol of MLC) | Contraction | ||||
|---|---|---|---|---|---|
| Rest | PE-stimulation | n | % of maximum | n | |
| MA | 0.21 ± 0.05 | 0.58 ± 0.02†* | 5 | 91 ± 3* | 5 |
| aorta | 0.19 ± 0.02 | 0.38 ± 0.03†* | 4 | 67 ± 3* | 4 |
Phosphorylation of 20 kDa MLC and contraction were measured at rest and at 30 s after PE stimulation in either artery type. The phosphorylation was determined using the method of isoelectric focusing/SDS polyacrylamide (2-D) gel electrophoresis and colloidal gold staining (see Methods). Symbols † and * represent, respectively, the significant difference (P < 0.05) from that of rest in the same vessel type and from that of MA under the same conditions.
To elucidate the mechanisms for the distinct effects of PKC inhibitors on PE-induced CPI-17 phosphorylation and contraction between small mesenteric artery and large aorta and to determine the physiological significance of CPI-17 phosphorylation in small mesenteric artery, the quantitative amounts of CPI-17 expression and phosphorylation were determined using given amounts of phosphorylated recombinant CPI-17 protein. The total CPI-17 content was about 12 μm in small mesenteric artery and 5 μm in aorta (Table 2). Cellular levels of active (phosphorylated) CPI-17 of small mesenteric artery at 30 s after PE stimulation were increased from less than 0.2 μm at rest to about 4 μm, which correspond to about 34% of total CPI-17, while in aorta, active CPI-17 was increased to only 0.3 μm, which corresponds to only 6% of the total. Direct activation of PKC with 1 μm PDBu for 5 min in aorta generated 95 ± 7% of peak PE-induced contraction. The PDBu-induced contraction was almost totally abolished by the presence of 3 μm GF109203X but not 10 μm Gö-6976, and the same concentration of PDBu dramatically increased CPI-17 phosphorylation by 9 ± 1-fold (n= 3) above the control at 30 s after PE stimulation, which corresponds to 2.8 μm.
Table 2.
Quantitative measurements of total and phos-phorylated CPI-17 in rat mesenteric artery and aorta
| Total CPI-17 | Phosphorylated CPI-17 | Contraction | ||||
|---|---|---|---|---|---|---|
| Concentration (μm) | n | Concentration (μm) | n | % of maximum | n | |
| MA | 12.1 ± 1.0* | 13 | 4.1 ± 0.3* | 5 | 80 ± 2* | 12 |
| Aorta | 4.7 ± 0.3* | 12 | 0.3 ± 0.1* | 4 | 54 ± 2* | 11 |
Phosphorylation of CPI-17 and contraction were measured at a freezing point 30 s after PE stimulation in both artery types. To calculate the CPI-17 concentration in smooth muscle cells, the protein content of a mammalian cell was assumed to be 18% of the total cell weight (Dimopoulos et al. 2007).
Significant difference (P < 0.05) from that of MA.
Discussion
The major finding in this study is that α1-adrenoceptor-mediated signal transduction in arterial smooth muscle contraction varies with vessel size and time elapsed after receptor stimulation with the size-dependent differences mainly due to variations in Ca2+-sensitizing mechanisms (Table 3). In small resistance arteries, Ca2+-dependent and -independent PKC/CPI-17 Ca2+-sensitizing mechanisms downstream of the α1A-adrenoceptor subtype play a predominant role in the initial rising and late tonic phases, respectively, of α1-agonist-induced MLC phosphorylation and contraction. In large conduit arteries, in contrast, the ‘constitutively active’ ROCK–MYPT1-mediated Ca2+-sensitizing pathway, which is neither downstream of α1-adrenoceptors nor mediated by PKC, plays a major role in an increase in the basal Ca2+ sensitivity of MLC phosphorylation and contraction. In midsized muscular arteries both signalling pathways are partially involved. These differences are not primarily due to protein expression of kinases, phosphatases or MYPT1 and CPI-17, but rather to signal transduction efficiency in each artery segment.
Table 3.
Summary of effects of various inhibitors and antagonists on 30 μm PE-induced contraction in rat small resistance artery, midsized muscular artery and large conduit aorta
| Small resistance artery | Midsized muscular artery | Large conduit artery | ||||
|---|---|---|---|---|---|---|
| Inhibitors or antagonists | Initial rising phase | Late sustained phase | Initial rising phase | Late sustained phase | Initial rising phase | Late sustained phase |
| PKC inhibitors (GF109203X, calphostin C) | +++ | +++ | ++ | ++ | ± | + or ++ |
| Ca2+-dependent PKC inhibitors (Gö-6976, PKC downregulation) | +++ | + or ++ | + | + | ND | ND |
| ROCK inhibitors (Y27632, H1152, GSK429286) | ± | + | ± | ++ | ± | +++ |
| PKC & ROCK inhibitors (GF109203X + Y27632) | +++ | +++ | ++ | +++ | + | +++ |
| Inhibition of Ca2+ release (ryanodine treatment) | +++ | ± or + | +++ | + | +++ | ++ |
| Inhibition of Ca2+ influx (nicardipine) | + or ++ | +++ | ± | ++ | ± | ++ |
| Inhibition of both release & influx (ryanodine + nicardipine) | +++ | +++ | +++ | ++ or +++ | +++ | ++ |
| α1A-antagonist (RS-100329) | +++ | +++ | ++ | + or ++ | ± or + | + |
| α1D-antagonist (BMY-7378) | ± | ± | ± or + | ± or + | +++ | +++ |
+++ represents a strong inhibition; ++, medial; +, small; ±, no change; ND, not determined.
Here, a series of pharmacological approaches revealed the biphasic regulation of α1-agonist-induced contraction in vascular smooth muscle through a mutually complementary pair of Ca2+-rising and Ca2+-sensitizing mechanisms (Table 3). Most importantly, a lack of either mechanism essentially abolished α1-agonist-induced contraction in every rat artery size. SR Ca2+ release and Ca2+ influx through L-type voltage-dependent Ca2+ channels are the key means of increasing Ca2+ and are responsible, respectively, for the initial rising and late sustained phase of α1-agonist-induced contraction in arteries of all sizes. In contrast, the efficacy of inhibitors for Ca2+-sensitizing pathways downstream of α1-adrenoceptors largely varied with artery size. In small mesenteric, intrarenal and ovarian arteries, the inhibitory efficacy of 3 μm of the PKC inhibitor GF109203X (GF) was much greater than 10 μm of the ROCK inhibitor Y27632 (Y) in PE-induced contraction, and was effectively equal in midsized caudal and superior mesenteric arteries. In large thoracic aorta, however, GF inhibition was much less than Y. Since the effect of GF109203X, Y27632 and GSK-429286 on Ca2+ signals was small or rather minimal, these results suggest that the difference in the α1-adrenoceptor-mediated signalling pathways of systemic arteries is largely due to differences in Ca2+-sensitizing mechanisms. These results are in agreement with previous findings by Budzyn et al. (2006) for the steady state in rat aorta and superior and small mesenteric arteries, but do not agree with the steady-state findings of Mueed et al. (2004) in rat aorta and caudal arteries. While further study is needed to reconcile these discrepancies, one possible cause could be the timing of contractile measurement. It also remains to be determined whether the order of the inhibitory efficacy observed here also occurs in arterial segments from the pulmonary and cerebral circulatory systems and whether the PKC–CPI-17–MLCP signalling pathway also plays a key role in regulation of α1-agonist-induced contraction in small resistance arteries from different tissue origins. In the different-sized arteries tested, the effects of PKC and ROCK inhibitors on PE-induced contraction were additive in arteries of varying sizes (Budzyn et al. 2006; this study), suggesting that the two signalling pathways are independent. Simultaneous inhibition of both PKC and ROCK almost completely eliminated the late-sustained phase of PE-induced contraction in rat arteries of varying sizes, suggesting that, without the Ca2+-sensitizing mechanism, α1-agonists cannot maintain the tonic component of contraction. On the other hand, inhibition of both Ca2+ release and Ca2+ influx almost totally eliminated both the initial-rising and late-sustained phases of PE-induced contraction, indicating that in the absence of a Ca2+ increase the α1-agonist hardly produced a significant contraction at resting [Ca2+]i in rat arteries of varying sizes (Table 3). As seen in rabbit femoral artery (Dimopoulos et al. 2007), the pretreatment with a combination of ryanodine and nicardipine in rat mesenteric artery did not decrease the intracellular Ca2+ concentration, which was similar to or rather a little higher than the resting concentration possibly due to store-operated Ca2+ influx (Albert & Large, 2003). Under these conditions, PE in rabbit femoral artery slowly caused a contraction to 30% of control and increased phosphorylation levels of MLC and CPI-17 without an increase in [Ca2+]i. In rat mesenteric artery, endothelin-1 but not PE produced a significant level (35%) of contraction. These results suggest that agonists are tissue- and agonist-dependently able to produce a significant contraction at resting [Ca2+]i possibly through upregulation of the Ca2+-sensitizing mechanism.
The effects of different PKC inhibitors including PKC downregulation clearly indicate that the Ca2+-dependent and -independent PKC isoforms are mainly involved in, respectively, the initial-rising and late-sustained phases of α1-agonist-induced contraction in small resistance arteries. The order of inhibitory efficacy of GF109203X in PE-induced contraction among arteries of different sizes was: small resistance arteries > midsized muscular arteries > large conduit aorta, which is the same as that seen for the α1A-specific antagonist RS-100329 (Table 3). This is also in agreement with the finding that α1A-subtype expression in mice is much higher in peripheral than central conduit arteries (Rokosh & Simpson, 2002). Likewise, PE-induced contraction is much smaller in α1A-deficient than wild-type mesenteric arteries, whereas there is no significant difference between α1A-deficient and wild-type carotid arteries (Methven et al. 2009b). There is a small discrepancy between the inhibitory effect of Gö-6976 and PKC downregulation on the sustained phase of PE-induced contraction: the former inhibitor had a larger effect than the latter treatment at high concentrations of PE. The discrepancy may be mainly due to a different magnitude of Ca2+-dependent PKC inhibition. The PKC downregulation treatment significantly but only partially decreased Ca2+-dependent PKC expression in an isoform-dependent manner: PKCα was decreased to 14% of control with no change in expression of another Ca2+-dependent PKCβ, whereas Gö-6976 has been shown to equally and potently inhibit both PKCα and β (Martiny-Baron et al. 1993). The decrease in PKCα expression appears to cause a delay in the initial rise and possibly a reduction in the sustained level of contraction at low but not high concentrations of PE. Downregulation of PKCδ by half appears to have an inhibitory effect on the sustained phase of contractile response to low but not high concentrations of PE, suggesting that the decrease in content of the δ-isoform is not the rate-limiting step in 30 μm PE-induced contraction. These results suggest that, although the Ca2+-independent PKCs play a major role in maintenance of the sustained phase of the contractile response to PE, Ca2+-dependent PKCs are also significantly but partially involved in maintenance of the contractile tone.
The α1-adrenoceptor is comprised of three subtypes (α1A, α1B and α1D), each encoded by distinct genes, all of which are thought to mediate smooth muscle contraction via the Gq/11 G protein and phosphoinositide-specific PLCβ in humans and rodents (Rudner et al. 1999; Docherty, 2010). In mice and humans, the α1-adrenoceptor major subtype in small mesenteric artery is α1A (Phillips et al. 1998; Rokosh & Simpson, 2002; Methven et al. 2009b). In fact, the α1A-subtype-specific antagonist RS-100329 almost completely abolished PE-induced contraction at least for the first 60 s in small mesenteric arteries, even though this artery type also co-expresses the α1D-subtype (Methven et al. 2009a; see Docherty, 2010), suggesting that α1-agonist-mediated responses are mainly regulated by the coupling efficiency of receptors to downstream signalling but not receptor expression levels (Methven et al. 2009b). Even at a high concentration (3 μm), the strong PKC inhibitor GF109203X had no additional effect on the initial phase of PE-induced contraction in the presence of RS-100329 in arteries of all sizes, indicating that the inhibitory effect of GF109203X is not independent of, but instead is sequential to the antagonistic effect of RS100329. As for contraction, both RS-100329 and GF109203X reduced CPI-17 and MLC phosphorylation to negligible levels. Together, these results clearly demonstrate that both the Ca2+-dependent and -independent PKCs and their target CPI-17 are downstream of the α1A-adrenergic receptor subtype and play an indispensable role in α1-agonist-induced contraction in small resistance arteries. After prolonged stimulation with 30 μm PE for several minutes, the contractile level in the presence of 1 nm RS-100329 gradually increased as shown in Fig. 10A. However, a decrease in PE to 10 μm or an increase in RS-100329 to 3 nm (Supplemental Fig. S6) eliminated this slow phase of contraction seen in the presence of 1 nm antagonist with 30 μm agonist, suggesting that the gradual recovery of contraction in the presence of 1 nm RS-100329 is not due to PE-induced activation of different α1-subtypes, but rather α1A-receptors from which RS-100329 molecules were slowly dissociated. The ROCK inhibitor Y27632, in contrast, potently and additively suppressed PE-induced contraction in the presence of RS-100329 in caudal artery and aorta, suggesting that ROCK is not downstream of the α1A-adrenergic receptor subtype. This conclusion is supported by the fact that contraction induced by the α1A-specific agonist A-61603 was almost completely abolished by 3 μm GF109203X, whereas the potent ROCK inhibitor GSK-429286 at 1 μm had no significant effect (Supplemental Fig. S7). PE increased CPI-17 phosphorylation from negligible levels at rest to 4 μm within 10 s, which is well over the in situ MLCP concentration (∼1 μm; Dimopoulos et al. 2007). On the other hand, nitric oxide rapidly decreases PE-induced CPI-17 phosphorylation and contraction in rabbit femoral artery (Kitazawa et al. 2009), suggesting that CPI-17 is a physiological on-and-off messenger that rapidly regulates MLCP and vascular contraction. This study did not take into account the possible involvement of α1B-adrenoceptors in the PE-induced arterial contraction, since there was no effect of α1B knockout on PE-induced contraction in both mouse carotid and mesenteric arteries (Methven et al. 2009a,b) and no selective α1B-subtype-specific antagonist available (see Docherty, 2010). Neither α1A-specific antagonists nor PKC inhibitors significantly decreased MYPT1 phosphorylation during PE-induced contraction in small mesenteric artery. Taken together, these results clearly indicate that both the Ca2+-dependent and -independent PKCs–CPI-17–MLCP pathways, but not the ROCK–MYPT1–MLCP pathway, are the major Ca2+-sensitizing mechanism downstream of the α1A-adrenergic receptor in small resistance arteries and play a critical role in sympathetic nerve-mediated regulation of blood pressure. This is supported by the finding that RS-100329 reduced blood pressure responses of presser nerve stimulation by 70% in pithed rats (Docherty, 2011). In α1A-subtype knockout mice, the ‘basal’ blood pressure was reduced by 10% compared with that of wild-type and infusion of the α1A-specific agonist A-61603 failed to increase mean arterial pressure while a maximum dose of non-specific PE still increased the pressure response to 85% of wild-type with a rightward shift of the dose–response relationship (Rokosh & Simpson, 2002), suggesting that other α1-receptor subtypes are also involved in blood pressure regulation. In vitro, both α1A- and α1A/α1B-knockout mesenteric arteries similarly lost response to A-61603 and produced a contraction to only 10% of wild-type in response to PE (Methven et al. 2009b), which is similar to the results obtained here in the presence of RS-100329.
In large conduit artery, the potent PKC inhibitor GF109203X only partially suppressed α1-agonist-induced contraction, strikingly different from the effect in small resistance arteries (Table 3). The major α1-adrenergic receptor subtype in rat aorta is α1D, which, like the α1A subtype, is coupled to PLCβ to produce IP3 and DAG (Rudner et al. 1999; Docherty, 2010). α1-Agonists elicit a rapid increase in transient Ca2+ and contraction even in the absence of extracellular Ca2+ in the aorta (Sato et al. 1988; Supplemental Fig. S5). Inhibition of Ca2+ release with ryanodine abolished PE-induced contraction in the absence of extracellular Ca2+ (Supplemental Fig. S5) and, under normal conditions, markedly delayed the initial fast development of α1-agonist-induced contraction with a significant reduction of the sustained phase of contraction in aorta (Table 3). The initial transient contraction in response to PE in the presence of PKC and ROCK inhibitors was totally abolished by ryanodine treatment. These results suggest that IP3 is produced upon stimulation by α1-agonists and, thus, the PKC activator DAG is also generated in parallel with SR Ca2+ release. Indeed, DAG production with α1-agonist stimulation was shown in rat aorta (Danthuluri & Deth, 1986; Okumura et al. 1991). ROCK1/2, PKCα and MLCP expression levels were similar between aorta and small mesenteric artery. Although CPI-17 in the aorta was about 50% that of small mesenteric artery, the quantity of CPI-17 in aorta is still about 5 μm, which is sufficient to inhibit 1 μm MLCP in smooth muscle cells if a significant amount of protein is phosphorylated. CPI-17 phosphorylation rapidly increased within 10 s to the peak level, followed by development of contraction, in a similar fashion to that seen in small mesenteric artery. However, PE-induced contraction and CPI-17 phosphorylation in aorta was rather insensitive to GF109203X whereas 90% of phosphorylation and contraction was inhibited by the same concentration of GF109203X in small mesenteric artery. We found that only a small amount (0.3 μm) of CPI-17 was phosphorylated in aorta 30 s after maximal PE stimulation in contrast to 4 μm phosphorylated CPI-17 at the same time point in small mesenteric artery. Although it is interesting that this small amount of phosphorylated CPI-17 in aorta was significantly but partially inhibited by Y27632 but not GF109203X, these changes have little physiological meaning for in situ regulation of MLCP. Direct PKC activation with PDBu, on the other hand, increased CPI-17 phosphorylation to an extremely high (about 3 μm) level and generated a large contraction in rat aorta, suggesting that most CPI-17 in aorta is available for directly- but not α1-agonist-activated PKCs. The functional phenotypic diversity of the PKC signalling pathway among different-sized arteries thus cannot be explained solely by gene expression data. The detailed mechanism for the minimal level of CPI-17 phosphorylation and α1-agonist activation of PKCs in aorta awaits further investigation.
That the α1D-specific antagonist BMY-7378 at 0.1 μm almost completely suppressed both the initial and sustained phases of PE-induced aortic contraction suggests that the major α1-adrenoceptor subtype in aorta is α1D (Piascik et al. 1995). Depletion of Ca2+ stores and blocking Ca2+ influx abolished PE-induced contraction, suggesting that both Ca2+ release and Ca2+ influx are coupled to α1D-adrenoceptor activation in aorta. At this concentration, the α1D-antagonist had no effect on PE-induced contraction in small mesenteric artery (Piascik et al. 1995), supporting that the major α1-adrenergic receptor of mesenteric artery is not the α1D subtype. These results are consistent with the fact that α1D- and α1D/α1B-knockout markedly inhibit PE-induced contraction in carotid artery and aorta but not in mesenteric artery (Hosoda et al. 2005; Methven et al. 2009a). An increase in BMY7378 concentration to 1–3 μm, however, did significantly reduce both the initial and sustained phases of contraction in small mesenteric and caudal arteries. This inhibition may not be related to an α1D-specific effect, because at such high concentrations BMY-7378 could also reduce 5-HT- and histamine-induced contraction in arteries. Since the sustained phase of PE-induced contraction in aorta is known to be suppressed by ROCK inhibitors (Uehata et al. 1997) and Y27632 also markedly reduced MYPT1 phosphorylation, these results would argue that ROCK–MYPT1 signalling is probably downstream of the α1D-adrenergic receptor subtype. The capacity of ROCK-specific inhibitors GSK-429286 and Y27632 to significantly reduce PE-induced contraction in the presence of high BMY-7378 concentrations in mesenteric and caudal arteries where most α1D receptors are blocked, suggests that the antagonistic effect of BMY-7378 and the inhibitory effect of ROCK inhibitors are rather additive and thus ROCK signalling appears not to be downstream of α1D- and α1A-adrenoceptor subtypes.
Several G protein-coupled receptors with agonists such as thromboxane A2 and endothelin-1 have been shown to couple to G12/13 G protein to activate the RhoA–ROCK signalling pathway (Gohla et al. 2000; Somlyo & Somlyo, 2003). ROCK activation results in MYPT1 phosphorylation at Thr853 that in turn inhibits MLCP, which results in an increase in MLC phosphorylation and contraction without a Ca2+ rise (Kitazawa et al. 2003; Murányi et al. 2005). It has recently been demonstrated that α1-adrenoceptors, including all three subtypes, couple to Gαq/11 but not Gα12/13 G protein (Wirth et al. 2008; Docherty, 2010). Thus, a smooth muscle-specific deficiency in Gαq/11 but not Gα12/13 eliminated both PE-induced arterial contraction and pressure response, and lowered blood pressure in mice (Wirth et al. 2008). Nevertheless, Y27632 reduced PE-induced phosphorylation of MYPT1 and MLC as well as contraction in aorta. Curiously, MYPT1 resting phosphorylation levels were high (about 80%) compared with that of PE stimulation, suggesting that PE evokes only a small fraction of MYPT1 phosphorylation. Y27632 reduced MYPT1 phosphorylation to ∼20% regardless of PE stimulation (Dimopoulos et al. 2007; this study), suggesting that ROCK inhibition enhances MLCP activity to similar levels under both resting and stimulated conditions. The enhanced MLCP activity at rest produced by ROCK inhibition leads to a decrease in the ‘basal’ Ca2+ sensitivity, which induces a pseudo inhibition of α1-agonist-induced Ca2+ sensitization of MLC phosphorylation and contraction. ROCK inhibition and the α1D-antagonism in PE-induced contraction do not occur through the same signalling pathway and their effects are thus additive. The effectiveness of ROCK inhibitors may also not be specific to large arteries, but could instead apply to arteries of all sizes where the ROCK activity is elevated, such as in aorta under normal conditions, in arteries under hypertensive and vasospasmic conditions, or even in cultured mesenteric artery smooth muscle (Uehata et al. 1997; Loirand et al. 2006; Woodsome et al. 2006; Shimokawa & Rashid, 2007; Wirth et al. 2008; Huh et al. 2011). In contrast, PKC activity is quiescent under resting conditions since CPI-17 phosphorylation is negligible. α1-Agonists raise the levels of Ca2+ and DAG to activate first Ca2+-dependent and then Ca2+-independent PKCs, which increase CPI-17 phosphorylation to high levels to signal to downstream contractile proteins in small resistance arteries. PKC inhibitors thus only suppress a fraction of the MLC phosphorylation and contraction that is augmented by the α1-agonist, but do not reduce basal Ca2+ sensitivity as ROCK inhibitors do.
Although both Ca2+ release from the SR and Ca2+ influx through voltage-dependent Ltype Ca2+ channels are essential for PE-induced contraction in arteries of all sizes (Table 3), their detailed mechanisms do vary. Ryanodine treatment induced a delay of the onset of PE-induced Ca2+ rise and contraction in all artery sizes tested, suggesting that Ca2+ influx and/or Ca2+ sensitization occur with a delay and Ca2+ release is crucial for the rapid development of α1-agonist-induced contraction in these tissues. The inhibitory effect of ryanodine treatment on the late sustained phase of contraction, in contrast, was more potent in aorta and caudal artery compared with smaller mesenteric arteries, suggesting that Ca2+ release plays a more important role in the late sustained phase of contraction in larger arteries or instead the store-operated Ca2+ entry has a more significant role in smaller arteries after depletion of the Ca2+ store. The PKC inhibitors GF109203X (Dimopoulos et al. 2007; this study) and calphostin C (Nobe & Paul, 2001) both have little effect on the initial Ca2+ increase, with a partial inhibitory effect on the sustained phase of Ca2+ in response to PE, but markedly reduced both the initial rising and late sustained phases of contraction in small mesenteric artery. In contrast, in caudal artery and aorta, significant initial transient contraction remained in the presence of GF109203X, Y27632 or both. This transient contraction in aorta was abolished by ryanodine treatment (Fig. 9C), suggesting that SR Ca2+ release generates a transient contraction even in the presence of ROCK and PKC inhibitors in aorta and caudal artery. This is consistent with the fact that both PKC and ROCK inhibitors induced no significant delay in the initial rising phase of PE-induced contraction in aorta. On the other hand, only negligible transient contraction with a significant delay in the presence of PKC inhibitors in small mesenteric artery suggests that PE cannot evoke significant contraction through Ca2+ release in the absence of the PKC-mediated Ca2+-sensitizing mechanism. Collectively, these results suggest that Ca2+ release is indispensable for the development of the initial phase of PE-induced contraction in both large and small arteries, but the former is mainly through activation of the classical Ca2+–calmodulin–MLCK–MLC signalling pathway, whereas the latter is through activation of the novel Ca2+–cPKC–CPI-17 signalling pathway inhibiting MLCP together with the Ca2+–calmodulin–MLCK pathway to rapidly increase MLC phosphorylation and contraction.
Voltage-dependent Ca2+ influx is mainly involved in maintaining the tonic level of [Ca2+]i (Nobe & Paul, 2001; Dimopoulos et al. 2007; this study) and the sustained phase of contraction in arteries (Table 3). However, the pattern by which nicardipine inhibited PE-induced contraction varied with vessel size. Since nicardipine reduction of contraction was more potent in smaller mesenteric arteries compared with larger arteries, L-type Ca2+ channels may play a more critical role in the steady-state amplitude of α1-agonist-induced contraction in small resistance arteries. Furthermore, a reduction of contraction induced by PE in the presence of nicardipine was seen a few seconds after stimulation in small mesenteric artery, 10 s in caudal artery, and more than 20 s in aorta. These results suggest that the time required for opening of voltage-dependent Ca2+ channels as well as the quantity of opened channels varies with arterial size. This further suggests that the mechanism in membrane depolarization required for opening of Ca2+ channels during α1-agonist-induced contraction also varies with arterial size. In fact, several different mechanisms have been proposed for the induction of membrane depolarization in arterial smooth muscle cells, such as a Ca2+ release-activated Cl− channel (Pacaud et al. 1991; Yuan, 1997), IP3-activated non-selective cation (TRPC) channels (Tai et al. 2008; Xi et al. 2008; Adebiyi et al. 2010), and DAG with/without PKC-activated TRPCs (Large et al. 2009). However, whether these mechanisms that cause membrane depolarization vary with agonist type and/or arterial sizes remains to be investigated. It should be noted that a combination of GF109203X and Y27632 totally abolished the sustained phase of PE-induced contraction in all arteries tested, suggesting that Ca2+ influx in response to PE is not sufficient to develop a significant contraction without PKC and/or ROCK Ca2+-sensitizing pathways in all rat artery sizes tested.
Blocking both SR Ca2+ release and voltage-dependent Ca2+ influx abolished an increase in cytoplasmic Ca2+ in response to PE (Dimopoulos et al. 2007; this study) and almost completely inhibited both the initial fast-rising and late sustained phases of PE-induced contraction in small mesenteric artery (Table 3). This suggests that the Ca2+-sensitizing pathways alone stimulated with α1-agonist evoked no contraction at resting [Ca2+]i. PKC inhibitors alone also potently suppressed both initial rising and late sustained contraction. Together, these results further suggest an importance of the co-operative mechanism for Ca2+ rise and Ca2+ sensitization in α1-agonist-induced contraction, which fuses the two processes, i.e. the SR Ca2+–Ca2+-dependent PKC–CPI-17 Ca2+-sensitizing pathway (Dimopoulos et al. 2007) in small resistance arteries. In fact, CPI-17 was rapidly phosphorylated to a level much higher than the MLCP content in a manner that depends on both SR Ca2+ release and PKC. In midsized caudal artery and large aorta, PE in the presence of Ca2+ blockers induced only slow and small contractions to 6 and 8%, respectively, of control, which is similar to that of midsized rabbit femoral artery where the increase in CPI-17 phosphorylation was markedly reduced but MYPT1 phosphorylation was not inhibited (Dimopoulos et al. 2007), suggesting that, even in large arteries, the ROCK–MYPT1 Ca2+-sensitizing pathway alone plays a minor role in the generation of α1-agonist-induced contraction without Ca2+ rise.
In conclusion, our results define the time-dependent and vessel size-dependent roles specific for Ca2+ release, Ca2+ influx, PKC and ROCK in α1-agonist-induced contraction in rat arteries. A special emphasis is on Ca2+ sensitization through both Ca2+-dependent and Ca2+-independent PKCs and their downstream target CPI-17 in, respectively, the initial rising and late sustained α1-agonist-induced contraction in small resistance arteries, whereas neither PKC signalling pathway plays an important role in large conduit arteries. Whether the heterogeneous roles of these two Ca2+-sensitizing pathways in arteries of different sizes in the vascular tree are due to distinct blood pressure, flow rate, sympathetic nerve innervation, endothelial effect or all of the above is currently unclear and warrants further examination. Although humans and small rodents do differ in several key indexes of cardiovascular function, the PKC–CPI-17 signalling pathway may play an important role in autonomic vasoconstriction of human small resistance arteries. Our findings provide insights into the development of new therapeutic agents controlling the size-dependent vasoconstriction.
Acknowledgments
We are grateful to Dr Masumi Eto for his phospho[Thr38]- specific CPI-17 antibody, totally phosphorylated CPI-17 proteins and valuable comments, and to Dr Albert Wang for his proof-reading of the manuscript.
Glossary
- CPI-17
protein kinase C-potentiated protein phosphatase inhibitor protein 17 kDa
- DAG
diacylglycerol
- G12/13
Gα12 and Gα13 subunit of heterotrimeric G protein
- Gq
Gαq subunit of heterotrimeric G protein
- MLCK
Ca2+–calmodulin-dependent myosin light chain kinase
- MLC
myosin light chain
- MLCP
myosin light chain phosphatase
- MYPT1
myosin targeting subunit of MLCP
- PDBu
phorbol 12,13-dibutyrate
- PE
phenylephrine
- PLCβ
phospholipase Cβ
- RhoA
member A of Rho family of small GTPases
- ROCK
Rho-associated kinase
- SR
sarcoplasmic reticulum
Author contributions
T.K.: conception and design of the experimental protocol, collection, analysis and interpretation of data, drafting and revising the manuscript. K.K.: collection and analysis of data. The experiments were performed at Boston Biomedical Research Institute at Watertown, MA, USA. Both authors approved the final version of the manuscript.
Supplementary material
Supplemental Fig. S1
Supplemental Fig. S2
Supplemental Fig. S3
Supplemental Fig. S4
Supplemental Fig. S5
Supplemental Fig. S6
Supplemental Fig. S7
Supplemental Fig. S8
References
- Adebiyi A, Zhao G, Narayanan D, Thomas-Gatewood CM, Bannister JP, Jaggar JH. Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ Res. 2010;106:1603–1612. doi: 10.1161/CIRCRESAHA.110.216804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert AP, Large WA. Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium. 2003;33:345–356. doi: 10.1016/s0143-4160(03)00048-4. [DOI] [PubMed] [Google Scholar]
- Ashida T, Schaeffer J, Goldman WF, Wade JB, Blaustein MP. Role of sarcoplasmic reticulum in arterial contraction: comparison of ryanodines's effect in a conduit and a muscular artery. Circ Res. 1988;62:854–863. doi: 10.1161/01.res.62.4.854. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RF, Miller FD, Merriman RL, Howbert JJ, Heath WF, Kobayashi E, Takahashi I, Tamaoki T, Nakano H. Inhibition of protein kinase C by calphostin C is light-dependent. Biochem Biophys Res Commun. 1991;176:288–293. doi: 10.1016/0006-291x(91)90922-t. [DOI] [PubMed] [Google Scholar]
- Budzyn K, Paull M, Marley PD, Sobey CG. Segmental differences in the roles of rho-kinase and protein kinase C in mediating vasoconstriction. J Pharmacol Exp Ther. 2006;317:791–796. doi: 10.1124/jpet.105.100040. [DOI] [PubMed] [Google Scholar]
- Clelland LJ, Browne BM, Alvarez SM, Miner AS, Ratz PH. Rho-kinase inhibition attenuates calcium-induced contraction in ß-escin but not Triton X-100 permeabilized rabbit femoral artery. J Muscle Res Cell Motil. 2011;32:77–88. doi: 10.1007/s10974-011-9253-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthuluri NR, Deth RC. Acute desensitization to angiotensin II: evidence for a requirement of agonist-induced diacylglycerol production during tonic contraction of rat aorta. Eur J Pharmacol. 1986;126:135–139. doi: 10.1016/0014-2999(86)90749-1. [DOI] [PubMed] [Google Scholar]
- Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res. 2007;100:121–129. doi: 10.1161/01.RES.0000253902.90489.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty JR. Subtypes of functionalα1-adrenoceptor. Cell Mol Life Sci. 2010;67:405–417. doi: 10.1007/s00018-009-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty JR. Vasopressor nerve responses in the pithed rat, previously identified asα2-adrenoceptor mediated, may be α1D-adrenoceptor mediated. Eur J Pharmacol. 2011;658:182–186. doi: 10.1016/j.ejphar.2011.02.029. [DOI] [PubMed] [Google Scholar]
- Dunn SD. Effects of the modification of transfer buffer composition and the renaturation of proteins in gels on the recognition of proteins on Western blots by monoclonal antibodies. Anal Biochem. 1986;157:144–153. doi: 10.1016/0003-2697(86)90207-1. [DOI] [PubMed] [Google Scholar]
- Eto M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J Biol Chem. 2009;284:35273–35277. doi: 10.1074/jbc.R109.059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz AS, King HK, Ward SD, True TA, Rimele TJ, Saussy DL., Jr BMY 7378 is a selective antagonist of the D subtype ofα1-adrenoceptors. Eur J Pharmacol. 1995;272:R5–R6. doi: 10.1016/0014-2999(94)00751-r. [DOI] [PubMed] [Google Scholar]
- Gohla A, Schultz G, Offermanns S. Role for G12/G13 in agonist-induced vascular smooth muscle cell contraction. Circ Res. 2000;87:221–227. doi: 10.1161/01.res.87.3.221. [DOI] [PubMed] [Google Scholar]
- Guo Z, Su W, Ma Z, Smith GM, Gong MC. Ca2+-independent phospholipase A2 is required for agonist-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2003;278:1856–1863. doi: 10.1074/jbc.M211075200. [DOI] [PubMed] [Google Scholar]
- Güth K, and Wojciechowski R. Perfusion cuvette for the simultaneous measurement of mechanical, optical and energetic parameters of skinned muscle fibres. Pflugers Arch. 1986;407:552–557. doi: 10.1007/BF00657515. [DOI] [PubMed] [Google Scholar]
- Horiuti K, Somlyo AV, Goldman YE, Somlyo AP. Kinetics of contraction initiated by flash photolysis of caged adenosine triphosphate in tonic and phasic smooth muscles. J Gen Physiol. 1989;94:769–781. doi: 10.1085/jgp.94.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda C, Tanoue A, Shibano M, Tanaka Y, Hiroyama M, Koshimizu TA, Cotecchia S, Kitamura T, Tsujimoto G, Koike K. Correlation between vasoconstrictor roles and mRNA expression ofα1-adrenoceptor subtypes in blood vessels of genetically engineered mice. Br J Pharmacol. 2005;146:456–466. doi: 10.1038/sj.bjp.0706325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh YH, Zhou Q, Liao JK, Kitazawa T. ROCK inhibition prevents fetal serum-induced alteration in structure and function of organ-cultured mesenteric artery. J Muscle Res Cell Motil. 2011;32:65–76. doi: 10.1007/s10974-011-9252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, Narumiya S. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996;15:1885–1893. [PMC free article] [PubMed] [Google Scholar]
- Isotani E, Zhi G, Lau KS, Huang J, Mizuno Y, Persechini A, Geguchadze R, Kamm KE, Stull JT. Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc Natl Acad Sci U S A. 2004;101:6279–6284. doi: 10.1073/pnas.0308742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem. 1991;266:1708–1715. [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem. 2000;275:9897–9900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Khalequzzaman M. Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca2+ sensitization in rabbit smooth muscle. J Physiol. 2003;546:879–889. doi: 10.1113/jphysiol.2002.029306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Semba S, Huh YH, Kitazawa K, Eto M. Nitric oxide-induced biphasic mechanism of vascular relaxation via dephosphorylation of CPI-17 and MYPT1. J Physiol. 2009;587:3587–3603. doi: 10.1113/jphysiol.2009.172189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potentα1-adrenergic receptor agonist, selective for the α1A receptor subtype. J Pharmacol Exp Ther. 1995;274:97–103. [PubMed] [Google Scholar]
- Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1989;159:548–553. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- Konishi M, Olson A, Hollingworth S, Baylor SM. Myoplasmic binding of fura-2 investigated by steady-state fluorescence and absorbance measurements. Biophys J. 1988;54:1089–1104. doi: 10.1016/S0006-3495(88)83045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large WA, Saleh SN, Albert AP. Role of phosphoinositol 4,5-bisphosphate and diacylglycerol in regulating native TRPC channel proteins in vascular smooth muscle. Cell Calcium. 2009;45:574–582. doi: 10.1016/j.ceca.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Loirand G, Guérin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98:322–334. doi: 10.1161/01.RES.0000201960.04223.3c. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marmé D, Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- Methven L, McBride M, Wallace GA, McGrath JC. Theα1B/D-adrenoceptor knockout mouse permits isolation of the vascular α1A-adrenoceptor and elucidates its relationship to the other subtypes. Br J Pharmacol. 2009a;158:209–224. doi: 10.1111/j.1476-5381.2009.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methven L, Simpson PC, McGrath JC. α1A/B-knockout mice explain the native α1D-adrenoceptor's role in vasoconstriction and show that its location is independent of the other α1-subtypes. Br J Pharmacol. 2009b;158:1663–1675. doi: 10.1111/j.1476-5381.2009.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueed I, Bains P, Zhang L, Macleod KM. Differential participation of protein kinase C and Rho kinase inα1-adrenoceptor mediated contraction in rat arteries. Can J Physiol Pharmacol. 2004;82:895–902. doi: 10.1139/y04-086. [DOI] [PubMed] [Google Scholar]
- Murányi A, Derkach D, Erdődi F, Kiss A, Ito M, Hartshorne DJ. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: Inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005;579:6611–6615. doi: 10.1016/j.febslet.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Kurihara J, Miyajima Y, Ishii K, Kato H. Calcium antagonistic properties of nicardipine, a dihydropyridine derivative assessed in isolated cerebral arteries and cardiac muscle. Arzneimittelforschung. 1985;35:687–693. [PubMed] [Google Scholar]
- Nichols RJ, Dzamko N, Hutti JE, Cantley LC, Deak M, Moran J, Bamborough P, Reith AD, Alessi DR. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem J. 2009;424:47–60. doi: 10.1042/BJ20091035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiro N, Koga Y, Ikebe M. Agonist-induced changes in the phosphorylation of the myosin binding subunit of myosin light chain phosphatase and CPI17, two regulatory factors of myosin light chain phosphatase, in smooth muscle. Biochem J. 2003;369:117–128. doi: 10.1042/BJ20021040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobe K, Paul RJ. Distinct pathways of Ca2+ sensitization in porcine coronary artery: effects of Rho-related kinase and protein kinase C inhibition on force and intracellular Ca2+ Circ Res. 2001;88:1283–1290. doi: 10.1161/hh1201.092035. [DOI] [PubMed] [Google Scholar]
- Ohanian V, Ohanian J, Shaw L, Scarth S, Parker PJ, Heagerty AM. Identification of protein kinase C isoforms in rat mesenteric small arteries and their possible role in agonist-induced contraction. Circ Res. 1996;78:806–812. doi: 10.1161/01.res.78.5.806. [DOI] [PubMed] [Google Scholar]
- Okumura K, Nishiura T, Awaji Y, Kondo J, Hashimoto H, Ito T. 1,2-diacylglycerol content and its fatty acid composition in thoracic aorta of diabetic rats. Diabetes. 1991;40:820–824. doi: 10.2337/diab.40.7.820. [DOI] [PubMed] [Google Scholar]
- Pacaud P, Loirand G, Baron A, Mironneau C, Mironneau J. Ca2+ channel activation and membrane depolarization mediated by Cl− channels in response to noradrenaline in vascular myocytes. Br J Pharmacol. 1991;104:1000–1006. doi: 10.1111/j.1476-5381.1991.tb12540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JK, McLean AJ, Hill CE. Receptors involved in nerve-mediated vasoconstriction in small arteries of the rat hepatic mesentery. Br J Pharmacol. 1998;124:1403–1412. doi: 10.1038/sj.bjp.0701976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piascik MT, Guarino RD, Smith MS, Soltis EE, Saussy P, Perez DM. The specific contribution of the novelα-1D adrenoceptor to the contraction of vascular smooth muscle. J Pharmacol Exp Ther. 1995;275:1583–1589. [PubMed] [Google Scholar]
- Ratz PH, Miner AS, Barbour SE. Calcium-independent phospholipase A2 participates in KCl-induced calcium sensitization of vascular smooth muscle. Cell Calcium. 2009;46:65–72. doi: 10.1016/j.ceca.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokosh DG, Simpson PC. Knockout of theα1A/C-adrenergic receptor subtype: the α1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci U S A. 2002;99:9474–9479. doi: 10.1073/pnas.132552699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner XL, Berkowitz DE, Booth JV, Funk BL, Cozart KL, D’Amico EB, El-Moalem H, Page SO, Richardson CD, Winters B, Marucci L, Schwinn DA. Subtype specific regulation of human vascularα1-adrenergic receptors by vessel bed and age. Circulation. 1999;100:2336–2343. doi: 10.1161/01.cir.100.23.2336. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- Sato K, Ozaki H, Karaki H. Changes in cytosolic calcium level in vascular smooth muscle strip measured simultaneously with contraction using fluorescent calcium indicator fura 2. J Pharmacol Exp Ther. 1988;246:294–300. [PubMed] [Google Scholar]
- Shimokawa H, Rashid M. Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol Sci. 2007;28:296–302. doi: 10.1016/j.tips.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Tai K, Hamaide MC, Debaix H, Gailly P, Wibo M, Morel N. Agonist-evoked calcium entry in vascular smooth muscle cells requires IP3 receptor-mediated activation of TRPC1. Eur J Pharmacol. 2008;583:135–147. doi: 10.1016/j.ejphar.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Williams TJ, Blue DR, Daniels DV, Davis B, Elworthy T, Gever JR, Kava MS, Morgans D, Padilla F, Tassa S, Vimont RL, Chapple CR, Chess-Williams R, Eglen RM, Clarke DE, Ford AP. In vitroα1-adrenoceptor pharmacology of Ro 70-0004 and RS-100329, novel α1A-adrenoceptor selective antagonists. Br J Pharmacol. 1999;127:252–258. doi: 10.1038/sj.bjp.0702541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DP, Susnjar M, Kiss E, Sutherland C, Walsh MP. Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: Rho-associated kinase-mediated phosphorylation of MYPT1 at Thr-855, but not Thr-697. Biochem J. 2005;389:763–774. doi: 10.1042/BJ20050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Őrsy P, Horváth B, Maser-Gluth C, Greiner E, Lemmer B, Schütz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signalling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- Woodsome TP, Polzin A, Kitazawa K, Eto M, Kitazawa T. Agonist- and depolarization-induced signals for myosin light chain phosphorylation and force generation of cultured vascular smooth muscle cells. J Cell Sci. 2006;119:1769–1780. doi: 10.1242/jcs.02805. [DOI] [PubMed] [Google Scholar]
- Xi Q, Adebiyi A, Zhao G, Chapman KE, Waters CM, Hassid A, Jaggar JH. IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ Res. 2008;102:1118–1126. doi: 10.1161/CIRCRESAHA.108.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Koike K. α1-Adrenoceptor subtypes in the mouse mesenteric artery and abdominal aorta. Br J Pharmacol. 2001;134:1045–1054. doi: 10.1038/sj.bjp.0704350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan XJ. Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am J Physiol Lung Cell Mol Physiol. 1997;272:L959–L968. doi: 10.1152/ajplung.1997.272.5.L959. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.