

Abstract
Cancer is one of the major causes of death worldwide. In spite of achieving significant successes in medical sciences in the past few decades, the number of deaths due to cancer remains unchecked. The conventional chemotherapy and radiotherapy have limited therapeutic index and a plethora of treatment related side effects. This situation has provided an impetus for search of novel therapeutic strategies that can selectively destroy the tumour cells, leaving the normal cells unharmed. Viral oncotherapy is such a promising treatment modality that offers unique opportunity for tumour targeting. Numerous viruses with inherent anti-cancer activity have been identified and are in different phases of clinical trials. In the era of modern biotechnology and with better understanding of cancer biology and virology, it has become feasible to engineer the oncolytic viruses (OVs) to increase their tumour selectivity and enhance their oncolytic activity. In this review, the mechanisms by which oncolytic viruses kill the tumour cells have been discussed as also the development made in virotherapy for cancer treatment with emphasis on their tumour specific targeting.
Keywords: Apoptosis, oncolytic viruses, tumour targeting, viral oncotherapy
Introduction
Cancer is one of the leading causes of death worldwide. Despite significant progress made in cancer therapies, mortality rates for most malignancies remain distressingly high. The inhibition of cancer growth and progression is one of the major challenges faced by modern medicine. Cancer results either due to decreased cell death or increased cell birth. In other words, decreased propensity for apoptosis contributes to tumour formation1. The classical regimen of cancer therapy (chemotherapy, radiotherapy and immunotherapy) suffers with disadvantages such as narrow therapeutic index that further reduces as tumour evolves drug resistance and severe side-effects2. Another major problem of cancer therapy is the incomplete eradication of the invasive primary tumour mass or dissemination of tumour cells leading to recurrence of disease. The current goal for developing new therapies for the treatment of cancer is to design therapeutic agents that have a large therapeutic index (i.e. high potency against malignant cells) with little or no toxicities to normal cells. Future treatment modalities need to be more selective and specific, allowing higher and thus effective drug doses to reach at each tumour cell3. With the advent of modern biotechnology tools and better understanding of cancer biology and virology, it has become feasible to engineer viruses with increased tumour selectivity and enhanced oncolytic activity. Naturally occurring lytic viruses have evolved to infect, replicate and lyse cells. It is interesting that the replication cycle of many viruses exploits the same cellular pathways that are altered in cancer cells4. Specific targeting of tumour cell can be achieved by taking advantage of the fact that tumour cells have altered microenvironment, display certain tumour specific receptors and modified cellular pathways. Gene therapy and viral oncolysis represent treatment modalities that also offer unique opportunities for tumour targeting.
Viruses and apoptosis
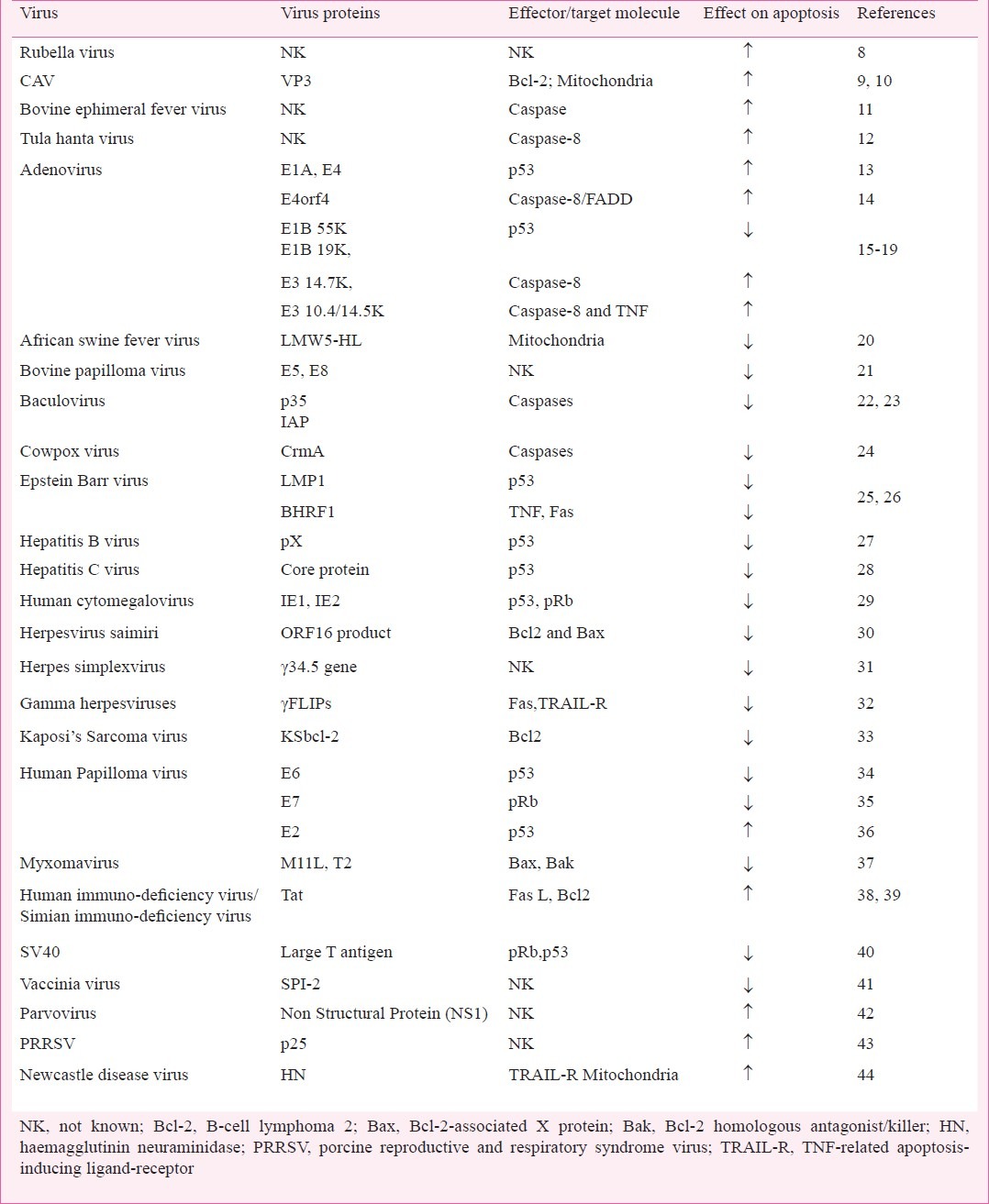
Pathogenesis of certain viruses is mediated by the modulation of apoptotic process (induction or suppression) in the homologous host5,6. Induction of apoptosis provides mechanism to evade the host cell immune response as during the process of apoptosis, the progeny virions along with the cellular contents of the cell are packaged into membrane enclosed apoptotic vesicles which are rapidly taken up by the neighbouring cells and thus the infection spreads undetected in the host. Virions enclosed within apoptotic vesicles are also protected from inactivation by host antibodies and proteases5. On the other hand, many viral gene products possess the ability to delay or suppress the apoptotic death of host cells. This offers several advantages to the virus since it provides time for replication and assembly of progeny virions which otherwise would severely limit the virus production and reduce or eliminate its spread in the host. Thus most viruses have evolved genes encoding proteins that can effectively mediate the induction or suppression of apoptosis by modulating host cell death pathways in order to facilitate their survival and spread (Table)7. Rubella virus induced apoptosis in various cell lines appears to be independent of p53 and Bcl-2 family proteins (B-cell lymphoma 2)8. Chicken anaemia virus encoded protein “apoptin” induces p53 independent, Bcl-2 dependent apoptosis in many transformed and tumour cell lines9,10. Bovine ephemeral fever virus was found to induce caspase dependent apoptosis in BHK-21 and Vero cells11. Li et al12 have reported that Tula hantavirus induces apoptosis in Vero E6 cells through caspase-8 activation. However, the apoptotic pathways and the targets within these pathways which viruses strike, vary between viruses and the type of viral proteins mediating it. Further, it also depends on the cell type6. For example, E1A of adenovirus associates with the pRb/p300 family of histone acetyl transferases and induces p53 dependent apoptosis in many cancer cells13; while adenovirus early region 4 open reading frame (E4orf4) induced apoptosis is p53 independent in mouse embryo fibroblast derived cells14. Further, the same proteins (E4orf4) induce apoptosis in 293T cells through caspase-8/(FADD) pathway15.
Table.
Viruses and their proteins having ability to modulate apoptosis ….
Apoptosis and cancer
Evasion of apoptosis is fundamental to tumour initiation, progression and maintenance. Despite the fact that many tumour cells have a defect in the decision machinery for apoptosis, these usually retain an intact execution system and hence if provided with an effective apoptotic signal, such tumour cells will die45. Unfortunately, various chemotherapeutic agents fail in inducing apoptosis because functional p53 is required for inducting apoptosis in tumour cells46. More than 50 per cent of the human tumours, including melanoma, lung cancer or colon carcinoma contain mutated p53 and patients with such tumours have a very low chance of responding to chemotherapy47. Likewise, overexpression of the anti-apoptotic proteins Bcl-2 and Bcl-XL or caspase inhibitors negatively influences the chemotherapeutic treatment of a large number of lymphomas48.
Considerable efforts are currently being made for the development of new anti-cancer therapies, which are based on the induction of apoptosis and are not hampered by the lack of functional p53 and/or overexpression of antiapoptotic genes49. Oncolytic virotherapy is a promising form of cancer therapy which employs nature's own agent to find and destroy malignant cells. It has a large therapeutic index but only limited pathogenicity to normal tissue. The anti-tumour activity of oncolytic viruses can be increased by arming these with therapeutic genes50.
Virotherapy for treatment of cancer
Viruses are nature's nanoparticle with a diameter ranging from 20 to 500 nm51. An oncolytic virus (OV) is a virus having the ability to specifically infect and lyse cancer cells, while leaving normal cells unharmed. Here, certain viral genes act as tumour destructive agent, and the viral capsid as a nano-sized nucleic acid delivery vehicle52. In general, oncolytic viruses derive their specificity by exploiting cell surface receptor or intracellular aberrations in gene expression that arise in malignancies during tumour development. The greatest advantage that the oncolytic virus offers over chemotherapeutic agent is its ability to be engineered by in vitro genetic manipulation in response to preclinical and clinical findings2. The first oncolytic virus used in clinical studies was a vaccine strain of rabies virus in 1950s for treatment of patients with melanomatosis, and 8 of 30 patients showed tumour regression53. Thereafter, the oncolytic activity of several other viruses was tested in various animal and human models for their anti-cancer potency, tumour specificity and safety54. These include West Nile virus strain Egypt 101, mumps, Newcastle disease, measles, autonomous parvo, adeno, reo, vesicular stomatitis, herpes simplex and entero viruses55. However, many viruses elicited side effects which ultimately ended the trials and interest in viruses as anti-cancer agents declined during the 1970s. With the advent of modern biotechnology, gene therapy and better understanding of cancer biology, there is resurgence of interest in viral therapy in cancer treatment44,56. The field has come a long way from the original pioneering research with Herpes simplex virus type-I to the 1st multiple clinical trials with adenovirus ONYX-01557. The world's first oncolytic virus approved by China's State Food & Drug Administration in 2005, was a genetically modified adenovirus-H101 type 552, in which E-1B-55 kD and partial E3 genes have been deleted. Though both DNA and RNA viruses have been used in oncolytic virotherapy, DNA viruses are more frequently used as these are more amenable to genetic manipulation56.
Criteria for selection of oncolytic viruses for cancer therapy
Although viral oncotherapy has great potential for cancer treatment, yet for successful application, the viruses have to meet stringent criteria for safety and efficacy.
Safety: To design a safe viral oncolytic agents, certain criteria should be given due consideration. These include cancer specificity, chances of regaining pathogenecity, possibility of transmission to healthy individual, undesired side effects and pre-existing immunity58. The use of non human viruses in viral oncotherapy would help in minimizing these risk factors.
Efficacy: Efficacy of OVs can be enhanced by developing strategies for efficient delivery of viruses and overcoming the host antiviral immune response. Approaches to evade antiviral response include serotype switching i.e. administration of different viral serotypes during treatment cycle59, and polymer coating (modification of amino groups by mixing viral particles with polymers such as poly [N-(2-hydroxypropyl)methacrylamide] (Phpma) bearing reactive 4-nitrophenyl esters on pendent diglycyl side chains) so that antibody cannot recognize the virus particle and use of cellular vehicles52.
Strategies for tumour targeting
Cancer cells distinguish themselves from their normal counterparts by alterations in cell physiology such as self sufficiency in growth signals, insensitivity to growth inhibition signals, evasion of apoptosis, limitless replication potential, sustained angiogenesis and tissue invasion and metastasis. These alterations make these a generous host for viruses and hence these properties can be utilized for selective replication of OVs in cancer cells. These cancer targeting mechanisms of viruses can be broadly achieved by two general approaches i.e. deletion of viral genes required for virus replication in normal cells but dispensable in tumour cells and use of tissue/tumour specific promoters for critical viral genes. The specific tumour targeting can be achieved by targeting various molecular steps/regulators of cell cycle. Some of these are discussed below:
(i) Pro apoptotic targeting: Many viruses delay apoptosis of infected cells in order to assist their replication. These encode certain proteins which alter the activity of important regulators of programmed cell death such as p53 and pRb60. Adenoviral proteins E1A and E1B inactivate pRb and p53 in normal cells, respectively, to delay premature apoptosis52,61. A virus having deletion in E1 can be rendered tumour specific. The examples of this type of mutants are ONYX-15 having mutation in E1B (dl 922-947) and HB101 having deletions in two viral genes- E1B and E362. Further, certain type of cancer cells express cellular E1A like activity and therefore, E1A deleted adenovirus can efficiently replicate in such cancers, e.g. HepG1 and Hep3B63. Some viral and non viral proteins are known to induce p53 independent apoptosis in tumour cells9. Proteins derived from viruses, i.e. chicken anaemia virus derived apoptosis-inducing protein (apoptin), E4orf4 and parvovirus-H1 derived non-structural protein 1 (NS1), torque teno virus derived protein TTV apoptosis inducing protein (TAIP), the human α-lactalbumin made lethal to tumour cells (HAMLET), which is present in human milk or the human cytokines melanoma differentiation-associated gene-7 (mda-7) and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), have the ability to induce tumour-selective apoptosis64.
(ii) Translational targeting: The response of a cell to virus infection, dsRNA and lipopolysacharide is production of Type I interferon (IFN). This shut downs the protein synthesis in the neighbouring cell, rendering these unfit for virus replication. However, most of the cancer cells have defective IFN signaling pathways, so one strategy to enhance tumour specificity is to mutate OVs to induce a more potent IFN response52. This will minimize the replication of such viruses in normal cells but the cancer cells will remain permissive. Also some viruses block IFN signaling by encoding protein which inhibits the translation of mRNA for IFN, e.g. matrix (M) protein of vesicular stomatitis virus (VSV). Such viruses can be rendered tumour specific by mutating the genes encoding the protein inhibiting IFN release. The non pathogenic VSVs can still multiply in tumour cells as these lack ability to produce and respond to IFN. The inherent oncolytic activity of Newcastle disease virus (NDV) was also believed to be derived from defective IFN signaling pathways in tumour cells65. However, recent studies suggested that the tumour specificity of NDV is independent of type I IFN response and is due to the tumour cell resistance to apoptosis66,67.
HSV-1 can be made tumour selective by mutating the γ134.5 gene (designated as R3616). The product of this gene (ICP34.5) binds with protein phosphatase-1 and inhibits phosphorylation of eukarykotic initiation factor-2 (eIF-2) by activated PKR (ds RNA induced protein kinase). This unphosphorylated eIF-2 cannot inhibit translation of viral transcripts unlike its phosphorylated counterpart. Cancer cells are resistant to the PKR activated inhibition of viral replication due to the high level of Ras activity which inhibits autophosphorylation of PKR. Thus mutant having deleted γ134.5 cannot multiply in normal cells but tumour cells remain permissive68. Tumour specificity can be further improved by a second mutation in the UL39 gene (G207, a neuroathenuated, replication competent HSV-1 with deletions in both copies of γ134.5 gene and UL39 gene), encoding the large subunit of the viral ribonucleotide reductase (ICP6). This further compels the virus to multiply in cancer cell with high endogenous ribonucleotide reductase.
(iii) Transcriptional targeting: Oncolytic viruses can be rendered tumour selective by placing essential viral gene under the regulation of tumour specific promoter69. However, this technique is limited to DNA viruses (excluding pox viruses). Certain tumour specific gene promoters like human telomerase reverse transcriptase (hTERT) and survivin are active in a variety of tumour types while others are specific for particular tumours, e.g. Prostrate specific antigen (PSA) for prostrate, foetoprotein for liver and tyrosinase for skin3. Activity of these promoters can be further augmented by binary regulatory system with recombinant Gal 4 fusion protein52.
Survivin is overexpressed in squamous carcinoma cell which makes it possible to use therapies specifically targeting this gene. Adenoviruses can be made tumour selective by placing essential viral gene (E1) under the control of human E2F-1 promoter60,61 which is selectively activated in tumour cells with a defect in Rb pathway. Replication-competent adenoviruses that have restricted expression of the E1A and E1B genes have been produced using prostate-specific promoters, such as the prostate-specific antigen (PSA) promoter, the probasin promoter and combinations of both (e.g., CV706 and CG0787) for prostate carcinomas70. Selective expression of E1A has been attempted in specific carcinomas, such as hepatocellular carcinoma (α-foetoprotein promoter) and breast carcinoma (mucin-1 promoter and estrogen-receptor promoter). In addition, the general characteristics of tumour cells, e.g. telomerase promoter and hypoxia-inducible factor (HIF) responsive elements have been used to design oncolytic adenoviruses71. In case of herpes viruses, transcriptional targeting is achieved by placing the γ134.1 gene under the control of tumour specific promoter72. The expression of the essential immediate-early ICP4 gene under control of the albumin-enhancer-promoter, limits replication of HSV to the liver and to hepatocellular carcinoma56. Similarly, the calponin promoter has been used to generate HSV mutants that replicate selectively in malignant human soft tissue and bone tumours. For retroviruses, the U3 gene which promotes enhancer region in the long terminal repeat has been replaced72.
(iv) Transductional targeting: The fact that tumour cells display high level of tumour specific receptors can be exploited for targeting of oncolytic viruses specifically to cancer cells73. For instance, many cancer cells overexpress intra cellular adhesion molecule-1 (ICAM-1) and decay accelerating factor (DAF), the receptors for coxsackievirus A21(CAV21)74. Another enterovirus, echovirus type 1, gains preferential entry to ovarian cancer cells due to overexpression of the I domain of integrin a2b175, and poliovirus infects cells expressing the CD155 receptor, abundant on many human cancer cell types76. However, for viruses which utilize receptors that are abundant on normal cells, the mechanisms that govern preferential replication in cancer cells are probably intracellular55. For example, despite that mumps viruses use sialic acid as their receptor and alphaviruses use heparin sulphate or ICAM-1, all abundantly expressed also on many normal cells, these viruses show high cancer cell selectivity due to defective interferon (IFN) signaling pathway in tumour cells77. Alternatively, tumour specificity of viruses can be reprogrammed by displaying single chain antibodies or other polypeptide binding ligands on the viral surface.
(v) Targeting strategies based on tumour microenvironment: To support uncontrolled growth and tissue invasion, tumour cells develop a modified microenvironment such as hypoxia, activation of certain proteases and angiogenesis. This can be harnessed for developing strategies for tumour targeting69. A dual regulated oncolytic Ad CNHK500 was developed in which the E1b gene is controlled by a hypoxia responsive promoter and the E1a gene is controlled by an human telomerase reverse transcriptase (hTERT) promoter56. Tumour selectivity might be further improved by incorporation of hypoxia-responsive elements into tumour-specific promoters to exploit the relatively hypoxic conditions within a tumour78. Vesicular stomatitis virus (VSV) is known to have an inherent capacity of replication under hypoxic tumour environment. Another approach to develop tumour specific oncolytic viruses is based on utilization of epithelial cell specific promoters to control the expression of important viral genes.
(vi) Targeting tumour using carrier cells as cellular vehicle for oncolytic viruses: Cancer cell secretes a number of chemokines which helps in trafficking of immune cells to tumour. These immune cells can be used as cellular vehicle for efficient delivery of OVs to tumour cells. Other types of cells such as stem cells (mesenchymal, endothelial progenitor cells) have also been developed as cellular vehicles to deliver OVs79. Cellular vehicles not only deliver the OVs to tumour site but also help in overcoming the problem of pre-existing antiviral immunity78. Specific delivery of HSV-1, adeno virus, VSV, parvovirus, measles virus and vaccinia virus has been achieved by utilizing carrier cells56,80. Manipulation of chemokine-chemokine receptor system can be harnessed for tumour targeting. The tumour derived chemokine RANTES (regulated and normal T cell expressed and secreted; also known as CCL5) (CCL5) attracts CD4+ T, CD8+ T and NK cells to tumour site56. T cells can be recruited to tumour site by genetic engineering of these cells to express chemokine receptors such as CXR2. Tumour cells can also be used as carriers to deliver an OV. This was first demonstrated in a regional delivery of replication-selective HSV-1 for the intraperitoneal therapy of epithelial ovarian cancer81. The tumour carrier cells can be inactivated by gamma-irradiation after infection with the OV. This does not affect the production and release of oncolytic parvovirus82.
Representative oncolytic viruses
Adenovirus: Adenoviruses were first isolated in 1953 from cultures of human adenoid tissues and were one of the first vector systems to be developed for gene delivery and expression. Adenovirus has emerged as one of the most potential viral therapeutic agent in cancer therapy. In the late 1950s, adenoviruses were reported to induce apoptosis in HeLa cells. Since then, clinical trials have begun on different experimental models both in vivo and in vitro83. Recently, genetically engineered mutants of the virus have been created, which are capable of killing tumour cells, sparing their normal counterparts. The adenoviral genome consists of linear, dsDNA of 26-45 kb. It is a non-enveloped, icosahedral virus with capsid of 65-80 nm diameter. The structural elements of capsid comprise penton, hexon and fibre proteins which play a crucial role in virus entry into the cells. Adenovirus entry into the cells occurs by receptor-mediated endocytosis involving different receptors. The capsid structural elements such as penton recognize and bind with the primary receptor, the coxsackie-adenovirus receptor (CAR) resulting in internalization of the virus and endocytosis by clathrin coated pits and subsequent lysis of the endosomes releases virus particles in the infected cell84. In this process, a series of viral proteins co-operate to promote efficient replication of the virus and its release. These major viral proteins include E1A, E1B-55kD, E1B-19kD, E3-11.6 kD and other associated proteins85. The two genes particularly, E1A and E1B (E1B-55kD) have been the targets of modification in order to create tumour-specific viruses. In normal circumstances, the products of these genes act in concert to force the host cell to enter S phase which is a prerequisite for the viral replication process. Thus, deletion of E1A will render the virus susceptible to the antiviral mechanisms of the retinoblastoma protein (Rb, a tumour suppressor protein), specifically by blocking the G1 to S transition. On the other hand, deletion of E1B, allows p53 to induce apoptosis in infected cells, aborting replication and spread of the virus. Therefore, the productive replication of adenoviral E1-deletion mutants can only take place in cells negative in Rb and p53. Most of the cancer cells fulfill these requirements and hence become selective targets for oncolysis by adenoviral E1-deletion mutants86.
A number of conditional replicating mutants have been engineered to target cancer cells that have mutations or defects in the p53 and Rb pathway. One such mutant of adenovirus, dl1520 (ONYX-015/ CI-1042) has shown promising results as anti-cancer agent in clinical trial conducted in mouse and humans and it was the first engineered replication selective virus to be used in humans87. It was proven to be a safe agent in phase I and II trials for the treatment of patients with squamous cell carcinoma of the head and neck83. These mutants have deletion of 827 bp in the E1B, an important adenovirus protein which helps in replication of the virus by suppressing the activity of p5388. Though these mutants have increased specificity for tumour cells, it is reported that these mutants also replicates in tumour cell line with wild type p53 activity indicating that p53 status may not play an important role in apoptosis and the most likely explanation may be due to late viral RNA export rather than p53 activity89. Another mutant dl 922-947 has deletions of 845 bp of the E1A proteins which normally play an essential role in binding and inhibiting the Rb group of proteins. This mutant primarily targets cancer cells defective in the Rb pathway and has shown greater efficacy than the similar mutant AdA-24 which contains a 24 bp deletion in E1A90. Studies conducted in mice have demonstrated improved efficacy of these mutants when combined with chemotherapy. Some engineered adenovirus constructs have been developed and many more are in the pipeline in different laboratories worldwide for use as oncolytic agents. Oncolytic adenoviruses have been used in clinical trials for various cancer types such as glioma, ovarian cancer, pancreatic cancer, prostrate cancer and colorecteal cancer91,92.
Chicken anaemia virus (CAV): CAV is a non enveloped virus which belongs to genus Gyrovirus of the family Circoviridae. It is one of the smallest known avian viruses with 23-25 nm size. The genome of the virus consists of long circular, single stranded minus-strand DNA of around 2319 bp93. It has three partially overlapping open reading frames (ORFs) - ORF-1/C1 (nucleotide position 853-2199), ORF-2/C2 (nucleotide position 380-1027) and ORF-3/C3 (nucleotide position 486-848) which on transcription produces an unspliced polycistronic mRNA of about 2 kb, encoding proteins of 51.6, 24.0 and 13.6 kDa proteins representing VP1, VP2 and VP3 proteins, respectively94. The VP3 also known as apoptin is a serine-threonine rich protein of 121 amino acids. Among the major proteins of CAV, VP2 and VP3 are known to induce apoptosis in infected cells. The apoptotic activity of VP2 is much weaker than VP3. Further, VP2 induces apoptosis both in normal and tumour cells but apoptin induces p53-independent apoptosis specifically in tumour cells95. Apoptin acts as a multimeric complex and forms super structures upon binding to DNA. In tumour cells, apoptin is phosphorylated and mainly nuclear whereas in normal cells it is unphosphorylated, cytoplasmic, and becomes readily neutralized62. Interestingly, apoptin phosphorylation, nuclear translocation, and apoptosis can transiently be induced in normal cells by co-transfecting SV40 large T oncogene, indicating that apoptin recognizes early stages of oncogenic transformation96. In cancer cells, apoptin appears to recognize survival signals, which redirect it into cell death impulses. Apoptin targets include DEDAF, Nur77, Nimi, Hippi, PML and the potential drug target anaphase promoting complex (APC1)62. The following properties of apoptin makes it a potent candidate for viral oncotherapy - (i) selective apoptotic activity against transformed and cancer cells but not in normal diploid ceils, implies that side-effects of apoptin treatment may be minor97, (ii) apoptin senses early stages of oncogenic transformation, (iii) apoptin mediated cell death is independent of death receptors, as cells deficient in Fas Associated Death Domain (FADD) or caspase-8, the key regulators of the extrinsic apoptotic pathway, remain sensitive to apoptin98, (iv) apoptin acts independently of p53, the most commonly mutated tumour suppressor gene in cancer, (v) its apoptotic activity is stimulated by antiapoptotic Bcl-2 protein99. Overexpression of antiapoptotic Bcl-2 and Bcl-XL probably occurs in more than half of all cancers leading to development of therapeutic resistance, and (vi) its apoptotic activity is insensitive to BCR-ABL99, indicating that apoptin can induce apoptosis in cases where present chemotherapeutic agents fail. Further, systemic delivery of apoptin reveals the safety and efficiency of apoptin as an anti-cancer therapeutic agent100. Olijslagers et al101 reported that chemotherapeutic agents when combined with apoptin resulted in enhanced cytotoxicity. Higher induction of an effective anti-tumor immune response and tumour regression was found when apoptin was used in combination with interleukin (IL)-18102. The anti-neoplastic effect of apoptin has been verified in Rous sarcoma virus induced tumour in chicken103. In our laboratory, we have found that the apoptin induced death of HeLa cells is mainly mediated by the intrinsic/mitochondrial pathway of apoptosis104.
Parvovirus: The genus Parvovirus comprised many important pathogens of human and animals that include B19 human parvovirus, feline panleukopenia virus (FPLV), canine parvovirus (CPV), mink enteritis virus (MEV), etc. The cell death in parvovirus infection is mainly due to apoptosis105. Members of Parvoviridae family like B-19, feline panleukopenia viruses, have an inherent property of oncolysis that could be used for cancer virotherapy106. Recent studies have shown that various species of parvovirus can induce apoptosis in vitro in different cell types such as haematopoietic cells, lymphocytes and glioblastoma cells107. Sol et al108 have reported that B19 human parvovirus induced apoptosis in erythroid cells can be attributed to the expression of non-structural proteins. Non-structural proteins from murine and human parvoviruses promote cell death when expressed in isolation in cell lines106. The non-structural proteins (NS1) of B19 human parvovirus, which are highly conserved among parvoviruses, promote apoptosis in erythroid cells through caspase-3 activation42. However, its mode of action remains unclear but it appears to be a nuclear protein capable of binding DNA which can alter the synthesis and phosphorylation of a number of cellular proteins. Parvovirus induced apoptotic cell death is one of the key pathogenic mechanisms in causing damage to digestive tract epithelia, lymphoid and haematopoietic tissues in parvovirus-infected cats and dogs109. The antineoplastic activity of parvoviruses appears to be linked to loss of genetic stability in tumour cells. Parvovirus DNA replication appears to select against cells bearing mutated or inactivated p53. It has been reported that SV40 virus transformed human fibroblasts were more susceptible to parvovirus infection and lysis than were normal control cells110.
It is reported that antineoplastic activity of oncolytic viruses is not only by direct killing of the cells but also by modulating the production of immunoregulatory molecules. Evidences suggest that in vivo H1 parvovirus infected irradiated tumour cells when administered parentally not only contribute to reduce tumour load by causing direct lysis of neoplastic cells but also by stimulating their recognition as therapeutic vaccine by the immune system111. It was also found that infection of human melanoma cells with this virus results in activation of co-cultured dendritic cells and ensuing cross-priming of cytolytic T-cell response112. It was also hypothesized that virus can activate tumour cells to reduce immuno-stimulating factors and/or kill these in a way that promote the uptake of tumour antigens113.
In a monocytic cell line, the apoptotic pathway triggered by H1 virus appears to share at least some steps of the pathway activated by tumour necrosis factor-α (TNF- α), which is accompanied by a rapid and marked downregulation of c-Myc expression prior to the appearance of apoptotic signs such as activation of caspase-3, cleavage of poly(ADP)-ribose-polymerase (PARP) and occurrence of apoptotic bodies. This suggests that c-Myc or factors regulated in a similar way could play a key role in the signaling of apoptosis induced by parvoviruses107.
Toolan et al113 first tested the oncolytic potential of H-1PV in two patients diagnosed with advanced osteosarcoma. An estimated dose of 109 plaque-forming units (PFU) was administered by intramuscular route. Later, a series of clinical trials were performed using H-1PV in cutaneous metastases from different solid tumors (breast cancers, melanomas, bronchial carcinoma, pancreatic adenocarcinoma and kidney leiomyosarcoma)114. Rommelaere et al115 have recently reviewed the oncolytic effect of parvoviruses.
Herpes simplex virus (HSV): HSV is an enveloped, dsDNA virus with 152 kb long genome. The transcription, replication and packaging of HSV take place in the nuclei of infected cells. In cells permissive for this virus, replication cycle is usually completed with in 20 h, releasing thousands of viral progeny on cell lysis. The virus enters into the cell by fusion of the viral envelope proteins with the host cell plasma membrane. The viral protein gC and gB are required for binding of the virus to host cells receptor whereas gD plays a major role in virus entry116. The genome of HSV-1 consists of three major gene regions- alpha, beta, and gamma and each of the genes act co-operatively to regulate viral entry, replication and multiplication in host cells. The alpha genes are transcribed in early infection in absence of de novo protein synthesis and these products regulate transcription of beta and gamma genes. The beta genes products are important in viral nucleic acid metabolism and are also required for viral DNA replication117. The HSV gamma genes have been divided into two subgroups, gamma 1 and gamma 2, which encode different proteins of each type. The gamma 1 genes are expressed in early infection and do not depend on viral DNA synthesis in contrast to gamma 2 genes that are lately expressed and are dependent on viral DNA synthesis and hence can be blocked by inhibitors of viral DNA synthesis. The HSV cycle is usually completed by assembly of virus followed by their release into extracellular space118.
To specifically target HSV replication to neoplastic cells, a variety of mutants have been designed with functional inactivation of the viral genes that encode for thymidine kinase, ribonucleotide reductase and infected cell protein 34.5 (ICP34.5)119. In quiescent cells, cellular forms of viral ribonucleotide reductase and thymidine kinase are not expressed but are upregulated only during the G1 and S phases of the cell cycle because these generate dNTPs required for DNA synthesis. This limits replication of HSV which is defective in such gene functions to rapidly proliferating cells, such as tumour cells120. The neurovirulence factor ICP34.5 has been characterized as an inhibitor of dsRNA dependent protein kinase (PKR). Therefore, PKR-induced shut-off of cellular protein synthesis following infection with HSV is circumvented by ICP34.5. However, Ras activity directly inhibits PKR-mediated effects, and thus the action of ICP34.5 is not required in cell lines that have a constitutively activated Ras signaling pathway67 and so to reduce the probability of the occurrence of wild-type revertants and to increase the safety margin, some viruses have been created which contain multiple mutations within their genomes55,121.
R3616 a deletion mutant of HSV-1 virus, has been constructed that is capable of replicating in cancer cells with high Ras activity65. Another mutant, G207 was developed in which mutation was done in the UL 39 gene which encodes for the large subunit of viral ribonucleotide reductase122. These mutants confirm the possibility of any viral or cellular mutation which might specifically eliminate the effects of ICP34.5 deletion and render the virus to replicate preferentially in proliferating cancer cells with high endogenous activity of ribonucleotide reductase. Phase I studies have demonstrated viral replication following direct injection of these mutants into malignant brain tumours, is also safe and there is some evidence of clinical efficacy, indicating this to be a potentially useful adjuvant therapy119,123.
Newcastle disease virus (NDV): Newcastle disease virus (NDV) also known as avian paramyxovirus-1 belongs to the genus Avulavirus of the family Paramyxoviridae. The NDV genome is 15,186 nucleotides long single stranded, negative-sense RNA which contains six genes encoding at least eight proteins- six structural (NP, P, M, F, HN, L) and two non-structural (V and W)124. NDV binds cells via the haemagglutinin neuraminidase (HN) protein, which attaches to sialic acid-containing host cell receptors125. Binding is followed by fusion of viral and cell-surface membranes, a process mediated by F protein. The viral RNA is then released into the cytoplasm and undergoes replication.
NDV is known to cause apoptosis in different cell types including chicken embryo fibroblast (CEF) cells and peripheral blood mononuclear cells126,127. The evidence of induction of apoptosis by NDV implies its role in the pathogenesis44. A study by Kommers et al128, based on the simultaneous positive staining for apoptosis and viral nucleoprotein in lymphoid tissues suggested that the lymphoid depletion observed in ND may be due to apoptosis mediated by NP. However, in another report, it was revealed that the HN protein alone is capable of upregulating the expression of interferon-alpha and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) in the target cells129. In our laboratory, we have demonstrated that HN protein, when expressed alone, causes apoptosis in CEF cells that requires activation of caspases, loss of mitochondrial membrane potential and augmentation of oxidative stress. Our study suggested that at early stage of ND infection, death receptor mediated pathways play role in inducing apoptosis in Vero cells which is shifted to mitochondria mediated pathways in later stage of infection. We also found that for NDV to cause apoptosis of Vero cells, de novo synthesis of protein after NDV infection is necessary130. Oncolytic potential of NDV and its HN gene on Rous sarcoma virus induced tumour in chicken indicated that NDV has anti-tumour effect which is stimulated when HN gene is given along with the intact virus indicating synergistic effect of NDV on tumour regression compared to either NDV or HN alone15,131. An attenuated strain of NDV, PV701 was used in phase I clinical trial and it was shown that when administered intravenously, PV701 replicates specifically in tumour cells and causes cytolysis of tumours of epithelial origin (carcinomas including breast, lung, prostate and colon) and of neuroectodermal (melanomas, glioblastmas and neuroblastomas) and mesenchymal origin (sarcomas). In all the cases, survival rate was encouraging with minimal side effects132. A strain of oncolytic Newcastle disease virus-NDV-HUJ was found to overcome the antiapoptotic effect of Livin (member of inhibitor of apoptosis protein family) in melanoma cells. The study suggested that NDV-HUJ is a potent inducer of apoptosis that activates caspases, causes cleavage of Livin converting it into the proapoptotic tLivin protein66. In future the use of such oncolytic viruses may provide a suitable alternative for management of cancers.
Conclusion and future perspectives
The revolution in molecular and cancer biology and better understanding of viruses and host cell interaction in the past decade has facilitated the development of rationally designed, targeted cancer therapies. The initial results of clinical trials using OVs are encouraging. There are many OVs currently under development. Tumour specific targeting remains a challenge but pre-clinical data indicate that transcriptional targeting might be achieved using tumour specific promoters. Microarray technology and proteomics will help in identifying novel tumour specific targets. Considering the heterogeneity of tumour in a patient and among patients a combination of OVs along with conventional therapy regimen of cancer such as radiation, chemotherapy, immunotherapy and gene therapy will be useful in combating cancer.
References
- 1.Zhivotovsky B, Orrenius S. Carcinogenesis and apoptosis: Paradigms and paradoxes. Carcinogenesis. 2006;27:1939–45. doi: 10.1093/carcin/bgl035. [DOI] [PubMed] [Google Scholar]
- 2.Bell JC, Litchy B, Stojdl D. Getting oncolytic virus therapies off the ground. Cancer cell. 2003;4:7–11. doi: 10.1016/s1535-6108(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 3.Dorer DE, Nettelbeck DM. Targeting cancer by transcriptional control in cancer gene therapy and viral oncolysis. Adv Drug Deliv Rev. 2009;61:554–71. doi: 10.1016/j.addr.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Ries SJ, Brandts CH. Oncolytic viruses for the treatment of cancer: current strategies and clinical trials. Drug Discov Today. 2004;9:759–68. doi: 10.1016/S1359-6446(04)03221-0. [DOI] [PubMed] [Google Scholar]
- 5.Roulston A, Marcellus RC, Branton PE. Viruses and apoptosis. Annu Rev Microbiol. 1999;53:1–60. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- 6.Hay S, Kannourakis G. A time to kill: viral manipulation of the cell death program. J Gen Virol. 2002;83:1547–64. doi: 10.1099/0022-1317-83-7-1547. [DOI] [PubMed] [Google Scholar]
- 7.Johnson DC, Huber MT. Directed egress of animal viruses promotes cell to cell spread. J Virol. 2002;76:1–8. doi: 10.1128/JVI.76.1.1-8.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann J, Pletz WR, Liebert UG. Rubella virus-induced cytopathic effect in vitro is caused by apoptosis. J Gen Virol. 1999;80:1657–64. doi: 10.1099/0022-1317-80-7-1657. [DOI] [PubMed] [Google Scholar]
- 9.Danen-van Oorschot AA, van Der Eb AJ, Noteborn MH. The chicken anemia virus-derived protein apoptin requires activation of caspases for induction of apoptosis human tumor cells. J Virol. 2000;74:7072–8. doi: 10.1128/jvi.74.15.7072-7078.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danen van Oorschot AA, Fischer DF, Grimbergen JM, Klein B, Zhuang SM, Falkenburg JHF, et al. Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc Natl Acad Sci USA. 1997;94:5843–7. doi: 10.1073/pnas.94.11.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang CJ, Shih WL, Yu FL, Liao MH, Liu HJ. Apoptosis induced by bovine ephemeral fever virus. J Virol Methods. 2004;122:165–70. doi: 10.1016/j.jviromet.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Li XD, Kukkonen S, Vapalahti O, Plyusnin A, Lankinen H, Vaheri A. Tula hantavirus infection of Vero E6 cells induces apoptosis involving caspase 8 activation. J Gen Virol. 2004;85:3261–7. doi: 10.1099/vir.0.80243-0. [DOI] [PubMed] [Google Scholar]
- 13.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nature Med. 2000;6:1134–9. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- 14.Marcellus RC, Teodoro JG, Wu T, Brough DE, Ketner G, Shore GC, et al. Adenovirus type 5 early region 4 is responsible for EIA-induced p53-independent apoptosis. J Virol. 1996;70:6207–15. doi: 10.1128/jvi.70.9.6207-6215.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravindra PV. Uttar Pradesh: Indian Veterinary Research Institute (Deemed University) Izatnagar; 2008. Elucidation of apoptotic pathways induced by Newcastle disease virus in cultured cells and assessment of its Oncolytic potential in experimentally induced tumor. Ph.D. thesis. [Google Scholar]
- 16.Han J, Sabbatini P, Perez D, Rao L, Modha D, White E. The E1B 19k protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996;10:461–77. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- 17.Farrow SN, White JHM, Martinou I, Raven T, Pun KT, Grinham CJ, et al. Cloning of a Bcl-2 homolog by interaction with adenovirus E1B 19k. Nature. 1995;374:731–3. doi: 10.1038/374731a0. [DOI] [PubMed] [Google Scholar]
- 18.Krajcsi P, Dimitrov T, Hermiston TW, Tollefson AE, Ranheim TS, Van de Pol SB, et al. The adenovirus E3-14±7k protein and the E3-10.4k,14.5k complex of proteins, which independently inhibit tumor necrosis factor (TNF)-induced apoptosis, also independently inhibit TNF-induced release of arachidonic acid. J Virol. 1996;70:4904–13. doi: 10.1128/jvi.70.8.4904-4913.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen P, Tian J, Kovesdi I, Bruder JT. Interaction of the adenovirus 14.7 kDa protein with FLICE inhibits Fas ligand-induced apoptosis. J Biol Chem. 1998;273:5815–20. doi: 10.1074/jbc.273.10.5815. [DOI] [PubMed] [Google Scholar]
- 20.Afonso CL, Neilan JG, Kutish GF, Rock DL. An African swine fever virus Bcl-2 homologue, 5-HL, suppresses apoptotic cell death. J Virol. 1996;70:4858–63. doi: 10.1128/jvi.70.7.4858-4863.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valle GF, Banks L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J Gen Virol. 1995;76:1239–45. doi: 10.1099/0022-1317-76-5-1239. [DOI] [PubMed] [Google Scholar]
- 22.Clem RJ, Hardwick JM, Miller LK. Anti-apoptotic genes of baculoviruses. Cell Death Differ. 1996;3:9–16. [PubMed] [Google Scholar]
- 23.Bertin J, Mendrysa SM, LaCount DJ, Gaur S, Krebs JF, Armstrong RC, et al. Apoptotic suppression by baculovirus P35 involves cleavage by and inhibition of a virus-induced CED-3/ICE-like protease. J Virol. 1996;70:6251–9. doi: 10.1128/jvi.70.9.6251-6259.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talley AK, Dewhurst S, Perry SW, Dollard SC, Gummuluru S, Fine SM, et al. Tumor necrosis factor alpha-induced apoptosis in human neuronal cells : protection by the antioxidant N-acetylcysteine and the genes Bcl-2 and crmA. Mol Cell Biol. 1995;15:2359–66. doi: 10.1128/mcb.15.5.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okan I, Wang YS, Chen F, Hu LF, Imreh S, Klein G, et al. The EBV-encoded LMP1 protein inhibits p53-triggered apoptosis but not growth arrest. Oncogene. 1995;11:1027–31. [PubMed] [Google Scholar]
- 26.Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Epstein-Barr virus-coded BHRF1 protein, a viral homolog of Bcl-2, protects human B-cells from programmed cell-death. Proc Natl Acad Sci USA. 1993;90:8479–83. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang XW, Gibson MK, Vermeulen W, Yeh H, Forrester K, Sturzbecher HW, et al. Abrogation of p53-induced apoptosis by the hepatitis B virus x gene. Cancer Res. 1995;55:6012–6. [PubMed] [Google Scholar]
- 28.Ray RB, Meyer K, Ray R. Suppression of apoptotic cell death by hepatitis C virus core protein. Virology. 1996;226:176–82. doi: 10.1006/viro.1996.0644. [DOI] [PubMed] [Google Scholar]
- 29.Zhu H, Shen YQ, Shenk T. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol. 1995;69:7960–70. doi: 10.1128/jvi.69.12.7960-7970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nava VE, Cheng EHY, Veliuona M, Zou SF, Clem RJ, Mayer ML, et al. Herpesvirus saimiri encodes a functional homolog of the human Bcl-2 oncogene. J Virol. 1997;71:4118–22. doi: 10.1128/jvi.71.5.4118-4122.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou J, Roizman B. The gamma-1 34.5 gene of herpes simplex virus-1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programmed cell-death in neuronal cells. Proc Natl Acad Sci USA. 1992;89:3266–70. doi: 10.1073/pnas.89.8.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thome M, Schneider P, Hofmann K, Fickenscher H, Meini E, Neipel F, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–21. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 33.Cheng EHY, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci USA. 1997;94:690–4. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheffner M, Werness BA, Huibregtse J M, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus type-16 and type-18 promotes the degradation of p53. Cell. 1990;63:1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 35.Pan HC, Griep AE. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Devel. 1995;9:2157–69. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- 36.Desaintes C, Demeret C, Goyat S, Yaniv M, Thierry F. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J. 1997;16:504–14. doi: 10.1093/emboj/16.3.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su J, Wang G, Barrett JW, Irvine TS, Gao X, McFadden G. Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of bax at the mitochondria. J Virol. 2006;80:1140–51. doi: 10.1128/JVI.80.3.1140-1151.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finkel TH, Tudorwilliams G, Banda NK, Cotton MF, Curiel T, Monks C, et al. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV-infected and SIV-infected lymph nodes. Nature Med. 1995;1:129–34. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 39.Sastry KJ, Marin MC, Nehete PN, McConnell K, Elnaggar AK, McDonnell TJ. Expression of human immunodeficiency virus type-1 Tat results in down-regulation of Bcl-2 and induction of apoptosis in hematopoietic cells. Oncogene. 1996;13:487–93. [PubMed] [Google Scholar]
- 40.McCarthy SA, Symonds HS, Van Dyke T. Regulation of apoptosis in transgenic mice by simian-virus-40 T-antigen-mediated inactivation of p53. Proc Natl Acad Sci USA. 1994;91:3979–83. doi: 10.1073/pnas.91.9.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobbelstein M, Shenk T. Protection against apoptosis by the vaccinia virus SPI-2 (B13R) gene product. J Virol. 1996;70:6479–85. doi: 10.1128/jvi.70.9.6479-6485.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morey AL, Ferguson DJP, Fleming KA. Ultrastructural features of fetal erythroid precursors infected with parvovirus-B19 in vitro evidence of cell-death by apoptosis. J Pathol. 1993;169:213–20. doi: 10.1002/path.1711690207. [DOI] [PubMed] [Google Scholar]
- 43.Suarez P, Diazguerra M, Prieto C, Esteban M, Castro JM, Nieto A, et al. Open reading frame-5 of porcine reproductive and respiratory syndrome virus as a cause of virus-induced apoptosis. J Virol. 1996;70:2876–82. doi: 10.1128/jvi.70.5.2876-2882.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravindra PV, Tiwari AK, Ratta B, Bais MV, Chaturvedi U, Palia SK, et al. Time course of Newcastle disease virus-induced apoptotic pathways. Virus Res. 2009;144:350–4. doi: 10.1016/j.virusres.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 45.Quintieri L, Fantin M, Vizler C. Identification of molecular determinants of tumor sensitivity and resistance to anticancer drugs. Adv Exp Med Biol. 2007;593:95–104. doi: 10.1007/978-0-387-39978-2_10. [DOI] [PubMed] [Google Scholar]
- 46.Daniel S. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2009;20:14–24. doi: 10.1016/j.tcb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez-Nieto S, Zhivotovsky B. Role of alterations in the apoptotic machinery in sensitivity of cancer cells to treatment. Curr Pharm Des. 2006;12:4411–25. doi: 10.2174/138161206779010495. [DOI] [PubMed] [Google Scholar]
- 48.Walensky LD. BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ. 2006;13:1339–50. doi: 10.1038/sj.cdd.4401992. [DOI] [PubMed] [Google Scholar]
- 49.Muthumani K, Choo AY, Hwang DS, Ugen KE, Weiner DB. HIV-1 Vpr: enhancing sensitivity of tumors to apoptosis. Curr Drug Deliv. 2004;1:335–44. doi: 10.2174/1567201043334614. [DOI] [PubMed] [Google Scholar]
- 50.Hermiston TW, Kuhn I. Armed therapeutic viruses: strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002;9:1022–35. doi: 10.1038/sj.cgt.7700542. [DOI] [PubMed] [Google Scholar]
- 51.Harrison SC. Principles of virus structure. In: Knipe DM, Howley PM, editors. Fields virology. Lippincott Williams and Wilkins; 2001. pp. 53–85. [Google Scholar]
- 52.Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28:326–33. doi: 10.1016/j.tips.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pack GT. Note on the experimental use of rabies vaccine for melanomatosis. AMA Arch Derm Syphilol. 1950;62:694–5. doi: 10.1001/archderm.1950.01530180083015. [DOI] [PubMed] [Google Scholar]
- 54.Ring CJA. Cytolitic viruses as potential anticancer agents. J Gen Virology. 2002;7:S293. doi: 10.1099/0022-1317-83-3-491. [DOI] [PubMed] [Google Scholar]
- 55.Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: Molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–31. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–9. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 58.Russell SJ. Replicating vectors for cancer therapy: a question of strategy. Semin Cancer Biol. 1994;5:437–43. [PubMed] [Google Scholar]
- 59.Novella IS, Gilbertson DL, Borrego B, Domingo E, Holland JJ. Adaptability costs in immune escape variants of vesicular stomatitis virus. Virus Res. 2005;107:27–34. doi: 10.1016/j.virusres.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 60.Bouchet BP, de Fromentel CC, Puisieux A, Galmarin CM. p53 as a target for anti-cancer drug development. Crit Rev Oncol Hematol. 2006;58:190–207. doi: 10.1016/j.critrevonc.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 61.Dobbelstein M. Replicating adenoviruses in cancer therapy. Curr Top Microbiol Immunol. 2004;273:291–334. doi: 10.1007/978-3-662-05599-1_9. [DOI] [PubMed] [Google Scholar]
- 62.Ko D, Lynda Hawkins L, Yu DC. Development of transcriptionally regulated oncolytic adenoviruses. Oncogene. 2005;24:7763–74. doi: 10.1038/sj.onc.1209048. [DOI] [PubMed] [Google Scholar]
- 63.Zhao T, Rao XM, Xie X, Li L, Thompson TC, McMasters KM, et al. Adenovirus with insertion-mutated E1A selectively propagates in liver cancer cells and destroys tumors in vivo. Cancer Res. 2003;63:3073–8. [PubMed] [Google Scholar]
- 64.Los M, Panigrahi S, Rashedi I, Mandal S, Stetefeld J, Essmann F, et al. Apoptin, a tumor-selective killer. Biochim Biophys Acta. 2009;1793:1335–42. doi: 10.1016/j.bbamcr.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 65.Stojdl DF, Lichty BD, Benjamin R, tenOever BR, Paterson JM, Power AT, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–75. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 66.Mansour M, Palese P, Zamarin D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J Virol. 2011;85:6015–23. doi: 10.1128/JVI.01537-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lazar I, Yaacov B, Shiloach T, Eliahoo E, Kadouri L, Lotem M, et al. The oncolytic activity of Newcastle disease virus NDV-HUJ on chemoresistant primary melanoma cells is dependent on the proapoptotic activity of the inhibitor of apoptosis protein Livin. J Virol. 2010;84:639–46. doi: 10.1128/JVI.00401-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarinella F, Calistri A, Sette P, Palu G, Parolin C. Oncolysis of pancreatic tumour cells by a gamma34.5-deleted HSV-1 does not rely upon Ras-activation, but on the PI 3-kinase pathway. Gene Ther. 2006;13:1080–7. doi: 10.1038/sj.gt.3302770. [DOI] [PubMed] [Google Scholar]
- 69.Palmer D H, Lawrence S Young, Vivien Mautner. Cancer gene-therapy: clinical trials. Trends Biotechnol. 2006;24:76–82. doi: 10.1016/j.tibtech.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 70.Chen L, Sapuntzakis MS, Duncan C, Sharifi R, Ghosh L, van Breemen R, et al. Oxidative DNA damage in prostate cancer patients consuming tomato sauce-based entrees as a whole-food intervention. J Natl Cancer Inst. 2001;93:1872–9. doi: 10.1093/jnci/93.24.1872. [DOI] [PubMed] [Google Scholar]
- 71.Post DE, Van Meir EG. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene. 2003;22:2065–72. doi: 10.1038/sj.onc.1206464. [DOI] [PubMed] [Google Scholar]
- 72.Dalba C, Klatzmann D, Logg CR, Kasahara N. Beyond oncolytic virotherapy: replication-competent retrovirus vectors for selective and stable transduction of tumors. Curr Gene Ther. 2005;5:655–67. doi: 10.2174/156652305774964659. [DOI] [PubMed] [Google Scholar]
- 73.Springfeld C, von Messling V, Frenzke M, Ungerechts G, Buchholz CJ, Cattaneo R. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metallo proteinases. Cancer Res. 2006;66:7694–700. doi: 10.1158/0008-5472.CAN-06-0538. [DOI] [PubMed] [Google Scholar]
- 74.Au GG, Lindberg AM, Barry RD, Shafren DR. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int J Oncol. 2005;26:1471–6. doi: 10.3892/ijo.26.6.1471. [DOI] [PubMed] [Google Scholar]
- 75.Shafren DR, Sylvester D, Johansson ES, Campbell IG, Barry RD. Oncolysis of human ovarian cancers by echovirus type 1. Int. J. Cancer. 2005;115:320–8. doi: 10.1002/ijc.20866. [DOI] [PubMed] [Google Scholar]
- 76.Ochiai H, Campbell SA, Archer GE, Chewning TA, Dragunsky E, Ivanov M, et al. Targeted therapy for glioblastoma multiforme neoplastic meningitis with intrathecal delivery of an oncolytic recombinant poliovirus. Clin. Cancer Res. 2006;12:1349–54. doi: 10.1158/1078-0432.CCR-05-1595. [DOI] [PubMed] [Google Scholar]
- 77.Krishnamurthy S, Takimoto T, Scroggs RA, Portner A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J Virol. 2006;80:5145–55. doi: 10.1128/JVI.02618-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ido A, Uto H, Moriuchi A, Nagata K, Onaga Y, Onaga M, Hori T, et al. Gene therapy targeting for hepatocellular carcinoma: selective and enhanced suicide gene expression regulated by a hypoxia-inducible enhancer linked to a human α-fetoprotein promoter. Cancer Res. 2001;61:3016–21. [PubMed] [Google Scholar]
- 79.Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006;5:755–66. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- 80.Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15:123–30. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 81.Coukos G, Makrigiannakis A, Kang EH, Caparelli D, Benjamin I, Kaiser LR, et al. Use of carrier cells to deliver a replication-selective herpes simplex virus-1 mutant for the intraperitoneal therapy of epithelial ovarian cancer. Clin Cancer Res. 1999;5:1523–37. [PubMed] [Google Scholar]
- 82.Raykov Z, Balboni G, Aprahamian M, Rommelaere J. Carrier cell-mediated delivery of oncolytic parvoviruses for targeting metastases. Int J Cancer. 2004;109:742–9. doi: 10.1002/ijc.20013. [DOI] [PubMed] [Google Scholar]
- 83.Ganly I, Kirn D, Eckhardt G, Rodriguez GI, Soutar DS, Otto R, et al. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res. 2000;6:798–806. [PubMed] [Google Scholar]
- 84.Meier O, Greber UF. Adenovirus endocytosis. J Gene Med. 2003;5:451–62. doi: 10.1002/jgm.409. [DOI] [PubMed] [Google Scholar]
- 85.White E, Gooding LR. Regulation of apoptosis by human adenoviruses. Curr Commun Cell Mol Biol. 1994;8:111–4. [Google Scholar]
- 86.Jiang H, Olson MV, Medrano DR, Lee OH, Xu J, Piao Y, et al. A novel CRM1-dependent nuclear export signal in adenoviral E1A protein regulated by phosphorylation. Faseb J. 2006;20:2603–5. doi: 10.1096/fj.06-6433fje. [DOI] [PubMed] [Google Scholar]
- 87.Barker DD, Berk AJ. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology. 1987;156:107–12. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- 88.Nemunaitis J, Khuri F, Ganly I, Arseneau J, Posner M, Vokes E, et al. Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neckcancer. J Clin Oncol. 2001;19:289–98. doi: 10.1200/JCO.2001.19.2.289. [DOI] [PubMed] [Google Scholar]
- 89.Shea CCO, Soria C, Bagus B, McCormick F. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell. 2005;8:61–74. doi: 10.1016/j.ccr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 90.Fueyo J, Manzano G, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 91.Jiang H, Gomez-Manzano C, Lang FF, Alemany R, Fueyo J. Oncolytic adenoviruses are emerging as a promising alternative therapy for glioma patients and are currently being tested in clinic. Curr Gene Ther. 2009;9:422–7. doi: 10.2174/156652309789753356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baird SK, Aerts JL, Eddaoudi A, Lockley M, Lemoine NR, McNeish IA. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene. 2008;27:3081–90. doi: 10.1038/sj.onc.1210977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Noteborn MH, de Boer GF, van Roozelaar DJ, Karreman C, Kranenburg O, Vos JG, et al. Characterization of cloned chicken anemia virus DNA that contains all elements for the infectious replication cycle. J Virol. 1991;65:3131–9. doi: 10.1128/jvi.65.6.3131-3139.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Todd D. Circoviruses: immunosuppressive threats to avian species: a review. Avian Pathol. 2000;29:373–94. doi: 10.1080/030794500750047126. [DOI] [PubMed] [Google Scholar]
- 95.Noteborn MH. Chicken anemia virus induced apoptosis: underlying molecular mechanisms. Vet Microbiol. 2004;98:89–94. doi: 10.1016/j.vetmic.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 96.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–71. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Danen-van Oorschot AA, Voskamp P, Seelen MC, van Miltenburg MH, Bolk MW, Tait SW, et al. Human death effector domain-associated factor interacts with the viral apoptosis agonist apoptin and exerts tumor-preferential cell killing. Cell Death Differ. 2004;11:564–73. doi: 10.1038/sj.cdd.4401391. [DOI] [PubMed] [Google Scholar]
- 98.Maddika S, Booy EP, Johar D, Gibson SB, Ghavami S, Los M. Cancer specific toxicity of apoptin is independent of death receptors but involves the loss of mitochondrial membrane potential and the release of mitochondrial cell-death mediators by a Nur77-dependent pathway. J Cell Sci. 2005;118:4485–93. doi: 10.1242/jcs.02580. [DOI] [PubMed] [Google Scholar]
- 99.Zhuang SM, Shvarts A, van Ormondt H, Jochemsen AG, van der Eb AJ, Noteborn MH. Apoptin, a protein derived from chicken anemia virus, induces p53- independent apoptosis in human osteosarcoma cells. Cancer Res. 1995;55:486–9. [PubMed] [Google Scholar]
- 100.Peng DJ, Sun J, Wang JYZ, Tian J, Zhang YH, Noteborn MH, et al. Inhibition of hepatocarcinoma by systemic delivery of Apoptin gene via the hepatic asialoglycoprotein receptor. Cancer Gene Ther. 2007;14:66–73. doi: 10.1038/sj.cgt.7700985. [DOI] [PubMed] [Google Scholar]
- 101.Olijslagers SJ, Zhang YH, Backendorf C, Noteborn MH. Additive cytotoxic effect of apoptin and chemotherapeutic agents paclitaxel and etoposide on human tumour cells. Basic Clin Pharmacol Toxicol. 2007;100:127–31. doi: 10.1111/j.1742-7843.2006.00016.x. [DOI] [PubMed] [Google Scholar]
- 102.Lian H, Jin N, Li X, Mi Z, Zhang J, Sun L, et al. Induction of an effective anti-tumor immune response and tumor regression by combined administration of IL-18 and Apoptin. Cancer Immunol Immunother. 2007;56:181–92. doi: 10.1007/s00262-006-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Natesan S, Kataria JM, Dhama K, Bhardwaj N, Sylvester A. Anti-neoplastic effect of chicken anemia virus VP3 protein (apoptin) in Rous sarcoma virus-induced tumours in chicken. J Gen Virol. 2006;87:2933–40. doi: 10.1099/vir.0.82085-0. [DOI] [PubMed] [Google Scholar]
- 104.Singh PK. 2010. Uttar Pradesh: Indian Veterinary Research Institute (Deemed University) Izatnagar; Elucidation of molecular mechanism of apoptosis involved in apoptin induced oncolysis in tumor cell line. M.V.Sc thesis. [Google Scholar]
- 105.Bauder B, Suchy A, Gabler C, Weissenbock H. Apoptosis in feline pauleukapenia and canine parvovirus enteritis. J Vet Med. 2000;47:775–84. doi: 10.1046/j.1439-0450.2000.00411.x. [DOI] [PubMed] [Google Scholar]
- 106.Ozawa K, Ayub J, Kajigaya S, Shimada T, Young N. The gene encoding the nonstructural protein of B19 (human) parvovirus may be lethal in transfected cells. J Virol. 1988;62:2884–9. doi: 10.1128/jvi.62.8.2884-2889.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rayet B, Lopez-Guerrero JA, Rommelaere J, Dinsart C. Induction of programmed cell death by parvovirus H-1 in U937 cells: connection with the tumor necrosis factor alpha-pathway. J Virol. 1998;72:8893–903. doi: 10.1128/jvi.72.11.8893-8903.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sol N, Junter JL, Vassias I, Freyssinier JM, Thomas A, Prigent AF, et al. Possible interactions between the NS-1 protein and tumor necrosis factor alpha pathways in erythroid cell apoptosis induced by human parvovirus B19. J Virol. 1999;73:8762–70. doi: 10.1128/jvi.73.10.8762-8770.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Anouja F, Wattiez R, Mousset S, Caillet-Fauquet P. The cytotoxicity of the parvovirus minute virus of mice-structural protein NS1 is related to changes in the synthesis in the synthesis and phosphorylation of cell proteins. J Virol. 1997;71:4641–78. doi: 10.1128/jvi.71.6.4671-4678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Telerman A, Tuynder M, Dupressoir T, Robaye B, Sigaux F, Shaulian E, et al. A model for tumor suppression using H-1 parvovirus. Proc Natl Acad Sci USA. 1993;90:8702–6. doi: 10.1073/pnas.90.18.8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zahari R, Svetlana G, Angel G, Ginette B, Ute K, Marc A, et al. Combined oncolytic and vaccination activities of parvovirus H-1 in a metastatic tumor model. Oncol Rep. 2007;17:1493–9. [PubMed] [Google Scholar]
- 112.Moehler MH, Zeidler M, Wilsberg V, Cornelis JJ, Woelfel T, Rommelaere J, et al. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation and cross-presentation by dendritic cells. Hum Gene Ther. 2005;16:996–1005. doi: 10.1089/hum.2005.16.996. [DOI] [PubMed] [Google Scholar]
- 113.Toolan HW, Saunders EL, Southam CM, Moore AE, Levin AG. H-1 virus viremia in the human. Proc Soc Exp Biol Med. 1965;119:711–5. doi: 10.3181/00379727-119-30278. [DOI] [PubMed] [Google Scholar]
- 114.Le Cesne A, Dupressoir T, Jamin N, Spielmann M, Le Chevalier T, Sancho-Garnier H, et al. Intra-lesional administration of a live virus, parvovirus H-1, in cancer patients: a feasibility study. Proc Am Soc Clin Oncol. 1993;12:297. [Google Scholar]
- 115.Rommelaere J, Geletneky K, Angelova AL, Daeffler L, Dinsart C, Kiprianova I, et al. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 2010;21:185–95. doi: 10.1016/j.cytogfr.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 116.Post DE, Fulci G, Chiocca EA, van Meir EG. Replicative oncolytic herpes simplex viruses in combination cancer therapies. Curr Gene Ther. 2004;4:41–51. doi: 10.2174/1566523044577988. [DOI] [PubMed] [Google Scholar]
- 117.Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–36. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 118.Norman KL, Farassati F, Lee PW. Oncolytic viruses and cancer therapy. Cytokine Growth Factor Rev. 2001;12:271–82. doi: 10.1016/s1359-6101(00)00024-1. [DOI] [PubMed] [Google Scholar]
- 119.Homa FL, Brown JC. Capsid assembly and DNA packaging in herpes simplex virus. Rev Med Virol. 1997;7:107–22. doi: 10.1002/(sici)1099-1654(199707)7:2<107::aid-rmv191>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 120.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–66. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 121.Yoon SS, Nakamura H, Carroll NM, Bode BP, Chiocca EA, Tanabe KK. An oncolytic herpes simplex virus type 1 selectively destroys diffuse liver metastases from colon carcinoma. FASEB J. 2000;14:301–11. [PubMed] [Google Scholar]
- 122.Cassady KA, Parker JN. Herpesvirus vectors for therapy of brain tumors. Open Virol J. 2010;4:103–8. doi: 10.2174/1874357901004010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Filippakis H, Spandidos DA, Sourvinos G. Herpesviruses: Hijacking the Ras signaling pathway. Biochim Biophys Acta. 2010;1803:777–85. doi: 10.1016/j.bbamcr.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 124.Krishnamurthy S, Samal SK. Nucleotide sequence of trailer nucleocapsid protein gene and intergenic region of Newcastle disease virus strain Beaudette, C and completion of the entire genome sequence. J Gen Virol. 1998;79:2419–24. doi: 10.1099/0022-1317-79-10-2419. [DOI] [PubMed] [Google Scholar]
- 125.Park MS, Garcia-Sastre A, Cos JF, Basler CF, Palese P. Newcastle disease virus V protein is a determinant of host range restriction. J Virol. 2003;77:9522–32. doi: 10.1128/JVI.77.17.9522-9532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lam KM. Growth of Newcastle disease virus in chicken macrophages. J Comp Pathol. 1996;115:253–63. doi: 10.1016/s0021-9975(96)80083-1. [DOI] [PubMed] [Google Scholar]
- 127.Ravindra PV, Tiwari AK, Ratta B, Chaturvedi U, Palia SK, Chauhan RS. Newcastle disease virus-induced cytopathic effect in infected cells is caused by apoptosis. Virus Res. 2009;141:13–20. doi: 10.1016/j.virusres.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 128.Kommers GD, King DJ, Seal BS, Brown CC. Pathogenesis of chicken passaged Newcastle disease viruses isolated from chickens and wild and exotic birds. Avian Dis. 2003;47:319–29. doi: 10.1637/0005-2086(2003)047[0319:POCNDV]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 129.Ravindra PV, Tiwari AK, Sharma B, Rajawat YS, Ratta B, Palia S, et al. HN protein of Newcastle disease virus causes apoptosis in chicken embryo fibroblast cells. Arch Virol. 2008;153:749–54. doi: 10.1007/s00705-008-0057-2. [DOI] [PubMed] [Google Scholar]
- 130.Ravindra PV, Tiwari AK, Ratta B, Chaturvedi U, Palia SK, Subudhi PK, et al. Induction of apoptosis in Vero cells by Newcastle disease virus requires viral replication, de-novo protein synthesis and caspase activation. Virus Res. 2008;133:285–90. doi: 10.1016/j.virusres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 131.Ravindra PV, Tiwari AK, Sharma B, Chauhan RS. Newcastle disease virus as an oncolytic agent. Indian J Med Res. 2009;130:507–13. [PubMed] [Google Scholar]
- 132.Lorence RM, Pecora AL, Major PP, Hotte SJ, Laurie SA, Roberts MS, et al. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Curr Opin Mol Ther. 2003;5:618–24. [PubMed] [Google Scholar]