

Abstract
The central goal of this overview article is to summarize recent findings in renal epithelial transport, focusing chiefly on the connecting tubule (CNT) and the cortical collecting duct (CCD). Mammalian CCD and CNT are involved in fine tuning of electrolyte and fluid balance through reabsorption and secretion. Specific transporters and channels mediate vectorial movements of water and solutes in these segments. Although only a small percent of the glomerular filtrate reaches the CNT and CCD, these segments are critical for water and electrolyte homeostasis since several hormones, e.g. aldosterone and arginine vasopressin, exert their main effects in these nephron sites. Importantly, hormones regulate the function of the entire nephron and kidney by affecting channels and transporters in the CNT and CCD. Knowledge about the physiological and pathophysiological regulation of transport in the CNT and CCD and particular roles of specific channels/transporters has increased tremendously over the last two decades. Recent studies shed new light on several key questions concerning the regulation of renal transport. Precise distribution patterns of transport proteins in the CCD and CNT will be reviewed, and their physiological roles and mechanisms mediating ion transport in these segments will be also covered. Special emphasis will be given to pathophysiological conditions appearing as a result of abnormalities in renal transport in the CNT and CCD.
Introduction
The major function of renal epithelial cells is reabsorption of solutes and water from the tubular fluid into the blood and secretion of wastes from blood into urine. The distal part of the nephron that is comprised of the distal convoluted tubule (DCT), the connecting tubule (CNT) and the collecting duct (CD) performs the final adjustment of renal excretion. Various transport proteins such as ion channels and transporters mediate these processes. The expression and activity of these transporters are regulated by specific hormones and different extra- and intracellular regulatory mechanisms. CNT and the cortical collecting duct (CCD) have been studied extensively for many years with respect to transport via these nephron segments and their roles in the development of various renal diseases (181, 401, 613). A large body of experimental evidence indicates that CNT and the initial connecting tubules (ICT) along with DCT avidly reabsorb sodium and chloride and secrete potassium. Sodium reabsorption and potassium secretion by the CCD are extremely significant, although quantitatively less important than transport rates in the preceding segments (543, 602, 643). Under certain conditions the CCD also secretes protons, and during metabolic alkalosis is capable of secreting bicarbonate. Furthermore, these segments are involved in regulation of Ca2+ and Mg2+ homeostasis, water and urea transport. In addition, it should be emphasized that functions of multiple channels and transporters are interconnected. The same signaling mechanism triggers various channels/transporters or activation of one channel/transporter and inhibition of another. Thus, the CCD and CNT to a certain extent mediate a variety of kidney functions by modulating combined efforts of channels and transporters. This overview article will cover some aspects of physiological regulation of Na+ reabsorption, K+ secretion, water and Cl− transport, Ca2+ homeostasis and acid base handling in these important segments.
Recently, significant progress has been made in our understanding of the cellular and molecular mechanisms responsible for epithelial water and electrolyte homeostasis. Multiple laboratories have employed electrophysiological, biochemical, microscopical, molecular and genetic methods to study the function of the CCD and CNT in both normal and pathological conditions. The development and application of various tools to study proteins mediating renal function revealed some of the intriguing physiological functions of the CNT and CCD. Research advances naturally resulted in cloning of multiple cDNAs and genes that are involved in water and solvents transport and thus mediate the physiological function of these segments (149). Proteomic analyses provide additional highlights into identification of essential proteins involved in the final adjustment of urine composition in these segments (29, 111, 430, 466, 684). In addition, new knowledge about the function of particular segments of the kidney, including the CNT and CCD, has accrued as a result of the development and application of gene deletion techniques, both conventional and cell specific gene knockouts. Furthermore, even though anatomic complexity of the kidney makes it difficult to select appropriate promoters that target a specific cell type along the nephron, genetic engineering in mice nowadays allows expressing transgenes in specific nephron segments and cells, including both principal and intercalated cells in the CCD. Recent review by Rubera and colleagues highlights in details segment-specific gene targeting and ablation in the kidney and their advantages and limitations (462). Although many factors that control functions of the CCD and CCT and transport whitin these segments have been identified and are becoming better understood, much remains to be investigated.
In summary, activity of specific ion channels and transporters underlie the properties of the CCD and CNT. Systemic hormones, such as aldosterone, vasopressin, angiotensin etc, are important signaling determinants of the water and solute transport of this segment. In addition, local/paracrine mechanisms contributes to this regulation (350). Several recent reviews specifically summarize different aspects of the epithelial transport in the CCD and CNT (46, 58, 149, 215, 334, 603, 610, 614). This article aims to overview the physiological function of the CCD and CNT based on variety of ion channels and transporters and their distinct properties and mechanisms of regulation.
I. Structure and function of the CNT and CCD
A. Basic structure of the nephron
Human kidney contains approximately one million nephrons, each capable of forming urine (223, 271, 432). Every nephron contains a renal corpuscle (glomerulus and Bowman’s capsule), a proximal tubule (proximal convoluted and straight tubules), a loop of Henle, a distal convoluted tubule (DCT), and CNT. The collecting duct system, which includes the initial collecting tubule (ICT), the cortical collecting duct (CCD), the outer medullary collecting duct (OMCD), and the inner medullary collecting duct (IMCD), is not considered as a part of the nephron because embryologically it arises from the ureteric bud. Moreover, one collecting duct may collect urine from many nephrons. However, all of the components of the nephron and the collecting duct system are functionally interconnected. Consecutive segments of the nephron are demonstrated in Fig. 1. This overview article will focus only on physiological role and function of the CNT and CCD (including ICT). These regions are highlighted in red in Fig. 1. Glomerular filtration and water and electrolyte transport in other nephron segments will be discussed in corresponding overview articles.
Figure 1.

Consecutive segments of the nephron. Tubules discussed in this overview article are highlighted in red.
CNT is the proximal part of the collecting duct system adjacent to the DCT. Cell composition and length of the CNT depends on the location of the nephron within the kidney and varies between species. As mentioned above, the collecting ducts are formed in the renal cortex by connection of several nephrons and are subdivided into the CCD, OMCD, and IMCD (565). Basic tubular segments of the nephron are presented in Fig. 2A.
Figure 2.

(A) Structure of the nephron. Abbreviations for the nephron segments are described in Fig. 1. The relative lengths of different segments are not drawn to scale. (B) The ICT and CCD are composed of principal and intercalated cells. Structure of the CCD shown as a cross-section and schematic presentation of principal and intercalated cells that comprise these segments. The connecting tubule cells and the principal cells have a polygonal shape. The intercalated cells have a rounded shape. Compared to intercalated cells, the connecting tubule cells and the principal cells have fewer mitochondria and only modestly develop invaginations of the basolateral membrane. Both types of cells develop apical microvilli. However, primary cilia is found only in principal cells.
B. Types of cells constituting the CNT and CCD
Epithelial lining of the segments beyond the thick ascending limb of Henle’s loop (TAL) – the DCT, CNT and CCD – displays distinct cellular heterogeneity. Basically, two distinct cell types are found in each of these segments. First cell type constitutes the majority of the cells in the given segment and is considered to be specific for it. The second type, intercalated cell, is interspersed among the segment-specific cells (68, 135, 259) (Fig. 2B). The CNT consists of two cell types: connecting tubule cells and intercalated cells. ICT and CCD are composed of principal and intercalated cells. Principal cells are also often refererred as collecting duct cells. At least three populations of intercalated cells, type A and type B, exist in the CNT, ICT, and CCD (274, 275). Briefly, type A intercalated cells secrete protons and reabsorb K+, whereas type B cells secrete HCO3− and reabsorb Cl−. A third type of intercalated cells (non A-non B type), which is ultrastructurally and immunologically distinct from the type A and type B intercalated cells, has been described in both the CNT and the CCD (9, 274, 563). Recent review by Brown et al. provides detailed overview of intercalated cells phenotypes in the kidney (68). Intercalated cells constitute approximately 30 % of the cells in the CNT, CCD, and OMCD and contribute to the renal control of the acid-base balance (65, 274, 351, 607). As demonstrated by Kim et al, significant differences exist in the prevalence and distribution of the different types of intercalated cells in the CNT, ICT, and CCD (274). Type A, type B, and non Anon B intercalated cells were observed in all of the three segments, with type A cells being the most prevalent in both rat and mouse. In the mouse, however, non A-non B cells constitute more than a half of the intercalated cells in the CNT, 39% in the ICT, and 22% in the CCD, compared to 14, 7, and 5%, respectively, in the rat. In contrast, type B intercalated cells accounted for only 8 to 16% of the intercalated cells in the three segments in the mouse compared to 26 to 39% in the rat (274).
Connecting tubule cells of the CNT and the principal cells of the ICT and CCD have a polygonal shape. Intercalated cells of both CNT and CCD have a rounded shape. Fig. 2B demonstrates a schematic representation of principal and intercalated cells that comprise the CCD. Compared to intercalated cells, connecting tubule cells and principal cells have fewer mitochondria and only modestly develop invaginations of the basolateral membrane (67, 352). The apical membrane of the connecting tubule cells and the principal cells is relatively smooth and contains microvilli. Intercalated cells also exhibit apical microvilli (602). Moreover, principal cells have primary cilium on the apical membrane. Ciliary defects can lead to a number of human diseases (now commonly referred to as ciliopathies) (116, 475, 560). Intercalated cells lack a central cilium on the apical membrane (146, 208, 431). One notable feature of the CNT is the morphological heterogeneity. Cell size, membrane amplification, and the number of mitochondria are highest in the beginning of the CNT and gradually decrease along the tubule (602). The structure of principal cells also gradually shifts along the collecting duct. The number of mitochondria and the extent of basolateral infolding decrease as collecting duct enters the outer medulla (447, 602).
Fig. 3 shows a representative immunohistochemical staining for water channel aquaporin 2 (AQP2) at 20x and 60x magnifications in a cortical section of the Sprague-Dawley rat kidney. Immunohistochemical analysis was performed as described previously (267, 314, 413). AQP2 is found predominantly in the apical cell membranes of the kidney’s collecting duct principal cells (388) and is often used as a gold marker of the CCD. In addition to identification of the CCD, AQP2 staining helps to discriminate between principal and intercalated cells, since AQP2 is expressed in the principal cells only (Figure 3).
Figure 3.

Representative immunohistochemical staining for AQP2 in the cortical sections of the Sprague-Dawley rat kidney. Original magnifications are 20x and 60x, scale bars are shown on the pictures. Negative controls (stained with secondary antibodies in the absence of primary antibodies or stained without primary and secondary antibodies) did not have any staining (data not shown). Several profiles of proximal tubules (PT), distal convoluted tubules (DCT), cortical collecting duct (CCD) and glomerulus (G) are marked by arrows at 20x. Representative examples of intercalated (IC; no staining) and principal (PC; stained for AQP2, shown in brown) cells are indicated on the close-up image. The kidney was fixed for 24 hrs in zinc formalin and processed for paraffin embedding as described previously (240, 314, 413). The kidney sections were cut, dried and deparaffinized for subsequent labeled streptavidin-biotin immunohistochemistry. All slides were counterstained with Mayer’s hematoxylin (Dako, Carpinteria, CA). Tissue sections were incubated for 45 min in a 1:200 concentration of anti-AQP2 (sc-28629; Santa Cruz Biotechnology).
C. Polarity of apical and basolateral plasma membranes proteins
Epithelial cells of the CNT and CCD are polarized and have a distinctive apical–basal axis of polarity for vectorial transport of ions and solutes across the epithelium. Cells in these segments, like many other epithelial cells, are organized into adherent groups that partition the kidney into discrete compartments and, thus, separate urine from blood. This organization provides a number of unique physiological properties, including functional apical–basal polarity. This conserved function of polarized epithelial cells requires membrane proteins to be sorted and retained in the correct apical or basolateral membrane domain. Several protein complexes, such as the Crumbs, the Scribble, and the Par complexes modulate kinase and small G protein activity, leading to segregation of the apical and basolateral membranes (419). Despite growing evidence of the importance of the Crumbs and the Scribble polarity complexes in apical–basal polarity of epithelial cells, their functions remain poorly understood (383). The Par proteins, including Par3, Par6, and aPKC, are fundamental players in cell polarization (185, 419). Small GTPases that function as molecular switches in many signaling pathways also seem to have a crucial role in cell polarization processes. Rho-family GTPases regulate many cellular events, including cytoskeletal organization, proliferation and gene expression (556). In addition, a role of small GTPases in regulation of ion channels, including those functioning in the CNT and CCD is shown in many studies (41, 424, 425, 427, 530). Recent data indicate that balanced activities of small GTPases RhoA and Rac1 or Cdc42 contribute to overall tissue stability via crosstalk with the PAR complex (238). The polarity complexes and cytoskeleton elements regulated by Rho-family proteins also serve as scaffolds to direct and retain proteins at the apical/basolateral membrane, or the tight junction. As the cell generates apical–basal polarity, the exocytic and endocytic pathways establish a complex interrelationship that regulates correct sorting of newly synthesized proteins to and between the apical and basolateral membrane domains (383). These pathways and their regulation in polarized cells are more complex than in nonpolarized cells (633).
There are also cases where cell signaling causes global alterations in localization of some proteins in the CNT and CCD under certain pathophysiological conditions. One of the best studied examples is mislocalization of the epidermal growth factor receptor (EGFR). EGFR is normally localized to the basolateral plasma membrane of the CCD. However, the EGFR axis has been found to be dysfunctional in many models of polycystic kidney disease (PKD) and in humans with PKD. In these models EGFR is inappropriately targeted to the luminal plasma membrane of epithelia lining the cysts (126, 446, 554, 641, 680). Similarly, abnormalities of apico-basal polarity of Na+/K+-ATPase have been observed in a variety of genotypic and phenotypic mouse models of both autosomal recessive and dominant PKD as well as in human (22, 452, 639, 641, 642). All channels and transporters at the CNT and CCD, except for the Na+/K+-ATPase during the development of PKD, have strict apical or basolateral localizations.
II. Physiology of the CNT and CCD
Major determinants of function of the CNT and CCD are ion channels and transporters. Fig. 4 and Fig. 5 demonstrate a schematic representation of transport processes in the CCD and major channels and transporters involved in water and electrolyte homeostasis in principal cells, respectively. Heterogenic vectorial transport in this segment is tightly controlled. This segment of the nephron is most sensitive to hormones and that oversees the fine control of plasma Na+ and K+. Variety of hormones, e.g. aldosterone, angiotensin II, vasopressin, atrial natriuretic peptide, and insulin are involved in this tight regulation of the CNT and CCD function. The late DCT, CNT and CCD are often referred to as the aldosterone-sensitive distal nephron (ASDN). Aldosterone, which is the final element of the renin-angiotensin-aldosterone system (RAAS), increases reabsorption of sodium and water and secretion of potassium ions. This increases blood volume and, therefore, blood pressure. Several stimuli, including volume depletion and hyperkalemia, mediate aldosterone release from the adrenal cortex.
Figure 4.

Primary transport characteristics of the cortical collecting duct. Principal and intercalated cells are in colored beige and green, respectively. Tight junctions are also schematically represented in between the cells.
Figure 5.

Major channels and transporters involved in water and electrolyte homeostasis in principal cells of the CCD.
Aldosterone-sensitivity is conferred by binding to the mineralocorticoid receptor (MR) that then translocates to the nucleus and upregulates transcription. Genomic actions of aldosterone are traditionally divided into an early and a late phase (506, 540, 598). It was suggested that the early phase responds to acute changes in salt and water balance to allow more rapid responses to impaired homeostasis. In comparison, it was predicted that the later phase of aldosterone action sets the capacity of transport epithelia for solute and water transport, and thus this phase may be considered as a chronic response (540).
MRs poorly distinguish between glucocorticoids and mineralocorticoids. Moreover, plasma concentrations of glucocorticoids greatly exceeds those of aldosterone. To avoid Na+ retention, under normal conditions the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) irreversibly converts cortisol into cortisone, an inactive metabolite with low affinity for mineralocorticoid or glucocorticoid (GR) receptors. Aldosterone, which is not metabolized, occupies MR and GR. Polymorphisms in HSD11B2 gene encoding this enzyme are associated with salt sensitivity of blood pressure in normotensives (26, 239, 294). 11β-HSD2 converts cortisol to cortisone in humans, while in rodents this enzyme converts corticosterone to 11-dehydrocorticosterone (153, 168). Fig. 6 demonstrates the cellular actions of aldosterone in principal cells of the CCD.
Figure 6.

Mechanism of action of aldosterone in principal cells of the CCD. Aldosterone (Aldo) binds to mineralocorticoid receptor (MR) that then translocates to the nucleus and upregulates transcription of aldosterone-induced proteins, which regulate sodium reabsorption and potassium secretion via affecting ENaC, ROMK and Na+/K+-ATPase. Effect of 11β-HSD2 that metabolizes cortisol to cortisone, which has little affinity for MR or glucocorticoid receptor (GR), is shown.
RAAS axis plays an important role in regulation of blood pressure via mediating water and electrolyte balance in the CNT and the CCD. In addition to aldosterone, renin, which is primarily released by the kidneys, stimulates formation of angiotensins in blood and tissues. Angiotensin II (ANGII) also stimulates sodium reabsorption. For instance, it was demonstrated that ANGII directly stimulates ENaC in the CCD via Angiotensin II receptors, type 1 (AT1 receptors) (417). Importantly, beyond its role in the regulation of renal sodium reabsorption in the ASDN, aldosterone may exert deleterious effects on the kidney and the cardiovascular system particularly in the presence of a high-salt diet (62, 63). Special overview articles will review mechanisms of action of aldosterone and the RAAS on water and electrolyte balance.
There is also a powerful feedback system for regulating plasma osmolarity and sodium concentration that alters renal excretion of water independently of the rate of solute excretion. A primary effector of this feedback is an antidiuretic hormone (ADH), also called vasopressin. This effect of vasopressin is mostly mediated through regulation of water transport. However, whilst vasopressin decreases water excretion to dilute plasma, it does this, in part, by promoting sodium reabsorption and consequently decreasing sodium excretion via the epithelial Na+ channel (ENaC) activated along the CNT and CCD (28, 73, 210, 439, 440, 541).
One interesting feature of the CNT is that connecting tubule cells produce and release renal kallikrein, a serine protease that converts kininogen to kinin, which then acts through kinin receptors such as bradykinin receptor. Therefore, CNT and CCD are exposed to large amounts of kallikrein in the lumen. The kallikrein–kinin system could be involved in the development and progression of cardiovascular diseases (366). Although tissue kallikrein is probably not a primary controller of blood pressure, low synthesis rate and urinary excretion of this enzyme have been linked to elevated blood pressure (365). Several ion channels in the CNT and CCD are regulated through these mechanisms. For instance, kallikrein activates ENaC by cleaving its gamma subunit (418). Using tissue kallikrein knockout mice, it was also shown that a unique kalliuretic factor protects against hyperkalemia after a dietary K+ load (134). Moreover, recent studies utilizing freshly isolated split opened CCD revealed that increased bradykinin production in response to kallikrein-kinin system activation acutely inhibits ENaC activity to adjust Na+- reabsorption in the CCD (678).
However, as seen in Fig. 4, these segments are not only responsible for Na+ and K+ transport. Principal cells reabsorb sodium and water from the lumen and secrete potassium. In addition, these cells reabsorb chloride and calcium. Intercalated cells reabsorb potassium and bicarbonate and secrete hydrogen and chloride. Moreover, paracellular chloride transport is also shown in these segments. Some details about these transport mechanisms will be provided below.
Studies of various portions of the collecting duct revealed heterogeneity of the electrical properties along the segments. The electrophysiologic properties of the CNT and the CCD, such as transepithelial voltage and resistance, vary widely. This variability is largely the result of the differences in the mineralocorticoid status of studied animals (397, 473, 543). As discussed by Muto, transepithelial voltage changes towards lumen-positive potentials from proximal to distal parts of the collecting duct (376). For instance, as demonstrated in isolated rabbit cortical ducts, transepithelial voltage (Vt) of CNT, CCD and inner strip portion of OMCD was −9.9±0.9, −7.4±0.7 and 7.5±1.6 mV, respectively. The transepithelial resistance RT increases progressively along the collecting duct, indicating values of 39.7±6.7, 111.7±6.8 and 293.5±37.6 Ω•cm2, respectively (378). Furthermore, there is a significant electrophysiological heterogeneity of principal and intercalated cells of these segments. It was described that specific basolateral membrane voltage (VB) of CNT cells allows distinguishing between principal cells (VB=−78.0±1.3 mV) and type A and type B intercalated cells (VB=−30.6±2.2 and −26.7±1.8 mV, respectively). Similar difference was found in CCD were VB values for these cell types were −79.6±2.0, −27.6±2.9 and −27.5±1.9 mV, respectively (378).
Heterogeneity in the conductive properties of these nephron segments is also demonstrated by the fractional apical membrane resistance (fRA). This parameter is defined as a ratio of the apical membrane resistance and sum of the apical and basolateral membranes resistances. Thus, for principal and intercalated cells of CNT fRA values were 0.48±0.03 and 0.92±0.01 whereas in CCD fRA=0.33±0.04 and 0.95±0.003, respectively (378). The fRA increases progressively along the CD, as does the RT. (376) Thus, electrophysiological heterogeneity in collecting duct structure reflects functional features performing barrier-transport functions.
III. Sodium Transport in the CNT and CCD
Sodium is freely filtered by the glomerulus. Thus, most of sodium has to be reabsorbed as the filtrate flows along the nephron. Briefly, this reabsorption is mediated by the Na+/H+ exchanger (NHE3) in the proximal tubule, Na+/K+/2Cl− cotransporter (NKCC) in the TAL, and by the Na+/Cl− cotransporter (NCC) in the DCT. The final adjustment of renal Na+ intake and excretion in the kidney is achieved in the ASDN, where sodium reabsorption via ENaC is regulated by aldosterone, vasopressin, and other hormonal and non-hormonal factors (334, 541). ENaC along with the thiazide-sensitive NCC co-transporter constitutes the predominant sodium transport systems in the ASDN (365, 621). Although the ASDN reabsorbs less than 10% of the filtered sodium load, this segment is finally crucial for the amount of Na+ that appears in the urine. Na+ reabsorption in the CNT and CCD is transcellular and is mediated by the connecting tubule cells and the principal cells. At the basolateral membrane of the principal cells Na+ extrusion is mediated by the Na+/K+ pump, which also provides the electrochemical driving force for the apical entry of Na+.
A. Na+/K+-ATPase
The Na+/K+ pump (or Na+/K+-ATPase) is located at the basolateral membrane of many epithelial cells in different segments of the kidney, but it is especially abundant in the TAL of the Henle’s loop, cells of the DCT, connecting tubule cells, and principal cells of the collecting duct (120, 602). Na+/K+-ATPase was either not detected in the intercalated cells (405) or detected at significantly lower levels than that of the principal cells (148, 478). This indicates that principal cells, but not intercalated cells, are involved in transepithelial Na+ transport (602).
Na+/K+-ATPase functions as a heterodimeric protein complex comprised of catalytic α- and auxillary β-subunits. The α-subunit has ten transmembrane segments and mediates active transport. The β-subunit, which has only one transmembrane segment, is essential for proper assembly and membrane targeting of the complex. Na+/K+-ATPase heterodimers are normally targeted to basally directed intracellular trafficking vesicles and inserted into basolateral membranes where they are stabilized by the membrane cytoskeletal proteins ankyrin and spectrin (383, 384).
With each cycle, the Na+/K+ pump couples the extrusion of three Na+ ions and uptake of two K+ ions with the intracellular hydrolysis of one ATP molecule (see Fig. 5). Thus, the Na+/K+ pump is electrogenic and provides the primary active transport mechanism for Na+ and K+. This sodium extrusion keeps low intracellular Na+, and high intracellular K+. In addition, Na+/K+-ATPase might also play a role in regulation of actin dynamics, control of cell movement, and cell signaling, regulation of tight junction structure and function, and induction of polarity. These functions appear to be modulated by Na+/K+-ATPase enzyme activity as well as protein-protein interactions of the α and β subunits (436).
B. Epithelial Na+ channel (ENaC)
The apical entry of Na+ in the CNT and the CCD is regulated by ENaC. ENaC is a highly Na+-selective channel found at the apical membrane of salt-reabsorbing tight epithelia of such tissues as the distal nephron, the distal colon, the salivary and sweat glands and the lung. In the kidney, discretionary Na+ reabsorption in response to endocrine input to the ASDN is an important determinant of the pressure-natriuresis relationship and ENaC activity is the rate-limiting step for this discretionary Na+ reabsorption (21). Within the scope of this overview, only some aspects of ENaC regulation will be provided. Different aspects of ENaC regulation are subjects of excellent recent reviews by Loffing and Korbmacher (334), Schild (487), Butterworth et al. (76), and Soundararajan et al. (523).
1. Structure of ENaC
ENaC belongs to the Deg/ENaC family of ion channels (12, 486). Functional ENaC, as many other ion channels, is a heteromeric protein complex containing several distinct channel subunits. Four ENaC subunits have been identified: α, β, γ and δ (78, 80, 606). The δ-ENaC subunit localizes to brain and reproductive tissues where it is believed to be a substitute for α-ENaC in the functional channel (606, 634). Functional ENaC in the kidney is comprised of three homologous subunits (α, β and γ ENaC) which share ~30–40% identity in their amino acid sequences (35, 78, 80). Each subunit has two transmembrane domains with short cytoplasmic NH2- and COOH-termini and an extra large cytoplasmic loop between them (79, 443, 517). This membrane topology of ENaC was predicted from the primary amino acid sequence and confirmed by several different approaches (173). All three ENaC subunits form the channel resident in the luminal plasma membrane of epithelial cells. Consistent with this idea are findings showing that co-expression of only two out of three subunits produces little to no current in heterologous expression systems (80, 244, 362, 529).
It was previously proposed that ENaC channel might be either a tetramer with 2α:1β:1γ stoichiometry (16, 106, 122, 156, 293) or a higher ordered channel with possibly a 3α:3β:3γ stoichiometry (94, 138, 516, 529). However, as we had previously demonstrated using electrophysiology and combination of fluorescence intensity ratio analysis with total internal reflection fluorescence microscopy (528) and as was later confirmed by crystal structure of an acid sensing ion channel 1 (ASIC1) (187, 248), which belongs to the same Deg/ENaC family, ENaC, ASICs, and degenerin ion channel proteins are trimers. Interestingly, recent studies utilizing atomic force microscopy revealed that subunit combinations also produce higher-order structures containing two or three individual trimers (539). As proposed by the authors, the trimer-of-trimers organization would probably account for earlier reports that ENaC contains 8–9 subunits (539).
The structure of ENaC awaits elucidation, but much can be inferred from the known structure of chicken ASIC1 (187, 248). Fig. 7 demostrates structure of human α-subunit and a channel’s trimer predicted on the basis of the chicken ASIC1 structure (187, 248, 542). Modeling of each ENaC subunit based on the ASIC1 monomer allowed to make several predictions about ENaC secondary structure (542). ENaC subunits likely contain many of the same secondary structures and domains identified in the cASIC1 monomer. Using the nomenclature of Jasti and colleagues (248), these higher ordered structures are as follows: 1) the palm domain (yellow) containing β1, β3, β6, β9, β10, β11, and β12 strands; 2) the β-ball (orange) containing β2, β4, β5, β7, and β8 strands; 3) the finger domain (magenta and blue) containing α1–3 helices; 4) the thumb domain (green) containing α4 and α5 helices; and 5) the knuckle domain (cyan) containing α6 and α7 helices (Fig. 7A, for details see (542)). Fig. 7B shows α-, β-, and γ-hENaC (red, yellow, and blue, respectively) modeled on oligomerized cASIC1 (542). Collier and Snyder recently proposed a model in which ENaC subunits assemble in the αγβ orientation (104). Moreover, in addition to our original hypotheses predicting ENaC structure in light of the resolved ASIC1 structure (542), two recent reviews also examine functional data, including ion selectivity, gating and amiloride block and provide further details related to structural mechanisms underlying the function of this channel (83, 268).
Figure 7.

Predicted structure for human ENaC (hENaC) based on the structure for cASIC1 (248). (A) Predicted subunit structure for α-subunit of hENaC. Predicted domain organization of adjusted α-hENaC modeled on the cASIC1 A monomer (using 2QTS coordinates). Secondary structure, domain labeling and coloring follows that used by Jasti and colleagues for cASIC1 (248): transmembrane domains TM1 and TM2 and linker regions red, palm is yellow, β-ball orange, knuckle cyan, and thumb green. The exception is that the finger domain is magenta and blue. Blue highlights areas of hENaC that likely have marked differences compared to cASIC1. Putative disulfide bridges are labeled 1–7 and shown as yellow sticks. The conserved Trp87 (green side chain) at the beginning of TM1 and Tyr391 (red side chain) within the putative coupling loop are shown. Conserved Ser115 and Glu538 possibly involved in intrasubunit H-bond formation are shown with red side chains. (B) View of the ribbon structure of the predicted heterotrimeric hENaC. Adapted α- (red), β- (yellow), and γ-(blue) hENaC modeled using the 2QTS structural coordinates for the A, B, and C subunits of the cASIC1 homotrimer. Figure is adapted from (542) with permission.
2. Human diseases associated with abnormalities in ENaC
Dysfunction and aberrant regulation of this channel leads to a spectrum of diseases ranging from hyper- and hypotension, Na+ retention or wasting and respiratory syndromes linked to cystic fibrosis (55, 235, 322, 354, 457). Gain of function mutations of both β- and γ-ENaC subunits leading to channel hyperactivity (Liddle’s syndrome) are two known forms of monogenic hypertension (202, 203, 321, 322). Most forms of monogenic hypertension, including apparent mineralocorticoid excess and glucocorticoid remediable aldosteronism, result from inappropriate regulation of ENaC activity and abnormal salt-water balance (321). Moreover, Lifton and colleagues demonstrated that a point mutation in the MR that causes progesterone to be perceived as an agonist instead of the normal antagonist is associated with pre-eclampsia (via increased Na+ reabsorption through ENaC) (174). Hyposecretion of aldosterone can be mimicked by loss of function mutations in ENaC subunits, which result in decreased channel activity (91, 197) and pseudohypoaldosteronism (PHA). PHA causes hypovolemia, hyperkalemia, salt wasting and in some instances hypotension. PHA also results from loss of function mutations in the MR (650). This is supported by the finding that MR knockout mice have PHA (38). ENaC knockout mice, as well, were used to show that aberrant ENaC activity is associated with an inability to clear the fetal lungs of fluid and results in neonatal death (30, 482).
Complete knockout of each of the ENaC subunits resulted in an early and lethal phenotype (30, 234, 361). Interestingly, mice with conditional knockout of the α-subunit of ENaC in the collecting duct but not in the late DCT and CNT survived well and were able to maintain sodium and potassium balance, even when challenged by salt restriction, water deprivation, or potassium loading (463). Thus, it was hypothesized that ENaC activity has an axial gradient along the nephron with the highest activity in the CNT. However, future studies are required to confirm this hypothesis.
Considerable experimental data also support the idea that there is a coordinated interaction between ENaC and the cystic fibrosis transmembrane conductance regulator Cl− channel (CFTR) suggesting the possibility that ENaC may play some role in cystic fibrosis and other disease processes associated with the dysfunctional CFTR (37, 74, 546, 547). This assertion is consistent with data showing that overexpression of β-subunit of ENaC in lung epithelia leads to the cystic fibrosis phenotype in mice (354). However, recent results obtained in pig model of CFTR demonstrated that lack of CFTR did not increase the transepithelial Na+ or liquid absorption or reduce periciliary liquid depth (92, 245). The interaction of ENaC and CFTR will be also discussed in the section describing chloride transport. Recently, ENaC dysfunction has also been demonstrated in renal epithelial cells from animals and humans with autosomal recessive polycystic kidney disease (ARPKD), which is a renal cystic disease confined to the distal nephron and associated with improper handling of NaCl (452, 594).
3. Regulation of ENaC by aldosterone and aldosterone-induced proteins
The activity of ENaC is positively regulated by aldosterone plasma levels and correlates with dietary intake. The RAAS is one of primary systems responsible for long-term control of Na+ transport, blood volume and blood pressure. Aldosterone-sensitive ENaC activity in the ASDN is an important effector of the RAAS. Thus, ENaC is central to Na+ homeostasis and blood pressure control.
ENaC activity is also mediated by a variety of factors that either activate or inhibit the activity of existing channels or modify the number of channels at the apical plasma membrane. The rate of Na+ absorption varies widely in response to conditions of Na+ deprivation and Na+ excess. ENaC activity, similar to other ion channels, can be regulated by two fundamental mechanisms – changes in channel gating (Po) or in the number of channels at the cell surface (76, 353, 456). Diverse signaling cascades play a role in regulating ENaC cellular localization and activity. The renal distal nephron principal cells are one of the primary targets for aldosterone. Here, aldosterone increases ENaC activity and this results in increased Na+-dependent fluid reabsorption. Aldosterone is found to upregulate the expression of several targets including but not limited to serum and glucocorticoid-inducible kinase 1 (SGK1), glucocorticoid-induced leucine zipper protein GILZ, with-no-lysine (WNK) kinase, phosphatidylinositol 3-kinase (PI 3-kinase), ubiquitin-specific protease Usp2-45, small GTPase K-Ras etc (11, 13, 45, 93, 141, 147, 226, 522, 524, 525, 570, 653).
4. Ubiquitination and deubiquitination of ENaC
Several studies have demonstrated that ENaC is ubiquitinated by the action of the E3 ligase, Nedd4-2 (77, 256, 504, 535–538, 598, 636). Physiological functions and mechanisms of action of Nedd4-2 were recently thoroughly reviewed by Rotin and Kumar (458) and Rotin and Staub (460). A major player for this Nedd4-2 antagonism of ENaC activity is SGK1 that has been shown to bind to Nedd4-2 and phosphorylate it, thereby providing docking sites for 14-3-3 proteins. The association of these 14-3-3 proteins with Nedd4-2 is then thought to prevent binding of Nedd4-2 to ENaC, and thereby to cause a reduction in ENaC ubiquitination leading to accumulation of ENaC at the plasma membrane (42, 114, 160, 359, 682). Similar to SGK1, protein kinase A (PKA) (518), G protein–coupled receptor kinase 2 (GRK2) (123, 469), Akt (309), AMP-activated kinase (AMPK) (44, 200) and IKKβ kinase (44, 133) are able to phosphorylate Nedd4-2, showing that this ubiquitin ligase also functions as a site of regulatory convergence.
Interestingly, several deubiquitinating enzymes that target ENaC were also identified. It was shown that ubiquitin-specific protease Usp2-45 deubiquitylates ENaC and stimulates ENaC-mediated sodium transport, and this effect is not additive to that of SGK1 (141). Another deubiquitinating enzyme, ubiquitin C-terminal hydrolase (UCH) isoform L3, is also involved in regulation of ENaC surface density by facilitating ENaC recycling as opposed to degradation (77). Eaton et al summarized recently some mechanisms related to regulation of ENaC by ubiquitination (130).
5. Proteolytic cleavage of ENaC
A number of studies are focused on the role of proteolysis in the regulation of ENaC. Vallet et al. demonstrated that channel-activating protease (CAP1) expressed in the kidney increases the activity of ENaC (583, 584). Recent findings indicate that ENaC is activated by the proteolytic release of inhibitory peptides from the α- and γ-subunits (70, 85, 232, 502, 583, 584, 599). Proteolysis of epithelial sodium channel subunits plays a key role in modulating epithelial sodium channel activity through changes in channel open probability (233). Specific proteases have been shown to activate ENaC by cleaving channel subunits at defined sites. Furthermore, pathophysiogical, proteolytic activation of ENaC has been demonstrated in proteinuric disease (409, 552). Several recent reviews provide details of this mechanism (281, 283, 408, 553).
6. Other important signaling mechanisms that regulate ENaC-mediated sodium reabsorption
Regualtion of ENaC by the RAAS system is extremely important. However, variety of other pathways, either in parallel with aldosterone or independently, regulate ENaC activity. A continually growing body of data is revealing new mechanisms that impact ENaC, not only confirming that this channel plays a central role in the regulation of Na+ reabsorption in aldosterone target epithelia, but also supporting the notion that its function is controlled by a network encompassing a number of pathways that are controlled by autocrine, paracrine, and hormonal inputs (43, 422, 459, 598).
For instance, phosphoinositides serve as important second messengers in many intracellular signaling cascades. Disruption of phosphoinositide regulation of ion channels can lead to various disease (e.g. Bartter’s, Andersen’s and long QT syndromes) (422). It was shown that phosphoinositides play key roles in physiologic control of ENaC (347, 348, 426, 428, 429, 532). Purinergic control of apical membrane PI(4,5)P2 levels is a major regulator of ENaC activity in renal epithelial cells (423). Regulation of ENaC by vasopressin (73, 541), endothelin (72, 290, 413), insulin and insulin-like growth factor 1 (IGF-1) (50, 52, 190, 532, 567), epidermal growth factors (314, 327, 357), metabolites of arachidonic acid (414, 549, 550, 625, 626) and other important hormones and mediators are also shown. Other proteins, enzymes, kinases etc may also contribute to regulation of ENaC as well as other ion channels and transporters. For example, several small GTPases, such as K-Ras, RhoA, Rac1, Rab11 etc modulate ENaC activity (264, 266, 267, 424, 427, 480, 481, 530, 531, 533, 557), and every small G protein has its own distinct mechanism of ENaC regulation. Thus, regulation of ENaC is an elaborated complex mediating both the number of channels at the plasma membrane and gating of this channel.
IV. Potassium Transport in the CNT and CCD
K+ is the most abundant cation in the intracellular fluid that is required for many functions of the cell. Mammalian CCD and CNT along with DCT play a dominant role in K+ handling by the nephron (376). Fine-tuning of renal potassium handling in these segments regulates whether the kidney retains K+ or excretes the excess. Indeed, this is the only part of the nephron where the handling of this critical ion is strictly regulated. To secrete K+ from the extracellular fluid to the tubular lumen, K+ is transported into the cell across the basolateral membrane via the Na+/K+-ATPase pump and secreted from the cell via apical K+ channels. Renal outer medullary K+ (ROMK) channel provides one of the major pathways for K+ secretion across kidney tubule epithelia under a typical westernized diet. As discussed above, aldosterone was shown to increase ROMK activity and abundance. Aldosterone through aldosterone-induced proteins also stimulates the expression of ENaC and activates basolateral Na+/K+- ATPase, and thus increases the electrochemical driving force for K+ excretion (see Fig. 6). In comparison, plasma aldosterone is suppressed and the abundance of ROMK is reduced due to increased internalization and degradation of the channel under low-K+ diet.
However, the kidney, and the CCD and CNT in particular, express multiple potassium channels participating in K+ secretion. For instance, both principal and intercalated cells express calcium-activated big-conductance (BK) potassium channel. There is accumulating evidence that this channel not only plays a role in flow-induced K+ secretion, but also in the maintenance of K+ homeostasis. Moreover, under certain conditions, such as K+-deficient diet, when potassium is retained by the kidney, the CCD reverses the transepithelial flux of K+ to yield net potassium reabsorption. This absorption occurs through the intercalated cells and depends on the apical electroneutral H+,K+-ATPase. Briefly summarizing, under normal circumstances, principal cells in the CCD secrete K+, whereas under K+ depletion, intercalated cells reabsorb K+ (376). However, recent in vivo and in vitro data provide a more comprehensive view of potassium transport in the ASDN. Several recent extensive reviews highlight various mechanisms of K+ transport in this part of the nephron, which are partially discussed in this manuscript, in a more detailed way (195, 406, 451, 614, 632).
A. Renal Outer Medullary K+ (ROMK) channels
Small-conductance K+ (SK), inwardly rectifying ROMK channel (also known as KCNJ1 or Kir1.1) is one of the most important potassium secretory channels in the kidney (208, 376, 613, 614, 632). Understanding the renal potassium handling was significantly improved since cDNA encoding ROMK channel was isolated from rat kidney about two decades ago by Ho et al (213). In the CCD, ROMK channels mediate K+ secretion, and in the TAL these channels mediate K+ recycling that enables efficient function of the NKCC cotransporter. Mutations in KCNJ1, encoding ROMK, are associated with antenatal Bartter syndrome, which is characterized by salt wasting, hypokalemic alkalosis, hypercalciuria, and low blood pressure (81, 118, 207, 513, 605). Mutations in ROMK channel associated with Bartter’s syndrome cause alterations in PKA phosphorylation, pH sensing, channel gating, proteolytic processing, and sorting towards the apical membrane (118, 158, 159, 212, 338, 496, 497, 527). Furthermore, recent studies provide evidence that polymorphisms in the KCNJ1 gene, that encodes ROMK, show associations with blood pressure alterations and thus, might underlie monogenic hypertension and hypotension in the general population (568).
1. ROMK structure
ROMK is the pore forming protein of the 30–35 pS K+ channel. The ROMK channel as well as other members of the Kir family of inward rectifying K+ channels have a tetrameric organization (312, 663). Four ROMK subunits assemble around a central pore. So far, heteromeric assemblies of ROMK with other members of the Kir family have not been reported. Each subunit contains two transmembrane domains, a conserved potassium selectivity filter, and cytoplasmic NH2- and COOH-terminal domains (208, 632) (Figs. 8A & B). Both the NH2- and COOH-termini provide regulatory domains that can be phosphorylated by kinases and interact with protons, nucleotides, and regulatory proteins (95, 96, 142, 208, 323, 331, 363, 371, 656, 671, 672). In addition, as demonstrated by the X-ray structure for the prokaryotic KirBac1.1 (125, 301), the amino terminus of one subunit might interact with the distal carboxy terminus of the adjacent subunit forming another type of interface that could be involved in subunit assembly (208).
Figure 8.

Structure and distribution of ROMK channels. (A) Schematic presentation of ROMK structure shows two characteristic transmembrane segments (TM1 and TM2; blue and green, respectively), NH2- and COOH-termini and an extracellular domain. (B) Predicted structure of ROMK subunit stoichiometry. (C) Distribution of ROMK isoforms expression along the nephron. TAL, thick ascending limb of Henle’s loop; DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct.
2. ROMK isoforms and distribution
The ROMK channel has several alternative splicing isoforms (ROMK1-6) (54, 208, 292, 632). The products translated from mRNA of ROMK4-6 isoforms are nearly identical as the ROMK2 protein (212). N-terminal splice variants of ROMK (ROMK1, 2, and 3) are differentially expressed along the nephron and impart unique regulatory features to the channel (208). Analysis of the nucleotide sequences of the ROMK isoforms indicates that molecular diversity of ROMK transcripts is due to alternative splicing at both the 5′-coding and 3′-noncoding regions (54). ROMK2 has the shortest NH2-terminus. ROMK1 and ROMK3 have 19 and 26 amino acid extensions at the beginning of the amino terminus, respectively. ROMK isoforms are widely expressed in the kidney, and especially in the cortex and outer medulla. As demonstrated by Beesley et al ROMK2 and ROMK3 mRNA expression was significantly higher than that of ROMK1 under both basal conditions and after administration of aldosterone for a period of 1 week (34). Hebert and colleagues determined that the TAL and DCT express ROMK2 and ROMK3 transcripts whilst principal cells in the CCD express ROMK1 and ROMK2. OMCD cells express only ROMK1 transcripts (see Fig. 8C) (54, 311). Thus, ROMK1 and ROMK2 isoforms are predominant in the CCD, and all three isoforms mediate potassium transport in the CNT. Immunohistochemical analysis shows that ROMK channels are specifically localized to the apical membrane of the epithelial cells (291, 396, 655, 658). Electrophysiological studies confirmed apical localization of ROMK channels (164, 167, 323, 325, 620, 627). K+ channel activity was absent in the apical membranes of either TAL or CCD of ROMK−/− mice (342). Interestingly, despite the loss of ROMK expression, the normokalemic null mice exhibited significantly increased kaliuresis. These data indicate alternative mechanisms for K+ absorption/secretion in the kidney (342).
3. Physiological determinants of ROMK function
ROMK channel expression and function is regulated by a wide variety of factors, including intracellular pH and both extracellular and cytosolic ATP (208, 212, 614, 632). K+ secretion typically depends on the apical Na+ permeability. However, there are also proposed Na+-independent mechanisms of potassium transport (see below). A number of well-known hormones and peptides are involved in regulation of ROMK function. For instance, ANG II inhibits ROMK-mediated K+ secretion in the CCD during dietary potassium restriction (628) via AT1 receptor (254). It was further shown that ANG II inhibits ROMK1 through stimulation of the protein tyrosine kinase c-Src following either phosphorylation of ROMK or disruption of SGK1’s inhibitory effect on WNK4, both resulting in endocytosis of ROMK (677). Another antidiuretic hormone, arginine vasopressin (AVP), is a major regulator of cell cAMP activity. AVP increases the activity of ROMK channels (88) and stimulates apical membrane driving force for K+ secretion in the CCD (484). Moreover, it was demonstrated that aldosterone reduces mRNA levels and activity of ROMK channels in immortalized mouse principal cells (161). Hovewer, other groups demonstrated that aldosterone has no direct effect on the ROMK channels in isolated CCD because neither infusion of aldosterone nor low-Na diet (a maneuver that stimulates aldosterone secretion) modulates the number of the SK channels in the CCD (402, 407, 612, 624).
B. BK channel
Although ROMK channels are thought to play a major role in K+ secretion under normal dietary K+ intake, other potassium channels, BK (also called Maxi-K or slo1) channels are observed in both CCD and CNT (140, 163, 196, 236, 400, 420). A functional BK channel contains four pore-forming α-subunits that are encoded by a single Slo1 gene (75) and four auxiliary β-subunits. There are four types of β subunits (β1–4); each type displays a distinct tissue-specific expression pattern and uniquely modifies gating properties of the channel (310). Different groups identified all four β-subunits in the kidney. Thus, Grimm and Sansom using RT-PCR analysis of the whole kidney have detected transcripts for the BK-β1, β2 and β4 (192). Morton et al. also utilizing RT-PCR identified β3 and β4-subunits (372). Satlin and colleagues performed RT-PCR studies in rabbit CCD and identified β2, β3 and β4 subunits (140, 380). Immunohistochemical staining showed that BK-β1 is localized in connecting tubule cells but not intercalated cells of the CNT (192, 420). BK-β4 is expressed in the TAL, DCT, and intercalated cells (192). As it was shown, BK channels are found at higher density in the intercalated cells of both CNT and CCD (164, 380, 400, 405) than in the connecting tubule cells of the CNT and principal cells of the CCD. Interestingly, it was shown that under Na+-deficient conditions, BK-α/β1 are expressed in the basolateral membrane of the CCD principal cells, where they may increase the driving force for Na+ reabsorption (196).
Each α-subunit is comprised of seven transmembrane segments and a large intracellular COOH-terminus. Transmembrane segments S1 to S4 form the voltage sensor; S5, S6, and the intervening amino acids form the pore and selectivity filter; and the COOH terminus forms the large cytoplasmic domain that consists of approximately 800 amino acids per subunit. This domain accounts for two thirds of the entire channel (Fig. 9). Recent studies by Yuan et al provide structure of the human BK channel at 3.0 Å resolution and, thus, offer new insights into the structure of this channel (675). In contrast to ROMK channels, BK channels have a low open probability. However, the single channel conductance of the BK channel is significantly higher compared to ROMK (approximately 250 versus 30–35 pS). Moreover, under certain conditions BK activity is increased. For instance, a high K+ diet and high flow both stimulate BK channels.
Figure 9.

The structure of the pore-forming α-subunit and regulatory β-subunit of the BK channel (A). α-subunit contains 7 putative transmembrane domains, S0–S6, a conserved K+-selective pore region between S5 and S6, and a long COOH-terminal cytosolic tail. β-subunit contains two transmembrane segments and short NH2- and COOH-termini. (B) Proposed model of BK channel. Four BK α-subunits co-assemble with four BK β-subunits to form the channel heteromultimer.
Furthermore, recent data obtained with genetic manipulations of BK α-, β1- and β4-subunits revealed a role of BK channels in hypertension (195). For instance, recent experiments utilizing mice lacking the β1-subunit of the BK channel demonstrated that Kcnmb1−/− mice (60) are beset with aldosteronism that is exacerbated with a high K+ diet. The authors suggest that elevated aldosterone concentration is due to an adrenal gland that is hypersensitive to an elevated plasma K+ concentration, which is explained by reduction in BK-mediated K+ secretion in the CNT (193, 195). The study by Fernández-Fernández et al (152) provides an evidence of that a gain-of-function BK channel β1-subunit variant (G352A) exerts a protective effect against diastolic hypertension in humans. Knock out of β4-subunit, which is localized in intercalated cells, also exhibits deficient K+ excretion, fluid retention and mild hypertension (218). Deletion of the pore-forming BK channel α-subunit leads to a significant blood pressure elevation resulting from accompanied hyperaldosteronism (448, 479). Rieg et al. suggested that upregulation of ROMK may compensate for the absence of functional BK channels (448). Thus, the loss of the BK channel subunits leads to an increase in vascular tone and hypertension. However, the BK channels in the CCD and CNT might only partially mediate this effect. Tissue specific knock out of BK subunits in the CCD should provide better details about involvement of BK channels in this nephron segment and its role in development of hypertension.
C. Other K+ channels expressed in the CCD and CNT
In addition to ROMK and BK channels, it was shown that CCD and CNT express other potassium channels. However, their physiological role in these nephron segments is still unclear. For instance, a two-pore domain K+ channel (KCNK1; also called TWIK) is expressed in the apical membrane of connecting tubule cells and principal cells (101, 386). RT-PCR analysis revealed that KCNK1 is expressed in cortical TAL, CNT, and CCD (399). In the collecting duct, immunofluorescence was intracellular or confined to the apical membrane and restricted to intercalated cells, i.e., in cells lacking AQP2, as shown by double immunofluorescence (101). KCNK1 deficient mice presented impaired regulation of phosphate transport in the proximal tubule and water transport in the medullary collecting duct (386). Moreover, the voltage gated K+ ether-à-go-go-related gene (ERG) channel was identified in the apical membranes of the CNT (87). ERG K+ channels belong to the superfamily of voltage-gated K+ channels (Kv) and the best-known function of these channels is their contribution to the repolarization of the heart action potential (129). Function of ERG channels in the kidney is also unclear.
KCNQ1, also known as Kv7.1 and KvLQT1, is the pore-forming α-subunit of a delayed rectifier voltage-gated K+ channel. KCNQ1 is able to associate with regulatory KCNE β-subunits, resulting in channel complexes with different electrical and pharmacological properties. This channel was also shown in the CNT and CCD (99, 681). Similar to ENaC, KCNQ1 K+ channel is shown to be regulated by Nedd4-2 via internalization from the plasma membrane and subsequent degradation (250), this effect being mediated by AMP-activated protein kinase (14). Moreover, SGK1-3 stimulate the KCNE1/KCNQ1 channel (136, 306). KCNQ1 surface expression is also regulated by signaling cascades involving protein kinase C and phosphoinositide 3-kinase (17). Thus, this channel displays similar upstream effectors as ENaC and ROMK channels.
K+ channels in the basolateral membrane of mouse CCD principal cells were identified with patch-clamp technique, RT-PCR, and immunohistochemistry (303). In cell-attached patches, three K+ channels with conductances of ~75, 40, and 20 pS were observed, but the K+ channel with intermediate conductance (40 pS) was predominant (303). It was proposed that Kir4.1/Kir5.1 channel is a major component of the K+ conductance in the basolateral membrane of mouse CCD principal cells (303). An inward rectifier K+ channel at the basolateral membrane of the mouse DCT also revealed similarities with Kir4-Kir5.1 channels (339). Kir4.1 (KCJN10) subunits form homotetrameric channels or co-assemble with Kir5.1 (KCNJ16) in heterotetramers with distinct physiological properties (211, 212, 578). Kir4.1 localizes to the basolateral membranes of epithelia of the DCT, CNT, and ICT (246). Two recent publications idenitified Kir4.1 as a channel playing an essential role in renal electrolyte homeostasis (53, 492). They demonstrate that mutations in KCNJ10, gene encoding Kir4.1, result in seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME)/epilepsy, ataxia, sensorineural deafness, and renal tubulopathy (EAST) syndrome, a complex disorder that includes salt wasting and hypokalemic alkalosis. Mutations detected in patients render the mutant channels less active or even inactive. Renal symptoms include hypokalemia, metabolic alkalosis and hypomagnesemia with hypocalciuria (53, 492). These findings reveal an importance of this basolateral potassium channel. Interestingly, disruption in mice of Kcnj16, encoding Kir5.1, induces a severe renal phenotype that, apart from hypokalemia, is the opposite of the phenotype seen in SeSAME/EAST syndrome (412).
D. Mechanisms of regulation of potassium transport
1. Effects of diets on potassium transport
As briefly discussed above, the potassium homeostasis system is tightly regulated. Recent review by Youn and McDonough highlights progress in understanding the individual components of the K+ homeostasis system in the kidney and interaction among these components (673). When dietary K+ intake is changed, the kidneys respond by appropriate increase or decrease of K+ excretion (612, 673). Similar to sodium reabsorbtion, fine tuning of potassium excretion is also mediated by ASDN, and especially by the CNT and CCD. It is well established that high K+ intake increases K+ secretion and low K+ intake suppresses K+ secretion in principal cells. The main mechanism by which high K+ intake enhances potassium secretion is the increase of the number of functional ROMK channels in the apical plasma membrane (403, 612). Recent studies by Wade et al utilizing new ROMK antibodies confirmed that high K+ diet causes a large increase in apical expression of ROMK in late DCT, CNT and CCD (601). High potassium diet induces increase in ROMK surface expression by both stimulating the trafficking of ROMK channels to the apical membrane and suppressing the endocytosis of channels from the plasma membrane. There are several mechanisms proposed explaining how high potassium intake stimulates ROMK channels. For example, autosomal recessive hypercholesterolemia (ARH) adaptor protein, WNK, SGK, Src-family and PI3-kinases change the number of ROMK channels in the plasma membrane (95, 98, 143, 323, 672, 676, 677). More recently it was reported that microRNA (miR-802) mediated the stimulatory effect of high K+ diet on ROMK channels activity by suppressing caveolin-1 expression, which leads to increased surface expression of ROMK channels (324). Moreover, consumption of a high potassium diet for several days enhances flow (165, 218, 448) which, in turn, activates BK channels (329, 629, 646). Thus, multiple mechanisms are involved in regulation of potassium secretion by a diet. However, some details of these mechanisms, as well as involvement of other pathways, are still uncovered.
2. Flow-mediated potassium transport
The flow rate of tubular fluid was suggested as one of several factors, which may influence potassium secretion, more than forty years ago. Early micropuncture studies by Kunau et al demonstrated that, as the flow rate of distal tubular fluid was increased by acute infusion of either an isotonic saline-bicarbonate or a hypertonic mannitol solution, there was an associated increase in distal tubular potassium secretion (299). While basal K+ secretion is mostly controled by ROMK channels, flow-induced K+ secretion is mediated by BK channels (646). Both BK α- and β1-subunit knockout mice do not exhibit flow-induced K+ secretion (194, 448). Interestingly, flow also increases ENaC activity (10, 84, 86, 265, 391, 477, 581). Thus, flow upregulates both sodium absorption and potassium secretion in these segments.
Flow-mediated activation of potassium secretion via BK channels requires an elevation of intracellular calcium [Ca2+]i. Ca2+ binding by BK channels is essential for activation of this channel since Ca2+ shifts the voltage-dependent gating of the BK channels to allow activation within the physiological range of membrane potentials (60, 75). Stimulus-induced increases in [Ca2+]i are mediated by Ca2+ influx mainly through plasma membrane Ca2+ channels and/or release from intracellular stores. The flow-induced increase in [Ca2+]i in the microperfused rabbit CCD appears to be, at least in part, due to both mobilization of internal stores and external Ca2+ influx (330). Prevention of the flow-induced [Ca2+]i transient by pretreatment of cells with BAPTA-AM to chelate intracellular Ca2+ or thapsigargin or 2-APB to inhibit internal Ca2+ store release abolished flow-stimulated K secretion (328). Thus, BK channel-mediated, flow-stimulated K+ secretion requires an increase in [Ca2+]i due to luminal Ca2+ entry and Ca2+ release from the endoplasmic reticulum. The identity of the apical Ca2+ entry pathway remains to be defined. Among the candidates that might mediate this increase of [Ca2+]i are several TRP channels, including TRPV4, TRPC3, TRPC6, and TRPP2. For instance, the TRPV4 is a mechanosensitive, swell-activated cation channel that is abundant in the renal distal tubules (320, 364, 508, 648). As demonstrated by Taniguchi et al TRPV4−/− mice reveal no flow dependence of net K+ and Na+ transports on CCD (562). Although TRPV4 is a preferable candidate for flow-mediated increases in [Ca2+]i required for activation of BK channels, other channels or channel complexes can be involved in this process. For example, it was proposed that TRPP2 utilizes TRPV4 to form a mechano- and thermosensitive molecular sensor in the cilium (295).
3. ENaC-independent K+ secretion
In the CNT and the CCD, aldosterone, by increasing both the activity and number of Na+/K+ pumps, K+ and Na+ channels, leads to increased capacity and efficiency of sodium reabsorption and K+ secretion. The driving force for the passive efflux of K+ is generated largely by depolarization of the apical membrane resulting from the conductance of apical membrane Na+ channels (165, 167). Under normal conditions K+ excretion is almost entirely dependent on Na+ channel activity (165). However, recent studies by Frindt and Palmer identified a new, ENaC-independent, pathway. The authors demonstrated that under some conditions, an amiloride-insensitive pathway can become a significant, and in some cases predominant, factor in K+ elimination (165). Thus, other renal mechanisms might be involved in the control of K+ balance. The authors proposed several candidate mechanisms, including reduced K+ reabsorption by farther proximal segments, involvement of intercalated cells, and basolateral Na+ entry through Na+/H+ exchange (NHE) (165). Consistently with previous data, Muto et al. revealed that basolateral NHE in the CCD plays an important role in maintaining potassium secretion whilst diminished sodium supplies and BK channels contribute to potassium secretion (377). Furthermore, recent studies by Frindt et al. defined that under combined restriction of Na+ and K+, rats on a low-Na, low-K diet for one week did not excrete more Na+ than those on a low-Na, control-K diet for the same period of time (166).
4. WNK kinases regulate potassium transport
WNK (with no K = lysine) kinases are a recently discovered family of serine/threonine protein kinases (652). Four WNK kinases are expressed in the kidney: WNK1, kidney-specific short form of WNK1 (KS-WNK1), WNK3, and WNK4; they are most abundant along the ASDN (219). KS-WNK1 and WNK1 converge in a pathway to regulate the ROMK since dietary potassium loading increases the relative abundance of KS-WNK1 to L-WNK1 transcript and protein in the kidney, indicating that physiologic up-regulation of ROMK channel activity involves a WNK1 isoform switch and KS-WNK1-mediated release from WNK1 inhibition (600). Mutations in the genes encoding two family members, WNK1 and WNK4, cause a chloride-dependent, thiazide-sensitive inherited syndrome of hypertension and hyperkalemia (257, 548). WNKs have been established as a key regulator of the balance between NaCl absorption and K+ secretion and are involved in modulating the apical activities of both ROMK and NCC (219, 257, 548, 616). WNK4 inhibits the ROMK channel. This inhibition is independent of WNK4 kinase activity and is mediated by clathrin-dependent endocytosis of ROMK; this mechanism is distinct from those that characterize WNK4 inhibition of NCCT (257). Similar to WNK4, WNK1 is able to suppress total current directly through ROMK by causing a reduction in its surface expression by accelerating endocytosis (105). WNK1 and WNK4 interact with endocytic scaffold protein intersectin and these interactions are crucial for stimulation of endocytosis of ROMK1 by WNKs (206). It was shown that ROMK directly binds to the clathrin adaptor protein ARH, causing a rare inherited form of hypercholesterolemia. ARH protein consecutively recruits ROMK to clathrin-coated pits for constitutive and WNK1-stimuated endocytosis (143). PI 3-kinase activating hormones, such as insulin and insulin growth factor IGF-1, phosphorylate WNK1 by Akt1 and SGK1, and this phosphorylation results in inhibition of ROMK by enhancing its endocytosis from the apical plasma membrane (95). Moreover, recent studies demonstrate that WNK4 inhibits BK activity by reducing BK protein at the membrane (683). Interestingly, inhibition of the BK channel is not due to an increase in clathrin-mediated endocytosis of BK, but more likely is due to enhancing its lysosomal degradation (683).
5. Other important signaling mechanisms mediating potassium transport in the CNT and CCD
Aldosterone, dietary intake, flow stimulation and WNKs are only a part of important factors regulating K+-transport in the CNT and CCD. In addition, regulation of BK and ROMK channels by ANG II (254, 628), SGK (228, 306, 585), mitogen-activated protein (MAP) (24, 317) and Src family receptor tyrosine kinases (623, 624), arachidonic acid metabolites, such as 11,12-EET and prostaglandin E (PGE2) (253, 551), intracellular and extracellular magnesium (467, 664) and other mechanisms were shown. Thus, we are just beginning to elucidate the variety of physiological regulators that modulate the activities of K+ channels. Moreover, the functional and molecular interactions between K+ channels in the CNT and CCD are still unclear. Several excellent reviews provide more details about signaling mechanisms mediating potassium transport (195, 365, 376, 451, 612, 614, 616, 631, 632).
V. Acid-base handling in the CNT and CCD
Acid-base homeostasis is an important function of the kidney. When protons are produced during a variety of metabolic reactions, they are eventually buffered by extracellular HCO3−. As emphasized by Al-Awqati and Beauwens, the task of the kidney in regulating acid-base balance is to regenerate the HCO3− (6). This is accomplished by proton secretion into the urine, which results in generation of new HCO3−. An additional task of the kidney is the reabsorption of filtered HCO3− and this is achieved by the same process, proton secretion into the urine (6). The proximal tubule is the major segment responsible for reabsorption of filtered bicarbonate (618). The NHE3 plays a prominent role in acid-base transport in the proximal tubule (179), where metabolic acidosis acutely increases the kinetic activity of NHE3 through direct pH effects and phosphorylation (368), while chronic acidosis increases the number of NHE3 transporters (15, 179). However, the fine regulation of acid-base transport and bicarbonate levels, particularly, also occurs in the CNT and the collecting duct. In the CNT and the CCD, urinary acidification is mainly performed by the intercalated cells.
Efficient proton secretion also depends on the presence of phosphates and concomitant excretion of ammonia (NH3) and positively charged ammonium (NH4+). Approximately 60% of the generated HCO3− is the product of net ammonium excretion. The collecting duct participates in the final step of ammonia/ammonium excretion. In the collecting duct, the major site of this excretion is the OMCD (151, 285, 603). However, during metabolic acidosis, a strong increase in ammonia and ammonium transport is also found in the CNT, CCD and IMCD (285, 603).
A. Intercalated cells specificity
The intercalated cells are subdivided into three subtypes of cells (type A, type B and non-A, non-B cell types) that differ functionally and morphologically (6, 64, 274, 603, 604). Sometimes type B and non-A, non-B cell types are referenced as non-type A intercalated cells. The classic view of acid base handling is that type A intercalated cells secrete protons whereas non-type A intercalated cells are responsible for bicarbonate excretion. Type A intercalated cells secrete H+ into the urine via a V-type H+-ATPase in the apical plasma membrane and transport HCO3− in exchange for Cl− via basolateral Cl−/HCO3− exchangers including the AE1 anion exchanger. Type B intercalated cells exhibit an inverse functional polarity to that of type A intercalated cells (66). Protons are transported into interstitium across the basolateral plasma membrane via the H+-ATPase, while HCO3− is secreted into the lumen by an apical Cl−/HCO3− exchanger. Thus, intercalated cells excrete or reabsorb net H+ equivalents depending on whether the H+-ATPase is expressed on the apical or basolateral membranes. Bicarbonate is produced from CO2 and H2O catalyzed by carbonic anhydrase II (603). A third type of intercalated cells (non-A, non-B) co-express Cl−/HCO3− exchangers such as pendrin and H+-ATPase in the apical plasma membrane. The function of this type is yet to be identified. Fig. 10 illustrates these types of intercalated cells and demonstrates major transport proteins involved in acid-base homeostasis in type A, type B and non-A, non-B cell types. Type A intercalated cells are dispersed from the late DCT to the IMCD. Non-type A cells are mostly expressed in the late DCT and CNT and less in the CCD. As described recently by Wagner et al the relative abundance of type A and non-type A intercalated cells may be tightly regulated (603).
Figure 10.

Types of intercalated cells. Type A intercalated cells secrete H+ via a V-type H+-ATPase in the apical plasma membrane and transport HCO3− in exchange for Cl− via basolateral Cl−/HCO3− exchangers including the AE1 anion exchanger. Type B intercalated cells exhibit an inverse functional polarity to that of type A intercalated cells. A non-A, non-B type co-express Cl−/HCO3− exchangers such as pendrin and H+-ATPase in the apical plasma membrane. Schematic representations of the V-ATPse, pendrin and AE-2 are shown in Figs. 11 and 12.
B. H-ATPases
The vacuolar H+-ATPase (V-ATPase) is a key player in acidification of intracellular organelles and regulation of extracellular pH (61). The V-ATPase is best known for its role in acidifying various intracellular organelles e.g. endosomes, lysosomes, trans-Golgi network. However, the V-ATPase is also expressed in the plasma membranes of several specialized cells that are involved in extracellular pH regulation, such as intercalated cells (68, 180). The V-ATPase is also expressed at the cell surface and in intracellular membranes of other cell types in the nephron (67). For instance, role of the V-ATPase in regulation of bicarbonate reabsorption in the proximal tubule is shown (68, 180).
V-ATPases are composed of at least 14 different subunits that are organized into an ATP-hydrolytic domain (V1) and a proton-translocation domain (V0) that work together as a rotary machine (162). Most subunits occur in different isoforms that may be specific for different species, organs, cell types, or subcellular organelles (603). The Vo domain of the V-ATPase contains transmembrane-spanning subunits. The V1 domain subunits have no transmembrane domains but are anchored to the membrane via interaction with components of the Vo domain (68). The precise arrangement of many of the subunits in relation to one another is not entirely clear and several slightly different schemes are proposed (68, 162, 474, 635). Fig. 11 demonstrates a schematic representation of the V-ATPse structure. In brief, the V1 domain is composed of eight cytosolic subunits (some of these domains are present in multiple copies) whereas the transmembrane Vo domain contains six subunits. The V1 domain is a peripheral complex of 650 kDa responsible for ATP hydrolysis. The V0 domain is a membrane-embedded complex of 260 kDa that is involved in proton translocation across the membrane (162). The V-ATPases operate by a rotary mechanism (241), and ATP hydrolysis is required for rotation (162). The conversion between ATP, and ADP and phosphate, plays a key role in the regulation of V-ATPase.
Figure 11.
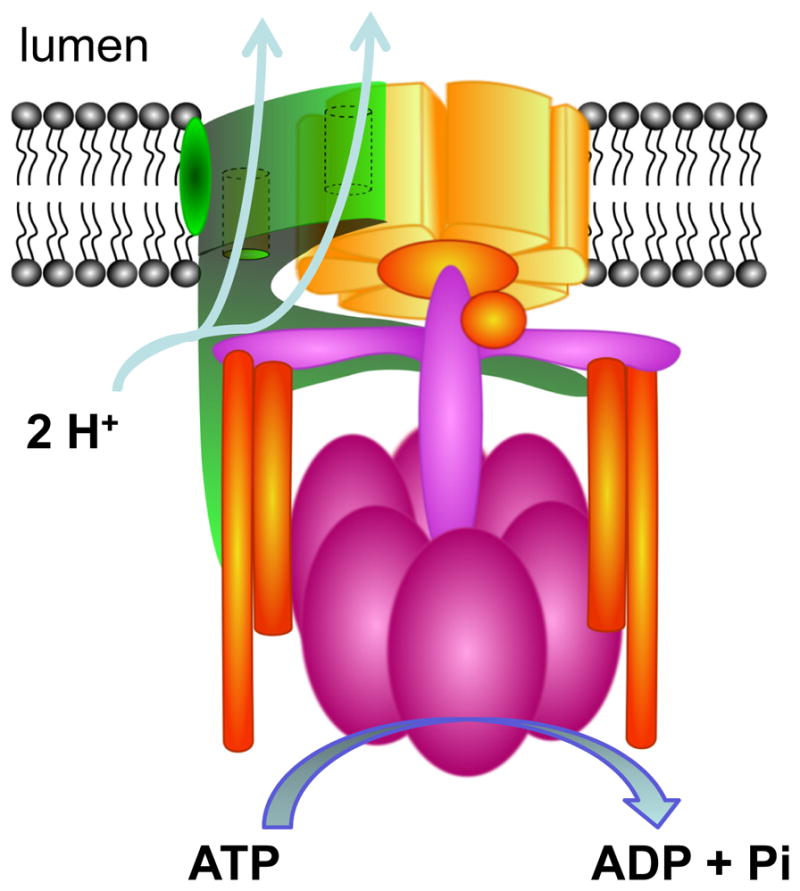
Scheme of the V-type H+-ATPase. H+-ATPases use the energy released by the hydrolysis of ATP to move protons against their concentration gradients. The V0 domain is involved in translocation of the protein. The V1 domain is involved in ATP-hydrolysis. The precise subunits composition is not entirely clear and several slightly different schemes are proposed. For details see recent excellent reviews providing details about subunits and domains of V-type H+-ATPase (68, 162, 474, 635).
Many morphological and functional studies (33, 410, 476, 498, 499, 667) showed that intercalated cells respond to acidosis or alkalosis by the regulation of V-ATPase molecules trafficking between intracellular compartment and cell surface (68). Acidosis results in V-ATPase accumulation in the apical plasma membrane of intercalated cells. In contrast, alkalosis causes recycling of V-ATPases into sub-apical vesicles (68, 465). Similarly, anion exchanger AE1 expression is elevated in metabolic acidosis and substantially reduced in metabolic alkalosis (465). Therefore, acute metabolic acidosis produces changes consistent with increased activity of type A intercalated cells and decreased activity of non-type A intercalated cells, whereas acute metabolic alkalosis produces changes corresponding to increased activity of non-type A and decreased activity of type A intercalated cells (465).
The functional importance of the V-ATPase in humans was revealed in patients harboring mutations of β1- and α4-subunits. Mutations in ATP6B1, encoding the β1-subunit of the V1 domain of V-ATPase complex, cause distal renal tubular acidosis, a condition characterized by impaired renal acid secretion resulting in metabolic acidosis, i.e. increased concentration of hydrogen ions in the blood (262, 514). Similarly, defects in the α4-subunit gene ATP6V0A4 of the Vo transmembrane pore complex cause autosomal recessive distal renal tubular acidosis (261, 544). Impairment of net proton secretion by collecting duct intercalated cells was proposed to be the leading cause of the disease. However, the exact mechanisms by which these mutations cause inhibition of the V-ATPase are still unknown (61).
C. Cl−/HCO3− exchangers
Several anion exchangers including members of the SLC4 and SLC26 transporter families are expressed in the collecting duct (124, 373, 454). However, the role of most of these transporters in the collecting duct is not known yet. Of this variety of transporters, AE1 and pendrin are the two mostly studied ones. The functions of AE1 and pendrin are also highlighted by genetic disorders in humans caused by mutations in these transporters.
The human SLC26 gene family consists of at least 11 members, with SLC26A10 likely being a pseudogene (124). Members of this family encode anion exchangers capable of transporting a wide variety of monovalent and divalent anions, including chloride, sulfate, iodide, formate, oxalate, hydroxyl ion and bicarbonate (124, 373, 521). The first identified and cloned member of the SLC26 family was a Na+-independent SO42− transporter SLC26A1 (49). The SLC26 transporters are large proteins comprised of 700–1000 amino acids, and the individual family members have 21–43 amino acid identity (124). Fig. 12A and 12B respectively demonstrate predicted topology of anion exchanger AE1 (SLC4A1) and pendrin (SLC26A4), two of the best studied members of this family, which is involved in regulation of acid-base transport in the CNT and CCD.
Figure 12.

Proposed schematics of Cl−/HCO3− exchangers. The structures of anion exchanger AE1 (SLC4A1) (A) and pendrin (SLC26A4) (B) are shown. As seen from these schemes, both NH2- and COOH-termini of AE1 are intracellular. In contrast, COOH-terminus of pendrin is extracellular.
Pendrin is an apical anion exchanger found in type B and non-A, non-B intercalated cells that is involved in bicarbonate secretion (461, 608). Perfused CCD tubules isolated from alkali-loaded SLC26A4−/− mice failed to secrete bicarbonate (461). Moreover, Cl− absorption was not observed in the CCD isolated from SLC26A4−/− mice. After moderate NaCl restriction, urinary volume and Cl− excretion were increased in pendrin knockout mice (609). These studies reveal that pendrin is critical for the maintenance of acid-base balance and in the renal conservation of Cl− and water during NaCl restriction (609). In the mouse kidney, pendrin-positive non-A, non-B cells are prevalent in the CNT, and pendrin-positive type B cells are prevalent in the CCD (274). In addition to the CNT and CCD, lower levels of pendrin mRNA are detected in the proximal tubule (520). Mutations in SLC26A4 are linked to Pendred syndrome, which is associated with deafness and goiter (108, 501). However, the physiological significance of pendrin in the human kidney is not obvious since affected individuals have no known acid-base or fluid and electrolyte abnormalities (89).
Recent studies on isolated CCD provided a novel role for Na+-driven Cl−/HCO3− exchanger (NDCBE/SLC4A8) in mediating Na+ reabsorption in the CCD and suggest a role for this transporter in the regulation of fluid homeostasis in mice. The authors proposed that the parallel action of the NDCBE and the Na+-independent Cl−/HCO3− exchanger pendrin accounted for the electroneutral thiazide-sensitive sodium and chloride reabsorption (315).
AE1 belongs to the SLC4 family of bicarbonate transporters. As noted above, AE1 is expressed at the basolateral plasma membrane and its presence characterizes type-A intercalated cells (see Fig. 12A). AE1 secretes newly generated bicarbonate into the interstitium by exchanging bicarbonate for extracellular chloride. Similar to pendrin, AE1 mediates 1:1 electroneutral exchange of Cl− and HCO3−. It was proposed that AE1 is a dimer or tetramer in the membrane (8). Current topographical model of the AE1 monomer is shown in Fig. 12A.
Mutations in the Cl−/HCO3− AE1 exchanger are associated with dominantly inherited renal tubular acidosis (69, 263, 464, 500). Primary distal renal tubular acidosis is characterized by reduced ability to acidify urine, variable hyperchloremic hypokalemic metabolic acidosis, nephrocalcinosis, and nephrolithiasis (263). However, while mutations in Cl−/HCO3− AE1 exchanger cause autosomal dominant distal renal tubular acidosis, defects in this gene are not responsible for the recessive disease (263).
D. H+,K+-ATPase
In addition to V-type H-ATPase, H+,K+-ATPase also contributes to luminal acidification and HCO3− transport in the collecting duct (199, 510, 511). H+,K+-ATPase is known to be localized to the apical membrane of intercalated cells. H+,K+-ATPases are composed of a catalytic α-subunit (110 kDa) and regulatory β-subunit (~35 kDa). The catalytic α-subunit is homologous to the other ion-motive P-type ATPases, such as the Na+,K+-ATPase and the sarcoplasmic reticulum Ca2+-ATPase (SERCA) (71). At least two H+,K+-ATPase α-subunits are expressed in the collecting duct: HKα1 and HKα2 (346). The H+,K+-ATPase α-subunits have primary structures very similar to the Na+,K+-ATPase α-subunits (>60% identity), and electron microscopy and structural analysis support that they share similar three dimensional structures (1, 2). Both the gastric (HKα1) and colonic (HKα2) H+,K+-ATPases have been identified in the kidney and are known to be highly regulated in response to acid-base and electrolyte disturbances (102). The catalytic subunit HKα1 appears to be restricted to the gastric parietal cells and the renal collecting duct. The nongastric colonic (HKα2) H+,K+-ATPase has a much wider tissue distribution. HKα2 mRNA and protein are expressed at low levels in the renal medulla but abundantly in the distal colon (110, 128). HKα1 and HKα2 H+,K+-ATPases are distinct pharmacologically. For instance, the HKα1 H+,K+-ATPase is inhibited by Sch-28080 (337), but the HKα2 H+,K+-ATPase is inhibited by ouabain (a classic inhibitor of the Na+-pump) and is completely insensitive to Sch-28080 (102).
H+,K+-ATPase mRNA and protein expression are increased with dietary K+ depletion (5, 128, 199, 358, 382). Moreover, intercalated cells from the NaCl-restricted rats have an increased rate of H+/K+ exchange compared to controls (509). Lynch et al. using mice lacking expression of HKα1, HKα2, or both H+,K+-ATPases (HKα1,2) demonstrated that HKα1 and HKα2 contribute to H+ secretion in both A-type and B-type intercalated cells in animals fed a normal diet (346). Recent studies from the same laboratory revealed that deoxycorticosterone pivalate (DOCP)-treated HKα1,2−/− mice exhibit a lower level of HCO3− in blood and less Na+ and K+ retention than either wild type or HKα1−/− mice (191). Thus, H+,K+-ATPases play an important role in the effects of mineralocorticoids on acid-base and Na+/K+ balance. It was also recently shown that CNT and CCD isolated from tissue kallikrein deficient mice exhibit net transepithelial K+ absorption because of an abnormal activation of the HKα2 H+,K+-ATPase in intercalated cells. In CCD isolated from tissue kallikrein deficient mice and microperfused in vitro, the addition of tissue kallikrein to the perfusate caused a 70% inhibition of H+,K+-ATPase activity (134). The upregulation of the HKα1 H+-K+-ATPase in the kidneys of NHE-3 knock-out mice also suggests that this H+-K+-ATPase contributes to the enhanced compensatory response in NHE-3-deficient animals (381). Thus, the collecting duct of the nephron is the primary renal location for the H+,K+-ATPases and these transporters play an important role in maintaining of K+ and acid-base homeostasis in the kidney.
E. Regulation of acid base homeostasis in the CNT and CCD
Aldosterone may indirectly stimulate proton secretion by enhancing Na+ reabsorption in the CNT and CCD via upregulation of ENaC activity, thus increasing the lumen negative voltage potential. Similarly, K+ depletion in response to mineralocorticoids stimulation may result in increased H+ secretion. Wagner and colleagues used several genetic mouse models to identify the exact region where ENaC-mediated electrogenic Na+ reabsorption promotes proton secretion by creating a more lumen-negative voltage (297). In mice with a kidney-specific inactivation of α-subunit of ENaC in the CD, but not in the CNT, furosemide induced normal H+-ATPases-mediated urinary acidification. Loss of ENaC channels in the CD does not affect this acidification. Thus, the authors concluded that functional expression of ENaC channels in the CNT is sufficient for furosemide-stimulated urinary acidification and identifies the CNT as a major segment in electrogenic urinary acidification (297).
Moreover, aldosterone directly stimulates H+ secretion in the CNT and collecting duct by increasing the activity of the H+-ATPases and Cl−/HCO3− exchangers. For instance, the acidification defect in a model of selective aldosterone deficiency with hyperkalemia and hyperchloremic metabolic acidosis appears to be at least partially a result of an impaired transfer from the loop of Henle to collecting duct and reduction in the rate of H+ secretion by the collecting duct (127). Similarly aldosterone increases H+ secretion in the CNT and the CCD in adrenalectomized rabbits by increasing the activity of H+-ATPase in these segments (172). Interestingly, non-genomic effect of aldosterone on the V-ATPase was also shown. Short-term (15 min) application of aldosterone increases V-ATPase activity 2- to 3-fold. Neither inhibition of mineralocorticoid receptors nor of transcription and protein synthesis prevented aldosterone-induced stimulation of the V-ATPase (645).
F. A role of acidosis in regulation of other channels expressed in the CNT and CCD
The metabolic acidosis that results from the renal tubular acidosis may be caused by insufficient secretion of H+ ions by intercalated cells. During the metabolic acidosis the urinary pH becomes acidic to compensate for the decreased systemic pH (356). Acid-base abnormalities have significant effects on potassium transport in the CNT and the CCD. Metabolic acidosis increases extracellular K+ concentration, whereas metabolic alkalosis decreases extracellular fluid K+ concentration. Wang et al. demonstrated two decades ago that changes in intracellular pH could be a signal for cells to regulate K+ secretion and this effect is mediated by both Na+/K+ pump and the apical K+ channels (617). Changing bath pH facing cytoplasmic side of inside-out patches from 7.4 to 6.9 reversibly reduced channel open probability of K+ channels at the apical plasma membrane of the CCD (615). Schlatter et al confirmed that cellular acidification in rat CCD principal cells down-regulates K+ conductances and thus reduces K+ secretion by direct inhibition of K+ channel activity (489).
Moreover, several lines of evidence indicate that transient receptor potential vanilloid 5 (TRPV5) channel, constituting the apical entry gate in the CNT, is sensitive to pH. It was shown that acid-based homeostasis affects renal Ca2+ handling as reflected by altered Ca2+ excretion in the kidneys during chronic acidosis or alkalosis (305, 392). Intra- and extracellular pH directly regulate the activity of TRPV5. Acidification inhibited, whereas alkalinization stimulated, TRPV5 activity. These effects are likely mediated by conformational changes of the channel pore helix (113, 595, 668–670). Upon extracellular alkalinization, a pool of TRPV5-containing vesicles is recruited to the cell surface and this recruitment is accompanied by increased TRPV5 activity. Conversely, upon subsequent extracellular acidification, vesicles are retrieved from the plasma membrane, simultaneously resulting in decreased TRPV5 activity (305). Furthermore, it was reported that polycystin-2, the product of the human PKD2 gene (member of TRPP family of ion channels) can be regulated by pH (188).
VI. Vasopressin-regulated water and urea transport in the CNT and CCD
Water reabsorption in the collecting duct is the key event for maintenance of body water balance. The production of the concentrated urine requires high water permeability in that nephron segment, allowing for the osmotically driven movement of water from the lumen to the interstitium (149). Though the CCD is normally relatively impermeable to water, it becomes highly permeable in the presence of the ADH, also known as arginine vasopressin (AVP). The physiological action of vasopressin in collecting duct has always been one of the most studied processes in the kidney. As shown in pioneering perfusion studies in isolated collecting ducts, addition of vasopressin caused reversible trafficking of AQP2 water channel from the intracellular pool to the apical membrane, and this process occurred in association with a fivefold increase in water permeability (387). Briefly, vasopressin increases the water reabsorption by binding to the basolateral V2 receptor (V2R). This binding results in activation of adenylate cyclase (AC) by the V2R-coupled α-subunit of Gs heteromeric G protein. This transiently increases intracellular cAMP levels and leads to activation of multiple mechanisms, including but not limited to activation of PKA and subsequent phosphorylation of AQP2. Thus, V2R-mediated signaling stimulates insertion of AQP2 channels into the apical plasma membrane and regulates AQP2 gene expression (389). Moreover, in addition to its direct regulation of water transport, AVP also regulates in the collecting duct sodium and urea transport via ENaC and UT-A1 transporter, respectively (471, 541, 619).
In humans and animals, dietary protein intake highly exceeds that necessary for the support of anabolic processes. Thus, surplus generated urea needs to be excreted. The process of urea accumulation is mostly dependent on facilitated urea transport across the epithelium of the IMCD (149, 286). This urea transport process and its regulation have been reviewed extensively (149, 150, 286, 470, 472) and recent updates are discussed in the corresponding overview article (151).
Interestingly, one of the major differences between the CNT and the CCD tubule segments is the lower CNT water permeability. In early CNT the luminal solution is hypotonic, with a K+ concentration less than that of plasma. It is predicted that osmotic equilibration requires the whole length of CNT, to end with a nearly isotonic fluid, whose K+ concentration is several fold greater than plasma (630).
A. Water transport in the CNT and CCD (aquaporins)
Eight types of aquaporin proteins are localized to various segments of the renal tubule of the kidney (186). Knocking out genes and identification of mutations in these proteins relevant for the development of various diseases provided important information on the role of AQPs in normal and pathological physiology (56, 395). AQP2 in the apical and combination of AQP3 and AQP4 in the basolateral membranes, respectively, are responsible for water transport in the collecting duct. AQP2 is abundantly expressed in the cells of all segments beyond the DCT, including connecting tubule cells, principal cells of the CCD and OMCD, and IMCD cells (149). Apart from the apical plasma membrane, AQP2 is found in the intracellular vesicles and in the basolateral plasma membrane (especially in the IMCD) (149, 389). AQP3 and 4 are localized to the basolateral membrane of principal cells and are absent in intercalated cells (131, 242). AQP4 is localized mostly to the basolateral plasma membrane of the OMCD and is more abundant in the IMCD cells than AQP3. Apparently, expression profiles of water channels in the kidney collecting duct are interconnected. For example, AQP4 and AQP2 are virtually absent from the CNT and CCD in AQP3 null mice (276).
One of interesting features of AQP3 is that this channel is not only permeable to water but also efficiently functions as a glycerol transporter (149). Yang and Verkman determined single channel permeabilities for AQP1-AQP5 channels. Surprisingly, they reported that significant glycerol permeability was found only for AQP3 (349, 661). Thus, AQP3 is an aquaglyceroporin protein as several other transport proteins (204, 433, 679).
cDNA for AQP2 was isolated from the collecting duct almost two decades ago (170). Since then, significant progress in understanding of mechanisms regulating AQP2 was made. AQP2 is expressed at the plasma membrane as a homotetramer and each subunit participates in forming the pore. Structurally, AQPs have six transmembrane domains with the NH2- and COOH-termini exposed to the cytoplasm (243) (see Fig. 13A). The activated V2R induces an increase of intracellular cAMP levels, which leads to activation of PKA and to phosphorylation of AQP2 channels (171, 269, 369, 450) (Fig. 13B). Protein phosphorylation and dephosphorylation are common dynamic posttranslational processes often involved in regulation of protein function and cellular distribution (369). The role of phosphorylation and dephosphorylation in the regulation of AQP2 has been a major focus of several studies and likely it is a key event in the modulation of AQP2 (369, 370, 559, 651). It appears that AQP2 phosphorylation by PKA is one of the main signals triggering AQP2 exocytosis. Phosphorylation of at least three of four monomers of the AQP2 tetramer is sufficient to redistribute AQP2 homotetramers from the storage vesicles to the apical membrane, rendering this membrane permeable to water (260, 450, 485, 586).
Figure 13.

Regulation of the aquaporin-2 (AQP2)-mediated water transport by arginine vasopressin (AVP). (A) Proposed topology of AQP2. An AQP2 monomer consists of six transmembrane domains connected by five loops. NH2- and COOH-termini are located intracellularly. (B) A scheme of water transport regulation by AVP. Vasopressin receptor (V2R), stimulatory GTP-binding protein (Gs), adenylate cyclase (AC), adenosine triphosphate (ATP), and cyclic adenosine monophosphate (cAMP) are indicated.
In addition to phosphorylation status, vasopressin-modulated AQP2 trafficking and expression can be modulated by several hormones. For instance, some of these hormones, such as ATP/UTP, dopamine and endothelin, are shown to counteract the action of vasopressin by reducing the expression of AQP2 at the apical plasma membrane (57, 280, 290, 319, 379). In contrast, ANG II enhanced vasopressin-modulated AQP2 trafficking and expression (316).
B. Pathologies associated with water transport impairments
The majority of the X-linked and most prominent form of congenital nephrogenic diabetes insipidus (NDI) is caused by loss-of-function mutations in the AVPR2 gene, encoding the V2R (336). Over 180 AVPR2 gene mutations have been described so far, of which many result in severe interference with receptor signaling, thus making the principal cells of the CCD insensitive to AVP. However, the molecular mechanism underlying this insensitivity differs among mutants (450).
There are, however, as many as 10% of cases of congenital NDI which are related to mutations in the AQP2 gene. As a result of NDI, the kidney loses its ability to concentrate urine, which may lead to severe dehydration and electrolyte imbalance. In contrast to the X-linked disease, the NDI caused by water channel mutations can be autosomal recessive or dominant (494). In contrast to the rare hereditary causes of NDI, acquired NDI is quite common. Many mutations in the AQP2 gene responsible for both autosomal recessive and dominant NDI have been identified so far (115, 308, 450). Most mutations in autosomal recessive NDI are found between the first and the last transmembrane domain (450) since this segment forms the AQP2 water pore (375, 485). The complete AQP2 knock-out mice developed recessive NDI and died postnatally (453). Transgenic mice expressing AQP2 selectively in the CNT but lacking AQP2 expression in the collecting duct were viable to adulthood, although demonstrated some abnormalities (453). Thus, AQP2 in CNT is sufficient for postnatal survival and AQP2 in collecting duct is essential for the regulation of body water balance and cannot be compensated by other mechanisms. This is most prominent taking into consideration that several generated mutant AQP2 knock-in mice showed a severely impaired urine-concentrating ability or failed to thrive and died within a couple of weeks after the birth (332, 360, 505, 519, 660, 662). Interestingly, it was recently demonstrated using mice lacking α-ENaC in the CD, that the absence of functional ENaC in the CD protects mice from lithium-induced NDI. Thus, the authors hypothesize that ENaC-mediated lithium entry into the CD principal cells contributes to the pathogenesis of lithium-induced NDI (97).
Moreover, AQP2 has a critical role in other pathologies associated with such water balance disorders as congestive heart failure (CHF) (345, 654). Water retention and hyponatremia are common and clinically important complications of CHF (390, 395, 493). The increase in AQP2 expression and enhanced plasma membrane targeting provide an explanation for the development of water retention and hyponatremia in severe CHF (390). These results indicate a major role for vasopressin-mediated upregulation of AQP2 water channels and water retention in CHF (390, 654).
VII. Calcium homeostasis in the CNT and CCD
Calcium (Ca2+) is the most abundant cation in the human body wherein the largest reservoir for Ca2+ is the bone (107). Under normal conditions, extracellular fluid calcium concentration in the kidney remains tightly controlled, but when it decreases the bone acts as a large reservoir and source of calcium. Though intracellular divalent cation concentration is low, calcium and magnesium ions play a fundamental role in many cellular processes. It is essential to maintain plasma Ca2+ and Mg2+ concentrations within a tight physiological range. The cytosolic free Ca2+ concentration is strictly controlled in the CNT and CCD cells as well as in other nephron segments and cell types. Active Mg2+ transport is confined to the DCT, whereas active Ca2+ reabsorption mainly occurs along the late DCT and CNT segments of the nephron (47). Magnesium homeostasis and pathophysiology of renal diseases connected to disturbances in this transport are covered by other insightful review articles (121, 154, 178, 434).
Calcium reabsorption in the DCT and CNT takes place against its chemical gradient, indicating that the transport is active (215, 441). In general, Transient Receptor Potential (TRP) channels mediate calcium influx across the apical plasma membrane. Intracellular carrier protein calbindin-D28K sequesters Ca2+ that is entering the cell through TRP channels, and the complex which is formed then diffuses towards the basolateral membrane where transporter proteins, such as Na+/Ca2+ exchanger (NCX1) and the plasma membrane ATPase type 1b (PMCA1b) extrude Ca2+ from the epithelial cells (47, 215). This active transcellular transport is regulated by an array of events, and mediated by hormones, including 1,25-dihydroxyvitamin D3, parathyroid hormone (PTH), and estrogen (58).
A. Transient Receptor Potential (TRP) superfamily
TRP superfamily consists of various cation-permeable channels involved in ion homeostasis and/or signal transduction (121, 215, 647, 649). TRP channels have been identified and are grouped into six subfamilies on the basis of amino acid sequence homology: TRPC (canonical), TRPM (melastatin), TRPV (vanilloid), TRPA (ankyrin), TRPML (mucolipin), and TRPP (or PKD) (polycystin) (100, 649). Many TRP channels are expressed in the kidney along different parts of nephron including the CNT and CCD. Table 1 summarizes members of this superfamily identified in the CNT and CCD and briefly highlights their functions in these segments.
Table 1.
Transient receptor potential (TRP) channels expressed in the CNT and the CCD.
| Transport protein | Expression/Function | References |
|---|---|---|
| TRPC3 | Principal cells of CCD/Ca2+ signaling | (182) |
| TRPC6 | Principal cells of CCD/Ca2+ signaling | (182) |
| TRPP2 (PKD2 or TRPP1) | Principal cells of CCD/Mechanosensing (in complex with PKD1); Ca2+ signaling | (638) |
| TRPV4 | CNT/osmotically and flow regulated cation channel | (562, 566, 648) |
| TRPV5 | CNT/calcium homeostasis; contribute to chronic hypercalciuria | (214, 216, 333, 335) |
| TRPV6 | CNT and CCD/calcium homeostasis | (393, 587, 588) |
The TRP channel family members share similar channel structure. Fig. 14A provides an example of TRP channel structure using the example of the TRPC channels. Typically, these channels contain six transmembrane domains, intracellular NH2- and COOH-terminal tails and a predicted pore region between transmembrane domains 5 and 6. TRP channels are mostly permeable for calcium. However, these channels are not only selective for Ca2+ and could under certain conditions conduct Na+ and K+ ions. Both NH2- and COOH-termini contain a variety of important domains, such as ankyrin binding, caveolin binding, coiled-coil, PDZ domains, as well as binding and phosphorylation sites, which interact with adaptor proteins and enzymes to modulate channel’s activity (121, 132, 176, 647). However, some of the TRP channels structurally deviate from this model. For instance, polycystin-1 (PKD1; also known as TRPP1) is a large membrane protein with a long, modular extracellular NH2-terminal component capable of protein-protein interactions, 11 transmembrane domains, and a short intracellular COOH-terminal domain (231) (Fig. 14B). However, since PKD1 and related proteins show very limited sequence similarity with TRP channels, many researchers do not consider these proteins to be members of the TRP superfamily.
Figure 14.

Transmembrane topology of TRP channels. TRP channels belong to the large superfamily of cation channels with six transmembrane-spanning segments forming a transmembrane domain with a pore loop inserted between TM5 and TM6 and NH2- and COOH-intracellular termini (A). In contrast, polycystin 1 (PKD1 or PC1) has eleven transmembrane domains and a large extracellular NH2 domain (B).
TRP channels require the assembly of four subunits to form a common central pore. Most mammalian TRP channels function as homomers. However, TRP channel subunits may assemble into heteromeric tetramers. Heteromeric channel assembly may give rise to additional diversity of resulting cation conductances in terms of the regulatory and biophysical properties (483, 534). Several protein–protein interactions have been reported previously among TRP channels in the kidney, most of them are known to occur between members of the same group. For instance, TRPP1 and TRPP2 assemble into a polycystin complex (175, 176, 201, 385). Moreover, polycystins interact with TRP channel subunits TRPC1 and TRPV4 (295, 575). It was shown that TRPP2 and TRPV4 form a mechanical and thermal sensor in cilia and it was suggested that TRPV4 is the mechanically sensitive component of the complex (295). The TRPP2/TRPC1 heteromeric channel is activated in response to G protein-coupled receptor (GPCR) activation and shows a pattern of single-channel conductance, amiloride sensitivity and ion permeability distinct from that of TRPP2 or TRPC1 alone (25). In the TRPV family, TRPV5 and TRPV6 combine into heterotetramers with properties that are distinct from homomeric channels (217). However, most of these studies were performed in heterologous expression systems and the precise molecular organization of TRP channels within native epithelia remains to be clarified.
Discovery of the TRP family and consequent identification and characterization of the proteins mediating Ca2+ transport in the kidney greatly increased our understanding of the molecular and cellular processes involved in Ca2+ and Mg2+ homeostasis. The readers may refer to previous excellent reviews that describe properties and physiological and pathophysiological mechanisms mediating TRP channels in the kidney (3, 47, 58, 121, 576, 647).
B. TRPC channels
TRPC channels form Ca2+-permeable cation channels and there is currently no doubt about their involvement in Ca2+ influx in various regions of the kidney. It is also becoming evident that TRPC channels are important players in the pathogenesis of renal and cardiovascular diseases (3, 279, 622, 647). For instance, several mutations associated with FSGS were identified in TRPC6 channel (209, 442, 644). However, little is known about the exact role of TRPC channels in the collecting duct.
Both TRPC3 and TRPC6 are expressed in principle cells of the collecting duct. TRPC3 and TRPC6 are shown to colocalize with AQP2, but not with the Na+/Ca2+ exchanger or peanut lectin. In polarized cultures of M-1 and IMCD-3 collecting duct cells, TRPC3 was localized exclusively in the apical plasma membrane, whereas TRPC6 was found in both the basolateral and apical membranes (182). Studies in polarized MDCK cells revealed endogenous expression of TRPC3 at the apical plasma membrane and TRPC6 in both locations (27). Interestingly, the authors also identified TRPC1 at the basolateral membrane (27).
It was proposed that TRPC3 plays a major role in the transepithelial Ca2+ flux in principal cells of the renal collecting duct (183) and trafficking of this channel is mediated by vasopressin. Immunofluorescence analysis revealed that TRPC3, but not TRPC6, colocalized with AQP2 in intracellular vesicles and vasopressin causes the insertion and accumulation of TRPC3 and AQP2 in the apical membrane but has no effect on the subcellular distribution of TRPC6 (183). AVP-induced translocation of TRPC3 channel to the apical plasma membrane is modulated by the inhibition or activation of the AC/cAMP/PKA signaling cascade that is required for vasopressin-stimulated activation of AQP2 (see Fig. 13B) (184).
C. TRPV channels
TRPV channels are Ca2+ permeable channels activated by physical and chemical stimuli. TRPV5 channel is expressed at the apical membrane of the DCT and CNT cells. It was shown that this channel can contribute to chronic hypercalciuria. Genetic ablation of TRPV5 in the mouse demonstrated that mice lacking TRPV5 display diminished active Ca2+ reabsorption despite enhanced vitamin D levels, that causing severe hypercalciuria (216, 444, 445). Besides hypercalciuria, the TRPV5 knockout mice demonstrate bone degradation, decreased bone mineral density, polyuria and increased urinary acidification. Regarding the crucial importance of TRPV5 in body calcium homeostasis and, furthermore, the renal calcium wasting observed in the knockout mice, TRPV5 is an important candidate gene for renal hypercalciuria (444). Genotyping of TRPV5 in renal hypercalciuria patients revealed three non-synonymous and five synonymous polymorphisms. However, electrophysiological characterization of the TRPV5 mutants did not reveal significant functional changes compared to wild-type TRPV5 channel recordings (444). Critical role of the TRPV5 channel in active Ca2+ reabsorption was also revealed by double-knockout mice of TRPV5 and calbindin-D28K (177). The authors proposed that TRPV5 represent the rate-limiting step in active Ca2+ reabsorption, unlike calbindin-D28K, which possibly is compensated by calbindin-D9K (177).
TRPV6 is an epithelial Ca2+ channel that is highly homologues to TRPV5 (587, 588). In contrast to TRPV5, distribution of TRPV6 extends to the CCD, where it is localized to the apical domain of principal and intercalated cells, which are not generally implicated in active Ca2+ reabsorption (393). In DCT2 and CNT, TRPV6 co-localizes with other known Ca2+ transport proteins, including TRPV5 and calbindin-D28K. Thus, these data suggest a role for TRPV6 in 1,25(OH)2D3-stimulated Ca2+ reabsorption in these segments (393). It was reported that a significant increase in TRPV6 and calbindin-D28K, but not TRPV5, was observed in Wnk4D561A/+ knock-in mice, a model of human PHA type II (665).
Another important protein that participates in regulation of TRPV channels is Klotho, an anti-aging protein (227, 302). Reduced production of this protein has been observed in patients with renal failure. Acute kidney injury (AKI) and chronic kidney disease (CKD) are states of systemic Klotho deficiency, making Klotho a very sensitive biomarker of impaired renal function (224, 225). Klotho hydrolyzes extracellular sugar residues on TRPV5, entrapping the channel in the plasma membrane. This maintains channel activity and membrane calcium permeability in kidney (90). In the kidney, klotho KO mice exhibited increased expression of TRPV5 and decreased expression of the calbindin-D28K, implying a failure to absorb Ca2+ through the DCT/CNT via TRPV5 (7). Interestingly, effect of Klotho is specific since it was reported that this enzyme significantly increases the activity of TRPV5 and TRPV6, but had no effect on TRPV4 and TRPM6 (343).
TRPV4, a nonselective cation channel, is gated by hypotonicity (562, 566, 648). This channel is abundant in the renal distal tubules and is expressed in the tALH, TAL, DCT and CNT (566). TRPV4 displays a mechanosensitive nature with activation properties consistent with a molecular sensor of both fluid flow and osmolality, or a component of a sensor complex, in flow-sensitive renal CCD (648). As it was discussed above (part of the review related to the potassium secretion), TRPV4 most likely mediates flow stimulated calcium flux required for activation of BK cahnnels (329, 330, 355, 562). Recent studies in freshly isolated split-opened CNT and CCD demonstrated that ATP uniformly increases intracellular Ca2+ in a PLC-sensitive manner and TRPV4 having a major role in this sustained calcium rise (355). However, TRPC3 and/or TRPC6 might also be involved in this mechanism.
D. TRPP channels
TRPP family of TRP channels is divided structuraly and functionally into two subfamilies: putative 11 transmembrane domains polycystin 1 (PKD1; also called PC1) and 6 transmembrane domains polycystin 2 subfamily (PKD2 or PC2) (see Fig. 14). Both PKD1 and PKD2 subfamilies contain several homologous proteins. PKD2-like subfamily contains three proteins, PKD2, PKD2L1 and PKD2L2, which are now referred to as TRPP1, TRPP2 and TRPP3, respectively (649).
It is generally accepted that PKD1 and PKD2 are interacting partners within a signaling pathway. It was initially demonstrated for Caenorhabditis elegans, that lov-1 and PKD-2, the C. elegans homologues of PKD1 and PKD2, respectively, function in the same pathway (31, 32). Later it was shown that these proteins assemble to form a functional complex (201). Co-expression of PKD1 and PKD2 in CHO cells generated time-independent, slightly outwardly rectifying currents. In contrast, whole-cell currents from cells expressing either PKD1 or PKD2 alone were indistinguishable from those observed in Mock-transfected cells (201). These data suggest that PKD1 is required both to bring PKD2 to the plasma membrane and to co-assemble a polycystin complex, enabling polycystin-2 to function as an ion channel (201). Another study suggested that polycystin-2 is highly expressed in the endoplasmic reticulum of epithelial cells and functions as a calcium release channel (296). Interestingly, several studies indicate that polycystin-2 can form an ion channel by itself (189, 344, 593).
The inherited polycystic kidney diseases (PKD), which are caused by mutations in single genes, include autosomal dominant and autosomal recessive polycystic kidney disease (638). In PKD cysts originate as expansions of the renal tubule. Defects in both PKD1 and PKD2 are a major cause of autosomal dominant polycystic kidney disease (ADPKD), whose manifestation entails the development of fluid-filled cysts in the kidney. All typical cases of autosomal recessive polycystic kidney disease (ARPKD) are linked to PKHD1, gene encoding fibrocystin (39, 398). Only brief overview of TRPP channels and their role in the collecting duct is provided here since the pathophysiology of polycystic kidney disease and mechanisms of regulation of these important proteins have been covered by numerous excellent review articles (205, 411, 571–574, 638, 640) and will be also discussed in a separate overview article.
As seen in Fig. 15A, in ADPKD, cystic outpushings arise in every tubule segment and rapidly close off from the nephron of origin. By contrast, in ARPKD, cysts are derived from collecting duct tubules, which remain connected to the nephron of origin (638). Cystic epithelial cells are markedly different from normal principal cells of the CCD. Fig. 15B and Fig. 15C demonstrate normal cortex section of the Sprague-Dawley rat and examples of cysts in the PCK rat, an orthologous model of ARPKD (51, 270, 304, 569, 680), respectively.
Figure 15.

Mutations in genes encoding proteins functionally expressed in CCD cause severe kidney disorders. (A) Mechanisms of Cyst Formation in autosomal dominant (ADPKD) and recessive (ARPKD) polycystic kidney diseases. Normal tubule is also shown. Representative images of kidney cortical sections of Sprague-Dawley (B) and PCK (C) rats. PCK rat, a model of ARPKD, demonstrates abundant formation of cysts. Original magnifications are 40x. Scale bar is presented.
Briefly summarizing, there are several TRP channels expressed in the CNT and CCD, which mediate distinct functions and regulated by variety of mechanisms. Moreover, these proteins regulate two processes. First, active transcellular calcium reabsorption is mediated in the CNT and is critical in the maintenance of the Ca2+ balance. Second, maintaining of intracellular calcium is extremely important and even small changes trigger different signaling processes. The main question not only for these segments of the nephron, but also for other cell types is how all of these TRP channels communicate with each other in order to facilitate optimal Ca2+ balance.
VIII. Cl− transport
Cl− reabsorption involves both paracellular and transcellular pathways. For a long time Cl− channels have been considered little more than a background conductance that passively follows cation transport (155). The mechanisms by which Cl− moves across the CNT and CCD still remain defined incompletely. Within the CCD, transepithelial transport of Cl− occurs primarily across intercalated rather than principal cells (488). Cl−/HCO3− exchangers AE1 and pendrin seems to play an important role in Cl− transport in the intercalated cells of the CNT and CCD. In addition, role for ClC-K channels in Cl− transport in the CNT and CCD is also shown (298, 580). Moreover, epithelial cells in these segments regulate Cl− secretion by modulating CFTR Cl−-channel activity through several signaling mechanisms, including those involving phosphorylation and regulation of the CFTR trafficking (40, 198, 255).
Chloride was also shown to be an inhibitor of ENaC activity (103, 104). Extracellular Cl− inhibited ENaC and the response to Cl− was modulated by changes in extracellular pH; acidic pH increased and alkaline pH reduced ENaC inhibition by Cl− (103). Cl− regulated ENaC activity in part through enhanced Na+ self-inhibition, a process by which extracellular Na+ inhibits ENaC. Thus, increase in Cl− in intercalated cells might provide a potential mechanism to modulate the epithelial Na+ absorption by changes in extracellular Cl−.
A. Role of Cl−/HCO3− exchangers in regulation of Cl− transport in the CNT and CCD
The Cl−/HCO3− exchanger pendrin (Fig. 12B) that was described above in the acid-base homeostasis section also was shown to mediate Cl− absorption. Thus, Cl− reabsorption via pendrin is linked molecularly to bicarbonate secretion. Mouse models of Pendred syndrome (Slc26a4−/−), during either NaCl restriction or after the administration of deoxycorticosterone pivalate (DOCP) and a high NaCl diet, develop lower blood pressure and greater Cl− excretion than wild type mice (597, 609). Aldosterone enhances absorption of both Na+ and Cl− through increased protein abundance and elevated function of ENaC and such Cl− transporters as pendrin. It is quite interesting that although pendrin and ENaC are localized in different cell types, ENaC protein abundance and function are among other regulatory pathways modulated through a pendrin-dependent mechanism (277). Pendrin knockout mice have lower blood pressure than wild type and show the reduced ENaC expression and function that contribute to this lower blood pressure and reduce ability to conserve Na+ during NaCl restriction (277). Another study revealed that pendrin modulates ENaC, at least in part, by increasing luminal HCO3− and/or pH (415). In contrast, Vallet et al demonstrated that major changes in pendrin protein expression were found only in experimental models that are associated with altered renal chloride transport, whereas no significant changes were detected in pendrin protein abundance in models with altered aldosterone secretion (582). Angiotensin II, another important peptide playing a critical role in the fluid and electrolyte balance and, consequently, in the regulation of systemic blood pressure, also increases transcellular Cl− absorption in the CCD through a pendrin-dependent process (416).
Similarly, Cl− also enters across the basolateral membrane of type A intercalated cells via another Cl−/HCO3− exchanger, AE1 (see Fig. 12A). This exchanger plays an important role in maintaining acid/base balance and participates in Cl− transport as well. However, the mechanisms that administer Cl− movement through AE1 in the CNT and CCD require further studies.
B. ClC Cl− channels in the CNT and CCD
The ClC proteins are members of a large family of chloride transport proteins, which are involved in a variety of physiological processes (249, 298, 580). The ClC gene family encodes 9 different ClC chloride channels in mammals. Human and mouse genetic studies have identified clear roles for ClC channels in the regulation of Cl− resorption in the kidney, since mutations in genes encoding ClC channels are known to cause diverse diseases such as myotonia (muscle stiffness), Bartter syndrome (renal salt loss) with or without deafness, Dent’s disease (proteinuria and kidney stones), osteopetrosis and neurodegeneration, and possibly epilepsy (249). ClC-K channel is a kidney-specific member of the ClC family (the “K” references to kidney expression) that is thought to play an essential role in the transepithelial renal chloride transport, especially in the TAL (298). Two highly homologous ClC channels, ClC-Ka and ClC-Kb, are found to be predominantly expressed in the kidney. The sequences of ClC-Ka and ClC-Kb (ClC-K1 and ClC-K2, respectively, in rodents) are about 90% identical (4, 249, 273). However, their expression pattern along the nephron and mechanisms of regulation are different. In addition to the kidney, ClC-Ka and ClC-Kb were also identified in epithelial cells of the inner ear (48, 139, 288, 580).
ClC-K2 Cl− channel was found in basolateral membranes of the type A intercalated cells residing along the collecting duct (273, 288, 289). Moreover, ClC-K2 is expressed in the basolateral membranes of the connecting tubule cells, suggesting that ClC-K2 imparts a potential basolateral Cl− conductance in these cells (288, 289). Loss-of-function mutations in the CLCNKB gene, the human homologue of ClC-K2, are found to cause Bartter’s syndrome type III, an autosomal recessive form of severe intravascular volume depletion resulting in low blood pressure together with hypokalemic alkalosis (298, 490, 512). In contrast, ClC-K1 was found to be localized only in the TAL, which has the highest chloride permeability among nephron segments (579). Human diseases associated with an isolated defect of gene encoding CLC-Ka have yet been described.
Palmer and Frindt using functional electrophysiological measurements characterized and quantified Cl− conductance in principal and intercalated cells of the rat CNT and CCD and proposed that ClC-K2 makes major contribution into this conductance (404). Cl− currents were much larger in intercalated cells compared to principal cells of the CCD and were also larger in the CNT compared to the CCD. Observed Cl− conductances were diminished by lowering the extracellular pH to 6.4 (404), which is also consistent with the previously published characteristics of this channel (298). Similarly, a 10-pS Cl− channel that is located mainly in the intercalated cells and has properties similar to ClC-K2 was reported in the mouse CNT and CCD (394).
Functional expression of both ClC-Ka and ClC-Kb channels requires the small accessory β-subunit barttin (139). Barttin is an accessory subunit that modifies protein stability, subcellular distribution, and voltage-dependent gating of ClC-K channels; mutations in this accessory subunit are also capable of causing renal diseases (157, 247, 491). Barttin stimulates chloride flux through ClC channels by modifying their gating properties (157). Similar to ENaC, at its COOH-terminus barttin harbors a PY site (139), which is important for the binding of WW-domain-containing ubiquitin ligases, such as Nedd4-2 (515). Coexpression of Nedd4-2 significantly decreased inwardly rectifying current through ClC-Ka/barttin (137). Consequently, the coexpression of SGK1 significantly stimulated the current (137). Thus, ENaC and barttin, and consequently ClC-K channels, have similar mechanisms of regulation. Therefore, sodium and chloride transports, similarly to Na+ reabsorption and K+ secretion, are also interconnected.
Interestingly, ClC family members were originally considered to be Ca2+-activated Cl− channels (CaCCs) (155). However, subsequent studies demonstrated that this is not true (20, 229). Recent studies identified transmembrane protein TMEM16A protein as CaCC (82, 495, 666). However, in the kidney, dense immunoreactivity was observed mainly in the epithelia of proximal and weakly in distal tubules (666). Therefore, identity of proteins mediating Ca2+-dependent Cl− transport in the CNT and CCD is unclear yet.
Although studies on ClC channels have greatly clarified the physiological roles of Cl− channels in the kidney, and it was shown that members of this family are expressed in the CNT and CCD, their role in these segments is not completely clear yet. It was hypothesized that Cl− channels optimize HCO3− flux mediated by Cl−/HCO3− exchange (394). At this scenario, the ClC channels help the AE1 exchanger to extrude HCO3− toward the interstitium by recycling Cl− across the basolateral membrane of intercalated cells, thus ultimately favoring H+ extrusion into the tubule lumen (394).
C. Cystic fibrosis transmembrane conductance regulator
1. Role of CFTR in the CNT and CCD
CFTR is a cAMP-dependent, PKA-regulated Cl− channel that is highly expressed in the apical membrane of all segments of the mammalian nephron, including the CNT, CCD and IMCD (109, 119, 237, 503, 591). The CFTR gene was identified more than two decades ago as genetic basis of cystic fibrosis, a common recessive genetic disease (272, 449, 455). There have been identified more than 1,500 loss-of-function mutations in CFTR that cause cystic fibrosis, with approximately 90% of patients having the ΔF508 mutation in one or both CFTR gene alleles (596). CFTR activity in epithelial cells lining renal cysts is in part responsible for progressive fluid accumulation and cyst enlargement seen in ADPKD (112, 596). Small-molecule CFTR inhibitors slowed cyst expansion in both in vitro and in vivo models of PKD (596, 659).
The role of the CFTR in the kidney under normal conditions remained unclear because there was no major disruption of renal function in cystic fibrosis patients (591). However, considerable evidence supports the role for CFTR in mediating Cl− secretion by the distal tubule and principal cells of the CCD and IMCD (237, 313, 341, 526, 592). Recent paper by Lu et al. provides direct evidence that mouse principal cells from freshly isolated CCD exhibit PKA-activated CFTR Cl− channels on their apical membranes (341). Comparing Cl− channel activity and characteristics in principal cells of wild-type mice, ΔF508-CFTR transgenic mice, and in Xenopus laevis oocytes expressing mouse CFTR, the authors provide definitive evidence for CFTR functioning as an apical Cl− channel in the mammalian kidney (341). In addition, as noted above, CFTR is responsible for Cl− secretion into the lumen of cysts in polycystic kidneys and, therefore, contributes to cyst enlargement (526, 558, 564, 611).
2. Interaction of CFTR with other channels in the CNT and CCD
In addition to participation in Cl− transport, a possible role for CFTR could be based on the finding that phosphorylation of CFTR functions as a switch modulating other ion channels or transporters. It was proposed that CFTR may interact with and regulate the activity of a variety of apical membrane proteins. For instance, it was shown that CFTR is required for PKA-regulated ATP sensitivity of ROMK in the TAL (340). In a CFTR-null strain and a ΔF508-CFTR strain, ATP and glibenclamide sensitivities were drastically reduced or eliminated, consistent with CFTR as a critical subunit for the ROMK channel. Moreover, in wild-type mice expressing CFTR, elevation of PKA activity also eliminated the sensitivity of ROMK to cytosolic concentrations of ATP (340). The authors proposed that CFTR regulates ROMK channel by providing a PKA-regulated functional switch that determines the distribution of open and ATP-inhibited K+ channels in the apical membrane. In addition to the TAL, the state of PKA phosphorylation rapidly modulated the functional interaction of ROMK and CFTR with regard to Mg-ATP sensitivity in the CCD. They also hypothesize that in the water diuretic state, when AVP levels are low, CFTR and ROMK interact, producing a functional complex wherein ROMK is highly sensitive to cell ATP and therefore become inactive. In contrast, in the antidiuretic state, when AVP is high, CFTR no longer interacts with ROMK, and the channel is insensitive to cytosolic ATP and become fully active. Thus, CFTR can act as a functional switch that modulates ROMK channel activity between the water diuretic and antidiuretic states. Interestingly, in mCFTR−/− mice ROMK activity was increased compared to wild-type mice (340). The latter is consistent with the requirement of CFTR expression in apical membranes for the regulation of ROMK activity. However, in ROMK−/− mice, CFTR Cl− channel activity was unchanged in comparison with wild-type mice (341). Thus, the authors proposed that the functional interaction of CFTR and ROMK is unidirectional: the absence of CFTR alters the functional expression of ROMK, but the absence of ROMK does not affect the functional expression of CFTR (341).
Apart from the regulation of ROMK, CFTR also colocalizes and has functional interactions with ENaC (36, 300). However, the molecular mechanism and physiological relevance of a regulatory relationship between ENaC and CFTR remain a subject of considerable controversy and may vary in different tissues (36). CFTR downregulates ENaC in most epithelial and non-epithelial cells (313, 326, 546, 547), whereas ENaC activates CFTR (252). In contrast, activation of CFTR in the sweat duct increases ENaC-mediated sodium transport (438). The functional interaction between CFTR and ENaC was initially demonstrated in MDCK cells. Expression of cDNAs for ENaC alone generated large amiloride-sensitive sodium currents that were stimulated by cAMP, whereas coexpression of CFTR with ENaC generated smaller basal sodium currents that were inhibited by cAMP (546). Later it was demonstrated that negative regulation of ENaC by CFTR reflects an effect on ENaC activity rather than ENaC number (547). In contrast, single Cl− channel analyses showed that both the channel open probability and the number of CFTR channels increased when they were co-expressed with ENaC (252). Moreover, CFTR−/− pigs showed increased amiloride-sensitive voltage and current, but lack of CFTR did not increase transepithelial Na+ (92). The authors proposed that the lack of apical Cl− conductance caused the change, not increased Na+ transport (92, 245).
It was reported that COOH-terminus of the β-ENaC subunit was required for the functional interaction between activated CFTR and ENaC, while the NH2-termini of the β- and γ-subunits were critical for the down-regulation of ENaC by quiescent CFTR (251). It is likely that coordinated regulation of CFTR and ENaC involves a large dynamic signaling complex that is composed of several proteins and kinases. However, details of this complex are not clear yet. Furthermore, other mechanisms could be involved in regulation of ENaC by CFTR. For instance, one of the possible mechanisms is that CFTR modulates ATP secretion (59), and ATP then acts via purinergic receptors to inhibit ENaC activity (421, 423).
In some patients with cystic fibrosis disease, mutations cannot be identified on both CFTR alleles. However, it was recently demonstrated that atypical cystic fibrosis patients harbor mutations in their ENaC genes (23) that change ENaC currents in overexpression systems (230, 437). While these findings are indirect, they highlight an importance of ENaC-CFTR interactions.
In addition to its role as a regulator of ROMK and ENaC, CFTR also mediates other transport proteins, including anion exchangers, sodium-bicarbonate transporters, and aquaporin water channels (198). For instance, it was shown that CFTR activates several Cl−/HCO3− exchangers in the CLC26 family, including Slc26a4 (pendrin) (287).
IX. Paracellular transport
The transport of electrolytes and water across epithelial barriers occurs not only through movement of fluid across the plasma membranes of the cells that comprise the epithelial layer (i.e. transcellular transport), but also involves paracellular transport (Figs. 16A and 16B, respectively). The paracellular pathway involves the movement of ions through the intercellular spaces between epithelial cells. The gatekeeper of the paracellular pathway is the tight junction, which is located at apical cell-cell interactions of adjacent epithelial cells. The tight junction, first identified approximately 50 years ago (144, 145), separates apical domain from basolateral and provides a barrier to paracellular movement of water and ions. Fig. 16C demonstrates a three-dimensional structure of tight junctions. As summarized by Tsukita and colleagues, two tight-junction strands in the membrane of adjacent cells associate with each other to form paired tight-junction strands, where the intercellular space is obliterated (577). Paracellular transport through the tight junctions is passive and is driven by electro-osmotic gradients produced by transcellular transport. The distal nephron, including the CNT and CCD, is considered a tight epithelium with high transepithelial resistance and low passive ion permeability. For instance, in the proximal tubule, which is considered to be a leaky epithelium because of low transepithelial resistance and high paracellular transport, one-third of total fluid reabsorption occurs through the paracellular pathway (117). Thus, in the CCD and CNT, the paracellular pathway is relatively impermeable and acts mostly as a barrier to solutes, so that these ion gradients are not dissipated (318). However, the paracellular pathway in the collecting duct might also play a role in transepithelial chloride reabsorption. In this case, Cl− is actively reabsorbed by pendrin localized to the apical membrane of the type B intercalated cell (see Fig. 10). Thereafter Cl− exits this cell via a basolateral Cl− channel and diffuses passively down electrochemical gradients via the paracellular pathway.
Figure 16.

Modes of transepithelial transport and major proteins involved in the paracellular transport. Schemes of the transcellular (A) and paracellular (B) epithelial transport. (C) Schematic three-dimensional structure of tight junctions. Proposed structures of claudin (D) and occludin (E).
A. Claudins
Claudins are key integral proteins that provide the barrier function and permit selective paracellular transport (18, 19, 117, 589, 590). Claudins were named from the Latin claudere, “to close”. Epithelial cells typically express multiple claudin isoforms. Several claudins, including claudins-3, -4, -6, -7, -8, and -10 are identified in the CCD and CNT (117, 222, 278, 282, 307). Claudins range from 20 to 27 kDa and have four transmembrane helices: a short internal amino-terminal sequence, two extracellular domains, and a longer and more variable cytoplasmic tail (Fig. 16D). Functional data provide considerable evidence that claudins determine the selectivity of paracellular transport and probably do so by forming paracellular pores through the tight junction (19). The study of paracellular transport, and especially role of claudins, is a rapidly evolving field. However, many questions remain unanswered. One of the limiting factors of these studies is that methods to study the paracellular transport are relatively restricted to provide sufficient details about mechanisms of this transport. Furthermore, more than 20 different claudins have now been discovered, and within the kidney, claudins exhibit distinct nephron segment-specific pattern of expression (18, 19, 117). In addition, function of claudins depends on the host cell. For instance, it was shown that knock down of claudin-4 or claudin-7 expression elevated the permeation of Na+ and enhanced the proclivity of the tight junction for cations in the MDCK cells. On the other hand, knockdown of claudin-4 or claudin-7 in LLC-PK1 cells depressed the permeation of Cl− and caused the tight junction to lose the anion selectivity (221). Therefore, despite that the paracellular transport is of considerable physiological importance, this pathway is poorly understood.
B. Occludins
In addition to claudin family members, there are several other tight junction transmembrane proteins, which might directly influence the adhesive barrier, including occludins, junctional adhesion molecules, and tricellulin (18). Among them, the most studied is occludin (169). Occludin is a tetraspan membrane protein with a molecular weight of approximately 60 kDa. This protein is comprised of four transmembrane domains, a short NH2-terminal cytoplasmic domain, a long COOH-terminal cytoplasmic domain, two extracellular loops, and one intracellular turn (Fig. 16E). One of the most characteristic aspects of its sequence is the high content of tyrosine and glycine residues in the first extracellular loop (468). It is clear that occludins are the main components of the tight junctions. For instance, Yu et al demonstrated that occludin, through its connection to RhoA and the actin cytoskeleton is intimately involved in dramatically restructuring the tight junction and cell border. However, the tight junction “fence” function was not impaired in occludin knock downed cells (674). Moreover, in occludin knockout mice, tight junctions themselves were not affected morphologically, and the barrier function was normal (468). Thus, the exact function of this protein is unclear yet.
C. Regulation of tight junction proteins and paracellular transport
As discussed above, aldosterone and WNK kinases regulate the transport of Na+ and K+ in the CNT and CCD. Interestingly, it was also demonstrated that aldosterone modulates tight junction properties in the CCD. This hormone promoted rapid and transient phosphorylation of endogenous claudin-4 on threonine residues, without affecting tyrosine or serine; this event was fully developed at 10 nM aldosterone and appeared specific for aldosterone because it was not observed after dexamethasone treatment and it was dependent on mineralocorticoid receptor occupancy (307). Furthermore, claudin-4 can be phosphorylated by the WNK4, protein kinase linked to PHA type II (637). As demonstrated by two independent studies, disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins in the CCD (258, 657). Similarly, as discussed above, Nedd4–2 plays a central role in the regulation of Na+ transport in the CCD and CNT (460, 515, 536). Recent studies revealed that Nedd4–2, via ubiquitination of occludin, may be also involved in the assembly and remodeling of tight junction and the regulation of paracellular conductance in the CCD (435). Moreover, it was also shown that LNX1p80, another E3 ubiquitin ligase, is involved in the dynamic remodeling of tight junctions by selective ubiquitylation, endocytosis, and degradation of claudins (555).
Thus, many proteins, including claudins and occludins are localized to the tight junction and might participate in the paracellular transport. Paracellular ion selectivities of different epithelia are most likely determined by varying combinations of different claudins. However, the stoichiometry of these proteins in a single tight junction and their oligomerization into heteromeric and heterotypic assemblies are not known yet. Elucidation of the composition of paracellular tight junction channels and the overall molecular architecture of the tight junction is a very challenging but essential step in understanding of paracellular ion transport (561). Recent studies identified that claudin-4 interacts with claudin-8 and their association is required for the Cl−-selective paracellular pathway in the collecting duct, suggesting a mechanism for coupling chloride reabsorption to sodium reabsorption in the CCD (220).
X. The role of channels and transporters expressed in the CNT and CCD in kidney disorders
As it is briefly discussed in this manuscript, there are several common mechanisms that regulate physiological functions of the CNT and CCD. However, this regulation is mediated by considerably distinct transport proteins, which as a whole system finely tune water and electrolyte balance in the kidney. Growing evidence suggests that various channels and transporters expressed in the CNT and CCD are involved in hereditary as well as acquired kidney diseases. Several examples of disorders that arise from mutations in transport proteins expressing and functioning in the CNT and CCD are summarized in Table 2. In addition to shown in this table proteins, other channels not listed in the Table 2 that express in the CNT and CCD, such as TRPV5 and TRPV4, are involved in maintaining of electrolyte homeostasis (214, 216, 295, 647). However, no mutations associated with human diseases in these channels have been yet identified. Furthermore, in addition to defects in channels and transporters, variety hereditary disorders are mediated by mutations in regulatory proteins, such as fibrocystin, WNK1 and WNK4, 11β-HSD2, carbonic anhydrase II etc. Thus, it is clear that these proteins are critical for human health and have tremendous potential in therapeutic settings. As more information on the in vivo role of channels and transporters and clinical data from patients carrying mutations in genes responcible for water and electrolyte transport become available, our knowledge of the role of these proteins in renal pathophysiology will expand considerably.
Table 2.
Human diseases mediated by mutations genes encoding of channels and transporters in the CNT and CCD. UCSC Genome Browser website (http://genome.cse.ucsc.edu/) was used as a source for gene/chromosome locations.
| Transport protein | Gene/Chromosome location (Human) | Diseases | References |
|---|---|---|---|
| TRPV5 | TRPV5; chromosome 7: 142,605,648- 142,630,820 | Chronic hypercalciuria, calcium hyperabsorption, polyuria and increased urinary acidification | (216) |
| α-subunit of ENaC | SCNN1A; chromosome 12: 6,456,011- 6,484,390 | Pseudohypoaldosteronism type 1 (PHA-1) | (91, 322) |
| β-subunit of ENaC | SCNN1B; chromosome 16: 23,313,591- 23,392,619 | Liddle’s syndrome, PHA-1 | (91, 197, 203, 322, 507) |
| γ-subunit of ENaC | SCNN1G; chromosome 16: 23,194,040- 23,228,200 | Liddle’s syndrome, PHA-1 | (202, 322, 545) |
| ROMK | KCNJ1; chromosome 11: 128,707,915–128,712,363 | Type II Bartter syndrome | (118, 207, 513) |
| BK β1- subunit | KCNMB1; chromosome 5: 169,805,167- 169,816,638 | Diastolic hypertension | (152) |
| Kir4.1 | KCNJ10; chromosome 1: 160,008,033- 160,039,961 | Hypokalemia, metabolic alkalosis and hypomagnesemia with hypocalciuria. | (53, 492) |
| β1-subunit of H+-ATPase | ATP6V1B1; chromosome 2: 71,162,998- 71,192,560 | distal renal tubular acidosis | (262, 514) |
| α4-subunit of H+-ATPase | ATP6V0A4; chromosome 7: 138,391,040- 138,458,782 | distal renal tubular acidosis | (261, 544) |
| Cl−/HCO3− AE1 exchanger | SLC4A1; chromosome 17: 42,325,759- 42,345,502 | distal renal tubular acidosis | (263, 464, 500) |
| Pendrin (Cl−−/HCO3− exchanger) | SLC26A4; chromosome 7: 107,301,080- 107,358,250 | Pendred syndrome and DFNB4 (hearing disorder) | (108, 501) |
| CLC-Kb | CLCNKB; chromosome 1: 16,370,247- 16,383,802 | Bartter’s syndrome type III | (490, 512) |
| Barttin (accessory β-subunit of CLC channels) | BSND; chromosome 1: 55,464,617- 55,474,464 | Bartter’s syndrome type IV | (48, 139) |
| CFTR | CFTR; chromosome 7: 117,120,017- 117,308,716 | Cystic fibrosis | (272, 455) |
| AQP2 | AQP2; chromosome 12: 50,344,524- 50,352,662 | Autosomal Dominant and Recessive forms of Nephrogenic Diabetes Insipidus (NDI) | (115, 450) |
| TRPC6 | TRPC6; chromosome 11: 101,322,296- 101,454,659 | Focal Segmental Glomerulosclerosis (FSGS) | (442, 644) |
| PC-1 (PKD1) | PKD1; chromosome 16: 2,138,711- 2,185,899 | Autosomal Dominant Polycystic Kidney Disease (ADPKD) | (231, 573, 638) |
| PC-2 (PKD2) | PKD2; chromosome 4: 88,928,820- 88,998,928 | ADPKD | (367, 573, 638) |
In the past two decades, considerable progress has been achieved in understanding the regulation of water and electrolyte homeostasis in the CNT and CCD; pathophysiological roles of channels and transporters mediating these transport processes have been significantly clarified, although the precise mechanisms by which these proteins are regulated have not been thoroughly defined. Understanding of these mechanisms is essential to develop therapies for renal and accompanying diseases resulting from their dysfunctions. Animal models must be developed and used to confirm the pathophysiological mechanisms hypothesized on the basis of cell biology research. Continued studies at both molecular and whole organism levels is required to develop potential drugs to treat conditions arising from the dysregulation of channels and transporters activity. In general, understanding of cellular defects in renal diseases opens exciting perspectives in the development of novel therapies.
XI Summary and conclusion
This overview article discusses in some details the transport characteristics of the CCD and CNT and the mechanisms by which aldosterone and other hormones maintain salt and water homeostasis in these nephron segments. Regulation of the epithelial transport in the CNT and CCD is not only relevant to the normal physiology of the kidney; dysfunctional regulation of channels and transporters in these segments has also been linked to various renal and cardiovascular diseases. Hence, understanding the hormonal and non-hormonal factors-mediated signaling pathways controlling channels and transporters of the CCD and CNT cells is important for understanding regulation of renal water and electrolyte transport in human health and disease. Furthermore, inhibition or activation of corresponding pathways may represent therapeutic targets for treating these diseases. Undoubtedly, many questions remain regarding regulation of renal transport in the CNT and CCD.
The CNT and CCD are the first and most important nephron segments to be mobilized when dietary intakes are modified and under control of various hormones sustain sufficient reabsorption and secretion rates to compensate for changes in dietary intakes. These segments express various channels and transporters, most of which are implicated in human diseases. The distinct distribution of these transport proteins along the nephron underlies their importance in the regulation of many renal and not only physiological processes. Growing evidence suggest that various channels and transporters are implicated in several disease processes including, but not limited to hypertension, CHF, NDI, PKD, and dysregulation of Na+, K+, Ca2+ and acid-base homeostasis. Sodium reabsorption, potassium secretion and water transport are among the most studied in these segments. However, the important role of the CNT and CCD in the control of acid-base balance, calcium homeostasis, chloride transport, and urea excretion should not be disregarded. Transport mechanisms, which mediate function of these nephron segments are strictly regulated and represent a complex rather than simple regulations of individual pathways.
Rapid advances in functional genomics and proteomics mean that majority of genes identified and mapped will be classified and have functions assigned to them (149, 284, 374, 462). Considerable knowledge can be obtained from the generation of animals carrying multiple mutations. Future studies are required to uncover the underlying cell and molecular mechanisms by which these proteins and their regulatory stimulus contribute to normal and pathological kidney physiology.
Acknowledgments
I apologize to the investigators of renal epithelial transport whose relevant publications were inadvertently not directly discussed. I am grateful to Drs. David L. Mattson (Medical College of Wisconsin), Oleh Pochynyuk (University of Texas Health Science Center at Houston), James D. Stockand (University of Texas Health Science Center at San Antonio), and Timo Rieg (University of California San Diego) for helpful discussions and critical reading of the manuscript. I also express my appreciation to Daria Ilatovskaya for her invaluable help in preparation of this manuscript and assistance in creating the figures. Work from the author’s laboratory was supported by Carl W. Gottschalk Research Scholar Grant from the American Society of Nephrology, American Society of Physiology S&R Foundation Ryuji Ueno and Lazaro J. Mandel Awards and American Diabetes Association grant 1-10-BS-168.
References
- 1.Abe K, Tani K, Fujiyoshi Y. Conformational rearrangement of gastric H+,K+-ATPase induced by an acid suppressant. Nat Commun. 2011;2:155. doi: 10.1038/ncomms1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abe K, Tani K, Nishizawa T, Fujiyoshi Y. Inter-subunit interaction of gastric H+,K+-ATPase prevents reverse reaction of the transport cycle. EMBO J. 2009;28:1637–1643. doi: 10.1038/emboj.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009;23:297–328. doi: 10.1096/fj.08-119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adachi S, Uchida S, Ito H, Hata M, Hiroe M, Marumo F, Sasaki S. Two isoforms of a chloride channel predominantly expressed in thick ascending limb of Henle’s loop and collecting ducts of rat kidney. J Biol Chem. 1994;269:17677–17683. [PubMed] [Google Scholar]
- 5.Ahn KY, Turner PB, Madsen KM, Kone BC. Effects of chronic hypokalemia on renal expression of the “gastric” H+,K+-ATPase alpha-subunit gene. Am J Physiol Renal Physiol. 1996;270:F557–F566. doi: 10.1152/ajprenal.1996.270.4.F557. [DOI] [PubMed] [Google Scholar]
- 6.Al-Awqati Q, Beauwens R. Cellular Mechanisms of H+ and HCO3− transport in tight urinary epithelia. Compr Physiol. 2011;(Supplement 25) doi: 10.1002/cphy.cp080108. Handbook of Physiology, Renal Physiology. [DOI] [Google Scholar]
- 7.Alexander RT, Woudenberg-Vrenken TE, Buurman J, Dijkman H, van der Eerden BCJ, van Leeuwen JPTM, Bindels RJ, Hoenderop JG. Klotho prevents renal calcium loss. J Am Soc Nephrol. 2009;20:2371–2379. doi: 10.1681/ASN.2008121273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 2009;212:1672–1683. doi: 10.1242/jeb.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alper SL, Natale J, Gluck S, Lodish HF, Brown D. Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. Proc Natl Acad Sci USA. 1989;86:5429–5433. doi: 10.1073/pnas.86.14.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Althaus M, Bogdan R, Clauss WG, Fronius M. Mechano-sensitivity of epithelial sodium channels (ENaCs): laminar shear stress increases ion channel open probability. FASEB J. 2007;21:2389–2399. doi: 10.1096/fj.06-7694com. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez de la Rosa D, Canessa CM. Role of SGK in hormonal regulation of epithelial sodium channel in A6 cells. Am J Physiol Cell Physiol. 2003;284:C404–C414. doi: 10.1152/ajpcell.00398.2002. [DOI] [PubMed] [Google Scholar]
- 12.Alvarez de la Rosa D, Canessa CM, Fyfe GK, Zhang P. Structure and regulation of amiloride-sensitive sodium channels. Annu Rev Physiol. 2000;62:573–594. doi: 10.1146/annurev.physiol.62.1.573. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez de la Rosa D, Zhang P, Naray-Fejes-Toth A, Fejes-Toth G, Canessa CM. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem. 1999;274:37834–37839. doi: 10.1074/jbc.274.53.37834. [DOI] [PubMed] [Google Scholar]
- 14.Alzamora R, Gong F, Rondanino C, Lee JK, Smolak C, Pastor-Soler NM, Hallows KR. AMP-activated protein kinase inhibits KCNQ1 channels through regulation of the ubiquitin ligase Nedd4-2 in renal epithelial cells. Am J Physiol Renal Physiol. 2010;299:F1308–F1319. doi: 10.1152/ajprenal.00423.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ambuhl PM, Amemiya M, Danczkay M, Lotscher M, Kaissling B, Moe OW, Preisig PA, Alpern RJ. Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney. Am J Physiol Renal Physiol. 1996;271:F917–F925. doi: 10.1152/ajprenal.1996.271.4.F917. [DOI] [PubMed] [Google Scholar]
- 16.Anantharam A, Palmer LG. Determination of epithelial Na+ channel subunit stoichiometry from single-channel conductances. J Gen Physiol. 2007;130:55–70. doi: 10.1085/jgp.200609716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen MN, Olesen SP, Rasmussen HB. Kv7.1 surface expression is regulated by epithelial cell polarization. Am J Physiol Cell Physiol. 2011;300:C814–C824. doi: 10.1152/ajpcell.00390.2010. [DOI] [PubMed] [Google Scholar]
- 18.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angelow S, Ahlstrom R, Yu AS. Biology of claudins. Am J Physiol Renal Physiol. 2008;295:F867–F876. doi: 10.1152/ajprenal.90264.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arreola J, Begenisich T, Nehrke K, Nguyen HV, Park K, Richardson L, Yang B, Schutte BC, Lamb FS, Melvin JE. Secretion and cell volume regulation by salivary acinar cells from mice lacking expression of the Clcn3 Cl− channel gene. J Physiol. 2002;545:207–216. doi: 10.1113/jphysiol.2002.021980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashkroft FM. Ion channels and Disease. San Diego, CA: Academic Press; 2000. pp. 1–502. [Google Scholar]
- 22.Avner ED, Sweeney WE, Jr, Nelson WJ. Abnormal sodium pump distribution during renal tubulogenesis in congenital murine polycystic kidney disease. Proc Natl Acad Sci USA. 1992;89:7447–7451. doi: 10.1073/pnas.89.16.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azad AK, Rauh R, Vermeulen F, Jaspers M, Korbmacher J, Boissier B, Bassinet L, Fichou Y, des GM, Stanke F, De BK, Dupont L, Balascakova M, Hjelte L, Lebecque P, Radojkovic D, Castellani C, Schwartz M, Stuhrmann M, Schwarz M, Skalicka V, de MI, Girodon E, Ferec C, Claustres M, Tummler B, Cassiman JJ, Korbmacher C, Cuppens H. Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum Mutat. 2009;30:1093–1103. doi: 10.1002/humu.21011. [DOI] [PubMed] [Google Scholar]
- 24.Babilonia E, Li D, Wang Z, Sun P, Lin DH, Jin Y, Wang WH. Mitogen-activated protein kinases inhibit the ROMK (Kir 1.1)-like small conductance K channels in the cortical collecting duct. J Am Soc Nephrol. 2006;17:2687–2696. doi: 10.1681/ASN.2006050426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008;9:472–479. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bailey MA, Craigie E, Livingstone DEW, Kotelevtsev YV, Al-Dujaili EAS, Kenyon CJ, Mullins JJ. Hsd11b2 haploinsufficiency in mice causes salt sensitivity of blood pressure. Hypertension. 2011;57:515–520. doi: 10.1161/HYPERTENSIONAHA.110.163782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bandyopadhyay BC, Swaim WD, Liu X, Redman RS, Patterson RL, Ambudkar IS. Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells. Role in apical Ca2+ influx. J Biol Chem. 2005;280:12908–12916. doi: 10.1074/jbc.M410013200. [DOI] [PubMed] [Google Scholar]
- 28.Bankir L, Bichet DG, Bouby N. Vasopressin V2 receptors, ENaC, and sodium reabsorption: a risk factor for hypertension? Am J Physiol Renal Physiol. 2010;299:F917–F928. doi: 10.1152/ajprenal.00413.2010. [DOI] [PubMed] [Google Scholar]
- 29.Barile M, Pisitkun T, Yu MJ, Chou CL, Verbalis MJ, Shen RF, Knepper MA. Large scale protein identification in intracellular Aquaporin-2 vesicles from renal inner medullary collecting duct. Mol Cell Proteomics. 2005;4:1095–1106. doi: 10.1074/mcp.M500049-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barker PM, Nguyen MS, Gatzy JT, Grubb B, Norman H, Hummler E, Rossier B, Boucher RC, Koller B. Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. J Clin Invest. 1998;102:1634–1640. doi: 10.1172/JCI3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr MM, DeModena J, Braun D, Nguyen CQ, Hall DH, Sternberg PW. The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr Biol. 2001;11:1341–1346. doi: 10.1016/s0960-9822(01)00423-7. [DOI] [PubMed] [Google Scholar]
- 32.Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature. 1999;401:386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 33.Bastani B, Purcell H, Hemken P, Trigg D, Gluck S. Expression and distribution of renal vacuolar proton-translocating adenosine triphosphatase in response to chronic acid and alkali loads in the rat. J Clin Invest. 1991;88:126–136. doi: 10.1172/JCI115268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beesley AH, Hornby D, White SJ. Regulation of distal nephron K+ channels (ROMK) mRNA expression by aldosterone in rat kidney. J Physiol. 1998;509:629–634. doi: 10.1111/j.1469-7793.1998.629bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benos DJ, Stanton BA. Functional domains within the degenerin/epithelial sodium channel (Deg/ENaC) superfamily of ion channels. J Physiol. 1999;520:631–644. doi: 10.1111/j.1469-7793.1999.00631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst. 2009;5:123–127. doi: 10.1039/b810471a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berdiev BK, Shlyonsky VG, Karlson KH, Stanton BA, Ismailov II. Gating of amiloride-sensitive Na+ channels: subunit-subunit interactions and inhibition by the cystic fibrosis transmembrane conductance regulator. Biophys J. 2000;78:1881–1894. doi: 10.1016/S0006-3495(00)76737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, Schutz G. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA. 1998;95:9424–9429. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergmann C, Senderek J, Kupper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schoneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Buttner R, Zerres K. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD) Hum Mutat. 2004;23:453–463. doi: 10.1002/humu.20029. [DOI] [PubMed] [Google Scholar]
- 40.Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial secretion. Am J Physiol Cell Physiol. 2003;285:C1–C18. doi: 10.1152/ajpcell.00554.2002. [DOI] [PubMed] [Google Scholar]
- 41.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 42.Bhalla V, Daidie D, Li H, Pao AC, LaGrange LP, Wang J, Vandewalle A, Stockand JD, Staub O, Pearce D. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol Endocrinol. 2005;19:3073–3084. doi: 10.1210/me.2005-0193. [DOI] [PubMed] [Google Scholar]
- 43.Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol. 2008;19:1845–1854. doi: 10.1681/ASN.2008020225. [DOI] [PubMed] [Google Scholar]
- 44.Bhalla V, Oyster NM, Fitch AC, Wijngaarden MA, Neumann D, Schlattner U, Pearce D, Hallows KR. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J Biol Chem. 2006;281:26159–26169. doi: 10.1074/jbc.M606045200. [DOI] [PubMed] [Google Scholar]
- 45.Bhalla V, Soundararajan R, Pao AC, Li H, Pearce D. Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol. 2006;291:F714–F721. doi: 10.1152/ajprenal.00061.2006. [DOI] [PubMed] [Google Scholar]
- 46.Bindels RJ. A molecularly guided tour along the nephron. Pflugers Arch. 2009;458:1–3. doi: 10.1007/s00424-009-0661-3. [DOI] [PubMed] [Google Scholar]
- 47.Bindels RJ. 2009 Homer W. Smith Award: Minerals in motion: from new ion transporters to new concepts. J Am Soc Nephrol. 2010;21:1263–1269. doi: 10.1681/ASN.2010010001. [DOI] [PubMed] [Google Scholar]
- 48.Birkenhager R, Otto E, Schurmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NV, Antignac C, Sudbrak R, Kispert A, Hildebrandt F. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- 49.Bissig M, Hagenbuch B, Stieger B, Koller T, Meier PJ. Functional expression cloning of the canalicular sulfate transport system of rat hepatocytes. J Biol Chem. 1994;269:3017–3021. [PubMed] [Google Scholar]
- 50.Blazer-Yost BL, Esterman MA, Vlahos CJ. Insulin-stimulated trafficking of ENaC in renal cells requires PI 3-kinase activity. Am J Physiol Cell Physiol. 2003;284:C1645–C1653. doi: 10.1152/ajpcell.00372.2002. [DOI] [PubMed] [Google Scholar]
- 51.Blazer-Yost BL, Haydon J, Eggleston-Gulyas T, Chen JH, Wang X, Gattone V, Torres VE. Pioglitazone Attenuates cystic burden in the PCK rodent model of polycystic kidney disease. PPAR Res. 2010;2010:274376. doi: 10.1155/2010/274376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blazer-Yost BL, Liu X, Helman SI. Hormonal regulation of ENaCs: insulin and aldosterone. Am J Physiol Cell Physiol. 1998;274:C1373–C1379. doi: 10.1152/ajpcell.1998.274.5.C1373. [DOI] [PubMed] [Google Scholar]
- 53.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t HW, Al MO, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, rcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boim MA, Ho K, Shuck ME, Bienkowski MJ, Block JH, Slightom JL, Yang Y, Brenner BM, Hebert SC. ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. Am J Physiol Renal Physiol. 1995;268:F1132–F1140. doi: 10.1152/ajprenal.1995.268.6.F1132. [DOI] [PubMed] [Google Scholar]
- 55.Bonny O, Hummler E. Dysfunction of epithelial sodium transport: from human to mouse. Kidney Int. 2000;57:1313–1318. doi: 10.1046/j.1523-1755.2000.00968.x. [DOI] [PubMed] [Google Scholar]
- 56.Boone M, Deen PMT. Congenital nephrogenic diabetes insipidus: what can we learn from mouse models? Experimental Physiology. 2009;94:186–190. doi: 10.1113/expphysiol.2008.043000. [DOI] [PubMed] [Google Scholar]
- 57.Boone M, Kortenoeven MLA, Robben JH, Tamma G, Deen PMT. Counteracting vasopressin-mediated water reabsorption by ATP, dopamine, and phorbol esters: mechanisms of action. Am J Physiol Renal Physiol. 2011;300:F761–F771. doi: 10.1152/ajprenal.00247.2010. [DOI] [PubMed] [Google Scholar]
- 58.Boros S, Bindels RJ, Hoenderop JG. Active Ca2+ reabsorption in the connecting tubule. Pflugers Arch. 2009;458:99–109. doi: 10.1007/s00424-008-0602-6. [DOI] [PubMed] [Google Scholar]
- 59.Braunstein GM, Roman RM, Clancy JP, Kudlow BA, Taylor AL, Shylonsky VG, Jovov B, Peter K, Jilling T, Ismailov II, Benos DJ, Schwiebert LM, Fitz JG, Schwiebert EM. Cystic fibrosis transmembrane conductance regulator facilitates ATP release by stimulating a separate ATP release channel for autocrine control of cell volume regulation. J Biol Chem. 2001;276:6621–6630. doi: 10.1074/jbc.M005893200. [DOI] [PubMed] [Google Scholar]
- 60.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 61.Breton S, Brown D. New insights into the regulation of V-ATPase-dependent proton secretion. Am J Physiol Renal Physiol. 2007;292:F1–F10. doi: 10.1152/ajprenal.00340.2006. [DOI] [PubMed] [Google Scholar]
- 62.Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6:261–273. doi: 10.1038/nrneph.2010.30. [DOI] [PubMed] [Google Scholar]
- 63.Briet M, Schiffrin EL. The role of aldosterone in the metabolic syndrome. Curr Hypertens Rep. 2011;13:163–172. doi: 10.1007/s11906-011-0182-2. [DOI] [PubMed] [Google Scholar]
- 64.Brown D, Breton S. Mitochondria-rich, proton-secreting epithelial cells. J Exp Biol. 1996;199:2345–2358. doi: 10.1242/jeb.199.11.2345. [DOI] [PubMed] [Google Scholar]
- 65.Brown D, Breton S, Ausiello DA, Marshansky V. Sensing, signaling and sorting events in kidney epithelial cell physiology. Traffic. 2009;10:275–284. doi: 10.1111/j.1600-0854.2008.00867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown D, Hirsch S, Gluck S. An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature. 1988;331:622–624. doi: 10.1038/331622a0. [DOI] [PubMed] [Google Scholar]
- 67.Brown D, Hirsch S, Gluck S. Localization of a proton-pumping ATPase in rat kidney. J Clin Invest. 1988;82:2114–2126. doi: 10.1172/JCI113833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown D, Paunescu TG, Breton S, Marshansky V. Regulation of the V-ATPase in kidney epithelial cells: dual role in acid-base homeostasis and vesicle trafficking. J Exp Biol. 2009;212:1762–1772. doi: 10.1242/jeb.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, Unwin RJ, Wrong O, Tanner MJ. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest. 1997;100:1693–1707. doi: 10.1172/JCI119694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J Biol Chem. 2007;282:6153–6160. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- 71.Bublitz M, Morth JP, Nissen P. P-type ATPases at a glance. J Cell Sci. 2011;124:2515–2519. doi: 10.1242/jcs.088716. [DOI] [PubMed] [Google Scholar]
- 72.Bugaj V, Pochynyuk O, Mironova E, Vandewalle A, Medina JL, Stockand JD. Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. Am J Physiol Renal Physiol. 2008;295:F1063–F1070. doi: 10.1152/ajprenal.90321.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol Renal Physiol. 2009;297:F1411–F1418. doi: 10.1152/ajprenal.00371.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burch LH, Talbot CR, Knowles MR, Canessa CM, Rossier BC, Boucher RC. Relative expression of the human epithelial Na+ channel subunits in normal and cystic fibrosis airways. Am J Physiol Cell Physiol. 1995;269:C511–C518. doi: 10.1152/ajpcell.1995.269.2.C511. [DOI] [PubMed] [Google Scholar]
- 75.Butler A, Tsunoda S, McCobb DP, Wei A, Salkoff L. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- 76.Butterworth MB, Edinger RS, Frizzell RA, Johnson JP. Regulation of the epithelial sodium channel by membrane trafficking. Am J Physiol Renal Physiol. 2009;296:F10–F24. doi: 10.1152/ajprenal.90248.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Butterworth MB, Edinger RS, Ovaa H, Burg D, Johnson JP, Frizzell RA. The deubiquitinating enzyme UCH-L3 regulates the apical membrane recycling of the epithelial sodium channel. J Biol Chem. 2007;282:37885–37893. doi: 10.1074/jbc.M707989200. [DOI] [PubMed] [Google Scholar]
- 78.Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 79.Canessa CM, Merillat AM, Rossier BC. Membrane topology of the epithelial sodium channel in intact cells. Am J Physiol Cell Physiol. 1994;267:C1682–C1690. doi: 10.1152/ajpcell.1994.267.6.C1682. [DOI] [PubMed] [Google Scholar]
- 80.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 81.Cantone A, Yang X, Yan Q, Giebisch G, Hebert SC, Wang T. Mouse model of type II Bartter’s syndrome. I. Upregulation of thiazide-sensitive Na-Cl cotransport activity. Am J Physiol Renal Physiol. 2008;294:F1366–F1372. doi: 10.1152/ajprenal.00608.2007. [DOI] [PubMed] [Google Scholar]
- 82.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 83.Carattino MD. Structural mechanisms underlying the function of epithelial sodium channel/acid-sensing ion channel. Curr Opin Nephrol Hypertens. 2011;20:555–560. doi: 10.1097/MNH.0b013e328348bcac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carattino MD, Liu W, Hill WG, Satlin LM, Kleyman TR. Lack of a role of membrane-protein interactions in flow-dependent activation of ENaC. Am J Physiol Renal Physiol. 2007;293:F316–F324. doi: 10.1152/ajprenal.00455.2006. [DOI] [PubMed] [Google Scholar]
- 85.Carattino MD, Sheng S, Bruns JB, Pilewski JM, Hughey RP, Kleyman TR. The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its α subunit. J Biol Chem. 2006;281:18901–18907. doi: 10.1074/jbc.M604109200. [DOI] [PubMed] [Google Scholar]
- 86.Carattino MD, Sheng S, Kleyman TR. Epithelial Na+ channels are activated by laminar shear stress. J Biol Chem. 2004;279:4120–4126. doi: 10.1074/jbc.M311783200. [DOI] [PubMed] [Google Scholar]
- 87.Carrisoza R, Salvador C, Bobadilla N, Trujillo J, Escobar L. Expression and immunolocalization of ERG1 potassium channels in the rat kidney. Histochem Cell Biol. 2010;133:189–199. doi: 10.1007/s00418-009-0658-1. [DOI] [PubMed] [Google Scholar]
- 88.Cassola AC, Giebisch G, Wang W. Vasopressin increases density of apical low-conductance K+ channels in rat CCD. Am J Physiol Renal Physiol. 1993;264:F502–F509. doi: 10.1152/ajprenal.1993.264.3.F502. [DOI] [PubMed] [Google Scholar]
- 89.Chang JH, Kim S. Role of pendrin in acid-base balance. Electrolyte Blood Press. 2009;7:20–24. doi: 10.5049/EBP.2009.7.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The β-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 91.Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 92.Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, Zabner J, Welsh MJ. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA. 1999;96:2514–2519. doi: 10.1073/pnas.96.5.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cheng C, Prince LS, Snyder PM, Welsh MJ. Assembly of the epithelial Na+ channel evaluated using sucrose gradient sedimentation analysis. J Biol Chem. 1998;273:22693–22700. doi: 10.1074/jbc.273.35.22693. [DOI] [PubMed] [Google Scholar]
- 95.Cheng CJ, Huang CL. Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J Am Soc Nephrol. 2011;22:460–471. doi: 10.1681/ASN.2010060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choe H, Zhou H, Palmer LG, Sackin H. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am J Physiol Renal Physiol. 1997;273:F516–F529. doi: 10.1152/ajprenal.1997.273.4.F516. [DOI] [PubMed] [Google Scholar]
- 97.Christensen BM, Zuber AM, Loffing J, Stehle JC, Deen PM, Rossier BC, Hummler E. αENaC-mediated lithium absorption promotes nephrogenic diabetes insipidus. J Am Soc Nephrol. 2011;22:253–261. doi: 10.1681/ASN.2010070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chu PY, Quigley R, Babich V, Huang CL. Dietary potassium restriction stimulates endocytosis of ROMK channel in rat cortical collecting duct. Am J Physiol Renal Physiol. 2003;285:F1179–F1187. doi: 10.1152/ajprenal.00150.2003. [DOI] [PubMed] [Google Scholar]
- 99.Cirovic S, Markovic-Lipkovski J, Todorovic J, Nesovic-Ostojic J, Jovic M, Ilic S, Tatic S, Cemerikic D. Differential expression of KCNQ1 K+ channel in tubular cells of frog kidney. Eur J Histochem. 2010;54:e7. doi: 10.4081/ejh.2010.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clapham DE, Julius D, Montell C, Schultz G. International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev. 2005;57:427–450. doi: 10.1124/pr.57.4.6. [DOI] [PubMed] [Google Scholar]
- 101.Cluzeaud F, Reyes R, Escoubet B, Fay M, Lazdunski M, Bonvalet JP, Lesage F, Farman N. Expression of TWIK-1, a novel weakly inward rectifying potassium channel in rat kidney. Am J Physiol Cell Physiol. 1998;275:C1602–C1609. doi: 10.1152/ajpcell.1998.275.6.C1602. [DOI] [PubMed] [Google Scholar]
- 102.Codina J, DuBose TD., Jr Molecular regulation and physiology of the H+,K+-ATPases in kidney. Semin Nephrol. 2006;26:345–351. doi: 10.1016/j.semnephrol.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 103.Collier DM, Snyder PM. Extracellular chloride regulates the epithelial sodium channel. J Biol Chem. 2009;284:29320–29325. doi: 10.1074/jbc.M109.046771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Collier DM, Snyder PM. Identification of epithelial Na+ channel (ENaC) intersubunit Cl− inhibitory residues suggests a trimeric αγβ channel architecture. J Biol Chem. 2011;286:6027–6032. doi: 10.1074/jbc.M110.198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cope G, Murthy M, Golbang AP, Hamad A, Liu CH, Cuthbert AW, O’Shaughnessy KM. WNK1 affects surface expression of the ROMK potassium channel independent of WNK4. J Am Soc Nephrol. 2006;17:1867–1874. doi: 10.1681/ASN.2005111224. [DOI] [PubMed] [Google Scholar]
- 106.Coscoy S, Lingueglia E, Lazdunski M, Barbry P. The Phe-Met-Arg-Phe-amide-activated sodium channel is a tetramer. J Biol Chem. 1998;273:8317–8322. doi: 10.1074/jbc.273.14.8317. [DOI] [PubMed] [Google Scholar]
- 107.Costanzo LS, Windhager EE. Renal Tubular Transport of Calcium. Compr Physiol. 2011;(Supplement 25) doi: 10.1002/cphy.cp080236. Handbook of Physiology, Renal Physiology. [DOI] [Google Scholar]
- 108.Coyle B, Coffey R, Armour JA, Gausden E, Hochberg Z, Grossman A, Britton K, Pembrey M, Reardon W, Trembath R. Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4. Nat Genet. 1996;12:421–423. doi: 10.1038/ng0496-421. [DOI] [PubMed] [Google Scholar]
- 109.Crawford I, Maloney PC, Zeitlin PL, Guggino WB, Hyde SC, Turley H, Gatter KC, Harris A, Higgins CF. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc Natl Acad Sci USA. 1991;88:9262–9266. doi: 10.1073/pnas.88.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Crowson MS, Shull GE. Isolation and characterization of a cDNA encoding the putative distal colon H+,K+-ATPase. Similarity of deduced amino acid sequence to gastric H+,K+-ATPase and Na+,K+-ATPase and mRNA expression in distal colon, kidney, and uterus. J Biol Chem. 1992;267:13740–13748. [PubMed] [Google Scholar]
- 111.Da SN, Pisitkun T, Belleannee C, Miller LR, Nelson R, Knepper MA, Brown D, Breton S. Proteomic analysis of V-ATPase-rich cells harvested from the kidney and epididymis by fluorescence-activated cell sorting. Am J Physiol Cell Physiol. 2010;298:C1326–C1342. doi: 10.1152/ajpcell.00552.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996;50:208–218. doi: 10.1038/ki.1996.304. [DOI] [PubMed] [Google Scholar]
- 113.de Groot T, Verkaart S, Xi Q, Bindels RJ, Hoenderop JG. The identification of Histidine 712 as a critical residue for constitutive TRPV5 internalization. J Biol Chem. 2010;285:28481–28487. doi: 10.1074/jbc.M110.117143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 2001;20:7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deen PM, Verdijk MA, Knoers NV, Wieringa B, Monnens LA, van Os CH, van Oost BA. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science. 1994;264:92–95. doi: 10.1126/science.8140421. [DOI] [PubMed] [Google Scholar]
- 116.Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MTF, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Bertheleme JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Ruther U, Schneider-Maunoury S, ttie-Bitach T, Saunier S. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 117.Denker BM, Sabath E. The biology of epithelial cell tight junctions in the kidney. J Am Soc Nephrol. 2011;22:622–625. doi: 10.1681/ASN.2010090922. [DOI] [PubMed] [Google Scholar]
- 118.Derst C, Konrad M, Kockerling A, Karolyi L, Deschenes G, Daut J, Karschin A, Seyberth HW. Mutations in the ROMK gene in antenatal Bartter syndrome are associated with impaired K+ channel function. Biochem Biophys Res Commun. 1997;230:641–645. doi: 10.1006/bbrc.1996.6024. [DOI] [PubMed] [Google Scholar]
- 119.Devuyst O, Guggino WB. Chloride channels in the kidney: lessons learned from knockout animals. Am J Physiol Renal Physiol. 2002;283:F1176–F1191. doi: 10.1152/ajprenal.00184.2002. [DOI] [PubMed] [Google Scholar]
- 120.DiBona DR, Mills JW. Distribution of Na+-pump sites in transporting epithelia. Fed Proc. 1979;38:134–143. [PubMed] [Google Scholar]
- 121.Dietrich A, Chubanov V, Gudermann T. Renal TRPathies. J Am Soc Nephrol. 2010;21:736–744. doi: 10.1681/ASN.2009090948. [DOI] [PubMed] [Google Scholar]
- 122.Dijkink L, Hartog A, van Os CH, Bindels RJ. The epithelial sodium channel (ENaC) is intracellularly located as a tetramer. Pflugers Arch. 2002;444:549–555. doi: 10.1007/s00424-002-0855-4. [DOI] [PubMed] [Google Scholar]
- 123.Dinudom A, Fotia AB, Lefkowitz RJ, Young JA, Kumar S, Cook DI. The kinase Grk2 regulates Nedd4/Nedd4-2-dependent control of epithelial Na+ channels. Proc Natl Acad Sci USA. 2004;101:11886–11890. doi: 10.1073/pnas.0402178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology. 2008;23:104–114. doi: 10.1152/physiol.00037.2007. [DOI] [PubMed] [Google Scholar]
- 125.Doyle DA, Morais CJ, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 126.Du J, Wilson PD. Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol Renal Physiol. 1995;269:C487–C495. doi: 10.1152/ajpcell.1995.269.2.C487. [DOI] [PubMed] [Google Scholar]
- 127.DuBose TD, Jr, Caflisch CR. Effect of selective aldosterone deficiency on acidification in nephron segments of the rat inner medulla. J Clin Invest. 1988;82:1624–1632. doi: 10.1172/JCI113774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.DuBose TD, Jr, Codina J, Burges A, Pressley TA. Regulation of H+-K+-ATPase expression in kidney. Am J Physiol. 1995;269:F500–F507. doi: 10.1152/ajprenal.1995.269.4.F500. [DOI] [PubMed] [Google Scholar]
- 129.Durdagi S, Subbotina J, Lees-Miller J, Guo J, Duff HJ, Noskov SY. Insights into the molecular mechanism of hERG1 channel activation and blockade by drugs. Curr Med Chem. 2010;17:3514–3532. doi: 10.2174/092986710792927886. [DOI] [PubMed] [Google Scholar]
- 130.Eaton DC, Malik B, Bao HF, Yu L, Jain L. Regulation of epithelial sodium channel trafficking by ubiquitination. Proc Am Thorac Soc. 2010;7:54–64. doi: 10.1513/pats.200909-096JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Echevarria M, Windhager EE, Tate SS, Frindt G. Cloning and expression of AQP3, a water channel from the medullary collecting duct of rat kidney. Proc Natl Acad Sci USA. 1994;91:10997–11001. doi: 10.1073/pnas.91.23.10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res. 2011;108:265–272. doi: 10.1161/CIRCRESAHA.110.225888. [DOI] [PubMed] [Google Scholar]
- 133.Edinger RS, Lebowitz J, Li H, Alzamora R, Wang H, Johnson JP, Hallows KR. Functional regulation of the epithelial Na+ channel by IκB kinase-β occurs via phosphorylation of the ubiquitin ligase Nedd4-2. J Biol Chem. 2009;284:150–157. doi: 10.1074/jbc.M807358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.El Moghrabi S, Houillier P, Picard N, Sohet F, Wootla B, Bloch-Faure M, Leviel F, Cheval L, Frische S, Meneton P, Eladari D, Chambrey R. Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci USA. 2010;107:13526–13531. doi: 10.1073/pnas.0913070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eladari D, Chambrey R, Frische S, Vallet M, Edwards A. Pendrin as a regulator of ECF and blood pressure. Curr Opin Nephrol Hypertens. 2009;18:356–362. doi: 10.1097/MNH.0b013e32832c91f4. [DOI] [PubMed] [Google Scholar]
- 136.Embark HM, Bohmer C, Vallon V, Luft F, Lang F. Regulation of KCNE1-dependent K+ current by the serum and glucocorticoid-inducible kinase (SGK) isoforms. Pflugers Arch. 2003;445:601–606. doi: 10.1007/s00424-002-0982-y. [DOI] [PubMed] [Google Scholar]
- 137.Embark HM, Bohmer C, Palmada M, Rajamanickam J, Wyatt AW, Wallisch S, Capasso G, Waldegger P, Seyberth HW, Waldegger S, Lang F. Regulation of CLC-Ka/barttin by the ubiquitin ligase Nedd4-2 and the serum- and glucocorticoid-dependent kinases. Kidney Int. 2004;66:1918–1925. doi: 10.1111/j.1523-1755.2004.00966.x. [DOI] [PubMed] [Google Scholar]
- 138.Eskandari S, Snyder PM, Kreman M, Zampighi GA, Welsh MJ, Wright EM. Number of subunits comprising the epithelial sodium channel. J Biol Chem. 1999;274:27281–27286. doi: 10.1074/jbc.274.38.27281. [DOI] [PubMed] [Google Scholar]
- 139.Estevez R, Boettger T, Stein V, Birkenhager R, Otto E, Hildebrandt F, Jentsch TJ. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- 140.Estilo G, Liu W, Pastor-Soler N, Mitchell P, Carattino MD, Kleyman TR, Satlin LM. Effect of aldosterone on BK channel expression in mammalian cortical collecting duct. Am J Physiol Renal Physiol. 2008;295:F780–F788. doi: 10.1152/ajprenal.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fakitsas P, Adam G, Daidie D, van Bemmelen MX, Fouladkou F, Patrignani A, Wagner U, Warth R, Camargo SM, Staub O, Verrey F. Early aldosterone-induced gene product regulates the epithelial sodium channel by deubiquitylation. J Am Soc Nephrol. 2007;18:1084–1092. doi: 10.1681/ASN.2006080902. [DOI] [PubMed] [Google Scholar]
- 142.Fakler B, Schultz JH, Yang J, Schulte U, Brandle U, Zenner HP, Jan LY, Ruppersberg JP. Identification of a titratable lysine residue that determines sensitivity of kidney potassium channels (ROMK) to intracellular pH. EMBO J. 1996;15:4093–4099. [PMC free article] [PubMed] [Google Scholar]
- 143.Fang L, Garuti R, Kim BY, Wade JB, Welling PA. The ARH adaptor protein regulates endocytosis of the ROMK potassium secretory channel in mouse kidney. J Clin Invest. 2009;119:3278–3289. doi: 10.1172/JCI37950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Farquhar MG, Palade GE. Cell junctions in amphibian skin. J Cell Biol. 1965;26:263–291. doi: 10.1083/jcb.26.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fejes-Toth G, Naray-Fejes-Toth A. Differentiation of intercalated cells in culture. Pediatr Nephrol. 1993;7:780–784. doi: 10.1007/BF01213359. [DOI] [PubMed] [Google Scholar]
- 147.Fejes-Toth G, Frindt G, Naray-Fejes-Toth A, Palmer LG. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol Renal Physiol. 2008;294:F1298–F1305. doi: 10.1152/ajprenal.00579.2007. [DOI] [PubMed] [Google Scholar]
- 148.Fejes-Toth G, Naray-Fejes-Toth A. Isolated principal and intercalated cells: hormone responsiveness and Na+-K+-ATPase activity. Am J Physiol Renal Physiol. 1989;256:F742–F750. doi: 10.1152/ajprenal.1989.256.4.F742. [DOI] [PubMed] [Google Scholar]
- 149.Fenton RA, Knepper MA. Mouse models and the urinary concentrating mechanism in the new millennium. Physiol Rev. 2007;87:1083–1112. doi: 10.1152/physrev.00053.2006. [DOI] [PubMed] [Google Scholar]
- 150.Fenton R. Essential role of vasopressin-regulated urea transport processes in the mammalian kidney. Pflugers Arch. 2009;458:169–177. doi: 10.1007/s00424-008-0612-4. [DOI] [PubMed] [Google Scholar]
- 151.Fenton RA, Praetorius J. Molecular physiology of the medullary collecting duct. Compr Physiol. 2011;1:1031–1056. doi: 10.1002/cphy.c100064. [DOI] [PubMed] [Google Scholar]
- 152.Fernandez-Fernandez JM, Tomas M, Vazquez E, Orio P, Latorre R, Senti M, Marrugat J, Valverde MA. Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J Clin Invest. 2004;113:1032–1039. doi: 10.1172/JCI20347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ferrari P. The role of 11β-hydroxysteroid dehydrogenase type 2 in human hypertension. Biochim Biophys Acta. 2010;1802:1178–1187. doi: 10.1016/j.bbadis.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 154.Ferre S, Hoenderop JG, Bindels RJ. Insight into renal Mg2+ transporters. Curr Opin Nephrol Hypertens. 2011;20:169–176. doi: 10.1097/MNH.0b013e3283435ee4. [DOI] [PubMed] [Google Scholar]
- 155.Ferrera L, Caputo A, Galietta LJ. TMEM16A protein: a new identity for Ca2+-dependent Cl channels. Physiology. 2010;25:357–363. doi: 10.1152/physiol.00030.2010. [DOI] [PubMed] [Google Scholar]
- 156.Firsov D, Gautschi I, Merillat AM, Rossier BC, Schild L. The heterotetrameric architecture of the epithelial sodium channel (ENaC) EMBO J. 1998;17:344–352. doi: 10.1093/emboj/17.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fischer M, Janssen AG, Fahlke C. Barttin activates ClC-K channel function by modulating gating. J Am Soc Nephrol. 2010;21:1281–1289. doi: 10.1681/ASN.2009121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Flagg TP, Tate M, Merot J, Welling PA. A mutation linked with Bartter’s syndrome locks Kir 1.1a (ROMK1) channels in a closed state. J Gen Physiol. 1999;114:685–700. doi: 10.1085/jgp.114.5.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Flagg TP, Yoo D, Sciortino CM, Tate M, Romero MF, Welling PA. Molecular mechanism of a COOH-terminal gating determinant in the ROMK channel revealed by a Bartter’s disease mutation. J Physiol. 2002;544:351–362. doi: 10.1113/jphysiol.2002.027581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Flores SY, Loffing-Cueni D, Kamynina E, Daidie D, Gerbex C, Chabanel S, Dudler J, Loffing J, Staub O. Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J Am Soc Nephrol. 2005;16:2279–2287. doi: 10.1681/ASN.2004100828. [DOI] [PubMed] [Google Scholar]
- 161.Fodstad H, Gonzalez-Rodriguez E, Bron S, Gaeggeler H, Guisan B, Rossier BC, Horisberger JD. Effects of mineralocorticoid and K+ concentration on K+ secretion and ROMK channel expression in a mouse cortical collecting duct cell line. Am J Physiol Renal Physiol. 2009;296:F966–F975. doi: 10.1152/ajprenal.90475.2008. [DOI] [PubMed] [Google Scholar]
- 162.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 163.Frindt G, Palmer LG. Ca-activated K channels in apical membrane of mammalian CCT, and their role in K secretion. Am J Physiol Renal Physiol. 1987;252:F458–F467. doi: 10.1152/ajprenal.1987.252.3.F458. [DOI] [PubMed] [Google Scholar]
- 164.Frindt G, Palmer LG. Apical potassium channels in the rat connecting tubule. Am J Physiol Renal Physiol. 2004;287:F1030–F1037. doi: 10.1152/ajprenal.00169.2004. [DOI] [PubMed] [Google Scholar]
- 165.Frindt G, Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol. 2009;297:F389–F396. doi: 10.1152/ajprenal.90528.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Frindt G, Houde V, Palmer LG. Conservation of Na+ versus K+ by the rat cortical collecting duct. Am J Physiol Renal Physiol. 2011;301:F14–F20. doi: 10.1152/ajprenal.00705.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Frindt G, Shah A, Edvinsson J, Palmer LG. Dietary K regulates ROMK channels in connecting tubule and cortical collecting duct of rat kidney. Am J Physiol Renal Physiol. 2009;296:F347–F354. doi: 10.1152/ajprenal.90527.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- 169.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fushimi K, Uchida S, Hara Y, Hirata Y, Marumo F, Sasaki S. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature. 1993;361:549–552. doi: 10.1038/361549a0. [DOI] [PubMed] [Google Scholar]
- 171.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem. 1997;272:14800–14804. doi: 10.1074/jbc.272.23.14800. [DOI] [PubMed] [Google Scholar]
- 172.Garg LC, Narang N. Effects of aldosterone on NEM-sensitive ATPase in rabbit nephron segments. Kidney Int. 1988;34:13–17. doi: 10.1038/ki.1988.139. [DOI] [PubMed] [Google Scholar]
- 173.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 174.Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, Meinke G, Tsai FT, Sigler PB, Lifton RP. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science. 2000;289:119–123. doi: 10.1126/science.289.5476.119. [DOI] [PubMed] [Google Scholar]
- 175.Giamarchi A, Feng S, Rodat-Despoix L, Xu Y, Bubenshchikova E, Newby LJ, Hao J, Gaudioso C, Crest M, Lupas AN, Honore E, Williamson MP, Obara T, Ong AC, Delmas P. A polycystin-2 (TRPP2) dimerization domain essential for the function of heteromeric polycystin complexes. EMBO J. 2010;29:1176–1191. doi: 10.1038/emboj.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Giamarchi A, Padilla F, Coste B, Raoux M, Crest M, Honore E, Delmas P. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006;7:787–793. doi: 10.1038/sj.embor.7400745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gkika D, Hsu YJ, van der Kemp AW, Christakos S, Bindels RJ, Hoenderop JG. Critical role of the epithelial Ca2+ channel TRPV5 in active Ca2+ reabsorption as revealed by TRPV5/calbindin-D28K knockout mice. J Am Soc Nephrol. 2006;17:3020–3027. doi: 10.1681/ASN.2006060676. [DOI] [PubMed] [Google Scholar]
- 178.Glaudemans B, Knoers NV, Hoenderop JG, Bindels RJ. New molecular players facilitating Mg2+ reabsorption in the distal convoluted tubule. Kidney Int. 2009;77:17–22. doi: 10.1038/ki.2009.358. [DOI] [PubMed] [Google Scholar]
- 179.Gluck SL. Acid sensing in renal epithelial cells. J Clin Invest. 2004;114:1696–1699. doi: 10.1172/JCI23864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gluck SL, Underhill DM, Iyori M, Holliday LS, Kostrominova TY, Lee BS. Physiology and biochemistry of the kidney vacuolar H+-ATPase. Annu Rev Physiol. 1996;58:427–445. doi: 10.1146/annurev.ph.58.030196.002235. [DOI] [PubMed] [Google Scholar]
- 181.Glynn IM. A hundred years of sodium pumping. Annu Rev Physiol. 2002;64:1–18. doi: 10.1146/annurev.physiol.64.081501.130716. [DOI] [PubMed] [Google Scholar]
- 182.Goel M, Sinkins WG, Zuo CD, Estacion M, Schilling WP. Identification and localization of TRPC channels in the rat kidney. Am J Physiol Renal Physiol. 2006;290:F1241–F1252. doi: 10.1152/ajprenal.00376.2005. [DOI] [PubMed] [Google Scholar]
- 183.Goel M, Sinkins WG, Zuo CD, Hopfer U, Schilling WP. Vasopressin-induced membrane trafficking of TRPC3 and AQP2 channels in cells of the rat renal collecting duct. Am J Physiol Renal Physiol. 2007;293:F1476–F1488. doi: 10.1152/ajprenal.00186.2007. [DOI] [PubMed] [Google Scholar]
- 184.Goel M, Zuo CD, Schilling WP. Role of cAMP/PKA signaling cascade in vasopressin-induced trafficking of TRPC3 channels in principal cells of the collecting duct. Am J Physiol Renal Physiol. 2010;298:F988–F996. doi: 10.1152/ajprenal.00586.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Goldstein B, Macara IG. The PAR proteins: fundamental players in animal cell polarization. Dev Cell. 2007;13:609–622. doi: 10.1016/j.devcel.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gomes D, Agasse A, Thiebaud P, Delrot S, Geros H, Chaumont F. Aquaporins are multifunctional water and solute transporters highly divergent in living organisms. Biochim Biophys Acta. 2009;1788:1213–1228. doi: 10.1016/j.bbamem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 187.Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. 2009;460:599–604. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gonzalez-Perrett S, Batelli M, Kim K, Essafi M, Timpanaro G, Moltabetti N, Reisin IL, Arnaout MA, Cantiello HF. Voltage dependence and pH regulation of human polycystin-2-mediated cation channel activity. J Biol Chem. 2002;277:24959–24966. doi: 10.1074/jbc.M105084200. [DOI] [PubMed] [Google Scholar]
- 189.Gonzalez-Perrett S, Kim K, Ibarra C, Damiano AE, Zotta E, Batelli M, Harris PC, Reisin IL, Arnaout MA, Cantiello HF. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci USA. 2001;98:1182–1187. doi: 10.1073/pnas.98.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gonzalez-Rodriguez E, Gaeggeler HP, Rossier BC. IGF-1 vs insulin: respective roles in modulating sodium transport via the PI-3 kinase/Sgk1 pathway in a cortical collecting duct cell line. Kidney Int. 2007;71:116–125. doi: 10.1038/sj.ki.5002018. [DOI] [PubMed] [Google Scholar]
- 191.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. Mineralocorticoids stimulate the activity and expression of renal H+,K+-ATPases. J Am Soc Nephrol. 2011;22:49–58. doi: 10.1681/ASN.2010030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Grimm PR, Foutz RM, Brenner R, Sansom SC. Identification and localization of BK-β subunits in the distal nephron of the mouse kidney. Am J Physiol Renal Physiol. 2007;293:F350–F359. doi: 10.1152/ajprenal.00018.2007. [DOI] [PubMed] [Google Scholar]
- 193.Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, Sansom SC. Hypertension of Kcnmb1−/− is linked to deficient K secretion and aldosteronism. Proc Natl Acad Sci USA. 2009;106:11800–11805. doi: 10.1073/pnas.0904635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Grimm PR, Sansom SC. BK channels in the kidney. Curr Opin Nephrol Hypertens. 2007;16:430–436. doi: 10.1097/MNH.0b013e32826fbc7d. [DOI] [PubMed] [Google Scholar]
- 195.Grimm PR, Sansom SC. BK channels and a new form of hypertension. Kidney Int. 2010;78:956–962. doi: 10.1038/ki.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Grimm PR, Irsik DL, Liu L, Holtzclaw JD, Sansom SC. Role of BKβ1 in Na+ reabsorption by cortical collecting ducts of Na+-deprived mice. Am J Physiol Renal Physiol. 2009;297:F420–F428. doi: 10.1152/ajprenal.00191.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP, Rossier BC. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J. 1997;16:899–907. doi: 10.1093/emboj/16.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol. 2006;7:426–436. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- 199.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol. 2010;298:F12–F21. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hallows KR, Mount PF, Pastor-Soler NM, Power DA. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol. 2010;298:F1067–F1077. doi: 10.1152/ajprenal.00005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 202.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel γ subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11:76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 203.Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. A de novo missense mutation of the β subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci USA. 1995;92:11495–11499. doi: 10.1073/pnas.92.25.11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hara-Chikuma M, Verkman A. Physiological roles of glycerol-transporting aquaporins: the aquaglyceroporins. Cell Mol Life Sci. 2006;63:1386–1392. doi: 10.1007/s00018-006-6028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.He G, Wang HR, Huang SK, Huang CL. Intersectin links WNK kinases to endocytosis of ROMK1. J Clin Invest. 2007;117:1078–1087. doi: 10.1172/JCI30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12:527–532. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 208.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev. 2005;85:319–371. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE. 2009;4:e7771. doi: 10.1371/journal.pone.0007771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Helman SI, Grantham JJ, Burg MB. Effect of vasopressin on electrical resistance of renal cortical collecting tubules. Am J Physiol. 1971;220:1825–1832. doi: 10.1152/ajplegacy.1971.220.6.1825. [DOI] [PubMed] [Google Scholar]
- 211.Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. 2004;279:44065–44073. doi: 10.1074/jbc.M405985200. [DOI] [PubMed] [Google Scholar]
- 212.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 213.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 214.Hoenderop JG, Muller D, van der Kemp AW, Hartog A, Suzuki M, Ishibashi K, Imai M, Sweep F, Willems PH, van Os CH, Bindels RJ. Calcitriol controls the epithelial calcium channel in kidney. J Am Soc Nephrol. 2001;12:1342–1349. doi: 10.1681/ASN.V1271342. [DOI] [PubMed] [Google Scholar]
- 215.Hoenderop JG, Nilius B, Bindels RJ. Calcium absorption across epithelia. Physiol Rev. 2005;85:373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- 216.Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, Merillat AM, Waarsing JH, Rossier BC, Vallon V, Hummler E, Bindels RJ. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest. 2003;112:1906–1914. doi: 10.1172/JCI19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hoenderop JG, Voets T, Hoefs S, Weidema F, Prenen J, Nilius B, Bindels RJ. Homo- and heterotetrameric architecture of the epithelial Ca2+ channels TRPV5 and TRPV6. EMBO J. 2003;22:776–785. doi: 10.1093/emboj/cdg080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Holtzclaw JD, Grimm PR, Sansom SC. Intercalated cell BK-α/β4 channels modulate sodium and potassium handling during potassium adaptation. J Am Soc Nephrol. 2010;21:634–645. doi: 10.1681/ASN.2009080817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol. 2011;22:605–614. doi: 10.1681/ASN.2010080827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci USA. 2010;107:18010–18015. doi: 10.1073/pnas.1009399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hou J, Gomes AS, Paul DL, Goodenough DA. Study of claudin function by RNA interference. J Biol Chem. 2006;281:36117–36123. doi: 10.1074/jbc.M608853200. [DOI] [PubMed] [Google Scholar]
- 222.Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci USA. 2010;107:18010–18015. doi: 10.1073/pnas.1009399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hoy WE, Hughson MD, Bertram JF, Douglas-Denton R, Amann K. Nephron number, hypertension, renal disease, and renal failure. J Am Soc Nephrol. 2005;16:2557–2564. doi: 10.1681/ASN.2005020172. [DOI] [PubMed] [Google Scholar]
- 224.Hu MC, Kuro-o M, Moe OW. Klotho and kidney disease. J Nephrol. 2010;23:S136–S144. [PMC free article] [PubMed] [Google Scholar]
- 225.Hu MC, Shi M, Zhang J, Quinones H, Griffith C, Kuro-o M, Moe OW. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124–136. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Huang CL, Kuo E. Mechanisms of disease: WNK-ing at the mechanism of salt-sensitive hypertension. Nat Clin Pract Nephrol. 2007;3:623–630. doi: 10.1038/ncpneph0638. [DOI] [PubMed] [Google Scholar]
- 227.Huang CL. Regulation of ion channels by secreted Klotho: mechanisms and implications. Kidney Int. 2010;77:855–860. doi: 10.1038/ki.2010.73. [DOI] [PubMed] [Google Scholar]
- 228.Huang DY, Wulff P, Volkl H, Loffing J, Richter K, Kuhl D, Lang F, Vallon V. Impaired regulation of renal K+ elimination in the sgk1-knockout mouse. J Am Soc Nephrol. 2004;15:885–891. doi: 10.1097/01.asn.0000120368.59693.a8. [DOI] [PubMed] [Google Scholar]
- 229.Huang P, Liu J, Di A, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ. Regulation of human CLC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 2001;276:20093–20100. doi: 10.1074/jbc.M009376200. [DOI] [PubMed] [Google Scholar]
- 230.Huber R, Krueger B, Diakov A, Korbmacher J, Haerteis S, Einsiedel J, Gmeiner P, Azad AK, Cuppens H, Cassiman JJ, Korbmacher C, Rauh R. Functional characterization of a partial loss-of-function mutation of the epithelial sodium channel (ENaC) associated with atypical cystic fibrosis. Cell Physiol Biochem. 2010;25:145–158. doi: 10.1159/000272059. [DOI] [PubMed] [Google Scholar]
- 231.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 232.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem. 2004;279:18111–18114. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 233.Hughey RP, Carattino MD, Kleyman TR. Role of proteolysis in the activation of epithelial sodium channels. Curr Opin Nephrol Hypertens. 2007;16:444–450. doi: 10.1097/MNH.0b013e32821f6072. [DOI] [PubMed] [Google Scholar]
- 234.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in α-ENaC-deficient mice. Nat Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 235.Hummler E, Horisberger JD. Genetic disorders of membrane transport. V. The epithelial sodium channel and its implication in human diseases. Am J Physiol Gastrointest Liver Physiol. 1999;276:G567–G571. doi: 10.1152/ajpgi.1999.276.3.G567. [DOI] [PubMed] [Google Scholar]
- 236.Hunter M, Lopes AG, Boulpaep EL, Giebisch GH. Single channel recordings of calcium-activated potassium channels in the apical membrane of rabbit cortical collecting tubules. Proc Natl Acad Sci USA. 1984;81:4237–4239. doi: 10.1073/pnas.81.13.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Husted RF, Volk KA, Sigmund RD, Stokes JB. Anion secretion by the inner medullary collecting duct. Evidence for involvement of the cystic fibrosis transmembrane conductance regulator. J Clin Invest. 1995;95:644–650. doi: 10.1172/JCI117709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Iden S, Collard JG. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol. 2008;9:846–859. doi: 10.1038/nrm2521. [DOI] [PubMed] [Google Scholar]
- 239.ikhani-Koupaei R, Fouladkou F, Fustier P, Cenni B, Sharma AM, Deter HC, Frey BM, Frey FJ. Identification of polymorphisms in the human 11beta-hydroxysteroid dehydrogenase type 2 gene promoter: functional characterization and relevance for salt sensitivity. FASEB J. 2007;21:3618–3628. doi: 10.1096/fj.07-8140com. [DOI] [PubMed] [Google Scholar]
- 240.Ilatovskaya DV, Pavlov TS, Levchenko V, Negulyaev YA, Staruschenko A. Cortical actin binding protein cortactin mediates ENaC activity via Arp2/3 complex. FASEB J. 2011;25:2688–2699. doi: 10.1096/fj.10-167262. [DOI] [PubMed] [Google Scholar]
- 241.Imamura H, Nakano M, Noji H, Muneyuki E, Ohkuma S, Yoshida M, Yokoyama K. Evidence for rotation of V1-ATPase. Proc Natl Acad Sci USA. 2003;100:2312–2315. doi: 10.1073/pnas.0436796100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Ishibashi K, Sasaki S, Fushimi K, Yamamoto T, Kuwahara M, Marumo F. Immunolocalization and effect of dehydration on AQP3, a basolateral water channel of kidney collecting ducts. Am J Physiol Renal Physiol. 1997;272:F235–F241. doi: 10.1152/ajprenal.1997.272.2.F235. [DOI] [PubMed] [Google Scholar]
- 243.Ishibashi K, Kondo S, Hara S, Morishita Y. The evolutionary aspects of aquaporin family. Am J Physiol Regul Integr Comp Physiol. 2011;300:R566–R576. doi: 10.1152/ajpregu.90464.2008. [DOI] [PubMed] [Google Scholar]
- 244.Ishikawa T, Marunaka Y, Rotin D. Electrophysiological characterization of the rat epithelial Na+ channel (rENaC) expressed in MDCK cells. Effects of Na+ and Ca2+ J Gen Physiol. 1998;111:825–846. doi: 10.1085/jgp.111.6.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Itani OA, Chen JH, Karp PH, Ernst S, Keshavjee S, Parekh K, Klesney-Tait J, Zabner J, Welsh MJ. Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. Proc Natl Acad Sci USA. 2011;108:10260–10265. doi: 10.1073/pnas.1106695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, Tomoike H, Kurachi Y. Immunolocalization of an inwardly rectifying K+ channel, KAB-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett. 1996;388:11–15. doi: 10.1016/0014-5793(96)00502-9. [DOI] [PubMed] [Google Scholar]
- 247.Janssen AG, Scholl U, Domeyer C, Nothmann D, Leinenweber A, Fahlke C. Disease-causing dysfunctions of barttin in Bartter syndrome type IV. J Am Soc Nephrol. 2009;20:145–153. doi: 10.1681/ASN.2008010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–323. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 249.Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl− channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- 250.Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baro I, Abriel H. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res. 2007;74:64–74. doi: 10.1016/j.cardiores.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 251.Ji HL, Chalfant ML, Jovov B, Lockhart JP, Parker SB, Fuller CM, Stanton BA, Benos DJ. The cytosolic termini of the β- and γ-ENaC subunits are involved in the functional interactions between cystic fibrosis transmembrane conductance regulator and epithelial sodium channel. J Biol Chem. 2000;275:27947–27956. doi: 10.1074/jbc.M002848200. [DOI] [PubMed] [Google Scholar]
- 252.Jiang Q, Li J, Dubroff R, Ahn YJ, Foskett JK, Engelhardt J, Kleyman TR. Epithelial sodium channels regulate cystic fibrosis transmembrane conductance regulator chloride channels in Xenopus oocytes. J Biol Chem. 2000;275:13266–13274. doi: 10.1074/jbc.275.18.13266. [DOI] [PubMed] [Google Scholar]
- 253.Jin Y, Wang Z, Zhang Y, Yang B, Wang WH. PGE2 inhibits apical K channels in the CCD through activation of the MAPK pathway. Am J Physiol Renal Physiol. 2007;293:F1299–F1307. doi: 10.1152/ajprenal.00293.2007. [DOI] [PubMed] [Google Scholar]
- 254.Jin Y, Wang Y, Wang ZJ, Lin DH, Wang WH. Inhibition of angiotensin type 1 receptor impairs renal ability of K conservation in response to K restriction. Am J Physiol Renal Physiol. 2009;296:F1179–F1184. doi: 10.1152/ajprenal.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Jouret F, Devuyst O. CFTR and defective endocytosis: new insights in the renal phenotype of cystic fibrosis. Pflugers Arch. 2009;457:1227–1236. doi: 10.1007/s00424-008-0594-2. [DOI] [PubMed] [Google Scholar]
- 256.Kabra R, Knight KK, Zhou R, Snyder PM. Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J Biol Chem. 2008;283:6033–6039. doi: 10.1074/jbc.M708555200. [DOI] [PubMed] [Google Scholar]
- 257.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 258.Kahle KT, MacGregor GG, Wilson FH, Van Hoek AN, Brown D, Ardito T, Kashgarian M, Giebisch G, Hebert SC, Boulpaep EL, Lifton RP. Paracellular Cl− permeability is regulated by WNK4 kinase: Insight into normal physiology and hypertension. Proc Natl Acad Sci USA. 2004;101:14877–14882. doi: 10.1073/pnas.0406172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kaissling B, Kriz W. Morphology of the loop of Henle, distal tubule, and collecting duct. Compr Physiol. 2011 doi: 10.1002/cphy.cp080103. Handbook of Physiology, Supplement 25: Renal Physiology. [DOI] [Google Scholar]
- 260.Kamsteeg EJ, Heijnen I, van Os CH, Deen PMT. The subcellular localization of an aquaporin-2 tetramer depends on the stoichiometry of phosphorylated and nonphosphorylated monomers. J Cell Biol. 2000;151:919–930. doi: 10.1083/jcb.151.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Karet FE, Finberg KE, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Medina JF, Lifton RP. Localization of a gene for autosomal recessive distal renal tubular acidosis with normal hearing (rdRTA2) to 7q33–34. Am J Hum Genet. 1999;65:1656–1665. doi: 10.1086/302679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di PA, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 263.Karet FE, Gainza FJ, Gyory AZ, Unwin RJ, Wrong O, Tanner MJ, Nayir A, Alpay H, Santos F, Hulton SA, Bakkaloglu A, Ozen S, Cunningham MJ, Di PA, Walker WG, Lifton RP. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci USA. 1998;95:6337–6342. doi: 10.1073/pnas.95.11.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Karpushev A, Pavlov T, Staruschenko A. Regulation of the epithelial sodium channels (ENaC) by small G proteins and phosphatidylinositides. Biol Membrany. 2009;26:265–279. [Google Scholar]
- 265.Karpushev AV, Ilatovskaya DV, Staruschenko A. The actin cytoskeleton and small G protein RhoA are not involved in flow-dependent activation of ENaC. BMC Res Notes. 2010;3:210. doi: 10.1186/1756-0500-3-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Karpushev AV, Levchenko V, Pavlov TS, Lam VY, Vinnakota KC, Vandewalle A, Wakatsuki T, Staruschenko A. Regulation of ENaC expression at the cell surface by Rab11. Biochem Biophys Res Commun. 2008;377:521–525. doi: 10.1016/j.bbrc.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Karpushev AV, Levchenko V, Ilatovskaya DV, Pavlov TS, Staruschenko A. Novel role of Rac1/WAVE signaling mechanism in regulation of the epithelial Na+ channel. Hypertension. 2011;57:996–1002. doi: 10.1161/HYPERTENSIONAHA.110.157784. [DOI] [PubMed] [Google Scholar]
- 268.Kashlan OB, Kleyman TR. ENaC structure and function in the wake of a resolved structure of a family member. Am J Physiol Renal Physiol. 2011 doi: 10.1152/ajprenal.00259.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Katsura T, Gustafson CE, Ausiello DA, Brown D. Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am J Physiol Renal Physiol. 1997;272:F816–F822. [PubMed] [Google Scholar]
- 270.Katsuyama M, Masuyama T, Komura I, Hibino T, Takahashi H. Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp Anim. 2000;49:51–55. doi: 10.1538/expanim.49.51. [DOI] [PubMed] [Google Scholar]
- 271.Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–108. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- 272.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 273.Kieferle S, Fong P, Bens M, Vandewalle A, Jentsch TJ. Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc Natl Acad Sci USA. 1994;91:6943–6947. doi: 10.1073/pnas.91.15.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kim J, Kim YH, Cha JH, Tisher CC, Madsen KM. Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J Am Soc Nephrol. 1999;10:1–12. doi: 10.1681/ASN.V1011. [DOI] [PubMed] [Google Scholar]
- 275.Kim J, Tisher CC, Madsen KM. Differentiation of intercalated cells in developing rat kidney: an immunohistochemical study. Am J Physiol Renal Physiol. 1994;266:F977–F990. doi: 10.1152/ajprenal.1994.266.6.F977. [DOI] [PubMed] [Google Scholar]
- 276.Kim SW, Gresz V, Rojek A, Wang W, Verkman AS, Frokiaer J, Nielsen S. Decreased expression of AQP2 and AQP4 water channels and Na,K-ATPase in kidney collecting duct in AQP3 null mice. Biol Cell. 2005;97:765–778. doi: 10.1042/BC20040148. [DOI] [PubMed] [Google Scholar]
- 277.Kim YH, Pech V, Spencer KB, Beierwaltes WH, Everett LA, Green ED, Shin W, Verlander JW, Sutliff RL, Wall SM. Reduced ENaC protein abundance contributes to the lower blood pressure observed in pendrin-null mice. Am J Physiol Renal Physiol. 2007;293:F1314–F1324. doi: 10.1152/ajprenal.00155.2007. [DOI] [PubMed] [Google Scholar]
- 278.Kirk A, Campbell S, Bass P, Mason J, Collins J. Differential expression of claudin tight junction proteins in the human cortical nephron. Nephrol Dial Transplant. 2010;25:2107–2119. doi: 10.1093/ndt/gfq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kiselyov K, Patterson RL. The integrative function of TRPC channels. Front Biosci. 2009;14:45–58. doi: 10.2741/3230. [DOI] [PubMed] [Google Scholar]
- 280.Kishore BK, Chou CL, Knepper MA. Extracellular nucleotide receptor inhibits AVP-stimulated water permeability in inner medullary collecting duct. Am J Physiol Renal Physiol. 1995;269:F863–F869. doi: 10.1152/ajprenal.1995.269.6.F863. [DOI] [PubMed] [Google Scholar]
- 281.Kitamura K, Tomita K. Regulation of renal sodium handling through the interaction between serine proteases and serine protease inhibitors. Clin Exp Nephrol. 2010;14:405–410. doi: 10.1007/s10157-010-0299-7. [DOI] [PubMed] [Google Scholar]
- 282.Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J Am Soc Nephrol. 2002;13:875–886. doi: 10.1681/ASN.V134875. [DOI] [PubMed] [Google Scholar]
- 283.Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem. 2009;284:20447–20451. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Knepper MA. Proteomics and the kidney. J Am Soc Nephrol. 2002;13:1398–1408. doi: 10.1097/01.asn.0000014782.37591.c7. [DOI] [PubMed] [Google Scholar]
- 285.Knepper MA, Packer R, Good DW. Ammonium transport in the kidney. Physiol Rev. 1989;69:179–249. doi: 10.1152/physrev.1989.69.1.179. [DOI] [PubMed] [Google Scholar]
- 286.Knepper MA, Star RA. The vasopressin-regulated urea transporter in renal inner medullary collecting duct. Am J Physiol Renal Physiol. 1990;259:F393–F401. doi: 10.1152/ajprenal.1990.259.3.F393. [DOI] [PubMed] [Google Scholar]
- 287.Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, Kim JY, Kim KH, Lee MG, Naruse S, Muallem S. A molecular mechanism for aberrant CFTR-dependent HCO3− transport in cystic fibrosis. EMBO J. 2002;21:5662–5672. doi: 10.1093/emboj/cdf580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kobayashi K, Uchida S, Okamura HO, Marumo F, Sasaki S. Human CLC-KB gene promoter drives the EGFP expression in the specific distal nephron segments and inner ear. J Am Soc Nephrol. 2002;13:1992–1998. doi: 10.1097/01.asn.0000023434.47132.3d. [DOI] [PubMed] [Google Scholar]
- 289.Kobayashi K, Uchida S, Mizutani S, Sasaki S, Marumo F. Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J Am Soc Nephrol. 2001;12:1327–1334. doi: 10.1681/ASN.V1271327. [DOI] [PubMed] [Google Scholar]
- 290.Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kohda Y, Ding W, Phan E, Housini I, Wang J, Star RA, Huang CL. Localization of the ROMK potassium channel to the apical membrane of distal nephron in rat kidney. Kidney Int. 1998;54:1214–1223. doi: 10.1046/j.1523-1755.1998.00120.x. [DOI] [PubMed] [Google Scholar]
- 292.Kondo C, Isomoto S, Matsumoto S, Yamada M, Horio Y, Yamashita S, Takemura-Kameda K, Matsuzawa Y, Kurachi Y. Cloning and functional expression of a novel isoform of ROMK inwardly rectifying ATP-dependent K+ channel, ROMK6 (Kir1.1f) FEBS Lett. 1996;399:122–126. doi: 10.1016/s0014-5793(96)01302-6. [DOI] [PubMed] [Google Scholar]
- 293.Kosari F, Sheng S, Li J, Mak DO, Foskett JK, Kleyman TR. Subunit stoichiometry of the epithelial sodium channel. J Biol Chem. 1998;273:13469–13474. doi: 10.1074/jbc.273.22.13469. [DOI] [PubMed] [Google Scholar]
- 294.Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, Seckl JR, Mullins JJ. Hypertension in mice lacking 11β-hydroxysteroid dehydrogenase type 2. J Clin Invest. 1999;103:683–689. doi: 10.1172/JCI4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 297.Kovacikova J, Winter C, Loffing-Cueni D, Loffing J, Finberg KE, Lifton RP, Hummler E, Rossier B, Wagner CA. The connecting tubule is the main site of the furosemide-induced urinary acidification by the vacuolar H+-ATPase. Kidney Int. 2006;70:1706–1716. doi: 10.1038/sj.ki.5001851. [DOI] [PubMed] [Google Scholar]
- 298.Kramer BK, Bergler T, Stoelcker B, Waldegger S. Mechanisms of disease: the kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat Clin Pract Neph. 2008;4:38–46. doi: 10.1038/ncpneph0689. [DOI] [PubMed] [Google Scholar]
- 299.Kunau RT, Jr, Webb HL, Borman SC. Characteristics of the relationship between the flow rate of tubular fluid and potassium transport in the distal tubule of the rat. J Clin Invest. 1974;54:1488–1495. doi: 10.1172/JCI107897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Kunzelmann K, Schreiber R, Nitschke R, Mall M. Control of epithelial Na+ conductance by the cystic fibrosis transmembrane conductance regulator. Pflugers Arch. 2000;440:193–201. doi: 10.1007/s004240000255. [DOI] [PubMed] [Google Scholar]
- 301.Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 302.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Yi. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 303.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel Sp, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol. 2008;294:F1398–F1407. doi: 10.1152/ajprenal.00288.2007. [DOI] [PubMed] [Google Scholar]
- 304.Lager DJ, Qian Q, Bengal RJ, Ishibashi M, Torres VE. The pck rat: A new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001;59:126–136. doi: 10.1046/j.1523-1755.2001.00473.x. [DOI] [PubMed] [Google Scholar]
- 305.Lambers TT, Oancea E, de Groot T, Topala CN, Hoenderop JG, Bindels RJ. Extracellular pH dynamically controls cell surface delivery of functional TRPV5 channels. Mol Cell Biol. 2007;27:1486–1494. doi: 10.1128/MCB.01468-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Lang F, Huang DY, Vallon V. SGK, renal function and hypertension. J Nephrol. 2010;23:S124–S129. [PMC free article] [PubMed] [Google Scholar]
- 307.Le Moellic C, Boulkroun S, Gonzalez-Nunez D, Dublineau I, Cluzeaud F, Fay M, Blot-Chabaud M, Farman N. Aldosterone and tight junctions: modulation of claudin-4 phosphorylation in renal collecting duct cells. Am J Physiol Cell Physiol. 2005;289:C1513–C1521. doi: 10.1152/ajpcell.00314.2005. [DOI] [PubMed] [Google Scholar]
- 308.Leduc-Nadeau A, Lussier Y, Arthus MF, Lonergan Ml, Martinez-Aguayo A, Riveira-Munoz E, Devuyst O, Bissonnette P, Bichet DG. New autosomal recessive mutations in aquaporin-2 causing nephrogenic diabetes insipidus through deficient targeting display normal expression in Xenopus oocytes. J Physiol. 2010;588:2205–2218. doi: 10.1113/jphysiol.2010.187674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Lee IH, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J Biol Chem. 2007;282:29866–29873. doi: 10.1074/jbc.M701923200. [DOI] [PubMed] [Google Scholar]
- 310.Lee US, Cui J. BK channel activation: structural and functional insights. Trends Neurosci. 2010;33:415–423. doi: 10.1016/j.tins.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Lee WS, Hebert SC. ROMK inwardly rectifying ATP-sensitive K+ channel. I. Expression in rat distal nephron segments. Am J Physiol Renal Physiol. 1995;268:F1124–F1131. doi: 10.1152/ajprenal.1995.268.6.F1124. [DOI] [PubMed] [Google Scholar]
- 312.Leng Q, MacGregor GG, Dong K, Giebisch G, Hebert SC. Subunit-subunit interactions are critical for proton sensitivity of ROMK: evidence in support of an intermolecular gating mechanism. Proc Natl Acad Sci USA. 2006;103:1982–1987. doi: 10.1073/pnas.0510610103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Letz B, Korbmacher C. cAMP stimulates CFTR-like Cl− channels and inhibits amiloride-sensitive Na+ channels in mouse CCD cells. Am J Physiol Cell Physiol. 1997;272:C657–C666. doi: 10.1152/ajpcell.1997.272.2.C657. [DOI] [PubMed] [Google Scholar]
- 314.Levchenko V, Zheleznova NN, Pavlov TS, Vandewalle A, Wilson PD, Staruschenko A. EGF and its related growth factors mediate sodium transport in mpkCCDc14 cells via ErbB2 (neu/HER-2) receptor. J Cell Physiol. 2010;223:252–259. doi: 10.1002/jcp.22033. [DOI] [PubMed] [Google Scholar]
- 315.Leviel F, Hubner CA, Houillier P, Morla L, El MS, Brideau G, Hatim H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest. 2010;120:1627–1635. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Li C, Wang W, Rivard CJ, Lanaspa MA, Summer S, Schrier RW. Molecular mechanisms of angiotensin II stimulation on aquaporin-2 expression and trafficking. Am J Physiol Renal Physiol. 2011;300:F1255–F1261. doi: 10.1152/ajprenal.00469.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Li D, Wang Z, Sun P, Jin Y, Lin DH, Hebert SC, Giebisch G, Wang WH. Inhibition of MAPK stimulates the Ca2+ -dependent big-conductance K channels in cortical collecting duct. Proc Natl Acad Sci USA. 2006;103:19569–19574. doi: 10.1073/pnas.0609555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Li J, Ananthapanyasut W, Yu AS. Claudins in renal physiology and disease. Pediatr Nephrol. 2011 doi: 10.1007/s00467-011-1824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Li L, Schafer JA. Dopamine inhibits vasopressin-dependent cAMP production in the rat cortical collecting duct. Am J Physiol Renal Physiol. 1998;275:F62–F67. doi: 10.1152/ajprenal.1998.275.1.F62. [DOI] [PubMed] [Google Scholar]
- 320.Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2003;100:14531–14536. doi: 10.1073/pnas.2235619100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Lifton RP. Genetic determinants of human hypertension. Proc Natl Acad Sci USA. 1995;92:8545–8551. doi: 10.1073/pnas.92.19.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 323.Lin D, Sterling H, Lerea KM, Giebisch G, Wang WH. Protein kinase C (PKC)-induced phosphorylation of ROMK1 is essential for the surface expression of ROMK1 channels. J Biol Chem. 2002;277:44278–44284. doi: 10.1074/jbc.M203702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Lin DH, Yue P, Pan C, Sun P, Wang WH. MicroRNA 802 stimulates ROMK channels by suppressing caveolin-1. J Am Soc Nephrol. 2011;22:1087–1098. doi: 10.1681/ASN.2010090927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Lin DH, Yue P, Pan CY, Sun P, Zhang X, Han Z, Roos M, Caplan M, Giebisch G, Wang WH. POSH stimulates the ubiquitination and the clathrin-independent endocytosis of ROMK1 channels. J Biol Chem. 2009;284:29614–29624. doi: 10.1074/jbc.M109.041582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ling BN, Zuckerman JB, Lin C, Harte BJ, McNulty KA, Smith PR, Gomez LM, Worrell RT, Eaton DC, Kleyman TR. Expression of the cystic fibrosis phenotype in a renal amphibian epithelial cell line. J Biol Chem. 1997;272:594–600. doi: 10.1074/jbc.272.1.594. [DOI] [PubMed] [Google Scholar]
- 327.Liu L, Duke BJ, Malik B, Yue Q, Eaton DC. Biphasic regulation of ENaC by TGF-α and EGF in renal epithelial cells. Am J Physiol Renal Physiol. 2009;296:F1417–F1427. doi: 10.1152/ajprenal.90337.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Liu W, Morimoto T, Woda C, Kleyman TR, Satlin LM. Ca2+ dependence of flow-stimulated K secretion in the mammalian cortical collecting duct. Am J Physiol Renal Physiol. 2007;293:F227–F235. doi: 10.1152/ajprenal.00057.2007. [DOI] [PubMed] [Google Scholar]
- 329.Liu W, Wei Y, Sun P, Wang WH, Kleyman TR, Satlin LM. Mechanoregulation of BK channel activity in the mammalian cortical collecting duct: role of protein kinases A and C. Am J Physiol Renal Physiol. 2009;297:F904–F915. doi: 10.1152/ajprenal.90685.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM. Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol. 2003;285:F998–F1012. doi: 10.1152/ajprenal.00067.2003. [DOI] [PubMed] [Google Scholar]
- 331.Liu Z, Wang HR, Huang CL. Regulation of ROMK channel and K+ homeostasis by kidney-specific WNK1 kinase. J Biol Chem. 2009;284:12198–12206. doi: 10.1074/jbc.M806551200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Lloyd DJ, Hall FW, Tarantino LM, Gekakis N. Diabetes insipidus in mice with a mutation in aquaporin-2. PLoS Genet. 2005;1:e20. doi: 10.1371/journal.pgen.0010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Loffing J, Kaissling B. Sodium and calcium transport pathways along the mammalian distal nephron: from rabbit to human. Am J Physiol Renal Physiol. 2003;284:F628–F643. doi: 10.1152/ajprenal.00217.2002. [DOI] [PubMed] [Google Scholar]
- 334.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflugers Arch. 2009;458:111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- 335.Loffing J, Loffing-Cueni D, Valderrabano V, Klausli L, Hebert SC, Rossier BC, Hoenderop JG, Bindels RJ, Kaissling B. Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol Renal Physiol. 2001;281:F1021–F1027. doi: 10.1152/ajprenal.0085.2001. [DOI] [PubMed] [Google Scholar]
- 336.Lolait SJ, O’Carroll AM, McBride OW, Konig M, Morel A, Brownstein MJ. Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature. 1992;357:336–339. doi: 10.1038/357336a0. [DOI] [PubMed] [Google Scholar]
- 337.Long JF, Chiu PJ, Derelanko MJ, Steinberg M. Gastric antisecretory and cytoprotective activities of SCH 28080. J Pharmacol Exp Ther. 1983;226:114–120. [PubMed] [Google Scholar]
- 338.Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 2002;34:933–944. doi: 10.1016/s0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- 339.Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J. An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol. 2002;538:391–404. doi: 10.1113/jphysiol.2001.012961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Lu M, Leng Q, Egan ME, Caplan MJ, Boulpaep EL, Giebisch GH, Hebert SC. CFTR is required for PKA-regulated ATP sensitivity of Kir1.1 potassium channels in mouse kidney. J Clin Invest. 2006;116:797–807. doi: 10.1172/JCI26961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lu M, Dong K, Egan ME, Giebisch GH, Boulpaep EL, Hebert SC. Mouse cystic fibrosis transmembrane conductance regulator forms cAMP-PKA-regulated apical chloride channels in cortical collecting duct. Proc Natl Acad Sci USA. 2010;107:6082–6087. doi: 10.1073/pnas.0902661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Lu M, Wang T, Yan Q, Yang X, Dong K, Knepper MA, Wang W, Giebisch G, Shull GE, Hebert SC. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. J Biol Chem. 2002;277:37881–37887. doi: 10.1074/jbc.M206644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lu P, Boros S, Chang Q, Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho exclusively activates the epithelial Ca2+ channels TRPV5 and TRPV6. Nephrol Dial Transplant. 2008;23:3397–3402. doi: 10.1093/ndt/gfn291. [DOI] [PubMed] [Google Scholar]
- 344.Luo Y, Vassilev PM, Li X, Kawanabe Y, Zhou J. Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol Cell Biol. 2003;23:2600–2607. doi: 10.1128/MCB.23.7.2600-2607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Lutken SC, Kim SW, Jonassen T, Marples D, Knepper MA, Kwon TH, Frøkiaer, Nielsen S. Changes of renal AQP2, ENaC, and NHE3 in experimentally induced heart failure: Response to angiotensin II AT1 receptor blockade. Am J Physiol Renal Physiol. 2009;297:F1678–F1688. doi: 10.1152/ajprenal.00010.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Lynch IJ, Rudin A, Xia SL, Stow LR, Shull GE, Weiner ID, Cain BD, Wingo CS. Impaired acid secretion in cortical collecting duct intercalated cells from H-K-ATPase-deficient mice: role of HKa isoforms. Am J Physiol Renal Physiol. 2008;294:F621–F627. doi: 10.1152/ajprenal.00412.2007. [DOI] [PubMed] [Google Scholar]
- 347.Ma HP, Eaton DC. Acute regulation of epithelial sodium channel by anionic phospholipids. J Am Soc Nephrol. 2005;16:3182–3187. doi: 10.1681/ASN.2005040434. [DOI] [PubMed] [Google Scholar]
- 348.Ma HP, Saxena S, Warnock DG. Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC) J Biol Chem. 2002;277:7641–7644. doi: 10.1074/jbc.C100737200. [DOI] [PubMed] [Google Scholar]
- 349.Ma T, Frigeri A, Hasegawa H, Verkman AS. Cloning of a water channel homolog expressed in brain meningeal cells and kidney collecting duct that functions as a stilbene-sensitive glycerol transporter. J Biol Chem. 1994;269:21845–21849. [PubMed] [Google Scholar]
- 350.MacKnight ADC. Ion and Water Transport in Toad Urinary Epithelia in Vitro. Compr Physiol. 2011 doi: 10.1002/cphy.cp080107. Handbook of Physiology, Supplement 25: Renal Physiology. [DOI] [Google Scholar]
- 351.Madsen KM, Tisher CC. Structural-functional relationships along the distal nephron. Am J Physiol Renal Physiol. 1986;250:F1–F15. doi: 10.1152/ajprenal.1986.250.1.F1. [DOI] [PubMed] [Google Scholar]
- 352.Madsen KM, Verlander JW, Tisher CC. Relationship between structure and function in distal tubule and collecting duct. J Electron Microsc Tech. 1988;9:187–208. doi: 10.1002/jemt.1060090206. [DOI] [PubMed] [Google Scholar]
- 353.Malik B, Price SR, Mitch WE, Yue Q, Eaton DC. Regulation of epithelial sodium channels by the ubiquitin-proteasome proteolytic pathway. Am J Physiol Renal Physiol. 2006;290:F1285–F1294. doi: 10.1152/ajprenal.00432.2005. [DOI] [PubMed] [Google Scholar]
- 354.Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 355.Mamenko M, Zaika O, Jin M, O’Neil RG, Pochynyuk O. Purinergic activation of Ca2+-permeable TRPV4 channels is essential for mechano-sensitivity in the aldosterone-sensitive distal nephron. PLoS ONE. 2011;6:e22824. doi: 10.1371/journal.pone.0022824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Markadieu N, Bindels RJ, Hoenderop JG. The renal connecting tubule: Resolved and unresolved issues in Ca2+ transport. Int J Biochem Cell Biol. 2011;43:1–4. doi: 10.1016/j.biocel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 357.Markadieu N, Crutzen R, Blero D, Erneux C, Beauwens R. Hydrogen peroxide and epidermal growth factor activate phosphatidylinositol 3-kinase and increase sodium transport in A6 cell monolayers. Am J Physiol Renal Physiol. 2005;288:F1201–F1212. doi: 10.1152/ajprenal.00383.2004. [DOI] [PubMed] [Google Scholar]
- 358.Marsy S, Elalouf JM, Doucet A. Quantitative RT-PCR analysis of mRNAs encoding a colonic putative H,K-ATPase α subunit along the rat nephron: effect of K+ depletion. Pflugers Arch. 1996;432:494–500. doi: 10.1007/s004240050161. [DOI] [PubMed] [Google Scholar]
- 359.McCormick JA, Bhalla V, Pao AC, Pearce D. SGK1: a rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology. 2005;20:134–139. doi: 10.1152/physiol.00053.2004. [DOI] [PubMed] [Google Scholar]
- 360.McDill BW, Li SZ, Kovach PA, Ding L, Chen F. Congenital progressive hydronephrosis (cph) is caused by an S256L mutation in aquaporin-2 that affects its phosphorylation and apical membrane accumulation. Proc Natl Acad Sci USA. 2006;103:6952–6957. doi: 10.1073/pnas.0602087103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB, Jr, Stokes JB, Welsh MJ, Williamson RA. Disruption of the beta subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proc Natl Acad Sci USA. 1999;96:1727–1731. doi: 10.1073/pnas.96.4.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.McNicholas CM, Canessa CM. Diversity of channels generated by different combinations of epithelial sodium channel subunits. J Gen Physiol. 1997;109:681–692. doi: 10.1085/jgp.109.6.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.McNicholas CM, MacGregor GG, Islas LD, Yang Y, Hebert SC, Giebisch G. pH-dependent modulation of the cloned renal K+ channel, ROMK. Am J Physiol Renal Physiol. 1998;275:F972–F981. doi: 10.1152/ajprenal.1998.275.6.F972. [DOI] [PubMed] [Google Scholar]
- 364.Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M, Zhang DX. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol. 2010;298:H466–H476. doi: 10.1152/ajpheart.00854.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol. 2004;287:F593–F601. doi: 10.1152/ajprenal.00454.2003. [DOI] [PubMed] [Google Scholar]
- 366.Meneton P, Bloch-Faure M, Hagege AA, Ruetten H, Huang W, Bergaya S, Ceiler D, Gehring D, Martins I, Salmon G, Boulanger CM, Nussberger J, Crozatier B, Gasc JM, Heudes D, Bruneval P, Doetschman T, Menard J, Alhenc-Gelas F. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc Natl Acad Sci USA. 2001;98:2634–2639. doi: 10.1073/pnas.051619598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 368.Moe OW. Acute regulation of proximal tubule apical membrane Na/H exchanger NHE-3: role of phosphorylation, protein trafficking, and regulatory factors. J Am Soc Nephrol. 1999;10:2412–2425. doi: 10.1681/ASN.V10112412. [DOI] [PubMed] [Google Scholar]
- 369.Moeller HB, Olesen ETB, Fenton RA. Regulation of the water channel aquaporin-2 by posttranslational modification. Am J Physiol Renal Physiol. 2011;300:F1062–F1073. doi: 10.1152/ajprenal.00721.2010. [DOI] [PubMed] [Google Scholar]
- 370.Moeller HB, Praetorius J, Rutzler MR, Fenton RA. Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc Natl Acad Sci USA. 2010;107:424–429. doi: 10.1073/pnas.0910683107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Moral Z, Dong K, Wei Y, Sterling H, Deng H, Ali S, Gu R, Huang XY, Hebert SC, Giebisch G, Wang WH. Regulation of ROMK1 channels by protein-tyrosine kinase and -tyrosine phosphatase. J Biol Chem. 2001;276:7156–7163. doi: 10.1074/jbc.M008671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Morton MJ, Hutchinson K, Mathieson PW, Witherden IR, Saleem MA, Hunter M. Human podocytes possess a stretch-sensitive, Ca2+-activated K+ channel: potential implications for the control of glomerular filtration. J Am Soc Nephrol. 2004;15:2981–2987. doi: 10.1097/01.ASN.0000145046.24268.0D. [DOI] [PubMed] [Google Scholar]
- 373.Mount D, Romero M. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 2004;447:710–721. doi: 10.1007/s00424-003-1090-3. [DOI] [PubMed] [Google Scholar]
- 374.Mullins LJ, Bailey MA, Mullins JJ. Hypertension, kidney, and transgenics: a fresh perspective. Physiol Rev. 2006;86:709–746. doi: 10.1152/physrev.00016.2005. [DOI] [PubMed] [Google Scholar]
- 375.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 376.Muto S. Potassium transport in the mammalian collecting duct. Physiol Rev. 2001;81:85–116. doi: 10.1152/physrev.2001.81.1.85. [DOI] [PubMed] [Google Scholar]
- 377.Muto S, Tsuruoka S, Miyata Y, Fujimura A, Kusano E, Wang W, Seldin D, Giebisch G. Basolateral Na+/H+ exchange maintains potassium secretion during diminished sodium transport in the rabbit cortical collecting duct. Kidney Int. 2009;75:25–30. doi: 10.1038/ki.2008.447. [DOI] [PubMed] [Google Scholar]
- 378.Muto S, Yasoshima K, Yoshitomi K, Imai M, Asano Y. Electrophysiological identification of α- and β-intercalated cells and their distribution along the rabbit distal nephron segments. J Clin Invest. 1990;86:1829–1839. doi: 10.1172/JCI114913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Nadler SP, Zimpelmann JA, Hebert RL. Endothelin inhibits vasopressin-stimulated water permeability in rat terminal inner medullary collecting duct. J Clin Invest. 1992;90:1458–1466. doi: 10.1172/JCI116013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Najjar F, Zhou H, Morimoto T, Bruns JB, Li HS, Liu W, Kleyman TR, Satlin LM. Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am J Physiol Renal Physiol. 2005;289:F922–F932. doi: 10.1152/ajprenal.00057.2005. [DOI] [PubMed] [Google Scholar]
- 381.Nakamura S, Amlal H, Schultheis PJ, Galla JH, Shull GE, Soleimani M. HCO3− reabsorption in renal collecting duct of NHE-3-deficient mouse: a compensatory response. Am J Physiol Renal Physiol. 1999;276:F914–F921. doi: 10.1152/ajprenal.1999.276.6.F914. [DOI] [PubMed] [Google Scholar]
- 382.Nakamura S, Wang Z, Galla JH, Soleimani M. K+ depletion increases. Am J Physiol Renal Physiol. 1998;274:F687–F692. doi: 10.1152/ajprenal.1998.274.4.F687. [DOI] [PubMed] [Google Scholar]
- 383.Nelson WJ. Remodeling epithelial cell organization: transitions between front-rear and apical-basal polarity. Cold Spring Harb Perspect Biol. 2009;1:a000513. doi: 10.1101/cshperspect.a000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Nelson WJ, Veshnock PJ. Ankyrin binding to (Na++K+)ATPase and implications for the organization of membrane domains in polarized cells. Nature. 1987;328:533–536. doi: 10.1038/328533a0. [DOI] [PubMed] [Google Scholar]
- 385.Newby LJ, Streets AJ, Zhao Y, Harris PC, Ward CJ, Ong AC. Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J Biol Chem. 2002;277:20763–20773. doi: 10.1074/jbc.M107788200. [DOI] [PubMed] [Google Scholar]
- 386.Nie X, Arrighi I, Kaissling B, Pfaff I, Mann J, Barhanin J, Vallon V. Expression and insights on function of potassium channel TWIK-1 in mouse kidney. Pflugers Arch. 2005;451:479–488. doi: 10.1007/s00424-005-1480-9. [DOI] [PubMed] [Google Scholar]
- 387.Nielsen S, Chou CL, Marples D, Christensen EI, Kishore BK, Knepper MA. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci USA. 1995;92:1013–1017. doi: 10.1073/pnas.92.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Nielsen S, DiGiovanni SR, Christensen EI, Knepper MA, Harris HW. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci USA. 1993;90:11663–11667. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Nielsen S, Frokiaer J, Marples D, Kwon TH, Agre P, Knepper MA. Aquaporins in the kidney: from molecules to medicine. Physiol Rev. 2002;82:205–244. doi: 10.1152/physrev.00024.2001. [DOI] [PubMed] [Google Scholar]
- 390.Nielsen S, Terris J, Andersen D, Ecelbarger C, Frokier J, Jonassen T, Marples D, Knepper MA, Petersen JS. Congestive heart failure in rats is associated with increased expression and targeting of aquaporin-2 water channel in collecting duct. Proc Natl Acad Sci USA. 1997;94:5450–5455. doi: 10.1073/pnas.94.10.5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Niisato N, Taruno A, Marunaka Y. Aldosterone-induced modification of osmoregulated ENaC trafficking. Biochem Biophys Res Commun. 2007;361:162–168. doi: 10.1016/j.bbrc.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 392.Nijenhuis T, Renkema KY, Hoenderop JG, Bindels RJ. Acid-base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol. 2006;17:617–626. doi: 10.1681/ASN.2005070732. [DOI] [PubMed] [Google Scholar]
- 393.Nijenhuis T, Hoenderop JG, van der Kemp AW, Bindels RJ. Localization and regulation of the epithelial Ca2+ channel TRPV6 in the kidney. J Am Soc Nephrol. 2003;14:2731–2740. doi: 10.1097/01.asn.0000094081.78893.e8. [DOI] [PubMed] [Google Scholar]
- 394.Nissant A, Paulais M, Lachheb S, Lourdel S, Teulon J. Similar chloride channels in the connecting tubule and cortical collecting duct of the mouse kidney. Am J Physiol Renal Physiol. 2006;290:F1421–F1429. doi: 10.1152/ajprenal.00274.2005. [DOI] [PubMed] [Google Scholar]
- 395.Noda Y, Sohara E, Ohta E, Sasaki S. Aquaporins in kidney pathophysiology. Nat Rev Nephrol. 2010;6:168–178. doi: 10.1038/nrneph.2009.231. [DOI] [PubMed] [Google Scholar]
- 396.Nusing RM, Pantalone F, Grone HJ, Seyberth HW, Wegmann M. Expression of the potassium channel ROMK in adult and fetal human kidney. Histochem Cell Biol. 2005;123:553–559. doi: 10.1007/s00418-004-0742-5. [DOI] [PubMed] [Google Scholar]
- 397.O’Neil RG, Sansom SC. Electrophysiological properties of cellular and paracellular conductive pathways of the rabbit cortical collecting duct. J Membr Biol. 1984;82:281–295. doi: 10.1007/BF01871637. [DOI] [PubMed] [Google Scholar]
- 398.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schoneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Orias M, Velazquez H, Tung F, Lee G, Desir GV. Cloning and localization of a double-pore K channel, KCNK1: exclusive expression in distal nephron segments. Am J Physiol Renal Physiol. 1997;273:F663–F666. doi: 10.1152/ajprenal.1997.273.4.F663. [DOI] [PubMed] [Google Scholar]
- 400.Pacha J, Frindt G, Sackin H, Palmer LG. Apical maxi K channels in intercalated cells of CCT. Am J Physiol Renal Physiol. 1991;261:F696–F705. doi: 10.1152/ajprenal.1991.261.4.F696. [DOI] [PubMed] [Google Scholar]
- 401.Palmer LG, Andersen OS. The two-membrane model of epithelial transport: Koefoed-Johnsen and Ussing (1958) J Gen Physiol. 2008;132:607–612. doi: 10.1085/jgp.200810149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Palmer LG, Antonian L, Frindt G. Regulation of apical K and Na channels and Na/K pumps in rat cortical collecting tubule by dietary K. J Gen Physiol. 1994;104:693–710. doi: 10.1085/jgp.104.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Palmer LG, Frindt G. Regulation of apical K channels in rat cortical collecting tubule during changes in dietary K intake. Am J Physiol Renal Physiol. 1999;277:F805–F812. doi: 10.1152/ajprenal.1999.277.5.F805. [DOI] [PubMed] [Google Scholar]
- 404.Palmer LG, Frindt G. Cl− channels of the distal nephron. Am J Physiol Renal Physiol. 2006;291:F1157–F1168. doi: 10.1152/ajprenal.00496.2005. [DOI] [PubMed] [Google Scholar]
- 405.Palmer LG, Frindt G. High-conductance K channels in intercalated cells of the rat distal nephron. Am J Physiol Renal Physiol. 2007;292:F966–F973. doi: 10.1152/ajprenal.00191.2006. [DOI] [PubMed] [Google Scholar]
- 406.Palmer LG, Frindt G. Na+ and K+ transport by the renal connecting tubule. Curr Opin Nephrol Hypertens. 2007;16:477–483. doi: 10.1097/MNH.0b013e32820ac850. [DOI] [PubMed] [Google Scholar]
- 407.Palmer LG, Frindt G. Aldosterone and potassium secretion by the cortical collecting duct. Kidney Int. 2000;57:1324–1328. doi: 10.1046/j.1523-1755.2000.00970.x. [DOI] [PubMed] [Google Scholar]
- 408.Passero CJ, Hughey RP, Kleyman TR. New role for plasmin in sodium homeostasis. Curr Opin Nephrol Hypertens. 2010;19:13–19. doi: 10.1097/MNH.0b013e3283330fb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the γ subunit. J Biol Chem. 2008;283:36586–36591. doi: 10.1074/jbc.M805676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Pastor-Soler N, Beaulieu Vr, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem. 2003;278:49523–49529. doi: 10.1074/jbc.M309543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens. 2009;18:99–106. doi: 10.1097/MNH.0b013e3283262ab0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel Sp, Teulon J, Tucker SJ. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc Natl Acad Sci USA. 2011;108:10361–10366. doi: 10.1073/pnas.1101400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Pavlov TS, Chahdi A, Ilatovskaya DV, Levchenko V, Vandewalle A, Pochynyuk O, Sorokin A, Staruschenko A. Endothelin-1 inhibits the epithelial Na+ channel through βPix/14-3-3/Nedd4-2. J Am Soc Nephrol. 2010;21:833–843. doi: 10.1681/ASN.2009080885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Pavlov TS, Ilatovskaya DV, Levchenko V, Mattson DL, Roman RJ, Staruschenko A. Effects of cytochrome P450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC) Am J Physiol Renal Physiol. 2011 doi: 10.1152/ajprenal.00597.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, Walp E, Kim YH, Sutliff RL, Bao HF, Eaton DC, Wall SM. Pendrin modulates ENaC function by changing luminal HCO3−. J Am Soc Nephrol. 2010;21:1928–1941. doi: 10.1681/ASN.2009121257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Pech V, Kim YH, Weinstein AM, Everett LA, Pham TD, Wall SM. Angiotensin II increases chloride absorption in the cortical collecting duct in mice through a pendrin-dependent mechanism. Am J Physiol Renal Physiol. 2007;292:F914–F920. doi: 10.1152/ajprenal.00361.2006. [DOI] [PubMed] [Google Scholar]
- 417.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT1 receptors. J Am Soc Nephrol. 2002;13:1131–1135. doi: 10.1097/01.asn.0000013292.78621.fd. [DOI] [PubMed] [Google Scholar]
- 418.Picard N, Eladari D, El MS, Planes C, Bourgeois S, Houillier P, Wang Q, Burnier M, Deschenes G, Knepper MA, Meneton P, Chambrey R. Defective ENaC processing and function in tissue kallikrein-deficient mice. J Biol Chem. 2008;283:4602–4611. doi: 10.1074/jbc.M705664200. [DOI] [PubMed] [Google Scholar]
- 419.Pieczynski J, Margolis B. Protein complexes that control renal epithelial polarity. Am J Physiol Renal Physiol. 2011;300:F589–F601. doi: 10.1152/ajprenal.00615.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Pluznick JL, Wei P, Grimm PR, Sansom SC. BK-β1 subunit: immunolocalization in the mammalian connecting tubule and its role in the kaliuretic response to volume expansion. Am J Physiol Renal Physiol. 2005;288:F846–F854. doi: 10.1152/ajprenal.00340.2004. [DOI] [PubMed] [Google Scholar]
- 421.Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, Stockand JD. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J Biol Chem. 2008;283:36599–36607. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens. 2008;17:533–540. doi: 10.1097/MNH.0b013e328308fff3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Pochynyuk O, Bugaj V, Vandewalle A, Stockand JD. Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am J Physiol Renal Physiol. 2008;294:F38–F46. doi: 10.1152/ajprenal.00403.2007. [DOI] [PubMed] [Google Scholar]
- 424.Pochynyuk O, Medina J, Gamper N, Genth H, Stockand JD, Staruschenko A. Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling. J Biol Chem. 2006;281:26520–26527. doi: 10.1074/jbc.M603716200. [DOI] [PubMed] [Google Scholar]
- 425.Pochynyuk O, Staruschenko A, Bugaj V, Lagrange L, Stockand JD. Quantifying RhoA facilitated trafficking of the epithelial Na+ channel toward the plasma membrane with total internal reflection fluorescence-fluorescence recovery after photobleaching. J Biol Chem. 2007;282:14576–14585. doi: 10.1074/jbc.M701348200. [DOI] [PubMed] [Google Scholar]
- 426.Pochynyuk O, Staruschenko A, Tong Q, Medina J, Stockand JD. Identification of a functional phosphatidylinositol 3,4,5-trisphosphate binding site in the epithelial Na+ channel. J Biol Chem. 2005;280:37565–37571. doi: 10.1074/jbc.M509071200. [DOI] [PubMed] [Google Scholar]
- 427.Pochynyuk O, Stockand JD, Staruschenko A. Ion channel regulation by Ras, Rho, and Rab small GTPases. Exp Biol Med. 2007;232:1258–1265. doi: 10.3181/0703-MR-76. [DOI] [PubMed] [Google Scholar]
- 428.Pochynyuk O, Tong Q, Medina J, Vandewalle A, Staruschenko A, Bugaj V, Stockand JD. Molecular determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. J Gen Physiol. 2007;130:399–413. doi: 10.1085/jgp.200709800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Pochynyuk O, Tong Q, Staruschenko A, Stockand JD. Binding and direct activation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. J Physiol. 2007;580:365–372. doi: 10.1113/jphysiol.2006.127449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Pradervand S, Zuber MA, Centeno G, Bonny O, Firsov D. A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Pflugers Arch. 2010;460:925–952. doi: 10.1007/s00424-010-0863-8. [DOI] [PubMed] [Google Scholar]
- 431.Praetorius HA, Spring KR. A physiological view of the primary cilium. Annu Rev Physiol. 2005;67:515–529. doi: 10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- 432.Puelles VG, Hoy WE, Hughson MD, Diouf B, Douglas-Denton RN, Bertram JF. Glomerular number and size variability and risk for kidney disease. Curr Opin Nephrol Hypertens. 2010 doi: 10.1097/MNH.0b013e3283410a7d. [DOI] [PubMed] [Google Scholar]
- 433.Qin H, Zheng X, Zhong X, Shetty AK, Elias PM, Bollag WB. Aquaporin-3 in keratinocytes and skin: Its role and interaction with phospholipase D2. Arch Biochem Biophys. 2011;508:138–143. doi: 10.1016/j.abb.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Quamme GA. Molecular identification of ancient and modern mammalian magnesium transporters. Am J Physiol Cell Physiol. 2010;298:C407–C429. doi: 10.1152/ajpcell.00124.2009. [DOI] [PubMed] [Google Scholar]
- 435.Raikwar NS, Vandewalle A, Thomas CP. Nedd4-2 interacts with occludin to inhibit tight junction formation and enhance paracellular conductance in collecting duct epithelia. Am J Physiol Renal Physiol. 2010;299:F436–F444. doi: 10.1152/ajprenal.00674.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Rajasekaran SA, Barwe SP, Rajasekaran AK. Multiple functions of Na,K-ATPase in epithelial cells. Semin Nephrol. 2005;25:328–334. doi: 10.1016/j.semnephrol.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 437.Rauh R, Diakov A, Tzschoppe A, Korbmacher J, Azad AK, Cuppens H, Cassiman JJ, Dotsch J, Sticht H, Korbmacher C. A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J Physiol. 2010;588:1211–1225. doi: 10.1113/jphysiol.2009.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Reddy MM, Light MJ, Quinton PM. Activation of the epithelial Na+ channel (ENaC) requires CFTR Cl− channel function. Nature. 1999;402:301–304. doi: 10.1038/46297. [DOI] [PubMed] [Google Scholar]
- 439.Reif MC, Troutman SL, Schafer JA. Sustained response to vasopressin in isolated rat cortical collecting tubule. Kidney Int. 1984;26:725–732. doi: 10.1038/ki.1984.208. [DOI] [PubMed] [Google Scholar]
- 440.Reif MC, Troutman SL, Schafer JA. Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J Clin Invest. 1986;77:1291–1298. doi: 10.1172/JCI112433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Reilly RF, Ellison DH. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev. 2000;80:277–313. doi: 10.1152/physrev.2000.80.1.277. [DOI] [PubMed] [Google Scholar]
- 442.Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, vila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Renard S, Lingueglia E, Voilley N, Lazdunski M, Barbry P. Biochemical analysis of the membrane topology of the amiloride-sensitive Na+ channel. J Biol Chem. 1994;269:12981–12986. [PubMed] [Google Scholar]
- 444.Renkema KY, Lee K, Topala CN, Goossens M, Houillier P, Bindels RJ, Hoenderop JG. TRPV5 gene polymorphisms in renal hypercalciuria. Nephrol Dial Transplant. 2009;24:1919–1924. doi: 10.1093/ndt/gfn735. [DOI] [PubMed] [Google Scholar]
- 445.Renkema KY, Nijenhuis T, van der Eerden BC, van der Kemp AW, Weinans H, van Leeuwen JP, Bindels RJ, Hoenderop JG. Hypervitaminosis D mediates compensatory Ca2+ hyperabsorption in TRPV5 knockout mice. J Am Soc Nephrol. 2005;16:3188–3195. doi: 10.1681/ASN.2005060632. [DOI] [PubMed] [Google Scholar]
- 446.Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J Clin Invest. 1998;101:935–939. doi: 10.1172/JCI2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Ridderstrale Y, Kashgarian M, Koeppen B, Giebisch G, Stetson D, Ardito T, Stanton B. Morphological heterogeneity of the rabbit collecting duct. Kidney Int. 1988;34:655–670. doi: 10.1038/ki.1988.230. [DOI] [PubMed] [Google Scholar]
- 448.Rieg T, Vallon V, Sausbier M, Sausbier U, Kaissling B, Ruth P, Osswald H. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int. 2007;72:566–573. doi: 10.1038/sj.ki.5002369. [DOI] [PubMed] [Google Scholar]
- 449.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 450.Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–F270. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- 451.Rodan AR, Cheng CJ, Huang CL. Recent advances in distal tubular potassium handling. Am J Physiol Renal Physiol. 2011;300:F821–F827. doi: 10.1152/ajprenal.00742.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Rohatgi R, Greenberg A, Burrow CR, Wilson PD, Satlin LM. Na transport in autosomal recessive polycystic kidney disease (ARPKD) cyst lining epithelial cells. J Am Soc Nephrol. 2003;14:827–836. doi: 10.1097/01.asn.0000056481.66379.b2. [DOI] [PubMed] [Google Scholar]
- 453.Rojek A, Fuchtbauer EM, Kwon TH, Frokier J, Nielsen S. Severe urinary concentrating defect in renal collecting duct-selective AQP2 conditional-knockout mice. Proc Natl Acad Sci USA. 2006;103:6037–6042. doi: 10.1073/pnas.0511324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Romero M, Fulton C, Boron W. The SLC4 family of HCO3− transporters. Pflugers Arch. 2004;447:495–509. doi: 10.1007/s00424-003-1180-2. [DOI] [PubMed] [Google Scholar]
- 455.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, RIordan JR, Lap-Chee T, Collins FS. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 456.Rossier BC. Hormonal regulation of the epithelial sodium channel ENaC: N or P(o)? J Gen Physiol. 2002;120:67–70. doi: 10.1085/jgp.20028638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Rossier BC, Pradervand S, Schild L, Hummler E. Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol. 2002;64:877–897. doi: 10.1146/annurev.physiol.64.082101.143243. [DOI] [PubMed] [Google Scholar]
- 458.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 459.Rotin D, Schild L. ENaC and its regulatory proteins as drug targets for blood pressure control. Curr Drug Targets. 2008;9:709–716. doi: 10.2174/138945008785132367. [DOI] [PubMed] [Google Scholar]
- 460.Rotin D, Staub O. Role of the ubiquitin system in regulating ion transport. Pflugers Arch. 2011;461:1–21. doi: 10.1007/s00424-010-0893-2. [DOI] [PubMed] [Google Scholar]
- 461.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA. 2001;98:4221–4226. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Rubera I, Hummler E, Beermann F. Transgenic mice and their impact on kidney research. Pflugers Arch. 2009;458:211–222. doi: 10.1007/s00424-008-0624-0. [DOI] [PubMed] [Google Scholar]
- 463.Rubera I, Loffing J, Palmer LG, Frindt G, Fowler-Jaeger N, Sauter D, Carroll T, McMahon A, Hummler E, Rossier BC. Collecting duct-specific gene inactivation of αENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest. 2003;112:554–565. doi: 10.1172/JCI16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Rungroj N, Devonald MAJ, Cuthbert AW, Reimann F, Akkarapatumwong V, Yenchitsomanus Pt, Bennett WM, Karet FE. A novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport function but is mistargeted in polarized epithelial cells. J Biol Chem. 2004;279:13833–13838. doi: 10.1074/jbc.M400188200. [DOI] [PubMed] [Google Scholar]
- 465.Sabolic I, Brown D, Gluck SL, Alper SL. Regulation of AE1 anion exchanger and H+-ATPase in rat cortex by acute metabolic acidosis and alkalosis. Kidney Int. 1997;51:125–137. doi: 10.1038/ki.1997.16. [DOI] [PubMed] [Google Scholar]
- 466.Sachs AN, Pisitkun T, Hoffert JD, Yu MJ, Knepper MA. LC-MS/MS analysis of differential centrifugation fractions from native inner medullary collecting duct of rat. Am J Physiol Renal Physiol. 2008;295:F1799–F1806. doi: 10.1152/ajprenal.90510.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Sackin H, Nanazashvili M, Li H, Palmer LG, Yang L. Modulation of kir1.1 inactivation by extracellular Ca and Mg. Biophys J. 2011;100:1207–1215. doi: 10.1016/j.bpj.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Sanchez-Perez A, Kumar S, Cook DI. GRK2 interacts with and phosphorylates Nedd4 and Nedd4-2. Biochem Biophys Res Commun. 2007;359:611–615. doi: 10.1016/j.bbrc.2007.05.134. [DOI] [PubMed] [Google Scholar]
- 470.Sands JM. Renal urea transporters. Curr Opin Nephrol Hypertens. 2004;13:525–532. doi: 10.1097/00041552-200409000-00008. [DOI] [PubMed] [Google Scholar]
- 471.Sands JM, Blount MA, Klein JD. Regulation of renal urea transport by vasopressin. Trans Am Clin Climatol Assoc. 2010;122:82–92. [PMC free article] [PubMed] [Google Scholar]
- 472.Sands JM, Layton HE. The physiology of urinary concentration: an update. Semin Nephrol. 2009;29:178–195. doi: 10.1016/j.semnephrol.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Sansom SC, O’Neil RG. Mineralocorticoid regulation of apical cell membrane Na+ and K+ transport of the cortical collecting duct. Am J Physiol Renal Physiol. 1985;248:F858–F868. doi: 10.1152/ajprenal.1985.248.6.F858. [DOI] [PubMed] [Google Scholar]
- 474.Saroussi S, Nelson N. The little we know on the structure and machinery of V-ATPase. J Exp Biol. 2009;212:1604–1610. doi: 10.1242/jeb.025866. [DOI] [PubMed] [Google Scholar]
- 475.Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123:499–503. doi: 10.1242/jcs.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Satlin LM, Schwartz GJ. Cellular remodeling of HCO3−-secreting cells in rabbit renal collecting duct in response to an acidic environment. J Cell Biol. 1989;109:1279–1288. doi: 10.1083/jcb.109.3.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Satlin LM, Sheng S, Woda CB, Kleyman TR. Epithelial Na+ channels are regulated by flow. Am J Physiol Renal Physiol. 2001;280:F1010–F1018. doi: 10.1152/ajprenal.2001.280.6.F1010. [DOI] [PubMed] [Google Scholar]
- 478.Sauer M, Dorge A, Thurau K, Beck FX. Effect of ouabain on electrolyte concentrations in principal and intercalated cells of the isolated perfused cortical collecting duct. Pflugers Arch. 1989;413:651–655. doi: 10.1007/BF00581816. [DOI] [PubMed] [Google Scholar]
- 479.Sausbier M, Arntz C, Bucurenciu I, Zhao H, Zhou XB, Sausbier U, Feil S, Kamm S, Essin K, Sailer CA, Abdullah U, Krippeit-Drews P, Feil R, Hofmann F, Knaus HG, Kenyon C, Shipston MJ, Storm JF, Neuhuber W, Korth M, Schubert R, Gollasch M, Ruth P. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation. 2005;112:60–68. doi: 10.1161/01.CIR.0000156448.74296.FE. [DOI] [PubMed] [Google Scholar]
- 480.Saxena SK, Horiuchi H, Fukuda M. Rab27a regulates epithelial sodium channel (ENaC) activity through synaptotagmin-like protein (SLP-5) and Munc13-4 effector mechanism. Biochem Biophys Res Commun. 2006;344:651–657. doi: 10.1016/j.bbrc.2006.03.160. [DOI] [PubMed] [Google Scholar]
- 481.Saxena SK, Kaur S. Regulation of epithelial ion channels by Rab GTPases. Biochem Biophys Res Commun. 2006;351:582–587. doi: 10.1016/j.bbrc.2006.10.087. [DOI] [PubMed] [Google Scholar]
- 482.Schaedel C, Marthinsen L, Kristoffersson AC, Kornfalt R, Nilsson KO, Orlenius B, Holmberg L. Lung symptoms in pseudohypoaldosteronism type 1 are associated with deficiency of the α-subunit of the epithelial sodium channel. J Pediatr. 1999;135:739–745. doi: 10.1016/s0022-3476(99)70094-6. [DOI] [PubMed] [Google Scholar]
- 483.Schaefer M. Homo- and heteromeric assembly of TRP channel subunits. Pflugers Arch. 2005;451:35–42. doi: 10.1007/s00424-005-1467-6. [DOI] [PubMed] [Google Scholar]
- 484.Schafer JA, Troutman SL, Schlatter E. Vasopressin and mineralocorticoid increase apical membrane driving force for K+ secretion in rat CCD. Am J Physiol Renal Physiol. 1990;258:F199–F210. doi: 10.1152/ajprenal.1990.258.1.F199. [DOI] [PubMed] [Google Scholar]
- 485.Schenk AD, Werten PJL, Scheuring S, de Groot BL, Mnller SA, Stahlberg H, Philippsen A, Engel A. The 4.5 A Structure of human AQP2. J Mol Biol. 2005;350:278–289. doi: 10.1016/j.jmb.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 486.Schild L. The epithelial sodium channel: from molecule to disease. Rev Physiol Biochem Pharmacol. 2004;151:93–107. doi: 10.1007/s10254-004-0023-7. [DOI] [PubMed] [Google Scholar]
- 487.Schild L. The epithelial sodium channel and the control of sodium balance. Biochim Biophys Acta. 2010;1802:1159–1165. doi: 10.1016/j.bbadis.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 488.Schlatter E, Greger R, Schafer JA. Principal cells of cortical collecting ducts of the rat are not a route of transepithelial Cl− transport. Pflugers Arch. 1990;417:317–323. doi: 10.1007/BF00370998. [DOI] [PubMed] [Google Scholar]
- 489.Schlatter E, Haxelmans S, Hirsch J, Leipziger J. pH dependence of K+ conductances of rat cortical collecting duct principal cells. Pflugers Arch. 1994;428:631–640. doi: 10.1007/BF00374587. [DOI] [PubMed] [Google Scholar]
- 490.Schlingmann KP, Konrad M, Jeck N, Waldegger P, Reinalter SC, Holder M, Seyberth HW, Waldegger S. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350:1314–1319. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- 491.Scholl U, Hebeisen S, Janssen AG, Muller-Newen G, Alekov A, Fahlke C. Barttin modulates trafficking and function of ClC-K channels. Proc Natl Acad Sci USA. 2006;103:11411–11416. doi: 10.1073/pnas.0601631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Schrier RW. Vasopressin and aquaporin 2 in clinical disorders of water homeostasis. Semin Nephrol. 2008;28:289–296. doi: 10.1016/j.semnephrol.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Schrier RW, Cadnapaphornchai MA. Renal aquaporin water channels: from molecules to human disease. Prog Biophys Mol Biol. 2003;81:117–131. doi: 10.1016/s0079-6107(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 495.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Schulte U, Hahn H, Konrad M, Jeck N, Derst C, Wild K, Weidemann S, Ruppersberg JP, Fakler B, Ludwig J. pH gating of ROMK (Kir1.1) channels: control by an Arg-Lys- Arg triad disrupted in antenatal Bartter syndrome. Proc Natl Acad Sci USA. 1999;96:15298–15303. doi: 10.1073/pnas.96.26.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Schwalbe RA, Bianchi L, Accili EA, Brown AM. Functional consequences of ROMK mutants linked to antenatal Bartter’s syndrome and implications for treatment. Hum Mol Genet. 1998;7:975–980. doi: 10.1093/hmg/7.6.975. [DOI] [PubMed] [Google Scholar]
- 498.Schwartz GJ, Al-Awqati Q. Regulation of transepithelial H+ transport by exocytosis and endocytosis. Annu Rev Physiol. 1986;48:153–161. doi: 10.1146/annurev.ph.48.030186.001101. [DOI] [PubMed] [Google Scholar]
- 499.Schwartz GJ, Barasch J, Al-Awqati Q. Plasticity of functional epithelial polarity. Nature. 1985;318:368–371. doi: 10.1038/318368a0. [DOI] [PubMed] [Google Scholar]
- 500.Shao L, Xu Y, Dong Q, Lang Y, Yue S, Miao Z. A novel SLC4A1 variant in an autosomal dominant distal renal tubular acidosis family with a severe phenotype. Endocrine. 2010;37:473–478. doi: 10.1007/s12020-010-9340-6. [DOI] [PubMed] [Google Scholar]
- 501.Sheffield VC, Kraiem Z, Beck JC, Nishimura D, Stone EM, Salameh M, Sadeh O, Glaser B. Pendred syndrome maps to chromosome 7q21-34 and is caused by an intrinsic defect in thyroid iodine organification. Nat Genet. 1996;12:424–426. doi: 10.1038/ng0496-424. [DOI] [PubMed] [Google Scholar]
- 502.Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol. 2006;290:F1488–F1496. doi: 10.1152/ajprenal.00439.2005. [DOI] [PubMed] [Google Scholar]
- 503.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 504.Shi PP, Cao XR, Sweezer EM, Kinney TS, Williams NR, Husted RF, Nair R, Weiss RM, Williamson RA, Sigmund CD, Snyder PM, Staub O, Stokes JB, Yang B. Salt-Sensitive Hypertension and Cardiac Hypertrophy in Mice Deficient in the Ubiquitin Ligase Nedd4-2. Am J Physiol Renal Physiol. 2008 doi: 10.1152/ajprenal.90300.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Shi PP, Cao XR, Qu J, Volk KA, Kirby P, Williamson RA, Stokes JB, Yang B. Nephrogenic diabetes insipidus in mice caused by deleting COOH-terminal tail of aquaporin-2. American Journal of Physiology - Renal Physiology. 2007;292:F1334–F1344. doi: 10.1152/ajprenal.00308.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Shibata S, Fujita T. The Kidneys and Aldosterone/Mineralocorticoid Receptor System in Salt-Sensitive Hypertension. Current Hypertension Reports. 2011;13:109–115. doi: 10.1007/s11906-010-0175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 508.Silva GB, Garvin JL. TRPV4 mediates hypotonicity-induced ATP release by the thick ascending limb. American Journal of Physiology - Renal Physiology. 2008;295:F1090–F1095. doi: 10.1152/ajprenal.90365.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Silver RB, Choe H, Frindt G. Low-NaCl diet increases H-K-ATPase in intercalated cells from rat cortical collecting duct. Am J Physiol. 1998;275:F94–102. doi: 10.1152/ajprenal.1998.275.1.F94. [DOI] [PubMed] [Google Scholar]
- 510.Silver RB, Frindt G. Functional identification of H-K-ATPase in intercalated cells of cortical collecting tubule. Am J Physiol. 1993;264:F259–F266. doi: 10.1152/ajprenal.1993.264.2.F259. [DOI] [PubMed] [Google Scholar]
- 511.Silver RB, Soleimani M. H+-K+-ATPases: regulation and role in pathophysiological states. Am J Physiol. 1999;276:F799–F811. doi: 10.1152/ajprenal.1999.276.6.F799. [DOI] [PubMed] [Google Scholar]
- 512.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 513.Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 514.Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- 515.Snyder PM. Down-regulating destruction: phosphorylation regulates the E3 ubiquitin ligase Nedd4-2. Sci Signal. 2009;2:e41. doi: 10.1126/scisignal.279pe41. [DOI] [PubMed] [Google Scholar]
- 516.Snyder PM, Cheng C, Prince LS, Rogers JC, Welsh MJ. Electrophysiological and biochemical evidence that DEG/ENaC cation channels are composed of nine subunits. J Biol Chem. 1998;273:681–684. doi: 10.1074/jbc.273.2.681. [DOI] [PubMed] [Google Scholar]
- 517.Snyder PM, McDonald FJ, Stokes JB, Welsh MJ. Membrane topology of the amiloride-sensitive epithelial sodium channel. J Biol Chem. 1994;269:24379–24383. [PubMed] [Google Scholar]
- 518.Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J Biol Chem. 2004;279:45753–45758. doi: 10.1074/jbc.M407858200. [DOI] [PubMed] [Google Scholar]
- 519.Sohara E, Rai T, Yang SS, Uchida K, Nitta K, Horita S, Ohno M, Harada A, Sasaki S, Uchida S. Pathogenesis and treatment of autosomal-dominant nephrogenic diabetes insipidus caused by an aquaporin 2 mutation. PNAS. 2006;103:14217–14222. doi: 10.1073/pnas.0602331103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burnham CE. Pendrin: an apical Cl−/OH−/HCO3− exchanger in the kidney cortex. Am J Physiol Renal Physiol. 2001;280:F356–F364. doi: 10.1152/ajprenal.2001.280.2.F356. [DOI] [PubMed] [Google Scholar]
- 521.Soleimani M, Xu J. SLC26 Chloride/Base Exchangers in the Kidney in Health and Disease. Seminars in Nephrology. 2006;26:375–385. doi: 10.1016/j.semnephrol.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 522.Soundararajan R, Melters D, Shih IC, Wang J, Pearce D. Epithelial sodium channel regulated by differential composition of a signaling complex. Proc Natl Acad Sci U S A. 2009;106:7804–7809. doi: 10.1073/pnas.0809892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem. 2010;285:30363–30369. doi: 10.1074/jbc.R110.155341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Soundararajan R, Wang J, Melters D, Pearce D. Differential activities of glucocorticoid-induced leucine zipper protein isoforms. J Biol Chem. 2007;282:36303–36313. doi: 10.1074/jbc.M707287200. [DOI] [PubMed] [Google Scholar]
- 525.Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem. 2005;280:39970–39981. doi: 10.1074/jbc.M508658200. [DOI] [PubMed] [Google Scholar]
- 526.Stanton BA. Cystic fibrosis transmembrane conductance regulator (CFTR) and renal function. Wien Klin Wochenschr. 1997;109:457–464. [PubMed] [Google Scholar]
- 527.Starremans PG, van der Kemp AW, Knoers NV, van den Heuvel LP, Bindels RJ. Functional implications of mutations in the human renal outer medullary potassium channel (ROMK2) identified in Bartter syndrome. Pflugers Arch. 2002;443:466–472. doi: 10.1007/s004240100708. [DOI] [PubMed] [Google Scholar]
- 528.Staruschenko A, Adams E, Booth RE, Stockand JD. Epithelial Na+ channel subunit stoichiometry. Biophys J. 2005;88:3966–3975. doi: 10.1529/biophysj.104.056804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Staruschenko A, Medina JL, Patel P, Shapiro MS, Booth RE, Stockand JD. Fluorescence resonance energy transfer analysis of subunit stoichiometry of the epithelial Na+ channel. J Biol Chem. 2004;279:27729–27734. doi: 10.1074/jbc.M404169200. [DOI] [PubMed] [Google Scholar]
- 530.Staruschenko A, Nichols A, Medina JL, Camacho P, Zheleznova NN, Stockand JD. Rho small GTPases activate the epithelial Na(+) channel. J Biol Chem. 2004;279:49989–49994. doi: 10.1074/jbc.M409812200. [DOI] [PubMed] [Google Scholar]
- 531.Staruschenko A, Patel P, Tong Q, Medina JL, Stockand JD. Ras activates the epithelial Na(+) channel through phosphoinositide 3-OH kinase signaling. J Biol Chem. 2004;279:37771–37778. doi: 10.1074/jbc.M402176200. [DOI] [PubMed] [Google Scholar]
- 532.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD. Acute Regulation of the Epithelial Na+ Channel by Phosphatidylinositide 3-OH Kinase Signaling in Native Collecting Duct Principal Cells. J Am Soc Nephrol. 2007;18:1652–1661. doi: 10.1681/ASN.2007010020. [DOI] [PubMed] [Google Scholar]
- 533.Staruschenko A, Pochynyuk OM, Tong Q, Stockand JD. Ras couples phosphoinositide 3-OH kinase to the epithelial Na+ channel. Biochim Biophys Acta. 2005;1669:108–115. doi: 10.1016/j.bbamem.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 534.Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 Interaction to the Single Channel Properties of the TRPA1 Channel. J Biol Chem. 2010;285:15167–15177. doi: 10.1074/jbc.M110.106153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, Horisberger JD, Schild L, Rotin D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. 2000;57:809–815. doi: 10.1046/j.1523-1755.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- 536.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 537.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Staub O, Verrey F. Impact of Nedd4 proteins and serum and glucocorticoid-induced kinases on epithelial Na+ transport in the distal nephron. J Am Soc Nephrol. 2005;16:3167–3174. doi: 10.1681/ASN.2005050454. [DOI] [PubMed] [Google Scholar]
- 539.Stewart AP, Haerteis S, Diakov A, Korbmacher C, Edwardson JM. Atomic force microscopy reveals the architecture of the epithelial sodium channel (ENaC) J Biol Chem. 2011 doi: 10.1074/jbc.M111.275289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Stockand JD. New ideas about aldosterone signaling in epithelia. Am J Physiol Renal Physiol. 2002;282:F559–F576. doi: 10.1152/ajprenal.00320.2001. [DOI] [PubMed] [Google Scholar]
- 541.Stockand JD. Vasopressin regulation of renal sodium excretion. Kidney Int. 2010;78:849–856. doi: 10.1038/ki.2010.276. [DOI] [PubMed] [Google Scholar]
- 542.Stockand JD, Staruschenko A, Pochynyuk O, Booth RE, Silverthorn DU. Insight toward epithelial Na+ channel mechanism revealed by the acid-sensing ion channel 1 structure. IUBMB Life. 2008;60:620–628. doi: 10.1002/iub.89. [DOI] [PubMed] [Google Scholar]
- 543.Stokes JB. Potassium secretion by cortical collecting tubule: relation to sodium absorption, luminal sodium concentration, and transepithelial voltage. Am J Physiol. 1981;241:F395–F402. doi: 10.1152/ajprenal.1981.241.4.F395. [DOI] [PubMed] [Google Scholar]
- 544.Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch ABS, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. Journal of Medical Genetics. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet. 1996;13:248–250. doi: 10.1038/ng0696-248. [DOI] [PubMed] [Google Scholar]
- 546.Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- 547.Stutts MJ, Rossier BC, Boucher RC. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. J Biol Chem. 1997;272:14037–14040. doi: 10.1074/jbc.272.22.14037. [DOI] [PubMed] [Google Scholar]
- 548.Subramanya AR, Yang CL, McCormick JA, Ellison DH. WNK kinases regulate sodium chloride and potassium transport by the aldosterone-sensitive distal nephron. Kidney Int. 2006;70:630–634. doi: 10.1038/sj.ki.5001634. [DOI] [PubMed] [Google Scholar]
- 549.Sun P, Lin DH, Wang T, Babilonia E, Wang Z, Jin Y, Kemp R, Nasjletti A, Wang WH. Low Na intake suppresses expression of CYP2C23 and arachidonic acid-induced inhibition of ENaC. Am J Physiol Renal Physiol. 2006;291:F1192–F1200. doi: 10.1152/ajprenal.00112.2006. [DOI] [PubMed] [Google Scholar]
- 550.Sun P, Lin DH, Yue P, Jiang H, Gotlinger KH, Schwartzman ML, Falck JR, Goli M, Wang WH. High Potassium Intake Enhances the Inhibitory Effect of 11,12-EET on ENaC. J Am Soc Nephrol. 2010;21:1667–1677. doi: 10.1681/ASN.2009111110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Sun P, Liu W, Lin DH, Yue P, Kemp R, Satlin LM, Wang WH. Epoxyeicosatrienoic acid activates BK channels in the cortical collecting duct. J Am Soc Nephrol. 2009;20:513–523. doi: 10.1681/ASN.2008040427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skott O. Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol. 2009;20:299–310. doi: 10.1681/ASN.2008040364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Svenningsen P, Friis UG, Bistrup C, Buhl KB, Jensen BL, Skott O. Physiological regulation of epithelial sodium channel by proteolysis. Curr Opin Nephrol Hypertens. 2011 doi: 10.1097/MNH.0b013e328348bcc7. [DOI] [PubMed] [Google Scholar]
- 554.Sweeney WE, Jr, Avner ED. Functional activity of epidermal growth factor receptors in autosomal recessive polycystic kidney disease. Am J Physiol. 1998;275:F387–F394. doi: 10.1152/ajprenal.1998.275.3.F387. [DOI] [PubMed] [Google Scholar]
- 555.Takahashi S, Iwamoto N, Sasaki H, Ohashi M, Oda Y, Tsukita S, Furuse M. The E3 ubiquitin ligase LNX1p80 promotes the removal of claudins from tight junctions in MDCK cells. J Cell Sci. 2009;122:985–994. doi: 10.1242/jcs.040055. [DOI] [PubMed] [Google Scholar]
- 556.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 557.Takemura Y, Goodson P, Bao HF, Jain L, Helms MN. Rac1-mediated NADPH oxidase release of OFormula regulates epithelial sodium channel activity in the alveolar epithelium. Am J Physiol Lung Cell Mol Physiol. 2010;298:L509–L520. doi: 10.1152/ajplung.00230.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Takiar V, Nishio S, Seo-Mayer P, King JD, Jr, Li H, Zhang L, Karihaloo A, Hallows KR, Somlo S, Caplan MJ. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A. 2011;108:2462–2467. doi: 10.1073/pnas.1011498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Tamma G, Robben JH, Trimpert C, Boone M, Deen PMT. Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. American Journal of Physiology - Cell Physiology. 2011;300:C636–C646. doi: 10.1152/ajpcell.00433.2009. [DOI] [PubMed] [Google Scholar]
- 560.Tammachote R, Hommerding CJ, Sinders RM, Miller CA, Czarnecki PG, Leightner AC, Salisbury JL, Ward CJ, Torres VE, Gattone VH, Harris PC. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum Mol Genet. 2009;18:3311–3323. doi: 10.1093/hmg/ddp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Tang VW, Goodenough DA. Paracellular ion channel at the tight junction. Biophys J. 2003;84:1660–1673. doi: 10.1016/S0006-3495(03)74975-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Taniguchi J, Tsuruoka S, Mizuno A, Sato Ji, Fujimura A, Suzuki M. TRPV4 as a flow sensor in flow-dependent K+ secretion from the cortical collecting duct. American Journal of Physiology - Renal Physiology. 2007;292:F667–F673. doi: 10.1152/ajprenal.00458.2005. [DOI] [PubMed] [Google Scholar]
- 563.Teng-umnuay P, Verlander JW, Yuan W, Tisher CC, Madsen KM. Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. J Am Soc Nephrol. 1996;7:260–274. doi: 10.1681/ASN.V72260. [DOI] [PubMed] [Google Scholar]
- 564.Terryn S, Ho A, Beauwens R, Devuyst O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. doi: 10.1016/j.bbadis.2011.01.011. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 565.The Renal Commission of the International Union of Physiological Sciences (IUPS) A standard nomenclature for structures of the kidney. American Journal of Physiology - Renal Physiology. 1988;254:F1–F8. doi: 10.1152/ajprenal.1988.254.1.F1. [DOI] [PubMed] [Google Scholar]
- 566.Tian W, Salanova M, Xu H, Lindsley JN, Oyama TT, Anderson S, Bachmann S, Cohen DM. Renal expression of osmotically responsive cation channel TRPV4 is restricted to water-impermeant nephron segments. Am J Physiol Renal Physiol. 2004;287:F17–F24. doi: 10.1152/ajprenal.00397.2003. [DOI] [PubMed] [Google Scholar]
- 567.Tiwari S, Sharma N, Gill PS, Igarashi P, Kahn CR, Wade JB, Ecelbarger CM. Impaired sodium excretion and increased blood pressure in mice with targeted deletion of renal epithelial insulin receptor. Proc Natl Acad Sci U S A. 2008;105:6469–6474. doi: 10.1073/pnas.0711283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Tobin MD, Tomaszewski M, Braund PS, Hajat C, Raleigh SM, Palmer TM, Caulfield M, Burton PR, Samani NJ. Common Variants in Genes Underlying Monogenic Hypertension and Hypotension and Blood Pressure in the General Population. Hypertension. 2008;51:1658–1664. doi: 10.1161/HYPERTENSIONAHA.108.112664. [DOI] [PubMed] [Google Scholar]
- 569.Togawa H, Nakanishi K, Mukaiyama H, Hama T, Shima Y, Sako M, Miyajima M, Nozu K, Nishii K, Nagao S, Takahashi H, Iijima K, Yoshikawa N. Epithelial-to-mesenchymal transition in cyst lining epithelial cells in an orthologous PCK rat model of autosomal-recessive polycystic kidney disease. American Journal of Physiology - Renal Physiology. 2011;300:F511–F520. doi: 10.1152/ajprenal.00038.2010. [DOI] [PubMed] [Google Scholar]
- 570.Tong Q, Booth RE, Worrell RT, Stockand JD. Regulation of Na+ transport by aldosterone: signaling convergence and cross talk between the PI3-K and MAPK1/2 cascades. Am J Physiol Renal Physiol. 2004;286:F1232–F1238. doi: 10.1152/ajprenal.00345.2003. [DOI] [PubMed] [Google Scholar]
- 571.Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 572.Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261:17–31. doi: 10.1111/j.1365-2796.2006.01743.x. [DOI] [PubMed] [Google Scholar]
- 573.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 575.Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc Natl Acad Sci U S A. 1999;96:3934–3939. doi: 10.1073/pnas.96.7.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Tsiokas L. Function and regulation of TRPP2 at the plasma membrane. Am J Physiol Renal Physiol. 2009;297:F1–F9. doi: 10.1152/ajprenal.90277.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–293. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 578.Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. pH Dependence of the Inwardly Rectifying Potassium Channel, Kir5.1, and Localization in Renal Tubular Epithelia. J Biol Chem. 2000;275:16404–16407. doi: 10.1074/jbc.C000127200. [DOI] [PubMed] [Google Scholar]
- 579.Uchida S, Sasaki S, Nitta K, Uchida K, Horita S, Nihei H, Marumo F. Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J Clin Invest. 1995;95:104–113. doi: 10.1172/JCI117626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Uchida S, Sasaki S. FUNCTION OF CHLORIDE CHANNELS IN THE KIDNEY. Annu Rev Physiol. 2004;67:759–778. doi: 10.1146/annurev.physiol.67.032003.153547. [DOI] [PubMed] [Google Scholar]
- 581.Vachugova DV, Morachevskaia EA. Mechanosensitivity of cationic channels of DEG/ENaC family. Tsitologiia. 2009;51:806–814. [PubMed] [Google Scholar]
- 582.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschenes G, Breton S, Meneton P, Loffing J, Aronson PS, Chambrey R, Eladari D. Pendrin Regulation in Mouse Kidney Primarily Is Chloride-Dependent. J Am Soc Nephrol. 2006;17:2153–2163. doi: 10.1681/ASN.2005101054. [DOI] [PubMed] [Google Scholar]
- 583.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–610. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 584.Vallet V, Pfister C, Loffing J, Rossier BC. Cell-surface expression of the channel activating protease xCAP-1 is required for activation of ENaC in the Xenopus oocyte. J Am Soc Nephrol. 2002;13:588–594. doi: 10.1681/ASN.V133588. [DOI] [PubMed] [Google Scholar]
- 585.Vallon V, Wulff P, Huang DY, Loffing J, Volkl H, Kuhl D, Lang F. Role of Sgk1 in salt and potassium homeostasis. Am J Physiol Regul Integr Comp Physiol. 2005;288:R4–10. doi: 10.1152/ajpregu.00369.2004. [DOI] [PubMed] [Google Scholar]
- 586.van Balkom BWM, Savelkoul PJM, Markovich D, Hofman E, Nielsen S, van der Sluijs P, Deen PMT. The Role of Putative Phosphorylation Sites in the Targeting and Shuttling of the Aquaporin-2 Water Channel. J Biol Chem. 2002;277:41473–41479. doi: 10.1074/jbc.M207525200. [DOI] [PubMed] [Google Scholar]
- 587.van de Graaf SF, Chang Q, Mensenkamp AR, Hoenderop JG, Bindels RJ. Direct interaction with Rab11a targets the epithelial Ca2+ channels TRPV5 and TRPV6 to the plasma membrane. Mol Cell Biol. 2006;26:303–312. doi: 10.1128/MCB.26.1.303-312.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.van de Graaf SF, Hoenderop JG, Bindels RJ. Regulation of TRPV5 and TRPV6 by associated proteins. Am J Physiol Renal Physiol. 2006;290:F1295–F1302. doi: 10.1152/ajprenal.00443.2005. [DOI] [PubMed] [Google Scholar]
- 589.Van Itallie CM, Anderson JM. The molecular physiology of tight junction pores. Physiology (Bethesda ) 2004;19:331–338. doi: 10.1152/physiol.00027.2004. [DOI] [PubMed] [Google Scholar]
- 590.Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- 591.Vandewalle A. Expression and function of CLC and cystic fibrosis transmembrane conductance regulator chloride channels in renal epithelial tubule cells: pathophysiological implications. Chang Gung Med J. 2007;30:17–25. [PubMed] [Google Scholar]
- 592.Vandorpe D, Kizer N, Ciampollilo F, Moyer B, Karlson K, Guggino WB, Stanton BA. CFTR mediates electrogenic chloride secretion in mouse inner medullary collecting duct (mIMCD-K2) cells. Am J Physiol. 1995;269:C683–C689. doi: 10.1152/ajpcell.1995.269.3.C683. [DOI] [PubMed] [Google Scholar]
- 593.Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, Babakhanlou H, Cruger G, Kanazirska M, Ye C, Brown EM, Hediger MA, Zhou J. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochem Biophys Res Commun. 2001;282:341–350. doi: 10.1006/bbrc.2001.4554. [DOI] [PubMed] [Google Scholar]
- 594.Veizis EI, Carlin CR, Cotton CU. Decreased amiloride-sensitive Na+ absorption in collecting duct principal cells isolated from BPK ARPKD mice. Am J Physiol Renal Physiol. 2004;286:F244–F254. doi: 10.1152/ajprenal.00169.2003. [DOI] [PubMed] [Google Scholar]
- 595.Vennekens R, Prenen J, Hoenderop JG, Bindels RJ, Droogmans G, Nilius B. Modulation of the epithelial Ca2+ channel ECaC by extracellular pH. Pflugers Arch. 2001;442:237–242. doi: 10.1007/s004240100517. [DOI] [PubMed] [Google Scholar]
- 596.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, Wall SM. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension. 2003;42:356–362. doi: 10.1161/01.HYP.0000088321.67254.B7. [DOI] [PubMed] [Google Scholar]
- 598.Verrey F, Fakitsas P, Adam G, Staub O. Early transcriptional control of ENaC (de)ubiquitylation by aldosterone. Kidney Int. 2008;73:691–696. doi: 10.1038/sj.ki.5002737. [DOI] [PubMed] [Google Scholar]
- 599.Vuagniaux G, Vallet V, Jaeger NF, Pfister C, Bens M, Farman N, Courtois-Coutry N, Vandewalle A, Rossier BC, Hummler E. Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J Am Soc Nephrol. 2000;11:828–834. doi: 10.1681/ASN.V115828. [DOI] [PubMed] [Google Scholar]
- 600.Wade JB, Fang L, Liu J, Li D, Yang CL, Subramanya AR, Maouyo D, Mason A, Ellison DH, Welling PA. WNK1 kinase isoform switch regulates renal potassium excretion. Proc Natl Acad Sci U S A. 2006;103:8558–8563. doi: 10.1073/pnas.0603109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. Differential Regulation of ROMK (Kir1.1) in Distal Nephron Segments by Dietary Potassium. American Journal of Physiology - Renal Physiology. 2011 doi: 10.1152/ajprenal.00592.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Wade JB, Stanton BA, Brown D. Structural Correlates of Transport in Distal Tubule and Collecting Duct Segments. New York, NY: Oxford University Press; 1992. [Google Scholar]
- 603.Wagner CA, Devuyst O, Bourgeois S, Mohebbi N. Regulated acid-base transport in the collecting duct. Pflugers Arch. 2009;458:137–156. doi: 10.1007/s00424-009-0657-z. [DOI] [PubMed] [Google Scholar]
- 604.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar H+-ATPase. Physiol Rev. 2004;84:1263–1314. doi: 10.1152/physrev.00045.2003. [DOI] [PubMed] [Google Scholar]
- 605.Wagner CA, Loffing-Cueni D, Yan Q, Schulz N, Fakitsas P, Carrel M, Wang T, Verrey F, Geibel JP, Giebisch G, Hebert SC, Loffing J. Mouse model of type II Bartter’s syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am J Physiol Renal Physiol. 2008;294:F1373–F1380. doi: 10.1152/ajprenal.00613.2007. [DOI] [PubMed] [Google Scholar]
- 606.Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem. 1995;270:27411–27414. doi: 10.1074/jbc.270.46.27411. [DOI] [PubMed] [Google Scholar]
- 607.Wall SM. Recent advances in our understanding of intercalated cells. Curr Opin Nephrol Hypertens. 2005;14:480–484. doi: 10.1097/01.mnh.0000168390.04520.06. [DOI] [PubMed] [Google Scholar]
- 608.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW. Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol. 2003;284:F229–F241. doi: 10.1152/ajprenal.00147.2002. [DOI] [PubMed] [Google Scholar]
- 609.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl− conservation. Hypertension. 2004;44:982–987. doi: 10.1161/01.HYP.0000145863.96091.89. [DOI] [PubMed] [Google Scholar]
- 610.Wall SM, Pech V. The interaction of pendrin and the epithelial sodium channel in blood pressure regulation. Curr Opin Nephrol Hypertens. 2008;17:18–24. doi: 10.1097/MNH.0b013e3282f29086. [DOI] [PubMed] [Google Scholar]
- 611.Wallace DP. Cyclic AMP-mediated cyst expansion. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. doi: 10.1016/j.bbadis.2010.11.005. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Wang W. Regulation of renal K transport by dietary K intake. Annu Rev Physiol. 2004;66:547–569. doi: 10.1146/annurev.physiol.66.032102.112025. [DOI] [PubMed] [Google Scholar]
- 613.Wang W, Hebert SC, Giebisch G. Renal K+ channels: structure and function. Annu Rev Physiol. 1997;59:413–436. doi: 10.1146/annurev.physiol.59.1.413. [DOI] [PubMed] [Google Scholar]
- 614.Wang WH, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch. 2009;458:157–168. doi: 10.1007/s00424-008-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Wang WH, Schwab A, Giebisch G. Regulation of small-conductance K+ channel in apical membrane of rat cortical collecting tubule. American Journal of Physiology - Renal Physiology. 1990;259:F494–F502. doi: 10.1152/ajprenal.1990.259.3.F494. [DOI] [PubMed] [Google Scholar]
- 616.Wang WH, Yue P, Sun P, Lin DH. Regulation and function of potassium channels in aldosterone-sensitive distal nephron. Curr Opin Nephrol Hypertens. 2010;19:463–470. doi: 10.1097/MNH.0b013e32833c34ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Wang W, Henderson RM, Geibel J, White S, Giebisch G. Mechanism of aldosterone-induced increase of K+ conductance in early distal renal tubule cells of the frog. Journal of Membrane Biology. 1989;111:277–289. doi: 10.1007/BF01871012. [DOI] [PubMed] [Google Scholar]
- 618.Wang X, Armando I, Upadhyay K, Pascua A, Jose PA. The regulation of proximal tubular salt transport in hypertension: an update. Curr Opin Nephrol Hypertens. 2009;18:412–420. doi: 10.1097/MNH.0b013e32832f5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Wang Y, Klein JD, Liedtke CM, Sands JM. Protein kinase C regulates urea permeability in the rat inner medullary collecting duct. Am J Physiol Renal Physiol. 2010;299:F1401–F1406. doi: 10.1152/ajprenal.00322.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Wang ZJ, Sun P, Xing W, Pan C, Lin DH, Wang WH. Decrease in dietary K intake stimulates the generation of superoxide anions in the kidney and inhibits K secretory channels in the CCD. American Journal of Physiology - Renal Physiology. 2010;298:F1515–F1522. doi: 10.1152/ajprenal.00502.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Warnock DG, Rossier BC. Renal sodium handling: the role of the epithelial sodium channel. J Am Soc Nephrol. 2005;16:3151–3153. doi: 10.1681/ASN.2005080886. [DOI] [PubMed] [Google Scholar]
- 622.Watanabe H, Murakami M, Ohba T, Ono K, Ito H. The pathological role of transient receptor potential channels in heart disease. Circ J. 2009;73:419–427. doi: 10.1253/circj.cj-08-1153. [DOI] [PubMed] [Google Scholar]
- 623.Wei Y, Bloom P, Gu R, Wang W. Protein-tyrosine phosphatase reduces the number of apical small conductance K+ channels in the rat cortical collecting duct. J Biol Chem. 2000;275:20502–20507. doi: 10.1074/jbc.M000783200. [DOI] [PubMed] [Google Scholar]
- 624.Wei Y, Bloom P, Lin D, Gu R, Wang WH. Effect of dietary K intake on apical small-conductance K channel in CCD: role of protein tyrosine kinase. Am J Physiol Renal Physiol. 2001;281:F206–F212. doi: 10.1152/ajprenal.2001.281.2.F206. [DOI] [PubMed] [Google Scholar]
- 625.Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, Falck JR, Wang WH. Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol. 2004;124:719–727. doi: 10.1085/jgp.200409140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Wei Y, Sun P, Wang Z, Yang B, Carroll MA, Wang WH. Adenosine inhibits ENaC via cytochrome P-450 epoxygenase-dependent metabolites of arachidonic acid. Am J Physiol Renal Physiol. 2006;290:F1163–F1168. doi: 10.1152/ajprenal.00301.2005. [DOI] [PubMed] [Google Scholar]
- 627.Wei Y, Wang Z, Babilonia E, Sterling H, Sun P, Wang W. Effect of hydrogen peroxide on ROMK channels in the cortical collecting duct. Am J Physiol Renal Physiol. 2007;292:F1151–F1156. doi: 10.1152/ajprenal.00389.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Wei Y, Zavilowitz B, Satlin LM, Wang WH. Angiotensin II inhibits the ROMK-like small conductance K channel in renal cortical collecting duct during dietary potassium restriction. J Biol Chem. 2007;282:6455–6462. doi: 10.1074/jbc.M607477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Weinbaum S, Duan Y, Satlin LM, Wang T, Weinstein AM. Mechanotransduction in the renal tubule. American Journal of Physiology - Renal Physiology. 2010;299:F1220–F1236. doi: 10.1152/ajprenal.00453.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Weinstein AM. A mathematical model of rat distal convoluted tubule. II. Potassium secretion along the connecting segment. Am J Physiol Renal Physiol. 2005;289:F721–F741. doi: 10.1152/ajprenal.00044.2005. [DOI] [PubMed] [Google Scholar]
- 631.Welling PA, Chang YP, Delpire E, Wade JB. Multigene kinase network, kidney transport, and salt in essential hypertension. Kidney Int. 2010;77:1063–1069. doi: 10.1038/ki.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol. 2009;297:F849–F863. doi: 10.1152/ajprenal.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Welling PA, Weisz OA. Sorting it out in endosomes: an emerging concept in renal epithelial cell transport regulation. Physiology (Bethesda ) 2010;25:280–292. doi: 10.1152/physiol.00022.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Wesch D, Miranda P, fonso-Oramas D, Althaus M, Castro-Hernandez J, Dominguez J, Morty RE, Clauss W, Gonzalez-Hernandez T, Alvarez de la Rosa D, Giraldez T. The neuronal-specific SGK1.1 kinase regulates d-epithelial Na+ channel independently of PY motifs and couples it to phospholipase C signaling. American Journal of Physiology - Cell Physiology. 2010;299:C779–C790. doi: 10.1152/ajpcell.00184.2010. [DOI] [PubMed] [Google Scholar]
- 635.Wieczorek H, Beyenbach KW, Huss M, Vitavska O. Vacuolar-type proton pumps in insect epithelia. Journal of Experimental Biology. 2009;212:1611–1619. doi: 10.1242/jeb.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Wiemuth D, Ke Y, Rohlfs M, McDonald FJ. Epithelial sodium channel (ENaC) is multi-ubiquitinated at the cell surface. Biochem J. 2007;405:147–155. doi: 10.1042/BJ20060747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Wilson FH, sse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human Hypertension Caused by Mutations in WNK Kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 638.Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151–164. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 639.Wilson PD. Apico-basal polarity in polycystic kidney disease epithelia. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbadis.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 640.Wilson PD, Goilav B. Cystic disease of the kidney. Annu Rev Pathol. 2007;2:341–368. doi: 10.1146/annurev.pathol.2.010506.091850. [DOI] [PubMed] [Google Scholar]
- 641.Wilson PD, Sherwood AC, Palla K, Du J, Watson R, Norman JT. Reversed polarity of Na(+) -K(+) -ATPase: mislocation to apical plasma membranes in polycystic kidney disease epithelia. Am J Physiol. 1991;260:F420–F430. doi: 10.1152/ajprenal.1991.260.3.F420. [DOI] [PubMed] [Google Scholar]
- 642.Wilson PD, Devuyst O, Li X, Gatti L, Falkenstein D, Robinson S, Fambrough D, Burrow CR. Apical Plasma Membrane Mispolarization of NaK-ATPase in Polycystic Kidney Disease Epithelia Is Associated with Aberrant Expression of the [beta]2 Isoform. The American Journal of Pathology. 2000;156:253–268. doi: 10.1016/s0002-9440(10)64726-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Wingo CS, Seldin DW, Kokko JP, Jacobson HR. Dietary modulation of active potassium secretion in the cortical collecting tubule of adrenalectomized rabbits. J Clin Invest. 1982;70:579–586. doi: 10.1172/JCI110650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 645.Winter C, Schulz N, Giebisch G, Geibel JP, Wagner CA. Nongenomic stimulation of vacuolar H+-ATPases in intercalated renal tubule cells by aldosterone. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2636–2641. doi: 10.1073/pnas.0307321101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Woda CB, Bragin A, Kleyman TR, Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol. 2001;280:F786–F793. doi: 10.1152/ajprenal.2001.280.5.F786. [DOI] [PubMed] [Google Scholar]
- 647.Woudenberg-Vrenken TE, Bindels RJ, Hoenderop JG. The role of transient receptor potential channels in kidney disease. Nat Rev Nephrol. 2009;5:441–449. doi: 10.1038/nrneph.2009.100. [DOI] [PubMed] [Google Scholar]
- 648.Wu L, Gao X, Brown RC, Heller S, O’Neil RG. Dual role of the TRPV4 channel as a sensor of flow and osmolality in renal epithelial cells. Am J Physiol Renal Physiol. 2007;293:F1699–F1713. doi: 10.1152/ajprenal.00462.2006. [DOI] [PubMed] [Google Scholar]
- 649.Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current Progress in the Mammalian TRP Ion Channel Family. Pharmacol Rev. 2010;62:381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Wyckoff JA, Seely EW, Hurwitz S, Anderson BF, Lifton RP, Dluhy RG. Glucocorticoid-remediable aldosteronism and pregnancy. Hypertension. 2000;35:668–672. doi: 10.1161/01.hyp.35.2.668. [DOI] [PubMed] [Google Scholar]
- 651.Xie L, Hoffert JD, Chou CL, Yu MJ, Pisitkun T, Knepper MA, Fenton RA. Quantitative analysis of aquaporin-2 phosphorylation. American Journal of Physiology - Renal Physiology. 2010;298:F1018–F1023. doi: 10.1152/ajprenal.00580.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 653.Xu BE, Stippec S, Chu PY, Lazrak A, Li XJ, Lee BH, English JM, Ortega B, Huang CL, Cobb MH. WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc Natl Acad Sci U S A. 2005;102:10315–10320. doi: 10.1073/pnas.0504422102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Xu DL, Martin PY, Ohara M, St John J, Pattison T, Meng X, Morris K, Kim JK, Schrier RW. Upregulation of aquaporin-2 water channel expression in chronic heart failure rat. J Clin Invest. 1997;99:1500–1505. doi: 10.1172/JCI119312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Xu JZ, Hall AE, Peterson LN, Bienkowski MJ, Eessalu TE, Hebert SC. Localization of the ROMK protein on apical membranes of rat kidney nephron segments. Am J Physiol. 1997;273:F739–F748. doi: 10.1152/ajprenal.1997.273.5.F739. [DOI] [PubMed] [Google Scholar]
- 656.Xu ZC, Yang Y, Hebert SC. Phosphorylation of the ATP-sensitive, inwardly rectifying K+ channel, ROMK, by cyclic AMP-dependent protein kinase. J Biol Chem. 1996;271:9313–9319. doi: 10.1074/jbc.271.16.9313. [DOI] [PubMed] [Google Scholar]
- 657.Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, Suda S, Hayama A, Sasaki S, Uchida S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4690–4694. doi: 10.1073/pnas.0306924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Yamauchi K, Yang SS, Ohta A, Sohara E, Rai T, Sasaki S, Uchida S. Apical localization of renal K channel was not altered in mutant WNK4 transgenic mice. Biochemical and Biophysical Research Communications. 2005;332:750–755. doi: 10.1016/j.bbrc.2005.04.169. [DOI] [PubMed] [Google Scholar]
- 659.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:1300–1310. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Neonatal Mortality in an Aquaporin-2 Knock-in Mouse Model of Recessive Nephrogenic Diabetes Insipidus. J Biol Chem. 2001;276:2775–2779. doi: 10.1074/jbc.M008216200. [DOI] [PubMed] [Google Scholar]
- 661.Yang B, Verkman AS. Water and Glycerol Permeabilities of Aquaporins 1GÇô5 and MIP Determined Quantitatively by Expression of Epitope-tagged Constructs inXenopus Oocytes. J Biol Chem. 1997;272:16140–16146. doi: 10.1074/jbc.272.26.16140. [DOI] [PubMed] [Google Scholar]
- 662.Yang B, Zhao D, Verkman AS. Hsp90 inhibitor partially corrects nephrogenic diabetes insipidus in a conditional knock-in mouse model of aquaporin-2 mutation. The FASEB Journal. 2009;23:503–512. doi: 10.1096/fj.08-118422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Yang J, Jan YN, Jan LY. Determination of the subunit stoichiometry of an inwardly rectifying potassium channel. Neuron. 1995;15:1441–1447. doi: 10.1016/0896-6273(95)90021-7. [DOI] [PubMed] [Google Scholar]
- 664.Yang L, Frindt G, Palmer LG. Magnesium modulates ROMK channel-mediated potassium secretion. J Am Soc Nephrol. 2010;21:2109–2116. doi: 10.1681/ASN.2010060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Yang SS, Hsu YJ, Chiga M, Rai T, Sasaki S, Uchida S, Lin SH. Mechanisms for Hypercalciuria in Pseudohypoaldosteronism Type II-Causing WNK4 Knock-In Mice. Endocrinology. 2010;151:1829–1836. doi: 10.1210/en.2009-0951. [DOI] [PubMed] [Google Scholar]
- 666.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 667.Yasoshima K, Satlin LM, Schwartz GJ. Adaptation of rabbit cortical collecting duct to in vitro acid incubation. Am J Physiol. 1992;263:F749–F756. doi: 10.1152/ajprenal.1992.263.4.F749. [DOI] [PubMed] [Google Scholar]
- 668.Yeh BI, Kim YK, Jabbar W, Huang CL. Conformational changes of pore helix coupled to gating of TRPV5 by protons. EMBO J. 2005;24:3224–3234. doi: 10.1038/sj.emboj.7600795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Yeh BI, Sun TJ, Lee JZ, Chen HH, Huang CL. Mechanism and molecular determinant for regulation of rabbit transient receptor potential type 5 (TRPV5) channel by extracellular pH. J Biol Chem. 2003;278:51044–51052. doi: 10.1074/jbc.M306326200. [DOI] [PubMed] [Google Scholar]
- 670.Yeh BI, Yoon J, Huang CL. On the role of pore helix in regulation of TRPV5 by extracellular protons. J Membr Biol. 2006;212:191–198. doi: 10.1007/s00232-006-0023-4. [DOI] [PubMed] [Google Scholar]
- 671.Yoo D, Fang L, Mason A, Kim BY, Welling PA. A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J Biol Chem. 2005;280:35281–35289. doi: 10.1074/jbc.M504836200. [DOI] [PubMed] [Google Scholar]
- 672.Yoo D, Kim BY, Campo C, Nance L, King A, Maouyo D, Welling PA. Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J Biol Chem. 2003;278:23066–23075. doi: 10.1074/jbc.M212301200. [DOI] [PubMed] [Google Scholar]
- 673.Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381–401. doi: 10.1146/annurev.physiol.010908.163241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Yu ASL, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Lynch RD, Schneeberger EE. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. American Journal of Physiology - Cell Physiology. 2005;288:C1231–C1241. doi: 10.1152/ajpcell.00581.2004. [DOI] [PubMed] [Google Scholar]
- 675.Yuan P, Leonetti MD, Pico AR, Hsiung Y, MacKinnon R. Structure of the human BK channel Ca2+-activation apparatus at 3.0 A resolution. Science. 2010;329:182–186. doi: 10.1126/science.1190414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Yue P, Lin DH, Pan CY, Leng Q, Giebisch G, Lifton RP, Wang WH. Src family protein tyrosine kinase (PTK) modulates the effect of SGK1 and WNK4 on ROMK channels. Proc Natl Acad Sci U S A. 2009;106:15061–15066. doi: 10.1073/pnas.0907855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Yue P, Sun P, Lin DH, Pan C, Xing W, Wang W. Angiotensin II diminishes the effect of SGK1 on the WNK4-mediated inhibition of ROMK1 channels. Kidney Int. 2011;79:423–431. doi: 10.1038/ki.2010.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Zaika O, Mamenko M, O’Neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. American Journal of Physiology - Renal Physiology. 2011 doi: 10.1152/ajprenal.00606.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Zelenina M, Tritto S, Bondar AA, Zelenin S, Aperia A. Copper Inhibits the Water and Glycerol Permeability of Aquaporin-3. J Biol Chem. 2004;279:51939–51943. doi: 10.1074/jbc.M407645200. [DOI] [PubMed] [Google Scholar]
- 680.Zheleznova NN, Wilson PD, Staruschenko A. Epidermal growth factor-mediated proliferation and sodium transport in normal and PKD epithelial cells. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2011 doi: 10.1016/j.bbadis.2010.10.004. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681.Zheng W, Verlander JW, Lynch IJ, Cash M, Shao J, Stow LR, Cain BD, Weiner ID, Wall SM, Wingo CS. Cellular distribution of the potassium channel KCNQ1 in normal mouse kidney. Am J Physiol Renal Physiol. 2007;292:F456–F466. doi: 10.1152/ajprenal.00087.2006. [DOI] [PubMed] [Google Scholar]
- 682.Zhou R, Snyder PM. Nedd4-2 phosphorylation induces serum and glucocorticoid-regulated kinase (SGK) ubiquitination and degradation. J Biol Chem. 2005;280:4518–4523. doi: 10.1074/jbc.M411053200. [DOI] [PubMed] [Google Scholar]
- 683.Zhuang J, Zhang X, Wang D, Li J, Zhou B, Shi Z, Gu D, Denson DD, Eaton DC, Cai H. WNK4 kinase inhibits Maxi K channel activity by a kinase-dependent mechanism. American Journal of Physiology - Renal Physiology. 2011 doi: 10.1152/ajprenal.00518.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.Zuber AM, Centeno G, Pradervand S, Nikolaeva S, Maquelin L, Cardinaux L, Bonny O, Firsov D. Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci U S A. 2009;106:16523–16528. doi: 10.1073/pnas.0904890106. [DOI] [PMC free article] [PubMed] [Google Scholar]