

Abstract
The autosomal dominant spinocerebellar ataxias (SCAs) are a genetically heterogeneous group of disorders exhibiting cerebellar atrophy and Purkinje cell degeneration whose subtypes arise from 31 distinct genetic loci. Our group previously published the locus for SCA26 on chromosome 19p13.3. In this study, we performed targeted deep sequencing of the critical interval in order to identify candidate causative variants in individuals from the SCA26 family. We identified a single variant that co-segregates with the disease phenotype that produces a single amino acid substitution in eukaryotic elongation factor 2. This substitution, P596H, sits in a domain critical for maintaining reading frame during translation. The yeast equivalent, P580H EF2, demonstrated impaired translocation, detected as an increased rate of −1 programmed ribosomal frameshift read-through in a dual-luciferase assay for observing translational recoding. This substitution also results in a greater susceptibility to proteostatic disruption, as evidenced by a more robust activation of a reporter gene driven by unfolded protein response activation upon challenge with dithiothreitol or heat shock in our yeast model system. Our results present a compelling candidate mutation and mechanism for the pathogenesis of SCA26 and further support the role of proteostatic disruption in neurodegenerative diseases.
INTRODUCTION
The autosomal dominant spinocerebellar ataxias (SCAs) are a genetically and clinically heterogeneous group of neurodegenerative disorders. To date, 31 unique subtypes attributed to distinct genetic loci have been identified, with a collective incidence of ∼50/100 000 (1). All subtypes share the common endpoint of Purkinje cell death and atrophy of the cerebellum (2). For 20 of the 31 subtypes, genetic bases have been identified (2–38). Six have been attributed to polyglutamine expansions in unrelated proteins (SCAs 1, 2, 3, 6, 7 and 17) (3–6,9,10,33), five to untranslated repeat expansions (SCAs 8, 10, 12, 31 and 36) (11–13,15,27,29), one to various deletions in a single gene (SCA15/16) (34), and eight to various point mutations in unrelated genes (SCAs 5, 11, 13, 14, 23, 27, 28 and 35) (7,8,14,16–21,23–26,28,30–32,35–38). The phenotypes can be grouped into two broad categories: The pure cerebellar ataxias and the cerebellar ataxias that involve additional neurological features (34). The immense genetic heterogeneity of these clinically overlapping diseases suggests that the unique functional complexity of cerebellar cell types makes them highly susceptible to a diverse array of molecular defects.
We previously reported genetic analysis of a six-generation kindred of Norwegian ancestry with a late-onset pure cerebellar ataxia that demonstrated linkage to a novel locus on chromosome 19p13.3, now known as SCA26 (OMIM: 609306) (39), that contains 104 known genes. Here, we report the identification of the putative disease gene and causal variant and provide functional studies that point to the potential disease mechanism.
RESULTS
Cerebellar pathology primarily in Purkinje cells
We previously described a six-generation kindred of late-onset SCA (SCA26) without extracerebellar features that mapped to a novel locus on chromosome 19p13.3 (39). We subsequently conducted post-mortem studies on two affected individuals who died of unrelated causes. One of these had subtle gait disorder since her 60's and died in her late 60's from ovarian cancer. The other had onset of ataxia and dysarthria at age 75 and died of cardiac arrest at age 79. Both individuals had a significant loss of Purkinje cells with minimal or no neuronal loss in other brain regions (Fig. 1). We counted 402 and 742 Purkinje cells in the vermis in each of the two SCA26 samples as compared with 2810 Purkinje cells in the normal control vermis. There were entire folia in the SCA26 samples without a single Purkinje cell. The cellular degeneration pattern is consistent with the disease phenotype, as seen in other forms of pure cerebellar ataxia (34).
Figure 1.

Cerebellar sections from an SCA26 patient show significant loss of Purkinje cells. (Left) Normal human cerebellar cortex with Purkinje cells at the boundary between the molecular and granular layers. (Right) SCA26 cerebellar cortex with loss of Purkinje cells and thinning of the molecular layer. Hematoxylin and eosin. Scale bar = 50 µm.
Identification of a variant that co-segregates with the disease phenotype
The locus for SCA26 was mapped to a 15.55 cM region with maximum logarithm of odds scores (6.06–14.61) between markers D19S886 and D19S894 (39), determined by obligate recombinants. Large-scale genomic rearrangements were not examined, but CAG repeat expansions were excluded (39) by repeat expansion detection (40). This region contained 3.5 Mb and 104 known genes. We designed an Agilent 1 M feature custom capture array to tile across all coding and non-coding non-repetitive bases within the critical interval and sequenced four individuals: two sibship pairs composed of affected/unaffected (without disease haplotype) individuals separated by six meioses (Supplementary Material, Fig. S1). Given the dominant mode of inheritance and the presumed low frequency of the causal allele, genotypes were filtered to identify heterozygous variants shared by both affecteds, but not present in either unaffected or dbSNP (build 131) or 1000 genomes project data (41). This process identified 38 variants that co-segregated with the disease phenotype (Table 1), including 10 non-coding single nucleotide polymorphisms (SNPs) and 24 non-coding indels in non-genic regions; 1 3′UTR indel; 1 synonymous variant in the coding region of HMHA1 and 2 non-synonymous variants, one in the minor histocompatibility HA-1 gene, HMHA1 (NM_012292), and the other in the eukaryotic elongation factor 2 gene, eEF2 (NM_001961). None of the concordant non-coding indels was longer than 13 bases and none overlapped an evolutionarily constrained element of a predicted cis-regulatory element. None of the concordant variants fell within 2 bp of a splice junction, and so putative effects on splicing were not examined. The HMHA1 variant generates a T1124I substitution at a highly variable residue, predicted by the SIFT (‘Sorting Tolerant from Intolerant’) algorithm (42) to be tolerated. The eEF2 variant is a C-to-A transversion at the highly evolutionarily constrained base chr19:3929097 (NCBI36/hg18) in exon 12 of the 15-exon coding region, producing a GERP (genomic evolutionary rate profiling) score (43) of 5.38 (i.e. is highly evolutionarily constrained; Fig. 2). The transversion generates a P596H substitution, which is predicted to be ‘damaging’ (SIFT) (42). This variant co-segregated with the disease phenotype in all 24 affected members of the family and was also found in two currently asymptomatic members of the family who bear the disease haplotype. Magnetic resonance brain imaging for these individuals was not available to detect cerebellar atrophy, as seen in pre-symptomatic individuals. Finally, we confirmed the presence of this genomic variant in transcribed mRNA, via polymerase chain reaction (PCR)-amplified cDNA from patient lymphoblastoid cell RNA (data not shown).
Table 1.
Four variants in genic regions were identified as co-segregating with the disease phenotype
| Hg18Pos Chr19 | REF | ALT | Gene name | Location | GERP | Amino acid | SIFT |
|---|---|---|---|---|---|---|---|
| Genic variants | |||||||
| 3929097 | G | T | eEF2 | Exon 12 | 5.38 | P596H | DAMAGING |
| 1036965 | C | T | HMHA1 | Exon 23 | 0.957 | T1124I | tolerated |
| 1033913 | G | A | HMHA1 | Exon 20 | 1.44 | SYN | |
| 3413773 | GTCTTCTTC | GTCTTC | NFIC | 3′UTR | NA | NA | |
Four family members were used for critical interval sequencing via custom capture array. The P596H amino acid substitution in eEF2 is the only highly evolutionarily conserved variant predicted to be ‘damaging’. REF, reference nucleotide; ALT, alternate found upon sequencing; GERP, genomic evolutionary rate profiling (43); SIFT, ‘Sorting Tolerant From Intolerant’ algorithm (42); SYN, synonymous variant.
Figure 2.
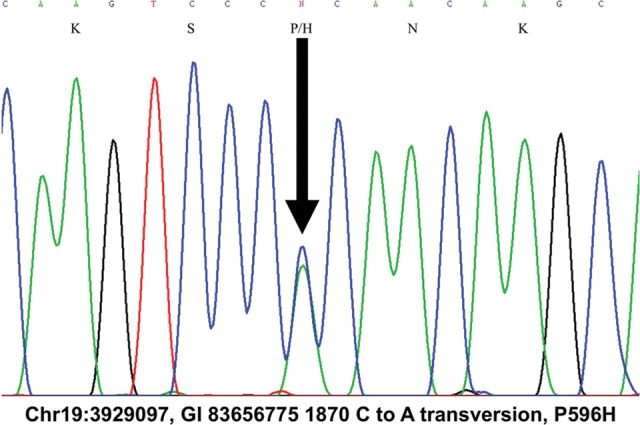
C-to-A transversion at Chr19:3929097 (GI 83656775 1870) co-segregates with disease in SCA26. The C-to-A transversion produces a P596H substitution in the eEF2 protein.
We investigated whether the C-to-A transversion in the eEF2 gene was a rare benign polymorphism. We sequenced exon 12 in 100 phenotypically normal individuals from the Center d'Etude du Polymorphisme Humain (CEPH) grandparent DNA collection and in 104 phenotypically normal individuals of Norwegian ancestry. Neither our SNP nor any other previously unreported SNPs were identified. The co-segregation with a disease phenotype, predicted functional impact and absence from publicly available and internally generated resequencing data in phenotypically normal individuals make the C-to-A transversion at chr19:3929097, producing a P596H substitution in eEF2, a likely candidate causal variant.
To test for other variants associated with SCA, we also sequenced exon 12 of 355 probands with unknown forms of an autosomal dominant cerebellar ataxia (ADCA) from European and American collections, but did not identify any other ataxia families with variants in exon 12. Although the data strongly support this as a candidate causal variant, the absence of other ADCA families with eEF2 mutations indicates that this is likely a rare cause of ataxia.
Characterization of the substituted protein
The mRNA produces a 95 kDa protein of 858 amino acids with six domains (NCBI protein ID AAI26260.1). The affected amino acid residue, P596, and its surrounding residues are highly conserved, from Arabidopsis thaliana to Homo sapiens (Fig. 3), indicating that the position has evolved under strong purifying selection. Given the predicted deleterious effect of the P596H variant, we tested whether eEF2 protein containing the P596H substitution was able to fold properly. cDNAs encoding reference and the P596H eEF2 were cloned, in-frame into the pCDNA3.1 vector with a C-terminal 3XFLAG™ epitope tag and transiently transfected into HEK293 cells. P596H and reference eEF2 proteins were expressed at comparable levels, with no evidence of unique degradation products, suggesting that the P596H protein is properly expressed and post-translationally processed (Supplementary Material, Fig. S2).
Figure 3.

Proline 596 (H. sapiens) is conserved throughout many eukaryotes. Alignment of the EF2 protein sequence demonstrates that P596 is highly conserved throughout eukaryotes.
The ubiquitously expressed eEF2 protein has been highly characterized. Its primary function is to facilitate the translocation of the peptidyl-tRNA from the A-site to the P-site in the ribosome (44,45). eEF2 is predominantly found near the rough endoplasmic reticulum (rER), consistent with its essential role in all protein translation. Several neurodegenerative diseases known to be caused by point mutations in specific proteins [familial amyotrophic lateral sclerosis (fALS), SCA14] (22,46) exhibit altered subcellular localization, misfolding and/or aggregates of the mutant protein in affected brain tissues, although the role of these aggregates in disease pathogenesis is still undefined. To investigate whether P596H eEF2 has altered subcellular localization or forms prominent intracellular aggregates, we first performed immunocytochemical staining for the eEF2 protein and the rER marker, calnexin, in lymphoblastoid cell lines derived from an SCA26 patient and a phenotypically normal genetically unrelated individual. In both control and patient cells, eEF2 and calnexin were highly co-localized, as shown by the cytofluorogram (Pearson's coefficients of 0.935 for wild-type (WT) and 0.946 for P596H eEF2 with calnexin), and there were no signs of aggregation (Supplementary Material, Fig. S3). We further tested for alterations in subcellular localization and signs of prominent intracellular aggregation by transiently transfecting human neuroblastoma (SH-SY5Y) cells with reference and P596H eEF2 fusion proteins. Co-transfected 3XHA-tagged WT protein (pKH3 vector) and 3XFLAG™-tagged P596H eEF2 were highly co-localized with each other and calnexin (Fig. 4). These data suggest that P596H eEF2 properly localizes to the rER.
Figure 4.

WT and P596H eEF2 co-localize with rER. WT and P596H eEF2 were expressed via transient transfection in SH-SY5Y cells. (Left) WT eEF2 with a C-terminal 3 × HA epitope tag co-localizes strongly with rER in SH-SY5Y cells upon staining for HA and calnexin (rER), 156×. (Middle) P596H eEF2 with a C-terminal 3 × FLAG tag co-localizes strongly with rER in SH-SY5Y cells upon staining for FLAG and calnexin (rER), 170×. (Right) WT eEF2 with a C-terminal 3 × HA epitope tag and P596H eEF2 with a C-terminal 3 × FLAG tag co-localize strongly together in SH-SY5Y cells upon staining for HA and FLAG, 220×
We also examined eEF2 subcellular localization within normal human and SCA26 cerebellum. Labeling with eEF2 antibody co-localizes with the endoplasmic reticulum (ER) marker calnexin in Purkinje cells in both normal human and SCA26 cerebella (data not shown), although the extent of labeling with eEF2 and calnexin is somewhat diminished in the SCA26 Purkinje cells, compared with normal human cerebellum. However, because the SCA26 Purkinje cells were extremely few in number, and most likely sick relative to the normal human Purkinje cells, this change may represent only an involution pattern on the part of the ER due to late stage disease. This is supported by the fact that our cell transfection studies that showed co-localization of the mutant and WT eEF2 proteins with ER markers exhibited no difference in the extent of the ER in the two types of transfected mammalian cells (Fig. 4).
Finally, to test the biological function of the P596H, we used a Saccharomyces cerevisiae model system, in which we generated the equivalent mutation at P580H. The yeast EF2 protein is 66% identical to the human eEF2 protein, with an additional 12% of the sequence consisting of conservative amino acid substitutions (Supplementary Material, Fig. S4), and is a well-established model for studying mammalian eEF2 function (47). As with all cells, yeast cells do not survive without EF2. We used a haploid yeast strain, TKY398, in which both non-allelic EF2 paralog genes have been deleted by homologous recombination (48) bearing the pURA3-EFT1 rescue plasmid (49). We found that TKY398 yeast cells bearing the YCP-LEU2 plasmid containing P580H EF2 as the only source of EF2 were viable, but exhibited a trend toward a slower growth rate, although not statistically significant (Supplementary Material, Fig. S5). Together, these results suggest that the EF2 P580H substitution does not cause gross destabilization or mislocalization of the protein and supports a degree of biological function that is compatible with life.
Yeast P580H EF2 demonstrates an increased rate of shifting into the −1 reading frame
In the yeast protein, P580 sits in a flexible loop near a functionally critical region known as the ‘anticodon mimicry’ domain that resembles the anticodon stem loop of tRNA (50,51). This domain interacts with the ribosomal decoding center during tRNA translocation (51–53) and contains a unique post-translational modification known as the diphthamide moiety that occurs on residues equivalent to yeast histidine 699 in other species of EF2 (54,55). The diphthamide moiety also serves as the pathological target of ADP-ribosylation by diphtheria toxin and Pseudomonas aeruginosa exotoxin A, which poisons the cell by inactivating the protein (54,55). Mutations disrupting the formation of the diphthamide result in an increased rate of −1 frameshifting (56), detected by means of a −1 programmed ribosomal frameshift (PRF) sequence between two reporter genes (56). PRFs are ‘sticky’ heptameric nucleotide sequences that serve to promote functional shifts in the reading frame (known as read-through) during translation, frequently to the +1 or −1 reading frame, in response to an intracellular biological signal to produce an alternate polypeptide product (57,58). According to the current model of PRF function, the PRF results in a brief pause during translation and, if the necessary biological signal is present, allows for the ribosomal machinery to shift the frame (59).
We next tested the hypothesis that the SCA26-associated EF2 variant generates an increase in PRF read-through. PRF read-through was assayed using a dual-luciferase reporter vector that contains a viral PRF flanked by coding regions for the Renilla and firefly luciferases (58). An increase in PRF read-through is detected as an increase in the downstream firefly luciferase relative to the upstream Renilla luciferase. As a positive control, we used the EF2 mutation H699Q, which lacks the diphthamide modification and generates an increased read-through rate of −1 PRF sequences (Fig. 5) (56). As predicted, we found that the P580H SCA26 variant also generated an increased rate of −1 PRF read-through, comparable with that seen with the H699Q substitution (Fig. 5). Moreover, PRF read-through was even higher in the double (P580H + H699Q) EF2 mutant. The increased read-through rate in these two mutants suggests that the anticodon mimicry domain is highly sensitive to changes in conformation. This perturbation appears to disrupt the normal mechanical processes involved in translocation, demonstrating that the P-to-H substitution impairs the reading frame maintenance function of the ‘anticodon mimicry’ domain.
Figure 5.

P580H EF2 shows a significant increase in the rate of frameshifting. The increase in −1 frameshifting is similar to that found in the positive control EF2 mutant, H699Q. The double mutant, P580H–H699Q, exhibits an even greater rate of frameshifting than either single mutation individually. *P < 5 × 10−4.
P580H EF2-bearing yeast is more susceptible to proteostatic insult than WT
We next sought to investigate the consequences of impaired translocation, which fall into three major categories. All three would ultimately affect the ability of the cell to meet demands of protein synthesis, which is critical in maintaining proteostasis (60,61). First, protein synthesis may be slowed; second, it may result in a global loss of translational fidelity, producing mistranslated proteins; and third, it may compromise the PRF mechanism of post-transcriptional gene expression regulation, resulting in a loss of translational regulation of transcripts bearing functional PRFs. Changes in PRF function, as well as an increase in nonsense polypeptides, would be virtually undetectable, due to the limitations of current knowledge and technical approaches. The consequence of any combination of these possible contributing factors would be disruption of proteostasis.
To assess whether the P580H substitution affects proteostasis, we looked at the ability of the cell to cope with the induction of misfolded proteins. Unfolded proteins activate the unfolded protein response (UPR) (62). Activation of the UPR results in alternate splicing of the HAC1 (XBP1 in mammalian cells) transcript that then produces a stable, functional transcription factor. The HAC1 protein binds the UPR element (UPRE) and drives transcription of other chaperones to combat the accumulation of unfolded proteins (62). We transformed the EF2 mutant yeast cells with pJC104 (63), a reporter plasmid containing four tandem UPRE repeats, driving expression of β-galactosidase to indicate UPR activation. Basal β-galactosidase expression (UPR activation) did not differ significantly between WT and P580H yeast (data not shown), possibly due to the high background in the assay. However, we challenged proteostasis using two well-known stressors associated with protein misfolding, dithiothreitol (DTT) and heat shock. After treatment with DTT, the P580H yeast exhibited more than two-fold greater induction of β-galactosidase expression than WT (Fig. 6). Furthermore, when incubated at 42°C to induce heat shock, P580H-expressing cells exhibited a 10-fold increase in β-galactosidase expression when compared with WT expressing yeast (Fig. 6). The more robust UPR response to DTT treatment and to heat shock in P580H-bearing yeast suggests that this mutant EF2 leads to proteostatic decompensation.
Figure 6.

P580H EF2-bearing yeast exhibit more robust unfolded protein response (UPR) induction in response to DTT and heat shock challenge. Yeast were transformed with pJC104, a plasmid containing four tandem repeats of the HAC1 binding element, that normally drives chaperone expression in response to UPR activation, driving β-galactosidase expression. Yeast were challenged either with 10 mm DTT or heat shock at 42°C for 15 min, and β -galactosidase activity was measured using the Promega Beta-glo assay. *P < 0.01.
DISCUSSION
We have identified a candidate causal variant in eukaryotic elongation factor 2 that co-segregates with the disease phenotype in SCA26. This variant was present in all 24 affected individuals in the family over the five generations sequenced, as well as in 2 asymptomatic individuals, consistent with the finding of incomplete penetrance in other autosomal dominant ataxias (2). The high degree of evolutionary conservation of this base (GERP 5.38) and of the P596 residue make it by far the most likely candidate causal variant when compared with any of the other variants we found, the most deleterious of which produced a GERP of 1.46. We note that, given the targeted resequencing strategy employed, it is unlikely that insertions or deletions, including repeat expansions, between 15 and 1000 bp would have been detected; however, the evolutionary constraint of the eEF2 variant provided compelling evidence to pursue it as a candidate causal variant. We subsequently demonstrated that this proline to histidine substitution at residue 580 (yeast equivalent of P596 in mammalian cells) produces a deficit in EF2 function. It results in an increased rate of shifting into the −1 reading frame during translation (detected over PRF sequences), and yeast cells bearing this P580H EF2 are more susceptible to challenges to proteostasis.
P580 sits in a critical region of the EF2 protein, adjacent to the diphthamide moiety, and contributes to the ability of EF2 to maintain the correct reading frame during translation. It has been hypothesized that a conformational shift of EF2 causes the diphthamide moiety to swing down into the ribosomal decoding center. Within the decoding center, two crucial adenines, AA1492 and AA1493, contribute vital H-bonds to the minor groove of the codon-anticodon pair. The diphthamide breaks and replaces these stabilizing hydrogen bonds during translocation (Fig. 7) (52). As the P580 and H699 side chains are only 10 Å apart, the substitution of histidine for proline at residue 580 likely contributes steric hindrance in a tightly packed interaction between EF2 and the ribosome, thereby impeding the diphthamide in its role of resetting AA1492 and AA1493.
Figure 7.
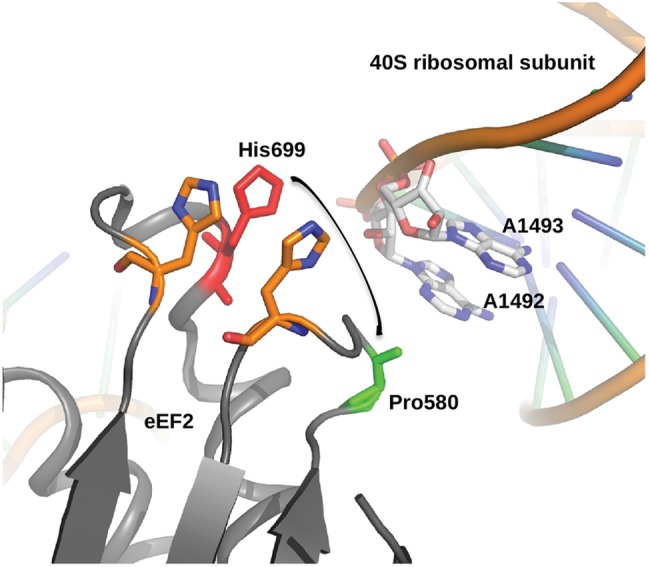
Yeast P580 lies in domain IV adjacent to the diphthamide-bearing moiety, H699. Diagram of the crystal structure of EF2 in complex with the yeast 40S ribosomal subunit (PDB: 1S1H), illustrating the proximity of P580 to both H699 and AA1492 and AA1493, all crucial elements in maintaining the fidelity of the translocation process. In a conformational shift of EF2 during translocation, the diphthamide moiety (essentially a 4-carbon chain extending from the imidazole ring of H699) swing down (arrow), breaking crucial hydrogen bonds between AA1492 and AA1493 and the minor groove of the codon-anticodon pair, stabilizing the codon–anticodon pair as it moves from the A- to the P-site within the ribosome, and allowing AA1492 and AA1493 to be available for the next codon–anticodon. P580 is only 10 Å from H699, highly suggesting that the bulky histidine side chain introduced in a P580H substitution would contribute steric hindrance and interfere with this delicate process. Figure courtesy of Rene Jørgensen, Statens Serum Institute, Copenhagen, Denmark.
One possible limitation of our dual-luciferase assay is that it specifically assesses changes in frameshifting over PRFs only, known hotspots for frameshifting (with a frequency ranging from 1 to 20% of the time) (58). Translational frameshifting errors do occur over non-PRF sequences, with a much lower estimated frequency of 3 × 10−5 to 1 × 10−4 (64). Although the mechanism by which programmed ribosomal frameshifting occurs involves the coordination of complex mRNA components (59,65), it seems likely that a structural defect in eEF2 impairing translocation over PRFs would also affect non-PRF frameshifting events, although perhaps with a lower frequency.
The disruption of translocation demonstrated here in yeast may be even more dramatic in mammalian eEF2, as yeast appear to tolerate H699 substitutions better than mammalian cells tolerate H715 substitutions. A random mutagenesis screen for diphtheria toxin-resistant EF2 in yeast produced four substitutions (L, Q, M and N) at H699 (48); targeted mutagenesis revealed that eight more possible substitutions were also fully functional, though yeast expressing only the substituted EF2s grew more slowly than WT (66). A random mutagenesis screen to identify diphtheria toxin resistant Chinese hamster ovary cells (haploid with only one copy of eEF2) failed to produce any viable substitutions of H715 (67), suggesting a more stringent requirement for the presence of the histidine at position 715.
Our dual-luciferase study demonstrates that the P580H yeast mutation produces a similar defect in the translocation mechanism as the loss of the diphthamide from H699 mutagenesis. The specific consequences of this disruption in translation are not certain. Impaired translocation may affect the rate at which the mRNA transcript moves through the ribosome, possibly altering ribosomal transit time or leading to nonsense-mediated decay. An artificially inserted, i.e. not biologically relevant, PRF led to increased activation of nonsense-mediate decay, possibly due to the pause effect of the PRF itself (68). Slowed ribosomal transit time could also simply slow protein translation, preventing the cell from meeting its anabolic demands. In previous studies, diphthamide loss not only led to an increased read-through rate of a −1 PRF, it also mildly affected the rate of protein synthesis and the overall growth of the yeast (56,66). The neurodegenerative disease vanishing white matter is caused by mutations in eukaryotic initiation factor 2B (69,70), part of the complex that regulates the rate-limiting step of initiating translation (45). Although the mechanism is not fully understood, the mutations very likely compromise protein synthesis, providing support for the disruption of the rate of global protein synthesis in neurodegeneration.
Frameshifting, especially over non-PRF sequences, may lead to mistranslation from the alternate reading frame, producing nonsense polypeptides unable to fold. The increase in frameshifting may also simply affect PRFs in mammalian cells. The biological relevance of PRFs to mammalian cells, and neurons, is a currently underdeveloped field, although there are a few examples. A +1 PRF is known to participate in a feedback loop to regulate levels of polyamines within cells (71–73). With future studies, altered post-transcriptional regulation of gene expression controlled by PRFs may prove to be a contributing factor in the pathological mechanism of SCA26.
The growing number of genes and mutational mechanisms that have been found to underlie different forms of hereditary SCA and Purkinje cell degeneration demonstrates the selective vulnerability of Purkinje neurons to a wide variety of molecular and cellular insults (74–79). Protein misfolding and altered proteostasis is well recognized as a molecular mechanism underlying various forms of neurodegeneration (74–79). It has been suggested that Purkinje cells may be vulnerable due to their large size, complex morphology, or their high metabolic and protein synthetic activity (74–79).
Dysregulation of PRFs specific to Purkinje cells or neurons in general could also potentially help explain the cell-type specificity of SCA26. Another possible contributing factor for cell-type specificity is the differences in proteostatic capacity, i.e. the capacity of a given cell to maintain the delicate balance between protein folding and the environmental and genetic forces promoting unfolding, known to vary between different cell types. Proteostatic capacity is influenced by genetic polymorphisms in chaperones and other components of translation (61,80–82), and variation from one cell type to another reflects the characteristic gene expression profile of that cell type (81). Within a given cell's lifetime, proteostatic capacity will also decline with age (61,80–82). This age-related decline in proteostatic capacity provides a plausible explanation to the adult-onset feature of SCA26.
One of the autosomal recessive ataxias is known to be caused by a genetic defect that results in disruption of proteostasis. Marinesco–Sjögren syndrome is due to a mutation in SIL1, a co-chaperone of BiP (83,84). Additionally, several mouse models of ataxia published in recent years highlight the unique susceptibility of neurons to the disruption of proteostasis. The ‘sticky’ mouse is a model of ataxia resulting from a point mutation in the proof-reading domain of the alanine–tRNA synthetase enzyme, leading to tRNAs mischarged with glycine and subsequent mistranslation of proteins (85). The ‘woozy’ mouse develops ataxia due to Purkinje cell degeneration from unfolded protein accumulation caused by a point mutation in SIL1 (86), just as in Marinesco–Sjögren syndrome. Furthermore, floxed knockouts of either atg5 or atg7, both crucial regulators of the autophagy process, disrupt basal levels of autophagy and, without any other environmental or genetic insult, resulted in neuronal protein aggregation, Purkinje cell loss and ataxia (87,88). These examples shed light on how a mutation in a ubiquitous protein can lead specifically to Purkinje cell loss.
The results of our DTT challenge experiments in yeast suggest that the P580H EF2 substitution, by disrupting the normal mechanics of translocation, impairs the overall proteostatic capacity of the cell, rendering it more susceptible to other insults. Recent evidence has demonstrated enhanced toxicity of an aggregating SOD1 mutant (fALS) due to mildly destabilizing polymorphisms (in the form of temperature sensitive mutations at the permissive temperature) that tax the proteostatic network in Caenorhabditis elegans (80,81). Insults to proteostasis have a compounding effect, in that less stable background genetic polymorphisms may not lead to decompensation in and of themselves, but they weaken the proteostatic network, promoting decompensation in the event of exogenous stresses that the cell might otherwise have been able to endure (80,81).
We attempted to model P596H eEF2 function in a mammalian cell culture system. We developed a lentivirus expressing human reference or P596H eEF2 under either the murine stem cell virus (MSCV) or elongation factor 1 (EF1) promoters (unpublished data). Transduction with either a high level of expression (MSCV) or a moderate level of expression (EF1) of exogenous eEF2 proved toxic to neuro2a cells. This fits with the existing evidence that EF2 expression is very tightly regulated, and overexpression is poorly tolerated (44). We additionally attempted a lentivirus-based shRNA knock-down (Sigma Aldrich Mission shRNA clone NM_007907.1-1133s1c1) of endogenous mouse eEF2 in neuro2a cells, followed by a subsequent rescue with our human eEF2 lentivirus constructs. These cells were also not viable. We were able to successfully generate stable HEK293 and PC12 cell lines expressing either reference or P596H eEF2-3XFLAG, from lentivirus transduction (unpublished data). Utilizing a large population of positively transduced cells with random sites of integration, we saw that P596H eEF2-3XFLAG was consistently expressed at a lower level than reference eEF2-3XFLAG, indicating that sufficiently high levels of P596H eEF2 expression were selected against while comparable levels of reference eEF2 were well tolerated. This illustrates that the variant is poorly tolerated in comparison with WT eEF2. Thus, P596H eEF2 modeling in a mammalian system will likely necessitate a knock-in or induced pluripotent stem cell strategy to generate the native ratios of the expression of reference and P596H eEF2 proteins.
Our findings lay the framework for a proposed pathogenic mechanism for SCA26: The P596H eEF2 substitution compromises the mechanics of translocation. This defect disrupts translation, thereby lessening the cell's overall proteostatic capacity, rendering it more susceptible to an array of exogenous stresses with which it would otherwise be able to cope. It is possible then that the natural accumulation of exogenous stresses over time, combined with age-related decline in proteostatic capacity, causes the cellular proteostatic machinery to fail, affecting the uniquely susceptible Purkinje cells first and resulting in ataxia. This framework for the pathogenesis of SCA26, with further validation in neuronal cells, would represent the first disease mechanism of impairment in proteostasis in an autosomal dominant ataxia. This novel mechanism further highlights the importance of the UPR in human neurodegeneration and suggests the potential therapeutic benefit of targeting the UPR to modulate neurodegenerative disease.
MATERIALS AND METHODS
Sequencing of CEPH and Norwegian samples and unknown ataxia probands
We performed Sanger sequencing on an ABI 3730 machine in a single direction over the exons examined. The data were analyzed with Sequencing Analysis 5.1.1 Sequencing within the family, and the genotyping of the unknown ataxia probands was performed with PCR amplification with exon-specific primers, followed by the standard Sanger sequencing (primers and PCR programs are available upon request.)
Custom capture array of SCA26 locus
Probes were designed following the method of Ng et al. (89). We generated 50–160 m of 100 bp paired reads per sample on an Illumina HiSeq2000. Reads were aligned to NCBI36/hg18 (bwa v0.5.9) (90), locally realigned around indels (GATK) (91), and Q scores were empirically recalibrated (GATK). The region was saturated: 2–4% of the target bases were not covered, and 5–10% of the target bases were covered <8-fold. The remaining bases were covered at least 8×. Samples were genotyped with the samtools mpileup genotyper (92).
RNA preparation and cDNA preparation
RNA was extracted from lymphoblastoid cell lines generated from SCA26 patient lymphoblasts using Triagent product (Sigma-Aldrich), as per the manufacturer's instructions. Synthesis of cDNA was performed using the Superscript III kit (Invitrogen).
Polymerase chain reaction
PCR was performed in an Eppendorf MasterCycler gradient machine, using 1 μg of cDNA and 2 mm of each forward and reverse primer in a total volume of 50 μl with Platinum High Fidelity Taq supermix (Invitrogen). PCR product (800 bp) was run on a 0.6% agarose gel with EtBr (1 μg/ml), extracted and purified using the Gel Extraction kit (Qiagen), as per manufacturer's instructions. Purified PCR product was sequenced with the forward primer using the standard Sanger dideoxy sequencing methods. All primer sequences available upon request.
Plasmids
eEF2 plasmids
eEF2 cDNA in pCR4-TOPO was purchased from Open Biosystems. The C-to-A transversion at nucleotide 1870 was introduced via Quikchange (Agilent), as per the manufacturer's instructions.
PCR was performed, as above (50 ng template), to add a BamHI site immediately upstream of the 5′UTR and a C-terminal 3XFLAG™ epitope tag followed by an XbaI site immediately downstream of the final amino acid, replacing the stop codon and generating an eEF2-3XFLAG™ fusion protein. The cDNA was then subcloned into the pcDNA3 vector.
PCR was performed, as above (50 ng template), to add a HindIII restriction site immediately upstream of the 5′UTR and an XbaI site immediately downstream of the last amino acid, replacing the stop codon. The cDNA was then subcloned into a pKH3 vector containing a C-terminal 3XHA epitope tag sequence, generating an eEF2–3XHA fusion protein.
Yeast plasmids
EFT2 (S. cerevisiae) and its promoter in the YCP vector (LEU2) were a kind gift from Dr Mythili Shastry (Merck). Point mutations were introduced via Quikchange (Agilent), as per the manufacturer's instructions.
The dual-luciferase reporter plasmids (pJD375 and pJD378) were a kind gift from the Dinman Laboratory, University of Maryland.
The 4XUPRE- β-galactosidase plasmid (pJC104) was a kind gift from the Walter Laboratory, UCSF.
Lymphoblastoid cell line production
Lymphoblast cell lines were created by transforming B lymphocytes with the Epstein–Barr virus (EBV) into a blastic stage, establishing a permanent cell line. The lymphocytes were separated from whole blood from patients using a Ficol-Paque™ Plus gradient. The lymphocytes were then set up in RPMI 1640 culture medium with 20% fetal bovine serum, 1% Glutamax™ and 1% gentamicin. EBV and cyclosporin were added to the culture medium to kill the T lymphocytes, whereas EBV transforms the B lymphocytes. The cell line was cultured in a 37°C, 5% CO2 incubator for ∼1 month before transformation occurred (evident by the formation of large clumps), and then it was frozen down for future use.
Culture
Lymphoblastoid cell lines were grown in the standard medium, as above. HEK293 cells were grown in the standard medium: Dulbecco's modified Eagle's medium + 10% fetal bovine serum, supplemented with 1× pen-strep and l-glutamine. SH-SY5Y cells were grown in the standard medium, 1:1 minimal Eagle's medium:F12K, supplemented with 1× pen-strep, 1× l-glutamine and glucose (final 0.5%).
Transfection
HEK293 cells were transfected using lipofectamine 2000 (Invitrogen), 1:3, at ∼70% confluency. SH-SY5Y cells were transfected using Fugene 6 (Roche), 1:6, at ∼80% confluency.
Immunostaining
Lymphoblastoid and SH-SY5Y cells were plate on 0.01% poly-l-lysine coated and uncoated coverslips, respectively. Cells were fixed and permeabilized in 1:1 methanol:acetone at −20°C for 20 min. Non-specific epitopes were blocked with goat serum for 1 h at room temperature. Primary antibodies in goat serum were added as follows: α-calnexin (Santa Cruz), 1:100; α-eEF2 (Cell Signaling Technology), 1:500; α-FLAG™ (Sigma Aldrich), 1:1000; α-HA (Roche), 1:500. Species-specific secondary antibodies (Abcam) in 1× phosphate buffered saline with 1% Tween-20 were added as appropriate (1:1000). Cells were visualized on a confocal Leica SP2 microscope under 63× objective with oil immersion.
Western blotting
Transfected HEK293 cells were collected 24–48 h post-transfection and lysed in 1× cell lysis buffer (Millipore) + 1 mm phenylmethanesulfonylfluoride (PMSF) + 1× protease inhibitor cocktail (Sigma Aldrich), and cell debris was pelleted by high-speed centrifugation. Western blotting was performed in the usual way (93), loading 30 μg of total cell lysate into a 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis gel. FLAG fusion proteins were detected with α-FLAG™ (Sigma-Aldrich), 1:1000. Secondary antibody was purchased from GE Healthcare, UK. Band densitometry was performed with ImageJ software.
Yeast
TKY398 (YEFD12h/pURA3-EFT1 MATa ade2 lys2 ura3 his3 leu2 trp1 eft1 Δ:HIS3 eft2 Δ:TRP1 pURA3-EFT1) was a kind gift from the laboratory of Dr Terri Kinzy, UMDNJ. Transformation of yeast was performed according to the standard protocol from Cold Spring Harbor Laboratory Manual Methods in Yeast Genetics (94). Positive transformants were selected for their respective prototrophies on drop-out media. For plasmid shuffling to introduce the point mutation-bearing EF2 plasmids, transformants containing the original WT EF2 rescue plasmid (URA3) and the LEU2 point mutation EF2 plasmid were grown in media containing uracil and plated on 5-fluoroorotic acid plates to select for the loss of the WT EF2-URA3 plasmid.
Doubling time was calculated by plotting a complete growth curve of the yeast (OD600 versus time) with time points every 30 min, performing a log transformation on the OD600 values and selecting the time points that best fit a linear regression (r2 > 0.990). Adjacent time points were used to generate the doubling time datum. All adjacent pairs of time points that fit the linear regression were utilized to calculate a single doubling time datum in this way.
Dual-luciferase assay
The dual-luciferase assays were performed as described previously, with the following modifications: yeast were grown to the mid-log phase (OD600 0.5–0.7) in a volume of 4 ml of culture. Yeast were spun down and resuspended in cold 1× phosphate-buffered saline supplemented with 1 mm PMSF and 1× protease inhibitor cocktail (Sigma-Aldrich). Yeast were then lysed using a Biospec Tissue Tearor 985370 tissue homogenizer for 45 s at ∼15 000 rpm. Cell debris were pelleted, and lysate was analyzed in triplicate for luciferase activity using the dual-luciferase assay kit (Promega), according to the manufacturer's instructions, in a Perkin Elmer Victor 3 dual-injector luminometer.
Dual-luciferase statistical analysis
The ratio of firefly luciferase activity to Renilla luciferase activity [firefly counts per second (CPS)/Renilla CPS] in a single well generated a single data point for a given EF2 variant and a given plasmid. Data sets were screened for outliers, as above, and the remaining data points were used to generate the mean, variance and standard deviation of the data set. Minimal sampling criteria were met using an error of 11% and α = 0.10. The data sets for pJD378 (−1 frameshift) were normalized to the in-frame control, pJD375. The degrees of freedom for this ratiometric mean were determined, and the statistical significance was set at P < 0.001.
UPR induction assay
Yeast were grown to the mid-log phase (OD600 0.5–0.7), then split into two cultures. One was treated with DTT in water at a final concentration of 10 mm, and the other with water as control, for 1 h. Alternatively, one tube was moved to a 42°C water bath for 15 min, and the second remained at 30°C as a control. After treatment, the OD600 was determined, and all cultures were diluted back to OD600 = 0.1 and mixed with an equal volume of Beta Glo reagent (Promega). The samples were analyzed as per the manufacturer's instructions in a Perkin Elmer Victor3 dual-injector luminometer. Statistical significance was determined with a two-sided type 2 Student's t-test, P < 0.01.
SUPPLEMENTARY MATERIAL
FUNDING
C.D.B. was supported by a Lilly-Life Sciences Research Foundation fellowship. K.E.H. was supported by the Graduate Training in Growth and Development program at the University of Chicago (T32 HD009007). G.-Y.Y. was supported by the Bob Allison Ataxia Research Center and the National Ataxia Foundation. This work was supported financially by the Verum Foundation, the ‘Association Connaitre les Syndromes Cérébelleux’ (France) and the European Union (6th PCRD call: EUROSCA), CTSA UL1 TR000430, and by NIH (R03-NS52582).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank James Bodley (University of Minnesota), Rod Merrill (University of Guelph), Rene Jørgensen (Statens Serum Institute) and Jonathan Dinman and Trey Belew (University of Maryland) for their expert guidance in developing this project. We would also like to thank Scott DeBoer for packaging the lentivirus; Devon Collins, Josh Aaker and KaReisha Robinson for their support in the laboratory, and Drs Alexandra Durr and Alexis Brice and the DNA and Cell bank of the CR-ICM (Paris, France) for sharing the DNA of patients from the SPATAX cohort. Lastly, we would also like to thank Terri Kinzy (UMDNJ) for her kind gift and her support, and Peter Walter (UCSF) for his kind gift.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Craig K., Keers S.M., Archibald K., Curtis A., Chinnery P.F. Molecular epidemiology of spinocerebellar ataxia type 6. Ann. Neurol. 2004;55:752–755. doi: 10.1002/ana.20110. doi:10.1002/ana.20110. [DOI] [PubMed] [Google Scholar]
- 2.Manto M.U. The wide spectrum of spinocerebellar ataxias (SCAs) Cerebellum. 2005;4:2–6. doi: 10.1080/14734220510007914. doi:10.1080/14734220510007914. [DOI] [PubMed] [Google Scholar]
- 3.Zoghbi H.Y., Orr H.T. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J. Biol. Chem. 2009;284:7425–7429. doi: 10.1074/jbc.R800041200. doi:10.1074/jbc.R800041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang S., Hong S. Molecular pathogenesis of spinocerebellar ataxia type 1 disease. Mol. Cells. 2009;27:621–627. doi: 10.1007/s10059-009-0095-y. doi:10.1007/s10059-009-0095-y. [DOI] [PubMed] [Google Scholar]
- 5.Lastres-Becker I., Rub U., Auburger G. Spinocerebellar ataxia 2 (SCA2) Cerebellum. 2008;7:115–124. doi: 10.1007/s12311-008-0019-y. doi:10.1007/s12311-008-0019-y. [DOI] [PubMed] [Google Scholar]
- 6.Riess O., Rub U., Pastore A., Bauer P., Schols L. SCA3: Neurological features, pathogenesis and animal models. Cerebellum. 2008;7:125–137. doi: 10.1007/s12311-008-0013-4. doi:10.1007/s12311-008-0013-4. [DOI] [PubMed] [Google Scholar]
- 7.Bauer P., Schols L., Riess O. Spectrin mutations in spinocerebellar ataxia (SCA) Bioessays. 2006;28:785–787. doi: 10.1002/bies.20443. doi:10.1002/bies.20443. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda Y., Dick K.A., Weatherspoon M.R., Gincel D., Armbrust K.R., Dalton J.C., Durr A., Zuhlke C., Burk K., Brent H.B., et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 2006;38:184–190. doi: 10.1038/ng1728. doi:10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- 9.Kordasiewicz H.B., Gomez C.M. Molecular pathogenesis of spinocerebellar ataxia type 6. Neurotherapeutics. 2007;4:285–294. doi: 10.1016/j.nurt.2007.01.003. doi:10.1016/j.nurt.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Lebre A.S., Brice A. Spinocerebellar ataxia 7 (SCA7) Cytogenet. Genome Res. 2003;100:154–163. doi: 10.1159/000072850. doi:10.1159/000072850. [DOI] [PubMed] [Google Scholar]
- 11.Mutsuddi M., Rebay I. Molecular genetics of spinocerebellar ataxia type 8 (SCA8) RNA Biol. 2005;2:49–52. doi: 10.4161/rna.2.2.1682. doi:10.4161/rna.2.2.1682. [DOI] [PubMed] [Google Scholar]
- 12.Lin X., Ashizawa T. Recent progress in spinocerebellar ataxia type-10 (SCA10) Cerebellum. 2005;4:37–42. doi: 10.1080/14734220510007897. doi:10.1080/14734220510007897. [DOI] [PubMed] [Google Scholar]
- 13.Lin X., Ashizawa T. SCA10 and ATTCT repeat expansion: Clinical features and molecular aspects. Cytogenet. Genome Res. 2003;100:184–188. doi: 10.1159/000072853. doi:10.1159/000072853. [DOI] [PubMed] [Google Scholar]
- 14.Houlden H., Johnson J., Gardner-Thorpe C., Lashley T., Hernandez D., Worth P., Singleton A.B., Hilton D.A., Holton J., Revesz T., et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat. Genet. 2007;39:1434–1436. doi: 10.1038/ng.2007.43. doi:10.1038/ng.2007.43. [DOI] [PubMed] [Google Scholar]
- 15.Holmes S.E., Hearn E.O., Ross C.A., Margolis R.L. SCA12: An unusual mutation leads to an unusual spinocerebellar ataxia. Brain Res. Bull. 2001;56:397–403. doi: 10.1016/s0361-9230(01)00596-2. doi:10.1016/S0361-9230(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 16.Stevanin G., Durr A., Benammar N., Brice A. Spinocerebellar ataxia with mental retardation (SCA13) Cerebellum. 2005;4:43–46. doi: 10.1080/14734220510007923. doi:10.1080/14734220510007923. [DOI] [PubMed] [Google Scholar]
- 17.Waters M.F., Minassian N.A., Stevanin G., Figueroa K.P., Bannister J.P., Nolte D., Mock A.F., Evidente V.G.H., Fee D.B., Muller U., et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 2006;38:447–451. doi: 10.1038/ng1758. doi:10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- 18.Figueroa K.P., Minassian N.A., Stevanin G., Waters M., Garibyan V., Forlani S., Strzelczyk A., Bürk K., Brice A., Dürr A., et al. KCNC3: Phenotype, mutations, channel biophysics-a study of 260 familial ataxia patients. Hum. Mutat. 2010;31:191–196. doi: 10.1002/humu.21165. doi:10.1002/humu.21165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevanin G., Hahn V., Lohmann E., Bouslam N., Gouttard M., Soumphonphakdy C., Welter M.L., Ollagnon-Roman E., Lemainque A., Ruberg M., Brice A., et al. Mutation in the catalytic domain of protein kinase C gamma and extension of the phenotype associated with spinocerebellar ataxia type 14. Arch. Neurol. 2004;61:1242–1248. doi: 10.1001/archneur.61.8.1242. doi:10.1001/archneur.61.8.1242. [DOI] [PubMed] [Google Scholar]
- 20.Miura S., Nakagawara H., Kaida H., Sugita M., Noda K., Motomura K., Ohyagi Y., Ayabe M., Aizawa H., Ishibashi M., Taniwaki T. Expansion of the phenotypic spectrum of SCA14 caused by the Gly128Asp mutation in PRKCG. Clin. Neurol. Neurosurg. 2009;111:211–215. doi: 10.1016/j.clineuro.2008.09.013. doi:10.1016/j.clineuro.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Nolte D., Klebe S., Baron R., Deuschl G., Muller U. Codon 101 of PRKCG, a preferential mutation site in SCA14. Mov. Disord. 2007;22:1831–1832. doi: 10.1002/mds.21654. doi:10.1002/mds.21654. [DOI] [PubMed] [Google Scholar]
- 22.Seki T., Takahashi H., Adachi N., Abe N., Shimahara T., Saito N., Sakai N. Aggregate formation of mutant protein kinase C gamma found in spinocerebellar ataxia type 14 impairs ubiquitin-proteasome system and induces endoplasmic reticulum stress. Eur. J. Neurosci. 2007;26:3126–3140. doi: 10.1111/j.1460-9568.2007.05933.x. doi:10.1111/j.1460-9568.2007.05933.x. [DOI] [PubMed] [Google Scholar]
- 23.Wieczorek S., Arning L., Gizewski E.R., Alheite I., Timmann D. Benign SCA14 phenotype in a German patient associated with a missense mutation in exon 3 of the PRKCG gene. Mov. Disord. 2007;22:2135–2136. doi: 10.1002/mds.21673. doi:10.1002/mds.21673. [DOI] [PubMed] [Google Scholar]
- 24.Dalski A., Mitulla B., Burk K., Schattenfroh C., Schwinger E., Zuhlke C. Mutation of the highly conserved cysteine residue 131 of the SCA14 associated PRKCG gene in a family with slow progressive cerebellar ataxia. J. Neurol. 2006;253:1111–1112. doi: 10.1007/s00415-006-0209-9. doi:10.1007/s00415-006-0209-9. [DOI] [PubMed] [Google Scholar]
- 25.Hiramoto K., Kawakami H., Inoue K., Seki T., Maruyama H., Morino H., Matsumoto M., Kurisu K., Sakai N. Identification of a new family of spinocerebellar ataxia type 14 in the Japanese spinocerebellar ataxia population by the screening of PRKCG exon 4. Mov. Disord. 2006;21:1355–1360. doi: 10.1002/mds.20970. doi:10.1002/mds.20970. [DOI] [PubMed] [Google Scholar]
- 26.Vlak M.H., Sinke R.J., Rabelink G.M., Kremer B.P., van de Warrenburg B.P. Novel PRKCG/SCA14 mutation in a Dutch spinocerebellar ataxia family: expanding the phenotype. Mov. Disord. 2006;21:1025–1028. doi: 10.1002/mds.20851. doi:10.1002/mds.20851. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi H., Abe K., Matsuura T., Ikeda Y., Hitomi T., Akechi Y., Habu T., Liu W., Okuda H., Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. doi:10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J.L., Yang X., Xia K., Hu Z.M., Weng L., Jin X., Jiang H., Zhang P., Shen L., Guo J.F., et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510–3518. doi: 10.1093/brain/awq323. doi:10.1093/brain/awq323. [DOI] [PubMed] [Google Scholar]
- 29.Sato N., Amino T., Kobayashi K., Asakawa S., Ishiguro T., Tsunemi T., Takahashi M., Matsuura T., Flanigan K.M., Iwasaki S., et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. doi:10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Bella D., Lazzaro F., Brusco A., Plumari M., Battaglia G., Pastore A., Finardi A., Cagnoli C., Tempia F., Frontali M., et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010;42:313–321. doi: 10.1038/ng.544. doi:10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- 31.van Swieten J.C., Brusse E., de Graaf B.M., Krieger E., van de Graaf R., de Koning I., Maat-Kievit A., Leegwater P., Dooijes D., Oostra B.A., Heutink P. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected] Am. J. Hum. Genet. 2003;72:191–199. doi: 10.1086/345488. doi:10.1086/345488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakalkin G., Watanabe H., Jezierska J., Depoorter C., Verschuuren-Bemelmans C., Bazov I., Artemenko K.A., Yakovleva T., Dooijes D., Van de Warrenburg B.P., et al. Prodynorphin mutations cause the neurodegenerative disorder spinocerebellar ataxia type 23. Am. J. Hum. Genet. 2010;87:593–603. doi: 10.1016/j.ajhg.2010.10.001. doi:10.1016/j.ajhg.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Roon-Mom W.M., Reid S.J., Faull R.L., Snell R.G. TATA-binding protein in neurodegenerative disease. Neuroscience. 2005;133:863–872. doi: 10.1016/j.neuroscience.2005.03.024. doi:10.1016/j.neuroscience.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 34.Marelli C., van de Leemput J., Johnson J.O., Tison F., Thauvin-Robinet C., Picard F., Tranchant C., Hernandez D.G., Huttin B., Boulliat J., et al. SCA15 due to large ITPR1 deletions in a cohort of 333 white families with dominant ataxia. Arch. Neurol. 2011;68:637–643. doi: 10.1001/archneurol.2011.81. doi:10.1001/archneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verbeek D.S., Knight M.A., Harmison G.G., Fischbeck K.H., Howell B.W. Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain. 2005;128:436–442. doi: 10.1093/brain/awh378. doi:10.1093/brain/awh378. [DOI] [PubMed] [Google Scholar]
- 36.Verbeek D.S., Warrenburg B.P., Hennekam F.A., Dooijes D., Ippel P.F., Verschuuren-Bemelmans C.C., Kremer H.P., Sinke R.J. Gly118Asp is a SCA14 founder mutation in the Dutch ataxia population. Hum. Genet. 2005;117:88–91. doi: 10.1007/s00439-005-1278-z. doi:10.1007/s00439-005-1278-z. [DOI] [PubMed] [Google Scholar]
- 37.Klebe S., Durr A., Rentschler A., Hahn-Barma V., Abele M., Bouslam N., Schöls L., Jedynak P., Forlani S., Denis E., et al. New mutations in protein kinase cgamma associated with spinocerebellar ataxia type 14. Ann. Neurol. 2005;58:720–729. doi: 10.1002/ana.20628. doi:10.1002/ana.20628. [DOI] [PubMed] [Google Scholar]
- 38.Alonso I., Costa C., Gomes A., Ferro A., Seixas A.I., Silva S., Cruz V.T., Coutinho P., Sequeiros J., Silveira I. A novel H101Q mutation causes PKCgamma loss in spinocerebellar ataxia type 14. J. Hum. Genet. 2005;50:523–529. doi: 10.1007/s10038-005-0287-z. doi:10.1007/s10038-005-0287-z. [DOI] [PubMed] [Google Scholar]
- 39.Yu G.Y., Howell M.J., Roller M.J., Xie T.D., Gomez C.M. Spinocerebellar ataxia type 26 maps to chromosome 19p13.3 adjacent to SCA6. Ann. Neurol. 2005;57:349–354. doi: 10.1002/ana.20371. doi:10.1002/ana.20371. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe M., Abe K., Aoki M., Kameya T., Itoyama Y., Shoji M., Ikeda M., Iizuka T., Hirai S. A reproducible assay of polymerase chain reaction to detect trinucleotide repeat expansion of Huntington's disease and senile chorea. Neurol. Res. 1996;18:16–18. doi: 10.1080/01616412.1996.11740370. [DOI] [PubMed] [Google Scholar]
- 41.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. doi:10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. doi:10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 43.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A. NISC Comparative Sequencing Program. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. doi:10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ortiz P.A., Kinzy T.G. Dominant-negative mutant phenotypes and the regulation of translation elongation factor 2 levels in yeast. Nucleic Acids Res. 2005;33:5740–5748. doi: 10.1093/nar/gki882. doi:10.1093/nar/gki882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ejiri S. Moonlighting functions of polypeptide elongation factor 1: From actin bundling to zinc finger protein R1-associated nuclear localization. Biosci. Biotechnol. Biochem. 2002;66:1–21. doi: 10.1271/bbb.66.1. doi:10.1271/bbb.66.1. [DOI] [PubMed] [Google Scholar]
- 46.Wang L., Popko B., Roos R.P. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2011;20:1008–1015. doi: 10.1093/hmg/ddq546. doi:10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perentesis J.P., Phan L.D., Gleason W.B., LaPorte D.C., Livingston D.M., Bodley J.W. Saccharomyces cerevisiae elongation factor 2. genetic cloning, characterization of expression, and G-domain modeling. J. Biol. Chem. 1992;15:1190–1197. [PubMed] [Google Scholar]
- 48.Phan L.D., Perentesis J.P., Bodley J.W. Saccharomyces cerevisiae elongation factor 2. mutagenesis of the histidine precursor of diphthamide yields a functional protein that is resistant to diphtheria toxin. J. Biol. Chem. 1993;268:8665–8668. [PubMed] [Google Scholar]
- 49.Jorgensen R., Carr-Schmid A., Ortiz P.A., Kinzy T.G., Andersen G.R. Purification and crystallization of the yeast elongation factor eEF2. Acta Crystallogr D. Biol. Crystallogr. 2002;58:712–715. doi: 10.1107/s0907444902003001. doi:10.1107/S0907444902003001. [DOI] [PubMed] [Google Scholar]
- 50.Stark H., Rodnina M.V., Wieden H.J., van Heel M., Wintermeyer W. Large-scale movement of elongation factor G and extensive conformational change of the ribosome during translocation. Cell. 2000;100:301–309. doi: 10.1016/s0092-8674(00)80666-2. doi:10.1016/S0092-8674(00)80666-2. [DOI] [PubMed] [Google Scholar]
- 51.Agrawal R.K., Penczek P., Grassucci R.A., Frank J. Visualization of elongation factor G on the Escherichia coli 70S ribosome: the mechanism of translocation. Proc. Natl Acad. Sci. USA. 1998;95:6134–6138. doi: 10.1073/pnas.95.11.6134. doi:10.1073/pnas.95.11.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorgensen R., Ortiz P.A., Carr-Schmid A., Nissen P., Kinzy T.G., Andersen G.R. Two crystal structures demonstrate large conformational changes in the eukaryotic ribosomal translocase. Nat. Struct. Biol. 2003;10:379–385. doi: 10.1038/nsb923. doi:10.1038/nsb923. [DOI] [PubMed] [Google Scholar]
- 53.Taylor D.J., Nilsson J., Merrill A.R., Andersen G.R., Nissen P., Frank J. Structures of modified eEF2 80S ribosome complexes reveal the role of GTP hydrolysis in translocation. EMBO J. 2007;26:2421–2431. doi: 10.1038/sj.emboj.7601677. doi:10.1038/sj.emboj.7601677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bodley J.W., Dunlop P.C., VanNess B.G. Diphthamide in elongation factor 2: ADP-ribosylation, purification, and properties. Methods Enzymol. 1984;106:378–387. doi: 10.1016/0076-6879(84)06040-7. doi:10.1016/0076-6879(84)06040-7. [DOI] [PubMed] [Google Scholar]
- 55.Oppenheimer N.J., Bodley J.W. Diphtheria toxin. site and configuration of ADP-ribosylation of diphthamide in elongation factor 2. J. Biol. Chem. 1981;256:8579–8581. [PubMed] [Google Scholar]
- 56.Ortiz P.A., Ulloque R., Kihara G.K., Zheng H., Kinzy T.G. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J. Biol. Chem. 2006;281:32639–32648. doi: 10.1074/jbc.M607076200. doi:10.1074/jbc.M607076200. [DOI] [PubMed] [Google Scholar]
- 57.Jacobs J.L., Belew A.T., Rakauskaite R., Dinman J.D. Identification of functional, endogenous programmed -1 ribosomal frameshift signals in the genome of Saccharomyces cerevisiae. Nucleic Acids Res. 2007;35:165–174. doi: 10.1093/nar/gkl1033. doi:10.1093/nar/gkl1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harger J.W., Dinman J.D. An in vivo dual-luciferase assay system for studying translational recoding in the yeast Saccharomyces cerevisiae. RNA. 2003;9:1019–1024. doi: 10.1261/rna.5930803. doi:10.1261/rna.5930803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dinman J.D. Programmed ribosomal frameshifting goes beyond viruses: organisms from all three kingdoms use frameshifting to regulate gene expression, perhaps signaling a paradigm shift. Microbe Wash DC. 2006;1:521–527. doi: 10.1128/microbe.1.521.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Voisine C., Pedersen J.S., Morimoto R.I. Chaperone networks: tipping the balance in protein folding diseases. Neurobiol. Dis. 2010;40:12–20. doi: 10.1016/j.nbd.2010.05.007. doi:10.1016/j.nbd.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balch W.E., Morimoto R.I., Dillin A., Kelly J.W. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. doi:10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 62.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. doi:10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 63.Cox J.S., Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. doi:10.1016/S0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 64.Atkins J.F., Weiss R.B., Thompson S., Gesteland R.F. Towards a genetic dissection of the basis of triplet decoding, and its natural subversion: programmed reading frame shifts and hops. Annu. Rev. Genet. 1991;25:201–228. doi: 10.1146/annurev.ge.25.120191.001221. doi:10.1146/annurev.ge.25.120191.001221. [DOI] [PubMed] [Google Scholar]
- 65.Plant E.P., Jacobs K.L., Harger J.W., Meskauskas A., Jacobs J.L., Baxter J.L., Petrov A.N., Dinman J.D. The 9-A solution: How mRNA pseudoknots promote efficient programmed -1 ribosomal frameshifting. RNA. 2003;9:168–174. doi: 10.1261/rna.2132503. doi:10.1261/rna.2132503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kimata Y., Kohno K. Elongation factor 2 mutants deficient in diphthamide formation show temperature-sensitive cell growth. J. Biol. Chem. 1994;269:13497–13501. [PubMed] [Google Scholar]
- 67.Foley B.T., Moehring J.M., Moehring T.J. Mutations in the elongation factor 2 gene which confer resistance to diphtheria toxin and pseudomonas exotoxin A. genetic and biochemical analyses. J. Biol. Chem. 1995;270:23218–23225. doi: 10.1074/jbc.270.39.23218. doi:10.1074/jbc.270.39.23218. [DOI] [PubMed] [Google Scholar]
- 68.Plant E.P., Wang P., Jacobs J.L., Dinman J.D. A programmed -1 ribosomal frameshift signal can function as a cis-acting mRNA destabilizing element. Nucleic Acids Res. 2004;32:784–790. doi: 10.1093/nar/gkh256. doi:10.1093/nar/gkh256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abbott C.M., Proud C.G. Translation factors: in sickness and in health. Trends Biochem. Sci. 2004;29:25–31. doi: 10.1016/j.tibs.2003.11.006. doi:10.1016/j.tibs.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 70.Leegwater P.A., Vermeulen G., Konst A.A., Naidu S., Mulders J., Visser A., Kersbergen P., Mobach D., Fonds D., van Berkel C.G., et al. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat. Genet. 2001;29:383–388. doi: 10.1038/ng764. doi:10.1038/ng764. [DOI] [PubMed] [Google Scholar]
- 71.Shigemoto K., Brennan J., Walls E., Watson C.J., Stott D., Rigby P.W., Reith A.D. Identification and characterisation of a developmentally regulated mammalian gene that utilises −1 programmed ribosomal frameshifting. Nucleic Acids Res. 2001;29:4079–4088. doi: 10.1093/nar/29.19.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Namy O., Rousset J.P., Napthine S., Brierley I. Reprogrammed genetic decoding in cellular gene expression. Mol. Cell. 2004;13:157–168. doi: 10.1016/s1097-2765(04)00031-0. doi:10.1016/S1097-2765(04)00031-0. [DOI] [PubMed] [Google Scholar]
- 73.Matsufuji S., Matsufuji T., Miyazaki Y., Murakami Y., Atkins J.F., Gesteland R.F., Hayashi S. Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell. 1995;80:51–60. doi: 10.1016/0092-8674(95)90450-6. doi:10.1016/0092-8674(95)90450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Orr H.T. Cell biology of spinocerebellar ataxia. J. Cell Biol. 2012;197:167–177. doi: 10.1083/jcb.201105092. doi:10.1083/jcb.201105092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matilla-Duenas A., Corral-Juan M., Volpini V., Sanchez I. The spinocerebellar ataxias: clinical aspects and molecular genetics. Adv. Exp. Med. Biol. 2012;724:351–374. doi: 10.1007/978-1-4614-0653-2_27. doi:10.1007/978-1-4614-0653-2_27. [DOI] [PubMed] [Google Scholar]
- 76.Matilla-Duenas A., Sanchez I., Corral-Juan M., Davalos A., Alvarez R., Latorre P. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum. 2010;9:148–166. doi: 10.1007/s12311-009-0144-2. doi:10.1007/s12311-009-0144-2. [DOI] [PubMed] [Google Scholar]
- 77.Verbeek D.S., van de Warrenburg B.P. Genetics of the dominant ataxias. Semin. Neurol. 2011;31:461–469. doi: 10.1055/s-0031-1299785. doi:10.1055/s-0031-1299785. [DOI] [PubMed] [Google Scholar]
- 78.Paulson H.L. The spinocerebellar ataxias. J. Neuroophthalmol. 2009;29:227–237. doi: 10.1097/WNO0b013e3181b416de. doi:10.1097/WNO0b013e3181b416de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carlson K.M., Andresen J.M., Orr H.T. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr. Opin. Genet. Dev. 2009;19:247–253. doi: 10.1016/j.gde.2009.02.009. doi:10.1016/j.gde.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gidalevitz T., Krupinski T., Garcia S., Morimoto R.I. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009;5:e1000399. doi: 10.1371/journal.pgen.1000399. doi:10.1371/journal.pgen.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gidalevitz T., Ben-Zvi A., Ho K.H., Brignull H.R., Morimoto R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. doi:10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 82.Ben-Zvi A., Miller E.A., Morimoto R.I. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl Acad. Sci. USA. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. doi:10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anttonen A.K., Lehesjoki A.E. Marinesco-Sjogren syndrome. In: Pagon R.A., Bird T.D., Dolan C.R., Stephens K., editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 84.Zhao L., Rosales C., Seburn K., Ron D., Ackerman S.L. Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjogren syndrome. Hum. Mol. Genet. 2010;19:25–35. doi: 10.1093/hmg/ddp464. doi:10.1093/hmg/ddp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee J.W., Beebe K., Nangle L.A., Jang J., Longo-Guess C.M., Cook S.A., Davisson M.T., Sundberg J.P., Schimmel P., Ackerman S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. doi:10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 86.Zhao L., Longo-Guess C., Harris B.S., Lee J.W., Ackerman S.L. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat. Genet. 2005;37:974–979. doi: 10.1038/ng1620. doi:10.1038/ng1620. [DOI] [PubMed] [Google Scholar]
- 87.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. doi:10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 88.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. doi:10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 89.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. doi:10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li H., Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. doi:10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. doi:10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. doi:10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu Y., Yao J., Liu Z., Liu X., Fu H., Ye K. Akt phosphorylates acinus and inhibits its proteolytic cleavage, preventing chromatin condensation. EMBO J. 2005;24:3543–3554. doi: 10.1038/sj.emboj.7600823. doi:10.1038/sj.emboj.7600823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amberg D.C., Burke D., Strathern J.N. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.