

Abstract
Ubiquitination plays a crucial role in neurodevelopment as exemplified by Angelman syndrome, which is caused by genetic alterations of the ubiquitin ligase-encoding UBE3A gene. Although the function of UBE3A has been widely studied, little is known about its paralog UBE3B. By using exome and capillary sequencing, we here identify biallelic UBE3B mutations in four patients from three unrelated families presenting an autosomal-recessive blepharophimosis-ptosis-intellectual-disability syndrome characterized by developmental delay, growth retardation with a small head circumference, facial dysmorphisms, and low cholesterol levels. UBE3B encodes an uncharacterized E3 ubiquitin ligase. The identified UBE3B variants include one frameshift and two splice-site mutations as well as a missense substitution affecting the highly conserved HECT domain. Disruption of mouse Ube3b leads to reduced viability and recapitulates key aspects of the human disorder, such as reduced weight and brain size and a downregulation of cholesterol synthesis. We establish that the probable Caenorhabditis elegans ortholog of UBE3B, oxi-1, functions in the ubiquitin/proteasome system in vivo and is especially required under oxidative stress conditions. Our data reveal the pleiotropic effects of UBE3B deficiency and reinforce the physiological importance of ubiquitination in neuronal development and function in mammals.
Introduction
Ubiquitination is a posttranslational protein modification that plays a key role in brain development. The ubiquitin/proteasome system (UPS) degrades damaged proteins and is part of the cellular protein quality control network.1–3 Polyubiquitination involves the sequential transfer of the 76 amino acid protein ubiquitin between a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). The specificity of this complex process is mainly determined by the E3 ligase, of which more than 600 are encoded in the human genome.4 HECT domain E3 ligases constitute a major group of E3 enzymes5 that not only are involved in substrate conjugation and subsequent degradation by the 26S proteasome but may also regulate the trafficking of specific receptors, channels, and transporters.6 Ubiquitination is involved in both gliogenesis and neurogenesis through its influence on cellular signaling cascades such as the Notch and Hedgehog pathways.1 Ubiquitination thereby contributes to regulating neuronal migration, neuritogenesis, and the formation and elimination of synapses. Consequently, alterations of the UPS can engender neuronal dysfunction and lead to neurological disease and intellectual disability (ID).2
A prototype disorder involving altered ubiquitination is the human neurodevelopmental disease Angelman syndrome (MIM 105830). Angelman syndrome is characterized by ID, speech delay, gait ataxia, and a characteristic behavior and is often associated with microcephaly and seizures.7 It is caused by mutations of the imprinted gene UBE3A (MIM 601623), which encodes the HECT domain-containing ubiquitin ligase E3A. Although some of the clinical signs of Angelman syndrome might be due to ubiquitin-independent functions of UBE3A, recent studies suggest that altered ubiquitination of synaptic proteins such as Arc and Ephexin5 contributes to the cognitive dysfunction that occurs in Angelman syndrome.8,9 Furthermore, maternal 15q duplications encompassing UBE3A lead to a predisposition for autism spectrum disorders, and an increased copy number of Ube3a may confer autism-related behaviors in mice,10,11 pointing to an important role of UBE3A gene dosage in brain development and function.
The application of classical and high-throughput genetic technologies has recently led to the identification of a large number of genes involved in the pathogenesis of nonsyndromic and syndromic forms of ID,12–14 and this approach has also been applied to the blepharophimosis-mental retardation (BMR) syndromes, with mutations of the histone acetyltransferase gene KAT6B (MIM 605880) causing the autosomal-dominant Say-Barber/Biesecker/Young-Simpson (SBBYS) type of Ohdo syndrome (MIM 603736).15 The clinically and genetically heterogeneous BMR or Ohdo-like syndromes16–19 (MIM 249620) are characterized—in addition to developmental delay and ID—by blepharophimosis, a narrowing and shortening of the palpebral fissures, and various congenital anomalies. Here, we have identified and characterized mutations of UBE3B (MIM 608047) in an ID syndrome of this clinical spectrum and we show that disruption of mouse Ube3b phenocopies some aspects of the human disorder.
Subjects and Methods
Subjects
The research project on the genetics of ID syndromes was performed with approval by the ethics committees of the Rabin Medical Center (Petah Tikva, Israel) and the University of Ulm (Ulm, Germany). Subjects were enrolled with parental written informed consent to participate in the study. DNA was extracted according to standard protocols.
Exome Sequencing
Genomic DNA was enriched for exonic and adjacent splice site sequences with the Agilent SureSelect Human All Exon 50 Mb kit and libraries were run on three lanes of the Illumina Genome Analyzer IIX Sequencer via the single end read 150 bp protocol. Further data analysis of the resulting filter-passed reads was performed with the in-house pipeline V1.4 with BWA-short20 in combination with SAMTOOLS 0.1.7 for SNP and indel detection.21 In-house developed scripts were applied to detect protein changes, affected splice sites, and overlaps to known variations (ENSEMBL build 61, 1000 Genomes project release 2010_03, in-house variation database containing >100 exomes). We searched for stretches of homozygosity by using SNPs (excluding indels) with a Phred score >100 and a coverage of >20-fold, and SNPs with an allele frequency >85% were considered homozygous.
Sanger Sequencing Validation and SNP Array Genotyping
Validation of variants detected by exome sequencing was performed by Sanger sequencing. Primer sequences are listed in Table S1 available online. PCR products were sequenced on an ABI 3730 DNA Analyzer with BigDye chemistry v3.1 (Applied Biosystems). Sequence traces were assembled, aligned, and analyzed with the Seqman software (DNASTAR Lasergene). Mutation nomenclature is based on GenBank transcript NM_130466.2, which contains 28 exons; the start codon is located in exon 3. DNA of 100 anonymous control individuals each of Israeli-Arab and Italian origin were tested for the relevant mutation(s) by direct sequencing. DNA from patient 4 and her parents was genotyped on an Affymetrix GeneChip Human Mapping 250K Nsp Array.
RT-PCR
Total RNA from peripheral white blood cells of affected individuals and controls was extracted from fresh EDTA blood by a standard trizol protocol. RT-PCR was performed with the QIAGEN OneStep RT-PCR kit with primers UBE3B-RT-PCR-F1 5′-TTGGAATGCCTGAACAATGA-3′ and UBE3B-RT-PCR-R1 5′-TGGAAGAGCACGCTAGGTTT-3′ (for the c.1741+2T>C mutation) and UBE3B-RT-PCR-F2 5′-GTATGTGTCCCTGGCTTGTTC-3′ and UBE3B-RT-PCR-R2 5′-ATCACGGCATCGATCTTGGTC-3′ (for the c.545−2A>G mutation). PCR products were cut out from a 2% agarose gel and directly sequenced after purification.
3D Structural Analysis
To assess the potential functional consequences of the p.Gln727Pro substitution, the HECT domain of UBE3B was modeled to several structurally characterized HECT domains, including the HECT domain of SMURF2,22 by using the PHYRE2 server.23
Analysis of Protein Degradation in C. elegans
Caenorhabditis elegans strains were maintained according to standard procedures and grown at 20°C.24 The strain PP563 unc-119(ed4)III; hhIs64 [unc-119(+); sur-5::UbV-GFP]III was used for analysis.
RNA interference was performed via the feeding method.25 The oxi-1 RNAi clone was obtained from the Ahringer RNAi library26 and the empty feeding vector was used as a control. Worms were applied at the L4 larval stage on standard RNAi plates with or without addition of paraquat (Sigma) and analyzed for the expression of GFP after 24 hr at 20°C.
Worms were treated with RNAi on control or oxi-1 RNAi plates without the addition of paraquat. qRT-PCR was performed as previously described.27
Worms were imaged under a Leica M165 FC stereomicroscope equipped with a Leica DFC 340 FX camera and handled with the Leica LAS V3.4.0 analysis software. Images were processed with Adobe Photoshop CS 4 software for figure assembly.
Immunoblotting was performed as previously described.27
Mouse In Situ Hybridization
Part of the Ube3b mRNA (nucleotides 3,666–4,464 of GenBank transcript NM_054093) was amplified from murine postnatal day (P) 46 brain cDNA. The PCR product was then cloned into the pCRII-TOPO vector (Invitrogen) and the insert was sequence verified. In situ hybridizations were performed on whole embryo sections at embryonic day (E) 12.5 and E14.5 and on brain (E16.5, P9, and P24) and head (P0) sections as previously described.28
Generation of Ube3b Genetrap Mice
Mutant mice were generated through blastocyst injection of mutant embryonic stem cells (ESCs) obtained from the International Gene Trap consortium; specifically BayGenomics gene trap ESC clone RRJ142 (129P2/OlaHsd). Chimeric mice were bred to CBA/Ca females, and the colony was expanded and maintained by intercrossing; control mice were wild-type littermates. Thus, these studies were carried out on a 129P2/Ola × CBA/Ca mixed genetic background. The strain has been deposited at the European Mutant Mouse Archive (EM:04922). Analyses were performed retrospectively on data generated by the Mouse Genetics Project at the Wellcome Trust Sanger Institute. The gene trap vector is inserted upstream of the majority of the exons encoding the HECT domain. Genotyping was performed by PCR on ear clip lysate to detect the wild-type (5′-CGGCACGTTCCACTGTCCC-3′ and 5′-TGCCAGCCCCACCAGCATC-3′; 664 bp) and mutant alleles (5′-CGGCACGTTCCACTGTCCC-3′ and 5′-GCACGCCATACAGTCCTCTTC-3′; 470 bp), as well as the E. coli lacZ cassette present in the gene trap vector (5′-ATCACGACGCGCTGTATC-3′ and 5′-ACATCGGGCAAATAATATCG-3′; 108 bp). Cycling conditions for the wild-type and mutant PCRs were 94°C 5 min; 35× (94°C 30 s, 58°C 30 s, 72°C 45 s); 72°C 5 min. For lacZ, the same conditions were used except the annealing temperature was shifted to 60°C.
We verified absence of Ube3b mRNA in Ube3b−/− mice by qRT-PCR. Frozen skin (pinnae) samples were homogenized and RNA was extracted with a QIAGEN fibrous tissue extraction kit. 1 μl (∼100–200 ng) of total RNA was used in a 10 μl reaction with a TaqMan RNA-to-CT One Step kit (Applied Biosystems). A TaqMan probe spanning exons 19–20 of Ube3b, flanking the gene trap insertion point (Applied Biosystems probe Mm00459868_m1), was used in a multiplex reaction with either a primer-limited B2m or a Gapdh endogenous control (Applied Biosystems). Reactions were performed in triplicate on an ABI Viia7 qPCR machine and analyzed with Viia7 1.1 software, with the relative quantification module. Reactions performed with the Gapdh endogenous probe gave a higher variation within the technical replicates than with the B2m probe. No Ube3b expression was detected with either endogenous control assay.
Mouse Phenotypic Analysis
Animals
The care and use of all mice in this study were in accordance with the UK Home Office regulations, UK Animals (Scientific Procedures) Act of 1986. Mice were bred and maintained in a specific-pathogen-free unit, room temperature and humidity regulated (21°C ± 2°C; 55% ± 10%), and 12/12 hr light/dark cycle with lights off at 19:30 hr and no twilight period. Mice were housed in individually ventilated cages (IVC) (Techniplast Seal Safe 1284L) receiving 60 air changes per hour, at a stocking density of 3–5 mice per cage (floor area = 530 cm2). Aspen bedding substrate and standard environmental enrichment of nestlet, cardboard tunnel, and three wooden chew blocks were provided. Mice were given water and breeding diet (5021, Labdiet) ad libitum.
Phenotyping
Female mice were weighed weekly between 9 and 14 weeks of age. At 9 weeks of age, grip strength was assessed (Bioseb grip strength meter). Three trials each of forepaws and all paws were performed in succession. Representative data are presented (all-paw trial 1). At 14 weeks of age, nonfasted mice were terminally anesthetized with Avertin (1.25% 2,2,2-Tribromoethanol, Sigma Aldrich) and blood was collected via the retro-orbital sinus into lithium/heparin-coated tubes. After centrifugation (5,000 × g, 10 min, 8°C), plasma was analyzed for low-density lipoprotein concentration with an Olympus AU400. After the terminal bleed, tissues were fixed in 4% paraformaldehyde, processed through to paraffin, and sectioned (5 μm). An automatic slide stainer (Leica ST5020) was used to generate hematoxylin and eosin (H&E)-stained sections of the eyes and brain. 14-week-old mice fed on high-fat diet (Western RD, 829100, Special Diets Services) from 4 weeks of age were anesthetized and high-resolution digital X-ray images were acquired with a MX20 (Faxitron X-ray Corporation). Brains from two 6-week-old male Ube3b−/− mice and nine age-, gender-, and background-matched controls were perfusion fixed in 4% PFA. Prior to sectioning, brains were placed in 30% sucrose/PBS solution for 24 hr. Brains were then sectioned coronally at 40 μm on a sliding microtome. Matched sections were mounted on electrostatic slides, Nissl stained, and digitally scanned to computer on an Epson V750 pro scanner at 6400 dpi. ImageJ freeware was subsequently used to take measurements of total brain section area, hippocampus area, and dentate gyrus length. To avoid introduction of bias, experimenters were blind to the genotype of the mice during all procedures. Furthermore, cages were processed randomly and different genotypes housed together. Auditory brainstem response (ABR) thresholds were recorded in anesthetized mice via subcutaneous pin electrodes on the head as described previously.29 Averaged ABR waveforms to broad-band clicks and tonepips presented in 3 dB steps were examined to determine the lowest intensity to give a recognizable response. After ABR, mice were culled, middle ears were opened and examined, and inner ears were removed, cleared, and examined.
Numbers of homozygous (Ube3b−/−; Hom), heterozygous (Ube3b+/−; Het) and WT mice used in the different phenotypic analyses are as follows: weight: 7 Hom, 15 Het, 9 WT; grip strength: 7 Hom, 15 Het, 9 WT; LDL cholesterol: 5 Hom, 14 Het, 4 WT; brain section area: 2 Hom, 0 Het, 9 WT; hippocampus area: 2 Hom, 0 Het, 9 WT; length of the dentate gyrus: 2 Hom, 0 Het, 9 WT; ABR at 3 months: 7 Hom, 10 Het, 14 WT; ABR at 6 months: 6 Hom, 7 Het, 9 WT; survival at 2 weeks: 31 Hom, 263 Het, 124 WT.
Statistical Methods
For the comparison of growth curves, statistical significance—in terms of differences in the rate of growth between the three genotypes—was assessed via the CompareGrowthCurve function of the statmod software package for R. The initial p values were adjusted for multiple testing by Holm’s method. For the comparison of LDL levels and grip strength, a mixed model data analysis was completed with a six step top-down methodology to build the final model as detailed in West et al.30 The process starts with a two level random intercept mixed model with assay date as a random effect and genotype, sex, body weight, and genotype∗sex interaction term as fixed effects. Within the six step procedure, a 0.05 significance threshold was used to assess the output of the hypothesis testing. ABR thresholds were significantly raised in homozygotes compared with wild-types via either the Kruskall Wallis ANOVA on ranks (p < 0.001) or Dunn’s method of multiple comparisons. Differences in brain section and hippocampus areas and the length of the dentate gyrus were assessed with t tests performed in R.
Results
Clinical Delineation of an Intellectual-Disability Syndrome
We initially studied three individuals from two unrelated families with an apparently identical BMR syndrome (Supplemental Data). Individual 1 from family 1 (Figures 1A and S1, Table 1) was a girl born to unaffected parents of Israeli-Arab origin who are first cousins. We noted a striking facial similarity and overlapping clinical features with the previously reported female and male siblings from family 2 (individuals 2 and 3, respectively; Figures 1B, 1C, and S1, Table 1)19 in which the unaffected parents of Italian descent are not consanguineous. Common clinical signs noted in the three affected individuals included characteristic facial dysmorphisms with blepharophimosis, ptosis, and mild upslanting of the palpebral fissures and epicanthus (Figures 1A–1C); ectodermal anomalies; developmental delay; and severe ID with absent speech (Supplemental Data). Proportionate growth retardation with a small head circumference/microcephaly, congenital malformations, muscular hypotonia, and anomalies on brain imaging with hypoplasia of the corpus callosum (Figure 1E) were variably present (Table 1). We refer to this syndrome as a blepharophimosis-ptosis-intellectual- disability (BPID) syndrome belonging to the heterogenous group of BMR syndromes.
Figure 1.
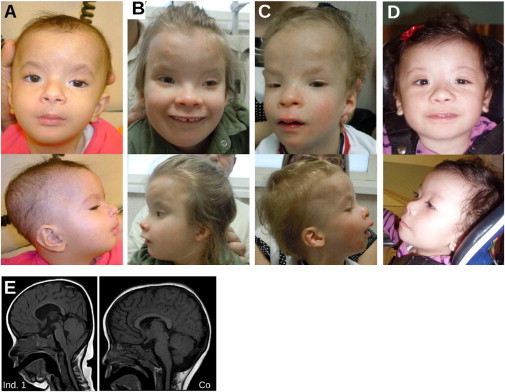
A Blepharophimosis-Ptosis-Intellectual-Disability Syndrome
(A–D) Individual 1 at the age of 1 year (A), individual 2 at the age of 7 years 7 months (B), individual 3 at the age of 3 years 1 month (C), and individual 4 at the age of 1 year 2 months (D). Note characteristic facial features with blepharophimosis and ptosis, sparse hair, sparse and arched eyebrows, long philtrum, anteverted nares, retrognathia, and low-set, posteriorly rotated ears. Individuals 2 and 3 also have upslanting palpebral fissures; this feature develops with age.
(E) Cranial MRI scan of individual 1 (Ind. 1) at age 1 year 10 months shows cerebellar tonsillar ectopia consistent with Chiari 1 malformation, thin corpus callosum, and an overall small head. A cranial MRI scan of a healthy age-matched control (Co) is also shown.
Table 1.
Clinical, Radiological, and Biochemical Features of Individuals with Biallelic UBE3B Mutations
|
Family 1 |
Family 2 |
Family 3 |
||
|---|---|---|---|---|
| Individual 1 | Individual 2 | Individual 3 | Individual 4 | |
| Origin | Israel | Italy | Tunisia | |
| Parental consanguinity | + (first cousins) | - | - | + (first cousins) |
| Sex | F | F | M | F |
| Pregnancy/Neonatal Period | ||||
| Oligohydramnios | - | - | + | - |
| Respiratory problems | stridor due to laryngomalacia | apnea | - | stridor due to laryngomalacia |
| Feeding difficulties/GER | + | (+) (improved at 5 years) | + | + |
| Frequent infections | recurrent otitis media | - | recurrent bronchitis until 1 year | recurrent respiratory infections |
| Growth | ||||
| Failure to thrive | + | + | + | + |
| Short stature | - | + | + | + |
| Microcephaly | + | + | + | + |
| Motor and Cognitive Development | ||||
| Delayed motor milestones | + | + | + | + |
| Absent speech | + (3 years 3 months) | + (7 years 7 months) | + (3 years 1 month) | + (17 months) |
| Severe intellectual disability | + | + | + | severe developmental delay |
| Craniofacial | ||||
| Blepharophimosis | + | + | + | + |
| Ptosis | + | + (left) | - | + |
| Upward-slanting palpebral fissures | - | + | + | - |
| Telecanthus | + | + | + | + |
| Hypertelorism | - | + | + | + |
| Depressed nasal bridge | + | + | + | + |
| Anteverted nares | + | + | + | + |
| Low-set/dysplastic ears | + | + | + | + |
| Palatal anomalies | high arched palate | + (submucous cleft palate) | - | - |
| Micrognathia | + | + | + | + |
| Small mouth | + | + | + | + |
| Neurological | ||||
| Abnormal brain image | chiari type I malformation, ventricular dilatation, hypoplastic CC | agenesis of the CC rostrum, anterior commissure not evident | reduced size of the pituitary gland, partial empty sella | hypoplastic CC |
| Axial hypotonia | + | + | - | + |
| Ocular impairment | + (astigmatism) | + (astigmatism, mild strabism, and myopia) | - | - |
| Hearing impairment | + (conductive) | - | - | - |
| Ectodermal Anomalies | ||||
| Sparse thin hair/eyebrows | + | + | + | + |
| Eczema | - | - | + | - |
| Thin skin | + | + | + | + |
| Cardiac | ||||
| Congenital heart disease | + (ASD, VSD, aortic coarctation) | - | - | + (ASD) |
| Gastrointestinal | ||||
| Gastrectasia | - | + | - | - |
| Constipation | - | (+) (resolved at 6y) | + | - |
| Distension of gall-bladder | - | + | - | - |
| Intestinal malrotation | + | - | - | - |
| Skeletal/Limb | ||||
| Clinodactyly of 5th fingers | - | + | + | - |
| Congenital dislocation of the hip | + | - | - | + |
| Urogenital abnormalities | mild left pyelectasis | double right kidney, right pyelectasis | - | Hypoplastic labia majora |
| Endocrine and Laboratory Anomalies | ||||
| Thyroid anomalies | - | elevated TSH (until 5 years) and reduced thyroid gland volume | - | - |
| Low growth hormone levels | NA | - | + | NA |
| Low ACTH level | NA | - | + | NA |
| Anomalies of cholesterol levels | low HDL cholesterol | low total cholesterol | - | low total cholesterol |
| Others | severe hyperlaxity | torticollis, delayed bone age | - | partial epilepsy |
| UBE3B mutation paternal allele | c.1741+2G>C | c.545−2A>G | c.545−2A>G | c.2180A>C (p.Gln727Pro) |
| UBE3B mutation maternal allele | c.1741+2G>C | c.2223_2224delAG (p.Arg741Serfs∗3) | c.2223_2224delAG (p.Arg741Serfs∗3) | c.2180A>C (p.Gln727Pro) |
Abbreviations: ASD, atrial septal defect; CC, corpus callosum; GER, gastresophageal reflux; HDL, high-density lipoprotein; NA, not available/not performed; VSD, ventricular septal defect.
Routine biochemical serum analysis revealed that HDL cholesterol was low in individual 1, and low total cholesterol was noted in individual 2, suggesting that a defect in cholesterol metabolism might be part of the syndrome. We therefore performed mass spectrometry-based analysis of patient serum samples, which showed a consistent downregulation of cholesterol synthesis in the three patients, including a low lathosterol/total sterol ratio (lathosterol/total sterol ratio in patients versus controls 0.46). In conclusion, the autosomal-recessive BPID syndrome affecting individuals 1–3 represents a clinically recognizable neurodevelopmental disorder.16,19
Exome Sequencing Identifies Truncating UBE3B Mutations
We sequenced the exomes of unrelated individuals 1 and 2 on an Illumina platform after enrichment of exonic and splice-site sequences. We mapped reads to the hg19 human reference genome and obtained a mean coverage of 124-fold for both subjects. Approximately 90% of target sequences were covered at least ten times (Table S2).
We observed no mutation in KAT6B, a gene recently shown to be mutated in the majority of the patients with the autosomal-dominant SBBYS type of Ohdo syndrome.15 Because of parental consanguinity in family 1 and the lack of consanguinity in family 2 and assuming an autosomal-recessive inheritance pattern and genetic homogeneity, we searched for a gene harboring a rare homozygous variant in individual 1 and rare compound heterozygous variants in individual 2. We considered variants if they were not annotated as single-nucleotide polymorphisms (SNPs) in dbSNP132, the 1000 Genomes data, and our in-house exome database and focused on missense, nonsense, small insertion/deletion, and splice site variants. We identified 29 autosomal regions of extended homozygosity in individual 1 (Table S3). Sixteen genes mapping to these regions harbored a rare homozygous nonsynonymous or splice site variant in individual 1 (Figure S2). Only 1 of these 16 genes, however, UBE3B, also carried two distinct rare variants in individual 2, consistent with compound heterozygous mutations. Thus, UBE3B was the only gene with rare or unique biallelic damaging mutations observed in both affected individuals.
Confirmation and Characterization of Biallelic UBE3B Mutations
We confirmed all three UBE3B mutations by capillary (Sanger) sequencing. Individual 1 was homozygous for a c.1741+2T>C mutation (Figure 2A) affecting the consensus splice donor site of exon 16. This mutation led to skipping of exon 16 and—to a lesser extent—skipping of exons 16 and 17 in RNA derived from blood of individual 1 (Figure 2B). The parents were both heterozygous carriers of this mutation. In family 2, both affected siblings were compound heterozygous for a maternally inherited 2 bp deletion (c.2223_2224delAG [p.Arg741Serfs∗3]) and a paternally inherited splice acceptor mutation (c.545−2A>G; Figure 2C). The latter mutation again caused exon skipping, namely skipping of exon 8 and skipping of exons 8 and 9 in RNA derived from blood of individual 2 (Figure 2D). All three UBE3B mutations introduce premature termination codons and are thus expected to result in nonsense-mediated mRNA decay and/or protein truncation. None of these mutations was present in 100 ethnically matched control individuals as shown by sequencing of the respective exons. The mutations were also not present in ∼7,000 European-American control alleles sequenced by the NHLBI Exome Sequencing Project.31
Figure 2.
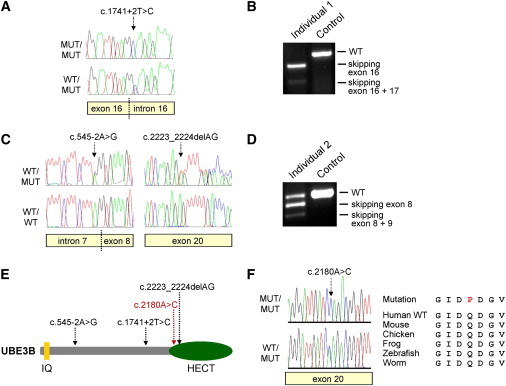
UBE3B Mutations Cause the BPID Syndrome
(A and B) Sequence chromatograms showing the homozygous (MUT/MUT) and heterozygous (WT/MUT) UBE3B splice donor mutation c.1741+2T>C identified in family 1 (A) that leads to skipping of exon 16 (119 bp; out-of-frame) and exons 16 and 17 (234 bp; in-frame) as shown by RT-PCR on RNA extracted from whole blood of individual 1 (B). Exon skipping was confirmed by sequence analysis of the respective RT-PCR products (not shown).
(C and D) Individuals 2 and 3 are compound heterozygotes for c.2223_2224delAG and the splice acceptor mutation c.545−2A>G (C), the latter leading to out-of-frame skipping of exon 8 (86 bp) and exons 8 and 9 (169 bp) as shown by RT-PCR (D) and sequence analysis (not shown).
(E) Protein diagram of human UBE3B depicting the N-terminal IQ domain (short calmodulin-binding motif containing conserved Ile [I] and Gln [Q] residues) and the C-terminal HECT (homologous to the E6-AP carboxyl terminus) domain. The positions of the three truncating mutations identified in individuals 1–3 are indicated by black arrows and the p.Gln727Pro missense alteration is shown in red.
(F) Sequence chromatograms showing the homozygous (MUT/MUT) and heterozygous (WT/MUT) UBE3B missense alteration p.Gln727Pro (c.2180A>C) identified in individual 4 and multiple sequence alignment of UBE3B orthologs surrounding the Gln727 position in the human protein.
UBE3B encodes the putative ubiquitin ligase E3B, a protein of 1,068 amino acids that contains an N-terminal IQ domain and a C-terminal HECT domain.32 HECT domains are crucial for the function of E3 ubiquitin ligases because they mediate both the binding of an E2 ubiquitin-conjugating enzyme through their large N-terminal subdomain and the transfer of ubiquitin to the substrate, which is mediated by the catalytic cysteine located in the small C-terminal subdomain.6 The three UBE3B mutations we report lead to protein truncation before or at the beginning of the HECT domain (Figure 2E), thus presumably completely abolishing the enzyme’s E3 ligase activity.
Identification of a Missense Substitution in the UBE3B HECT Domain in an Additional Individual with BPID Syndrome
To obtain additional evidence for the involvement of UBE3B mutations in BMR syndromes, we sequenced the gene in a girl (individual 4 from family 3; Figures 1D and S1, Table 1) with a BMR syndrome characterized by short stature, developmental delay, seizures, hypoplasia of the corpus callosum, sparse hair, and thin skin. She had minor facial dysmorphic features (Figure 1D), and the clinical picture strongly suggested that she was affected by the same BPID syndrome as individuals 1–3. In line with this assumption, her total cholesterol was <100 mg/dl (which is below the fifth centile for children and adolescents) with relatively low lathosterol, comparable to the findings in individuals 1–3. Individual 4 was born to unaffected parents of Tunisian ancestry who are first cousins, suggesting that a causative mutation would be present in the homozygous state. By sequencing the coding exons of UBE3B, we indeed detected a homozygous mutation, c.2180A>C, that was predicted to lead to a substitution of a highly conserved glutamine by proline (p.Gln727Pro; Figure 2F). Both parents were heterozygous mutation carriers and the mutation was located within a 35 Mb region of homozygosity (between rs2583147 and rs11059122) in the proband as shown by genome-wide SNP array genotyping, consistent with a large region of homozygosity-by-descent. The c.2180A>C mutation was absent from 100 Tunisian control individuals as ascertained by sequencing of UBE3B exon 20. It was also not present in the ∼10,000 alleles sequenced by the NHLBI Exome Sequencing Project.
Gln727 is located in the HECT domain of UBE3B and is strictly conserved in orthologs from 41 species for which Ube3b protein sequences are available at the UCSC multiple species sequence alignment, including mouse, chicken, zebrafish, the frog Xenopus tropicalis, and the worm Caenorhabditis elegans (Figure 2F and data not shown). Three-dimensional modeling of the UBE3B HECT domain to several structurally characterized HECT domains showed that the mutated Gln727 is not within reach of the catalytic site but is on the same face of the HECT domain (Figure S3) and might thus be involved in substrate recognition and binding.
The C. elegans Ortholog of UBE3B Plays a Role in the UPS under Conditions of Oxidative Stress
Although the primary protein sequence and domain architecture of UBE3B predict an E3 ligase function,32 experimental support for this assumption is lacking and specific ubiquitination targets of UBE3B have not been identified. We used a C. elegans in vivo assay27 to investigate and characterize a possible role of the orthologous gene oxi-1 in the UPS. The protein sequences of human UBE3B and worm OXI-1 are 37% identical and 58% similar overall and 62% identical in the HECT domain. Moreover, a phylogenetic analysis shows that OXI-1 is more closely related to human UBE3B than to UBE3A and UBE3C (Figure S4A), suggesting that OXI-1 represents the species ortholog of UBE3B. oxi-1 was originally identified as an oxidative stress-responsive gene because its expression is upregulated under high-oxygen conditions.33
We used an engineered ubiquitin fusion degradation (UFD) substrate in which a noncleavable ubiquitin is N-terminally fused to GFP (UbV-GFP) and expressed under the control of the sur-5 promoter to monitor protein degradation in vivo.27 In adult wild-type worms, the UbV-GFP protein is efficiently ubiquitinated and degraded by the 26S proteasome (see control RNAi in Figures 3A–3C). RNAi-mediated knockdown of hecd-1, a well-studied E3 ligase-encoding gene, leads to an impairment of protein degradation under basal conditions, as shown by a stabilization of the UbV-GFP substrate in hecd-1 (RNAi)-treated worms (Figure 3A). Knockdown of oxi-1 (Figure S4B) only minimally affected the degradation of UbV-GFP under basal conditions (Figures 3A–3C). Exposing wild-type worms to low doses of the oxidative stress-inducing agent paraquat resulted in stabilization of UbV-GFP, as shown by GFP fluorescence and immunoblotting.27 Notably, paraquat treatment led to a dose-dependent stronger stabilization of the UbV-GFP substrate in oxi-1 (RNAi)-treated worms in comparison to control RNAi (Figures 3B and 3C). Thus, the C. elegans ortholog of UBE3B is involved in protein degradation by the UPS and might be especially required under oxidative stress conditions.
Figure 3.
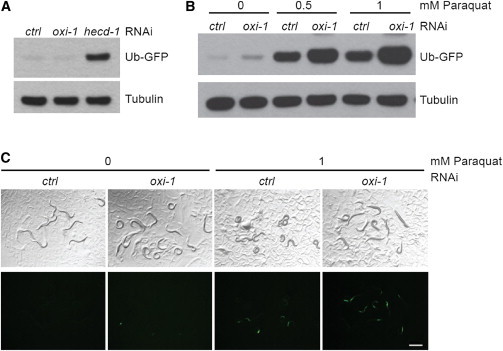
oxi-1, the Caenorhabditis elegans Ortholog of UBE3B, Is Involved in Ubiquitin/Proteasome-Mediated Protein Turnover In Vivo
(A) Immunoblot of lysates derived from worms after control (ctrl) RNAi or RNAi against oxi-1 or hecd-1.
(B) Immunoblot of lysates derived from worms after control or oxi-1 RNAi without (0) or with addition of 0.5 or 1 mM paraquat. The blot was performed with a GFP-specific antibody; tubulin serves as loading control.
(C) Images of C. elegans that express the UbV-GFP reporter to measure protein turnover. Worms were treated as in (B). Top, transmitted light; bottom, GFP fluorescence. Scale bar represents 0.5 mm.
Mouse Ube3b Is Expressed in the Central Nervous System
To characterize the spatial and temporal expression pattern of Ube3b, we performed in situ hybridizations of embryonic and postnatal mouse tissues, which revealed predominant expression in distinct anatomical structures of the developing central nervous system. These results confirm and extend high-throughput data generated by GenePaint, a digital atlas of gene expression patterns in the mouse.34 We detected Ube3b expression from embryonic day (E) 12.5 through E16.5 in the ventricular zone of the cerebral cortex (Figures 4A–4C) where neuronal progenitor cells undergo asymmetric cell divisions. Ube3b was also enriched at E12.5 in the cortical hem (Figure 4A), the hippocampal anlage. Moreover, we found Ube3b expression in the tectum (Figures 4A and 4B), the cortical plate (Figures 4B and 4C), the dorsal root ganglia (Figure 4B), and the preoptic neuroepithelium (Figure 4C). Interestingly, Ube3b expression was observed in craniofacial structures, namely in the epithelium of the medial nasal process (Figure 4A) and in the whiskers (Figures 4B). Postnatally, we detected prominent Ube3b expression in the upper layers of the cerebral cortex (Figures 4D–4F), the cerebellum (Figures 4E and 4F), and the hippocampus (Figure 4F). Taken together, Ube3b expression in the central nervous system and in craniofacial structures is consistent with the phenotypes observed in individuals with UBE3B-deficient BPID syndrome.
Figure 4.
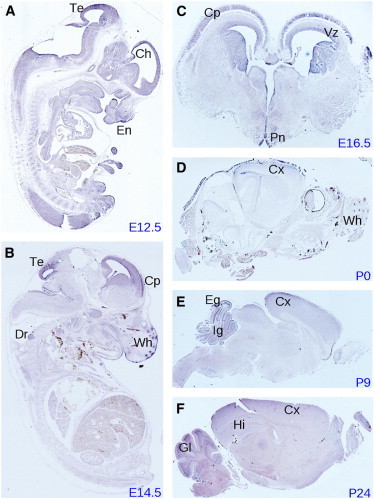
Expression Pattern of Ube3b in Mouse
In situ hybridization via a Ube3b antisense probe on sagittal (A, B, D–F) and coronal (C) sections at embryonic (E) and postnatal (P) days as indicated. Ch, cortical hem; Cp, cortical plate; Cx, cerebral cortex; Dr, dorsal root ganglia; Eg, external granular layer; En, epithelium of the medial nasal process; Gl, granular layer; Hi, hippocampus; Ig, internal granular layer; Pn, preoptic neuroepithelium; Te, tectum; Vz, ventricular zone; Wh, whiskers.
Disruption of Mouse Ube3b Recapitulates Clinical Aspects of the BPID Syndrome
To gain further insight into the consequences of UBE3B deficiency on an organismal level, we analyzed a Ube3b mutant mouse. Mice with a disrupted Ube3b locus were generated with a BayGenomics genetrap clone carrying the Ube3bGt(RRJ142)Byg allele and were maintained on a 129P2/Ola × CBA/Ca background. Phenotyping was conducted at the Wellcome Trust Sanger Institute by the Mouse Genetics Project (see the Sanger Mouse Resources Portal in the Web Resources). The structure of the genetrap allele was confirmed by 5′-RACE and by mapping and sequencing the genetrap integration site within a Ube3b intron (data not shown). We confirmed by quantitative PCR that Ube3b−/− mice do not express detectable levels of Ube3b mRNA (Figure S5).
Although some Ube3b−/− mice were born from heterozygous intercrosses, homozygotes represented only 7% of offspring at 2 weeks (Figure S6A), which is significantly below the expected Mendelian ratio (χ2 test, p < 0.0001) and indicates an elevated embryonic and/or perinatal lethality. Surviving Ube3b−/− mice could reproduce, although poorly (data not shown). Ube3b−/− mice had severely reduced body weight (Figure 5A), small body size (Figure 5B), and reduced brain section area (Figure 5C) with reduced sizes of the hippocampus (Figure 5D) and dentate gyrus (Figure S6B) but no obvious structural abnormalities on histopathological examination (Figure S6C). The reduced brain section size and hippocampus area of Ube3b−/− mice might in part be related to their low weight but this is unlikely to be the only reason. We verified this by conducting Pearson correlation tests in a pool of 29 age- and sex-matched WT animals. We found that although body weight correlated with brain weight (p < 0.05, r = 0.4), body weight did not correlate with hippocampus area (p = 0.274) or brain section area size (p = 0.267) in WT mice. Thus, Ube3b−/− mice have anomalies of brain size that may reflect the small head circumference/microcephaly seen in affected individuals with the BPID syndrome caused by UBE3B mutations.
Figure 5.
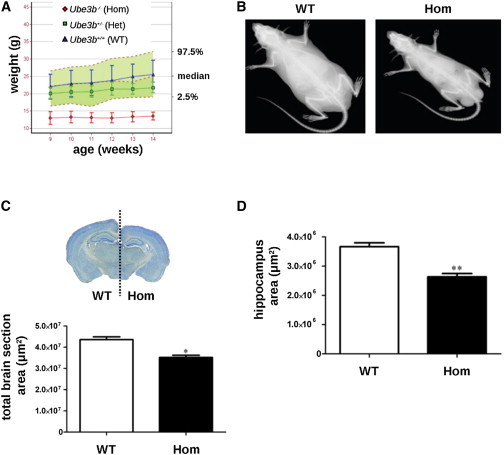
Reduced Growth of Ube3b Mutant Mice
(A) Homozygous Ube3b−/− mice (Hom) show a significantly reduced growth rate (weight curve comparison wild-type [WT] versus Hom: p < 0.001) than WT or heterozygote (Het) controls. The data are presented relative to a reference range that encompasses 95% of the natural variation, shown as the green shaded area, constructed with 134 WT mice from a variety of genetic backgrounds. Error bars represent SD.
(B) A representative Ube3b−/− mouse is smaller and thinner than a WT control.
(C and D) A significantly reduced brain section area (C) and a smaller hippocampus area (D) are also evident. ∗p = 0.0151, ∗∗p = 0.0049. Error bars represent SEM.
Ube3b−/− mice also displayed reduced grip strength (Figure 6A), possibly reflecting central or muscular hypotonia. Additionally, we observed reduced low-density lipoprotein (LDL) levels in mutants (Figure 6B) and a trend toward reduced total and HDL cholesterol levels (data not shown). Notably, mass spectrometry-based analysis of mouse plasma samples showed a significant reduction of total cholesterol and lathosterol in Ube3b−/− versus WT mice, recapitulating the cholesterol findings in the affected individuals. Mean total cholesterol was 1,642 μg/ml (range 744–2,633) in WT mice (n = 12), whereas it was 491 μg/ml (range 338–593) in Ube3b−/− mice (n = 5; p = 0.0004). The lathosterol/total sterol ratio was significantly lower in Ube3b−/− mice (0.025) than in WT mice (0.033; p = 0.045).
Figure 6.
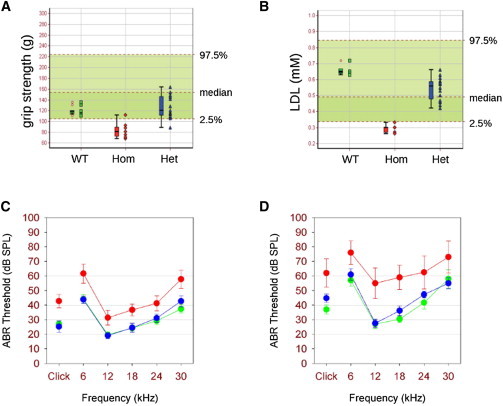
Neurosensory Abnormalities in Ube3b−/− Mice
(A and B) Ube3b−/− mice show reduced grip strength (A; p < 0.0001 WT versus Hom), and low serum low density lipoprotein (LDL) levels were also observed (B; p < 0.0001 WT versus Hom). The data are presented relative to a reference range that encompasses 95% of the natural variation, shown as the green shaded area, constructed with 132 (A) and 93 (B) WT mice from a variety of genetic backgrounds. Data are shown both as individual data points and as a five point box plot summary that indicates the median, first and third quartiles (box), and minimum and maximum values (whiskers).
(C and D) Auditory brainstem response (ABR) thresholds (±SEM) were significantly elevated in homozygotes (red) compared with heterozygotes (blue) and WT (green) littermates at 3 months (C) and at 6 months (D) (mean elevations cf. wild-types 15.0 ± 3.5 dB, Q = 4.427, p < 0.05 at 3 months; 22.8 ± 5.4 dB, Q = 4.671, p < 0.05 at 6 months).
In addition, homozygotes showed mild hearing impairment (Figures 6C and 6D), in line with previous reports of variable degrees of hearing impairment in Ohdo-like BMR syndromes.17 We detected no gross inner ear malformations or signs of middle ear inflammation (data not shown). Scanning electron microscopy of the organ of Corti also showed no obvious defect to account for the mild hearing impairment observed (data not shown). Finally, histopathological analysis of eye tissue sections revealed that 2/4 Ube3b−/− mice displayed acute inflammation, calcification, and dilated lymphovascular channels of the cornea (Figure S6D). Thus, Ube3b−/− mice phenocopy the UBE3B-deficient BPID syndrome in several key aspects, namely anomalies of body and brain size, weight, muscular strength, and cholesterol levels.
Discussion
Exome sequencing has greatly accelerated the identification of causative mutations and novel disease-associated genes in monogenic disorders.35 It can be combined with linkage analysis in autosomal-dominant, autosomal-recessive, and X-linked disorders or it can be performed in affected children and their unaffected parents in cases of sporadic disease caused by de novo mutations. However, even with these strategies aiming at reducing the number of candidate mutations, the analysis of exome-sequencing data remains challenging because of the sheer number of sequence variants identified. We have applied a stringent filtering strategy that takes advantage of pedigree information. Indeed, filtering for homozygous variants located in larger homozygous stretches in the exome of the affected child born to consanguineous parents and for possible compound heterozygous mutations in the same gene in the family in which the parents were unrelated left us with a single candidate gene, thereby greatly reducing the need for follow-up sequencing studies. Although this filtering strategy critically depended on the genetic homogeneity of the disorder, it is applicable to other recessive disorders, particularly when few patients are available for analysis.
Furthermore, our study illustrates the power of combining results from exome sequencing with the generation and phenotypic analysis of a mouse model. For example, observation of low cholesterol levels in two out of three affected individuals might have been attributed to chance or to dietary rather than genetic reasons. However, significantly reduced cholesterol levels in Ube3b−/− mice seem to argue in favor of an underlying defect in cholesterol metabolism. Our biochemical analyses in patients and mice have indeed consistently confirmed hypocholesterolemia and revealed low levels of cholesterol precursors. Because individuals with low dietary intake or increased intestinal loss of cholesterol have increased levels of cholesterol precursors, including lathosterol (unlike the affected individuals and mice reported here), the observed hypocholesterolemia is more probably due to decreased cholesterol synthesis. Ube3b has no recognized function in cholesterol synthesis, and low cholesterol levels in humans and mice deficient for Ube3b might be an indirect consequence of altered ubiquitination and degradation of cholesterol-metabolizing enzymes. Although the exact mechanism underlying the observed downregulation of cholesterol synthesis is unknown, the observation of altered cholesterol metabolism in a novel ID syndrome is interesting by itself. As a crucial component of membranes, cholesterol is a constituent of myelin sheaths and lipid rafts in neurons and astrocytes. Because circulating cholesterol derived from dietary uptake and liver synthesis cannot cross the blood-brain barrier, the brain is dependent on cholesterol synthesis of astrocytes and—to a lesser extent—neurons, making it particularly vulnerable to cholesterol deficiency.36 This vulnerability is paradigmatically illustrated in Smith-Lemli-Opitz syndrome (SLOS; MIM 270400), a clinically distinct autosomal-recessive neurodevelopmental disorder characterized by growth retardation, microcephaly, ID, and multiple malformations.37 SLOS is caused by deficiency of the enzyme 7-dehydrocholesterol reductase, which catalyzes the final step of cholesterol biosynthesis. Although SLOS and the UBE3B-deficient BPID syndrome share some rather unspecific features such as growth retardation, microcephaly, ptosis, and ID, it is evident from a clinical, dysmorphological, and biochemical point of view that they represent distinct disorders and that most probably there is no direct overlap in their molecular pathophysiology.
In addition to the well-known involvement in neurodevelopment, defects in protein degradation and ubiquitination have also been implicated in the pathogenesis of neurodegenerative conditions such as Alzheimer and Parkinson disease and amyotrophic lateral sclerosis.38 Furthermore, recent large-scale genome-wide studies of copy-number variations have implicated gene networks of neuronal cell adhesion and ubiquitination in the pathogenesis of autism.39 A homozygous p.Arg922Cys variant in the HECT domain of UBE3B (annotated as p.Arg40Cys by the authors with reference to a shorter transcript)40 was recently found to cosegregate with autism in a small family, but the pathogenicity of this variant and the possible clinical overlap with the BPID syndrome we describe are currently unknown.40 The study of monogenic disorders has shed additional light on the role of ubiquitination in the development and function of the central nervous system. Whereas alterations of the imprinted UBE3A gene cause Angelman syndrome, mutations of UBE2A (MIM 312180; encoding an E2 enzyme) and HUWE1 (MIM 300697; encoding a HECT domain E3 ligase)41 cause X-linked ID.42,43 The identification of mutations of UBE3B in a complex BMR syndrome comprising ID and multiple congenital anomalies further supports the proposed pivotal role of UPS-mediated protein degradation in organogenesis and in the developing central nervous system. The substrate specificity and biological function of UBE3B are currently unknown, but we have shown that its probable C. elegans ortholog is indeed involved in UPS-mediated protein degradation. It has long been debated whether reactive oxygen species generated by oxidative stress are a cause or a consequence of neurodegenerative diseases.44 Oxidative stress can cause protein damage and mitochondrial dysfunction and ultimately result in cell death. Our in vivo studies in C. elegans suggest that UBE3B orthologs are particularly important in the UPS under oxidative stress conditions. Thus, neuronal dysfunction associated with UBE3B deficiency might be a consequence of a genetically determined vulnerability toward oxidative stress.
Future studies will be directed at identifying the response to oxidative stress and the ubiquitination targets of Ube3b, particularly in the brain. The identification of target proteins of individual ubiquitin ligases is experimentally challenging, but recent large-scale approaches have revealed hundreds of ubiquitination substrates and thereby contributed to a better understanding of the regulation of the proteome.45 The closest UBE3B paralog, UBE3C, is associated with the human 26S proteasome through its N terminus46,47 and is enriched in the human postsynaptic density proteome,48 members of which are highly associated with ID. These data further link ubiquitin ligases to cognitive function in humans.
Taken together, our results implicate biallelic UBE3B loss-of-function mutations in a syndromic form of ID and Ube3b deficiency in multiple anomalies in the mouse and open up new research avenues into the complex contribution of ubiquitination and oxidative stress to mammalian neurodevelopment and brain function.
Acknowledgments
We thank the families for participating in this study and B. Wollnik for discussions. We thank A. Fire, the Caenorhabditis Genetics Center (funded by the NIH Center for Research Resources), the Dana-Farber Cancer Institute, and Geneservice Ltd for antibodies, plasmids, cDNAs, and strains. We also thank E. Delaney for ES cell work leading to the generation of the Ube3b mutant mice and auditory phenotyping. This work was supported by the Deutsche Forschungsgemeinschaft (FOR 885, Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases and DIP grant HO 2541/4-1 to T.H.), the Rubicon European Union Network of Excellence (to T.H.), the Israeli Ministry of Health Chief Scientist Foundation (grant no. 3-4963) and the Israeli Science Foundation (grant no. 558/09; both to L.B.-V.), the Wellcome Trust, the MRC, and the EC (EUMODIC). D.J.A. and M.J.A. are supported by Cancer Research UK. T.H. is an EMBO Young Investigator. G.B. was supported by the Deutsche Forschungsgemeinschaft.
Contributor Information
Lina Basel-Vanagaite, Email: basel@post.tau.ac.il.
Guntram Borck, Email: guntram.borck@uni-ulm.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Browser, http://browser.1000genomes.org/index.html
Compare groups of growth curves, http://bioinf.wehi.edu.au/software/compareCurves
GenePaint, http://www.genepaint.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Sanger Mouse Resources Portal, http://www.sanger.ac.uk/mouseportal
UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Kawabe H., Brose N. The role of ubiquitylation in nerve cell development. Nat. Rev. Neurosci. 2011;12:251–268. doi: 10.1038/nrn3009. [DOI] [PubMed] [Google Scholar]
- 2.Tai H.C., Schuman E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008;9:826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- 3.Komander D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009;37:937–953. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 4.Li W., Bengtson M.H., Ulbrich A., Matsuda A., Reddy V.A., Orth A., Chanda S.K., Batalov S., Joazeiro C.A. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huibregtse J.M., Scheffner M., Beaudenon S., Howley P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA. 1995;92:2563–2567. doi: 10.1073/pnas.92.7.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rotin D., Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009;10:398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 7.Mabb A.M., Judson M.C., Zylka M.J., Philpot B.D. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34:293–303. doi: 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greer P.L., Hanayama R., Bloodgood B.L., Mardinly A.R., Lipton D.M., Flavell S.W., Kim T.K., Griffith E.C., Waldon Z., Maehr R. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating Arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Margolis S.S., Salogiannis J., Lipton D.M., Mandel-Brehm C., Wills Z.P., Mardinly A.R., Hu L., Greer P.L., Bikoff J.B., Ho H.Y. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143:442–455. doi: 10.1016/j.cell.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogart A., Wu D., LaSalle J.M., Schanen N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 2010;38:181–191. doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith S.E., Zhou Y.D., Zhang G., Jin Z., Stoppel D.C., Anderson M.P. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 2011;3:103ra97. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ropers H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genomics Hum. Genet. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 13.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 14.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O’Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clayton-Smith J., O’Sullivan J., Daly S., Bhaskar S., Day R., Anderson B., Voss A.K., Thomas T., Biesecker L.G., Smith P. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verloes A., Bremond-Gignac D., Isidor B., David A., Baumann C., Leroy M.A., Stevens R., Gillerot Y., Héron D., Héron B. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am. J. Med. Genet. A. 2006;140:1285–1296. doi: 10.1002/ajmg.a.31270. [DOI] [PubMed] [Google Scholar]
- 17.Day R., Beckett B., Donnai D., Fryer A., Heidenblad M., Howard P., Kerr B., Mansour S., Maye U., McKee S. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin. Genet. 2008;74:434–444. doi: 10.1111/j.1399-0004.2008.01087.x. [DOI] [PubMed] [Google Scholar]
- 18.Brancati F., Bernardini L., Cavalcanti D.P., Romano C., Novelli A., Dallapiccola B. Genome rearrangements in patients with blepharophimosis, mental retardation and hypothyroidism, so-called Young-Simpson syndrome. Clin. Genet. 2009;76:210–213. doi: 10.1111/j.1399-0004.2009.01235.x. [DOI] [PubMed] [Google Scholar]
- 19.Dentici M.L., Mingarelli R., Dallapiccola B. The difficult nosology of blepharophimosis-mental retardation syndromes: report on two siblings. Am. J. Med. Genet. A. 2011;155A:459–465. doi: 10.1002/ajmg.a.33642. [DOI] [PubMed] [Google Scholar]
- 20.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogunjimi A.A., Briant D.J., Pece-Barbara N., Le Roy C., Di Guglielmo G.M., Kavsak P., Rasmussen R.K., Seet B.T., Sicheri F., Wrana J.L. Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol. Cell. 2005;19:297–308. doi: 10.1016/j.molcel.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 23.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 24.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mouysset J., Deichsel A., Moser S., Hoege C., Hyman A.A., Gartner A., Hoppe T. Cell cycle progression requires the CDC-48UFD-1/NPL-4 complex for efficient DNA replication. Proc. Natl. Acad. Sci. USA. 2008;105:12879–12884. doi: 10.1073/pnas.0805944105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamath R.S., Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 27.Segref A., Torres S., Hoppe T. A screenable in vivo assay to study proteostasis networks in Caenorhabditis elegans. Genetics. 2011;187:1235–1240. doi: 10.1534/genetics.111.126797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miró X., Zhou X., Boretius S., Michaelis T., Kubisch C., Alvarez-Bolado G., Gruss P. Haploinsufficiency of the murine polycomb gene Suz12 results in diverse malformations of the brain and neural tube. Dis. Model. Mech. 2009;2:412–418. doi: 10.1242/dmm.001602. [DOI] [PubMed] [Google Scholar]
- 29.Ingham N.J., Pearson S., Steel K.P. Using the auditory brainstem response (ABR) to determine hearing sensitivity in mutant mice. Curr. Protoc. Mouse Biol. 2011;1:279–287. doi: 10.1002/9780470942390.mo110059. [DOI] [PubMed] [Google Scholar]
- 30.West B., Welch K., Galecki A. Chapman & Hall/CRC; Boca Raton, FL: 2007. Linear Mixed Models: A Practical Guide Using Statistical Software. [Google Scholar]
- 31.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong T.W., Huang L., Warner S.J., Lomax M.I. Characterization of the human UBE3B gene: structure, expression, evolution, and alternative splicing. Genomics. 2003;82:143–152. doi: 10.1016/s0888-7543(03)00111-3. [DOI] [PubMed] [Google Scholar]
- 33.Yanase S., Ishi N. Cloning of the oxidative stress-responsive genes in Caenorhabditis elegans. J. Radiat. Res. (Tokyo) 1999;40:39–47. doi: 10.1269/jrr.40.39. [DOI] [PubMed] [Google Scholar]
- 34.Visel A., Thaller C., Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32(Database issue):D552–D556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 36.Benarroch E.E. Brain cholesterol metabolism and neurologic disease. Neurology. 2008;71:1368–1373. doi: 10.1212/01.wnl.0000333215.93440.36. [DOI] [PubMed] [Google Scholar]
- 37.Porter F.D., Herman G.E. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 2011;52:6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Q., Figueiredo-Pereira M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis. 2010;15:1292–1311. doi: 10.1007/s10495-010-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chahrour M.H., Yu T.W., Lim E.T., Ataman B., Coulter M.E., Hill R.S., Stevens C.R., Schubert C.R., Greenberg M.E., Gabriel S.B., Walsh C.A., ARRA Autism Sequencing Collaboration Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X., Heng J.I., Guardavaccaro D., Jiang R., Pagano M., Guillemot F., Iavarone A., Lasorella A. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 2008;10:643–653. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nascimento R.M., Otto P.A., de Brouwer A.P., Vianna-Morgante A.M. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am. J. Hum. Genet. 2006;79:549–555. doi: 10.1086/507047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Froyen G., Corbett M., Vandewalle J., Jarvela I., Lawrence O., Meldrum C., Bauters M., Govaerts K., Vandeleur L., Van Esch H. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 2008;82:432–443. doi: 10.1016/j.ajhg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersen J.K. Oxidative stress in neurodegeneration: cause or consequence? Nat. Med. 2004;10(Suppl):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 45.Emanuele M.J., Elia A.E., Xu Q., Thoma C.R., Izhar L., Leng Y., Guo A., Chen Y.N., Rush J., Hsu P.W. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.You J., Pickart C.M. A HECT domain E3 enzyme assembles novel polyubiquitin chains. J. Biol. Chem. 2001;276:19871–19878. doi: 10.1074/jbc.M100034200. [DOI] [PubMed] [Google Scholar]
- 47.Wang X., Chen C.F., Baker P.R., Chen P.L., Kaiser P., Huang L. Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry. 2007;46:3553–3565. doi: 10.1021/bi061994u. [DOI] [PubMed] [Google Scholar]
- 48.Bayés A., van de Lagemaat L.N., Collins M.O., Croning M.D., Whittle I.R., Choudhary J.S., Grant S.G. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.