

Abstract
Human genetic variation is distributed nonrandomly across the genome, though the principles governing its distribution are only partially known. DNA replication creates opportunities for mutation, and the timing of DNA replication correlates with the density of SNPs across the human genome. To enable deeper investigation of how DNA replication timing relates to human mutation and variation, we generated a high-resolution map of the human genome’s replication timing program and analyzed its relationship to point mutations, copy number variations, and the meiotic recombination hotspots utilized by males and females. DNA replication timing associated with point mutations far more strongly than predicted from earlier analyses and showed a stronger relationship to transversion than transition mutations. Structural mutations arising from recombination-based mechanisms and recombination hotspots used more extensively by females were enriched in early-replicating parts of the genome, though these relationships appeared to relate more strongly to the genomic distribution of causative sequence features. These results indicate differential and sex-specific relationship of DNA replication timing to different forms of mutation and recombination.
Introduction
A human genome undergoes more than 20 mitotic replications and 1 meiotic replication in its passage from one generation to the next and dozens of additional mitotic replications in giving rise to the cells of the human soma. Each of these genome replications presents opportunities for error. These opportunities for error are not distributed uniformly across the genome, because DNA replication involves a structured program in which genomic regions replicate in a specific temporal order (reviewed in Hiratani et al.1). Locus-specific replication timing was previously shown to correlate with evolutionary divergence and the density of SNPs in human populations, such that regions of the genome that replicate later during S phase have accumulated more mutations during human evolution.2–4 Late replication also associates with elevated mutation rates in mice,4,5 fruit flies,6 yeast,7,8 archaea,9 and bacteria.10 Recent reports also suggest an association of DNA replication timing with cancer mutations11–13 and with deletions and duplications in cancer cells14 and in Drosophila melanogaster.15
To better understand the human genome’s replication program and its relationship to these and other forms of human genome variation and mutation, we sought to generate a high-resolution map of human genome replication timing and to analyze the distributions of distinct forms of mutation and recombination across this map.
Materials and Methods
Replication Timing
We used six lymphoblastoid cell lines whose genomes have been fully sequenced (at >30× average coverage) by the 1000 Genomes Project.16 These cell lines are derived from father-mother-offspring trios, one of West African ancestry (YRI; individuals NA19239, NA19238, NA12940) and the other of European ancestry (CEU; individuals NA12891, NA12892, NA12878).
For each cell line, four parallel asynchronous cultures of ∼50 million cells each were fixed with 70% ethanol. Fixed cultures were subsequently washed with PBS and treated with Accutase (Innovative Cell Technologies) for 20 min. Cells were pelleted, suspended in 10 ml PBS, and treated with 2.5 ml 10 mg/ml RNaseA for 30 min at 37°C and 2.5 ml 1 mg/ml propidium iodide for 30 min (Sigma). More than 1 million S and G1 cells were sorted with a FACSAria cell sorter (Beckton Dickinson).
DNA was extracted with the DNeasy blood and tissue kit (QIAGEN) and 100-base, paired-end reads were sequenced on the Illumina HighSeq 2000 and aligned to the human genome reference hg18 with BWA.17
Data Processing
We combined the reads from all experiments into a single “consensus” data set. For the X chromosome, reads from male and female cell lines were considered separately; the female profile was used for all analyses besides the comparison with the cell line mutation data, most of which is based on male cell lines. We defined varying-size, equal-coverage chromosomal windows as segments covered by 200 reads in the G1 fraction and counted S phase reads in the same windows. The average size of these segments was ∼2 Kb. Data were normalized to 0 mean and 1 SD. Windows of 100 Kb along the chromosomes that contained data segments with an internal standard deviation larger than 1.1 were removed from further analysis (these corresponded predominantly to borders of centromeric gaps). Subsequently, we considered chromosomal fragments covering contiguous sequenced positions (i.e., fragmented the chromosomes to intergap fragments). Fragments containing fewer than 20 data windows were removed from further analysis. The remaining fragments were smoothed with a cubic smoothing spline via the Matlab function csaps with a parameter of 10−16.
The processed replication timing data are available from the author’s (S.A.M.) website.
Comparison to Replication Timing
p values were determined by the Kolmogorov-Smirnov test for differences between distributions.
Regression Analysis
In order to test whether replication timing has an additional impact on rates of the different genetic variation types beyond those caused by other confounding factors, we modeled rates of genetic variation events as linear functions of different factors by using Poisson regression. In order to do so we divided the genome into 100 Kb long windows. For every window, we calculated the number of the following response variables: transition mutations, transversion mutations, NAHR CNVs, NH CNVs, and female and male recombination hotspots. For each of the windows we calculated the following quantities: GC content, SNP density (data from the 1000 Genomes pilot project), replication timing, female and male recombination rates, and log distance to the telomere. Corrected variation rates were calculated with the R function glm (“generalized linear model”), with an exposure variable being the number of nucleotides in a given window for CNV and recombination events and the number of weak bases for W-to-S mutations or strong bases for S-to-W mutations.
Results
A High-Resolution Map of DNA Replication Timing
To better understand how DNA replication timing relates to mutations in humans, we first generated a DNA replication timing profile of high temporal and spatial resolution as a resource for human genetic studies. Extending a method we recently described for profiling replication of yeast genomes,18 we flow-sorted lymphoblastoid S and G1 phase cells, then generated chromosomal DNA replication profiles based on sequence read depth of S phase DNA relative to G1 DNA (Figure S1; Materials and Methods). This method reads out the genome’s temporal replication program as a distribution of locus-specific copy number, because early-replicating regions of the genome are overrepresented and late-replicating regions are underrepresented, in DNA from S phase cells. G1 phase cells serve as a control for other influences of sequence features on molecular abundance in a sequencing library.
As evidence of their quality, our data showed high autocorrelation along each chromosome, reflecting a genomically structured program (Figure S1). They also showed high reproducibility, with a Pearson r = 0.97 for biological repeats. The data were also broadly consistent with lower-resolution replication timing profiles available from earlier methods (r = 0.55–0.82; Figure S1).
We applied our method to six lymphoblastoid cell lines from two mother-father-offspring trios deeply sequenced by the 1000 Genomes Project (Materials and Methods). Interindividual correlations between the measured replication profiles were 0.91–0.97 (Figure S1). For the purpose of the current study, and justified by the high correlations across individuals, we combined data across individuals to generate a high-resolution “consensus” human genome DNA replication profile. Our data are more accurate and complete than previous data sets in two dimensions. One of these dimensions is spatial resolution, which was accomplished by both (1) high genome-wide sequence coverage (∼46× for all individuals and fractions combined [Table S1], more than two orders of magnitude more than previous studies) and (2) higher relative coverage of repetitive regions resulting from longer read lengths. These data also provided substantially improved temporal resolution, because our approach allows cells to be sampled across S phase, rather than simply comparing two specific S phase fractions. The use of samples with fully sequenced genomes enabled a high level of control for structural variation within the studied genomes when analyzing replication patterns.
Point Mutations Are Enriched in Late-Replicating Regions
Late-replicating regions of the human genome have previously been shown to harbor a greater density of SNPs in human populations.3 This relationship was also apparent in our data, showing (as expected from the earlier work) a statistically significant though quantitatively modest magnitude (r = −0.15, p < 10−100) (Figure S2).
SNP diversity data reflect both mutational processes operating on germline cells and evolutionary processes operating in human populations. We hypothesized that the replication program might relate more strongly to the distribution of mutations themselves, particularly if those mutations were ascertained in the same cell type and without the additional influences of population genetics. We therefore performed a direct comparison of replication timing to 5,740 cell line mutations in the same cell type. The mutations were ascertained from two sets of experiments analyzing deeply sequenced genomes of lymphoblastoid cell lines derived from families: (1) two father-mother-offspring trios (the same cell lines used to generate the replication timing profiles) in which mutations were ascertained as alleles unique to the offspring19 and (2) a novel data set of ten family quartets, each consisting of a father, mother, and pair of monozygotic twins, in which somatic mutations were ascertained as alleles unique to an offspring and not shared with the offspring’s twin or parents (Table S2; we note that most mutations originate from six individuals in this data set; J.J.M., J.S., unpublished data).
The rate of point mutations along chromosomes tightly followed the replication timing profile (Figure 1A). Intriguingly, the correlation of mutations with replication timing was far stronger than had been apparent from earlier comparisons of replication timing to the density of SNPs (r = −0.57, versus r = −0.15 for SNPs; Figure 1A). The most likely explanatory factor is that we have ascertained mutations and replication in the same cell type; additional contributing influences could in principle also come from the high quantitative resolution of our data and the ability to neutralize population-genetic noise by examining mutation directly rather than using standing variation (SNPs) as a proxy. We obtained comparable results when analyzing each mutation data set and each individual separately (Figure S2 and data not shown).
Figure 1.
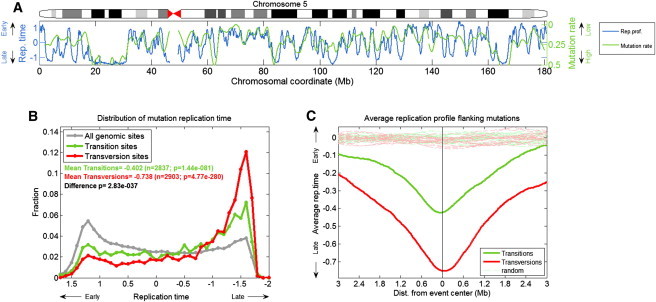
Transition Mutations, and Especially Transversion Mutations, Are Enriched in Late S Phase
(A) The replication time of chromosome 5 along with the density of cell line mutations. The density of mutations is shown in reverse scale (i.e., higher mutation rates are shown lower on the plot), as mutation rate per 100 Kb windows (smoothed over 40 windows) in all cell lines combined.
(B) Distribution of DNA replication timing for the entire genome (gray; mean set to 0) and for cell line mutation locations (green: transitions; red: transversions): transitions, and especially transversions, show a strong bias toward late S phase.
(C) The average replication timing structure in the region extending to 3 megabases of both sides of all mutation locations. In dashed lines are the same plots for 20 sets each of random genomic locations matching in number to the mutation events of the different types.
Point mutations consist of two types: transitions (from a purine [A or G] to another purine or from a pyrimidine [C or T] to another pyrimidine) and transversions (from a purine to a pyrimidine or vice versa). Our analysis revealed a strong association between DNA replication timing and the specific types of point mutations observed: transition mutations were >2-fold more abundant in late-replicating than in early-replicating parts of the genome, whereas transversion mutations showed a >6-fold increase (when comparing the mutation rich-most bins in early and late S phase; Figures 1B, 1C, and S2). Such a relationship had not been observed in earlier studies3 but was strong and prominent in our data, given the increase in power afforded by the high-resolution replication and cell line mutation maps. We also observed the difference between transitions and transversions in population-level SNP data, indicating that this relationship was not specific to cell lines (Figure S3). The difference between transitions and transversions was not explained by any one specific type of mutation, nor by mutations in CpG dinucleotides (Figure S4 and see Table S1). The effect of late replication on mutations was similar for transcribed and nontranscribed regions (data not shown), suggesting that interference or cooperativity between the replication and transcription machinery is not a major factor in mutagenesis.
Structural Mutations
Copy number variations (CNVs) such as deletions and duplication are a substantial source of human genome variation. Most CNVs are thought to form by one of two types of mechanism: (1) recombination-based mechanisms, particularly nonallelic homologous recombination (NAHR) mediated by flanking homologous sequences, or (2) nonhomology (NH)-based mechanisms such as nonhomologous end-joining (NHEJ) and microhomology-mediated break-induced replication (MMBIR) (reviewed in Hastings et al.20). The mechanism of formation of thousands of CNVs was recently inferred by the 1000 Genome Project based on analysis of their precise breakpoint sequences.21 CNVs show a strongly nonrandom distribution, with “hotspots” of multiple events, typically of the same formation mechanism, in close genomic proximity. This suggests that regional factors affect the probability that CNV arises at any particular site.
We therefore evaluated how CNV formation hotspots distribute across the genome’s replication program. Surprisingly, NAHR- and NH-mediated CNVs showed distinct relationships to DNA replication timing: hotspots of NAHR-mediated events were >4-fold enriched in early-replicating regions, whereas NH hotspots were ∼2-fold enriched in late-replicating regions (when comparing the CNV rich-most bins in early and late S phase; Figures 2A–2C). The difference between NAHR- and NH-mediated CNVs was also observed for nonhotspot CNV sites (Figure 2D), as well as for deletion and insertion/duplication events separately (data not shown). These patterns were observed even when removing events flanked by repetitive elements or overlapping any known functional elements (Figure S5), suggesting that these patterns do not arise from the distribution of high-copy repetitive elements nor from selection. There was no significant correlation between CNV allele frequency and replication timing (Figure S5), further supporting the idea that selection is not the underlying cause of these relationships.
Figure 2.
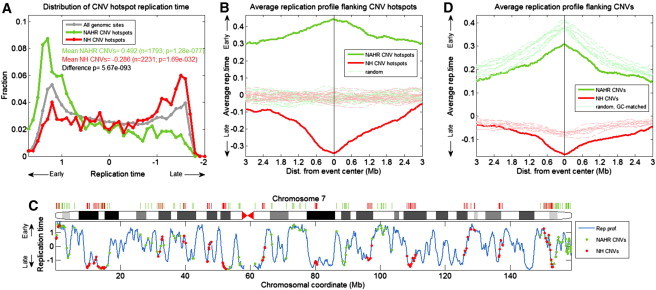
Hotspots of NAHR-Mediated CNVs Are Enriched in Early S Phase, whereas NH-Mediated CNV Hotspots Are Enriched in Late S Phase
(A) Distribution of DNA replication timing for the entire genome (gray) and hotspot locations of NAHR (green)-, and NH (red)-mediated CNVs. All CNV locations of each type were hierarchically clustered with a distance cutoff of 5 Mb (for NAHR) or 1 Mb (NH), and hotspots were defined as clusters with at least five events.
(B) The average replication timing structure surrounding CNV hotspot locations (as in Figure 1B).
(C) The replication profile of chromosome 7, with the locations of hotspots of NAHR (green)- and NH (red)-mediated events shown.
(D) The average replication timing structure surrounding all CNV locations compared to random locations matched for GC content (within a 0.01% range). The association of NH events with DNA replication timing is not due to GC effects. In contrast, when considering GC content, NAHR events occur in later-replicating regions than expected for genomic regions with the same GC content (and not early-replicating regions as suggested when ignoring GC content).
To further evaluate these relationships, we also analyzed the genomic distributions of historical deletions that have become fixed in the ancestors of all humans.22 Analysis of the genomic sequence flanking each CNV (in a manner analogous to the analysis of polymorphic CNVs in Mills et al.21) enabled separation of these human-specific deletions into those predicted to have occurred by NAHR and by NH mechanisms. Deletions consistent with formation by NAHR were enriched in the genomic regions that replicate early in S phase. In contrast, deletions consistent with an NH formation mechanism were enriched in late-replicating parts of the genome (Figure S6).
Meiotic Recombination
NAHR is intimately related to the normal process of allelic homologous recombination: both are mediated by homology, arise during meiosis from processing of double-strand breaks, and overlap in their preferred locations21,23–26 (reviewed in Sasaki et al.27). Meiotic recombination is distributed nonrandomly along the genome, with distinct hotspots at which the probability of recombination is much higher than elsewhere in the genome. Although the location of hotspots is determined to a large extent by binding sites for the histone methyltransferase PRDM9 (reviewed in Ségurel et al.28), recombination rate also varies on larger scales.29 It is unclear what determines these large-scale variations. Intriguingly, a recent study of recombination events in individual humans, based on analysis of more than 15,000 Icelandic parent-offspring pairs, revealed a difference between male and female recombination rates that spanned scales of several megabases and led to the identification of close to 9,000 hotspots that were observed to be utilized primarily by one sex or the other.30
We analyzed the sex-specific distribution of meiotic recombination events along the human DNA replication timing profile. In order to capture the full range of differences between the sexes, we calculated a “difference map” from the female and male recombination maps30 and analyzed hotspot regions in this differential map. Recombination events in females were substantially more biased toward early-replicating regions of the genome than were recombination events in males (Figures 3A and 3B). This was also observed when analyzing hotspots in each recombination map separately: recombination hotspots used more frequently by females were significantly biased toward early S phase, whereas male-preferred recombination did not show a strong bias (Figure 3C). This result was confirmed with a second, lower-resolution data set on sex-specific recombination31 (data not shown). We also analyzed the correlation between replication timing and recombination rates over a wide range of recombination scales and found that broad-scale female recombination rates also associate with DNA replication timing substantially more strongly than male recombination rates do (Figure 3D).
Figure 3.
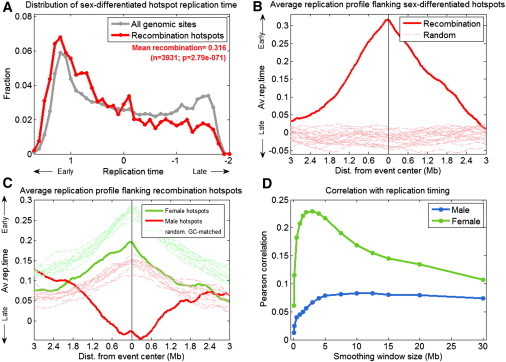
Sex-Specific Associations of Recombination Hotspots and De Novo CNVs with DNA Replication Timing
(A) Distribution of DNA replication timing for the entire genome (gray) and locations of hotspots of the sex-differentiated recombination rate, calculated by subtracting male recombination rates from female recombination rates.
(B) The average replication timing structure surrounding sex-differentiated recombination hotspots (as in Figure 1B).
(C) The replication timing structure surrounding male and female recombination hotspots separately, alongside control regions matched for GC content. When considering GC content, female recombination hotspots occur in later-replicating regions than expected for genomic regions with the same GC content (and not early replicating regions as suggested when ignoring GC content); male recombination hotspots occur in much later replicating regions than expected for regions with similar GC content (and hence do show an association with replication timing, in a negative direction, in contrast to the lack of association suggested when ignoring GC content).
(D) Large-scale correlation between male and female recombination and DNA replication timing.
Because recombination-based mechanisms mediate the formation of many CNVs, the above result predicts that CNVs arising in fathers will show a different genomic distribution than CNVs arising in mothers. We analyzed the distribution of 368 de novo copy number mutations that had been resolved with respect to parent-of-origin. To do this, we collated data from several recent studies of neuropsychological disease (see Figure S7 for references) that used SNP array-genotyped father-mother-offspring trios to find de novo copy number mutations and inferred the parental origin of the mutation through analysis of the SNP alleles that changed in copy number at each CNV. De novo CNVs associated with DNA replication timing in a manner consistent with sex-specific recombination: mutations occurring on maternally inherited segments tended to involve genomic regions that replicate early in S phase, whereas male mutation events showed a weaker relationship to replication timing (Figure S7). This sex difference in the distributions of copy number mutations could be due to a direct effect of DNA replication timing or to a difference in the relative frequency of NAHR versus NH in males and females.
Recombination-Dependent Events Associate with Local Sequence Composition
Meiotic recombination depends on the PRDM9 histone methyltransferase that binds a GC-rich motif;23 GC-rich sequences can also promote recombination by facilitating the formation of unstable structures such as G quadruplexes.32,33 In turn, GC content itself distributes nonrandomly with respect to DNA replication timing, with early-replicating regions having higher GC content than late-replicating regions. To better understand these relationships, we performed partial correlation and regression analyses of mutation rates, recombination rates, replication timing, and GC content, considering as additional predictors recombination rates, SNP density, and distance to the telomere (Figure S8 and Table S1). The relationships of cell line mutations and NH CNVs to replication timing remained strong and consistent when these covariates were included in the analysis. In contrast, NAHR CNVs and female recombination rates associated more strongly with the GC content of local sequence (at 10 kb scale) than to DNA replication timing itself; when adjusting for covariation with GC content, their relationship to replication timing reversed sign. Male recombination showed an association with late-replication timing that was stronger than that of female recombination. We confirmed these results by analyzing the replication structure around random locations in the genome that were matched to have a similar GC content as the tested events. NAHR CNVs (Figure 2D; note that NAHR and NH CNVs appeared to be biased toward late replication to similar extents) and female- and male-preferred recombination hotspots (Figure 3C) associated with genomic regions that replicate later in S phase than expected for regions with similar GC content. In contrast, the relationships of replication timing to the distributions of transition and transversion mutations was robust to the effects of GC content of local sequence (Figure S9). Therefore, whereas NAHR and female recombination events are strongly enriched in early-replicating parts of the genome, this relationship appears to be driven by local sequence content; the effect of replication timing itself appears toward increasing the likelihood of these events in late-replicating regions.
We also used these multivariate analyses to further scrutinize the distribution of substitution polymorphisms (SNPs); the result (Figure S8) revealed an even stronger influence of replication timing on SNP variation than had been apparent in our earlier unadjusted analysis. The relative strength of association of replication timing to new mutations, relative to the previously observed relationship to the distribution of SNPs, appears to be at least partly due to the latter relationship being influenced by the mutagenic effects of recombination.
To further evaluate whether any of the relationships found in this study were due to effects in subtelomeric or in centromeric regions, we repeated all analyses excluding 10 Mb regions from the ends and/or flanking the centromeres of each chromosome. The results were substantially consistent, indicating that these relationships persist across the entire length of chromosomes.
Discussion
Our results expand on earlier observations that replication timing correlates with the distribution of sequence mutations.3–8 In particular, we find that late replication differentially increases the chances of occurrence of different types of mutations and recombination, with a notably stronger relationship to the distribution of transversions than transition mutations (Figure 1). This observation may begin to distinguish among models to explain the earlier observation that SNP density is greater in late-replicating regions.3 In particular, this observation would not be expected under a model in which DNA is just generally more prone to damage during later stages of S phase;3 rather, specific biochemical aspect(s) of the replication process are likely to change during S phase with concomitant effects on the introduction or repair of DNA lesions. Possible factors include dNTP levels and/or balance,34,35 activity of alternative DNA polymerases (replicative or trans-lesion),36 and mismatch repair activity.37
Different types of mutational and recombination events have very different distributions along the genome (Figure 4), a finding that has implications for understanding the evolution of genome organization. For example, protein-coding genes are known to concentrate in the early-replicating parts of the genome, a distribution that may protect them from both somatic and germline mutations. To further explore these relationships with the high-resolution replication timing map generated in this study, we analyzed how functionally defined classes of genes relate to replication timing. Genes involved in the negative regulation of cell growth showed a particularly early average replication time, an organization that may be critical for protection of somatic cells from cancer.
Figure 4.
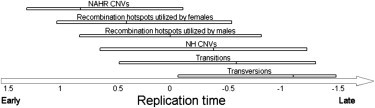
Summary of the Distribution of Various Mutational Events along the Replication Timing Program
Shown are the 25th, 50th, and 75th percentile of the replication time distribution of events of the indicated genetic variation type. The timing shown is the replication timing of genomic locations carrying these variation types, but not necessarily the time of occurrence of the relevant events (some of which, particularly meiotic recombination events, occur separately than DNA replication).
Another form of genome variation that varies along the replication profile is mobile element insertions: the positions of Alu insertions are strongly biased toward early-replicating regions, whereas the positions of L1 insertions are biased toward late-replicating regions38 (and further supported by our data). This is at least partially determined by the preferred AT-rich cleavage site of the LINE endonuclease and by selective forces,38 such as the tendency of Alu to insert and/or be maintained at genic loci (which are early replicating). Disentangling these effects would require the observation of a large number of new Alu mutation events.
New mutations are increasingly used as a gene discovery strategy in the study of cancer and other diseases, as the advent of high-throughput sequencing now makes it possible to identify new mutations in all protein-coding genes and to identify genes that harbor statistical excesses of mutations in diseased tissues and patients (e.g., Neale et al., 39 O’Roak et al.,40 Sanders et al.41). A critical feature of all such analyses is to identify genes that contain an excess of mutations (relative to the number observed in all other genes) that is statistically surprising, in a way that defines that specific gene as being of importance in the disease. It will be critical that such analyses are calibrated to the true mutation rate of each gene. The high-resolution replication map provided here will be a useful resource for such studies.
Finally, we report a sex difference for the association of recombination and copy-number mutations with DNA replication timing. This sex difference could result from the conjunction of two factors: first, an influence of replication timing on the subsequent likelihood that a sequence will sustain meiotic DSBs;42,43 second, a difference between the premeiotic replication timing programs of male and female germlines. The latter is likely, given previously observed differences in the replication timing programs of different tissues (up to ∼20% of the genome44,45). Further methodological development will be required to determine the replication programs of male and female germline cells; however, the possibility of a sex difference in the replication program would have far-reaching implications for understanding sequence evolution. In particular, given the strong association we describe between replication timing and point mutations, we predict that mutations that originate in the two sexes will ultimately be found to have different genomic distributions.
Acknowledgments
We thank David Reich for helpful comments. This work was supported by startup resources from the Harvard Medical School Department of Genetics, by a Harvard-Armenise Junior Faculty Award to S.A.M., and by the Stanley Center for Psychiatric Research at the Broad Institute of Harvard and MIT.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
McCarroll website, http://genepath.med.harvard.edu/mccarroll/datasets.html
Accession Numbers
Sequence data described in this paper have been deposited in the Sequence Read Archive under accession number SRA052697.
References
- 1.Hiratani I., Takebayashi S.-i., Lu J., Gilbert D.M. Replication timing and transcriptional control: beyond cause and effect—part II. Curr. Opin. Genet. Dev. 2009;19:142–149. doi: 10.1016/j.gde.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolfe K.H., Sharp P.M., Li W.-H. Mutation rates differ among regions of the mammalian genome. Nature. 1989;337:283–285. doi: 10.1038/337283a0. [DOI] [PubMed] [Google Scholar]
- 3.Stamatoyannopoulos J.A., Adzhubei I., Thurman R.E., Kryukov G.V., Mirkin S.M., Sunyaev S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009;41:393–395. doi: 10.1038/ng.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen C.-L., Rappailles A., Duquenne L., Huvet M., Guilbaud G., Farinelli L., Audit B., d’Aubenton-Carafa Y., Arneodo A., Hyrien O., Thermes C. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010;20:447–457. doi: 10.1101/gr.098947.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pink C.J., Hurst L.D. Timing of replication is a determinant of neutral substitution rates but does not explain slow Y chromosome evolution in rodents. Mol. Biol. Evol. 2010;27:1077–1086. doi: 10.1093/molbev/msp314. [DOI] [PubMed] [Google Scholar]
- 6.Weber C.C., Pink C.J., Hurst L.D. Late-replicating domains have higher divergence and diversity in Drosophila melanogaster. Mol. Biol. Evol. 2012;29:873–882. doi: 10.1093/molbev/msr265. [DOI] [PubMed] [Google Scholar]
- 7.Lang G.I., Murray A.W. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol. Evol. 2011;3:799–811. doi: 10.1093/gbe/evr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agier N., Fischer G. The mutational profile of the yeast genome is shaped by replication. Mol. Biol. Evol. 2012;29:905–913. doi: 10.1093/molbev/msr280. [DOI] [PubMed] [Google Scholar]
- 9.Flynn K.M., Vohr S.H., Hatcher P.J., Cooper V.S. Evolutionary rates and gene dispensability associate with replication timing in the archaeon Sulfolobus islandicus. Genome Biol. Evol. 2010;2:859–869. doi: 10.1093/gbe/evq068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp P.M., Shields D.C., Wolfe K.H., Li W.H. Chromosomal location and evolutionary rate variation in enterobacterial genes. Science. 1989;246:808–810. doi: 10.1126/science.2683084. [DOI] [PubMed] [Google Scholar]
- 11.Hodgkinson A., Chen Y., Eyre-Walker A. The large-scale distribution of somatic mutations in cancer genomes. Hum. Mutat. 2012;33:136–143. doi: 10.1002/humu.21616. [DOI] [PubMed] [Google Scholar]
- 12.Schuster-Böckler B., Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 13.Woo Y.H., Li W.-H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat. Commun. 2012;3:1004. doi: 10.1038/ncomms1982. [DOI] [PubMed] [Google Scholar]
- 14.De S., Michor F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat. Biotechnol. 2011;29:1103–1108. doi: 10.1038/nbt.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardoso-Moreira M., Emerson J.J., Clark A.G., Long M. Drosophila duplication hotspots are associated with late-replicating regions of the genome. PLoS Genet. 2011;7:e1002340. doi: 10.1371/journal.pgen.1002340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koren A., Soifer I., Barkai N. MRC1-dependent scaling of the budding yeast DNA replication timing program. Genome Res. 2010;20:781–790. doi: 10.1101/gr.102764.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conrad D.F., Keebler J.E.M., DePristo M.A., Lindsay S.J., Zhang Y., Casals F., Idaghdour Y., Hartl C.L., Torroja C., Garimella K.V., 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mills R.E., Walter K., Stewart C., Handsaker R.E., Chen K., Alkan C., Abyzov A., Yoon S.C., Ye K., Cheetham R.K., 1000 Genomes Project Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean C.Y., Reno P.L., Pollen A.A., Bassan A.I., Capellini T.D., Guenther C., Indjeian V.B., Lim X., Menke D.B., Schaar B.T. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature. 2011;471:216–219. doi: 10.1038/nature09774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myers S., Freeman C., Auton A., Donnelly P., McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 24.Lindsay S.J., Khajavi M., Lupski J.R., Hurles M.E. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am. J. Hum. Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raedt T.D., Stephens M., Heyns I., Brems H., Thijs D., Messiaen L., Stephens K., Lazaro C., Wimmer K., Kehrer-Sawatzki H. Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat. Genet. 2006;38:1419–1423. doi: 10.1038/ng1920. [DOI] [PubMed] [Google Scholar]
- 26.Berg I.L., Neumann R., Lam K.-W.G., Sarbajna S., Odenthal-Hesse L., May C.A., Jeffreys A.J. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasaki M., Lange J., Keeney S. Genome destabilization by homologous recombination in the germ line. Nat. Rev. Mol. Cell Biol. 2010;11:182–195. doi: 10.1038/nrm2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ségurel L., Leffler E.M., Przeworski M. The case of the fickle fingers: how the PRDM9 zinc finger protein specifies meiotic recombination hotspots in humans. PLoS Biol. 2011;9:e1001211. doi: 10.1371/journal.pbio.1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chowdhury R., Bois P.R.J., Feingold E., Sherman S.L., Cheung V.G. Genetic analysis of variation in human meiotic recombination. PLoS Genet. 2009;5:e1000648. doi: 10.1371/journal.pgen.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong A., Thorleifsson G., Gudbjartsson D.F., Masson G., Sigurdsson A., Jonasdottir A., Walters G.B., Jonasdottir A., Gylfason A., Kristinsson K.T. Fine-scale recombination rate differences between sexes, populations and individuals. Nature. 2010;467:1099–1103. doi: 10.1038/nature09525. [DOI] [PubMed] [Google Scholar]
- 31.Broman K.W., Murray J.C., Sheffield V.C., White R.L., Weber J.L. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao J., Bacolla A., Wang G., Vasquez K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De S., Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 2011;18:950–955. doi: 10.1038/nsmb.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar D., Abdulovic A.L., Viberg J., Nilsson A.K., Kunkel T.A., Chabes A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011;39:1360–1371. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burkhalter M.D., Roberts S.A., Havener J.M., Ramsden D.A. Activity of ribonucleotide reductase helps determine how cells repair DNA double strand breaks. DNA Repair (Amst.) 2009;8:1258–1263. doi: 10.1016/j.dnarep.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diamant N., Hendel A., Vered I., Carell T., Reissner T., de Wind N., Geacinov N., Livneh Z. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 2012;40:170–180. doi: 10.1093/nar/gkr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hombauer H., Srivatsan A., Putnam C.D., Kolodner R.D. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science. 2011;334:1713–1716. doi: 10.1126/science.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ashida H., Asai K., Hamada M. Shape-based alignment of genomic landscapes in multi-scale resolution. Nucleic Acids Res. 2012;40:6435–6448. doi: 10.1093/nar/gks354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.-F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borde V., Goldman A.S.H., Lichten M. Direct coupling between meiotic DNA replication and recombination initiation. Science. 2000;290:806–809. doi: 10.1126/science.290.5492.806. [DOI] [PubMed] [Google Scholar]
- 43.Kugou K., Fukuda T., Yamada S., Ito M., Sasanuma H., Mori S., Katou Y., Itoh T., Matsumoto K., Shibata T. Rec8 guides canonical Spo11 distribution along yeast meiotic chromosomes. Mol. Biol. Cell. 2009;20:3064–3076. doi: 10.1091/mbc.E08-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen R.S., Thomas S., Sandstrom R., Canfield T.K., Thurman R.E., Weaver M., Dorschner M.O., Gartler S.M., Stamatoyannopoulos J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryba T., Hiratani I., Lu J., Itoh M., Kulik M., Zhang J., Schulz T.C., Robins A.J., Dalton S., Gilbert D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.