

Abstract
The neuromuscular junction (NMJ) is a specialized synapse with a complex molecular architecture that provides for reliable transmission between the nerve terminal and muscle fiber. Using linkage analysis and whole-exome sequencing of DNA samples from subjects with distal hereditary motor neuropathy type VII, we identified a mutation in SLC5A7, which encodes the presynaptic choline transporter (CHT), a critical determinant of synaptic acetylcholine synthesis and release at the NMJ. This dominantly segregating SLC5A7 mutation truncates the encoded product just beyond the final transmembrane domain, eliminating cytosolic-C-terminus sequences known to regulate surface transporter trafficking. Choline-transport assays in both transfected cells and monocytes from affected individuals revealed significant reductions in hemicholinium-3-sensitive choline uptake, a finding consistent with a dominant-negative mode of action. The discovery of CHT dysfunction underlying motor neuropathy identifies a biological basis for this group of conditions and widens the spectrum of disorders that derive from impaired NMJ transmission. Our findings compel consideration of mutations in SLC5A7 or its functional partners in relation to unexplained motor neuronopathies.
Main Text
The distal hereditary motor neuronopathies (dHMNs) comprise a heterogeneous group of diseases that share a common feature of a length-dependent neuropathy resulting in progressive distal muscle wasting and weakness. Electrophysiology and electromyography (EMG) studies have revealed normal motor and sensory conduction velocities with reduced compound-motor-action-potential amplitudes and neurogenic changes, suggesting that pathology is at the level of the anterior horn cell. Harding described seven subtypes of dHMNs on the basis of clinical and genetic criteria.1 Isolation of the causative genes in the dominant forms of dHMNs has revealed genetic heterogeneity within subtypes, and mutations in six genes have been described to date and include DCTN1 (MIM 601143), GARS (MIM 600287), BSCL2 (MIM 606168), HSPB8 (MIM 608041), HSPB1 (MIM 602195), and HSPB3 (MIM 604624). These findings implicate processes such as protein misfolding, RNA metabolism, and axonal transport as pathologic mechanisms in this disorder.2 Several of these genes also cause forms of Charcot-Marie-Tooth disease type 2 (CMT2), which is categorized separately on the basis of sensory-nerve involvement.
We previously detailed the clinical features of members of a large UK family affected by dominantly transmitted dHMN type VII (dHMN-VII [MIM 158580]), distinguished by the presence of vocal-cord involvement in affected subjects, and we mapped the gene responsible to chromosomal region 2q14.3–5 Here, we report the identification of a mutation within SLC5A7 (MIM 608761) as the underlying cause of dHMN-VII. SLC5A7 encodes the presynaptic choline transporter (CHT), a critical determinant of synaptic acetylcholine (ACh) synthesis and release at the neuromuscular junction (NMJ). Mutations affecting genes coding for NMJ proteins are well known to cause a contrasting phenotype of congenital myasthenic syndrome, which is characterized by fatiguing muscle weakness and cranial-nerve and respiratory-muscle involvement.6 Our findings are thus unexpected and widen the phenotypical spectrum associated with NMJ dysfunction.
Our studies derive from the analysis of samples from 26 family members (14 affected and 12 unaffected) obtained with informed consent; the research protocol was approved by the Wandsworth Local Research Ethics Committee. In order to identify the disease-associated gene, we performed whole-exome sequencing of a single affected individual (VI:5) in this family (Figure 1). Coding regions were captured with SureSelect All Exons (50 Mb) and sequenced by Illumina HiSeq, yielding 9.8 Gb data (∼130 million reads) corresponding to 91% target coverage with a mean depth of 107× and identifying 52,806 variants (Tables S2 and S3, available online). After filtering, only one probably deleterious variant was identified within the critical region; this was a single base deletion (c.1497delG) in SLC5A7 (RefSeq accession number NM_021815.2), encoding the Na+/Cl− dependent, high-affinity CHT7–9 (Figure S1). Dideoxy sequence analysis, as well as Ssp1 restriction digestion (Figure 1), confirmed cosegregation of the variant with the disease phenotype, and the variant was found to be present in monocyte mRNA of the affected cases (not shown). The variant is not present in genomic databases or 150 UK control individuals. Three other unrelated individuals, who each had dHMN-VII-like features involving some degree of distal muscle wasting and vocal-cord paralysis, were investigated for SLC5A7 mutations; TRPV4 (MIM 605427) mutations, which might phenotypically overlap with mutations causing dHMN-VII, were previously excluded in each case. This revealed no other novel sequence variants, indicating that the individuals investigated might possess a clinically distinct condition or that there exists further genetic heterogeneity in this very rare condition.
Figure 1.
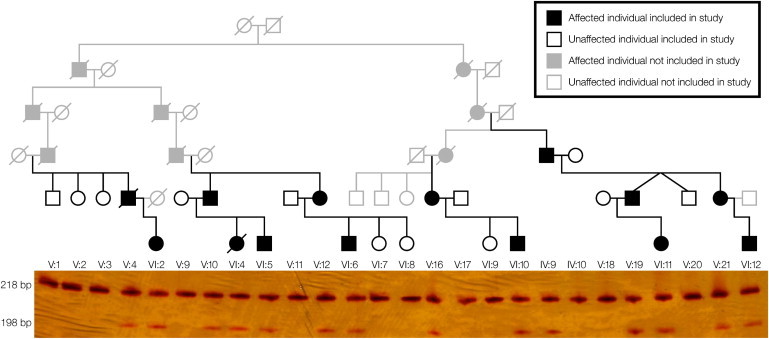
Family Pedigree and c.1497delG Cosegregation
The c.1497delG variant results in the creation of a novel SspI restriction site that facilitates cosegregation analysis by restriction digestion of exon 9 PCR products resolved by polyacrylamide gel electrophoresis (PCR primers used for amplification of genomic DNA were 5′-CCCTGGCTATTACCCTGATG-3′ and 5′-CACAAGTGCAAGTTCATCTAATTT-3′). The c.1497delG variant results in the digestion of the WT 218 bp product into fragments of 198 bp and 20 bp (not shown). All subjects with the 198 bp fragment are heterozygous and thus also have an undigested 218 bp band. These genotypings were also confirmed by dideoxy sequence analysis. For cross reference with the previously published pedigree, refer to Table S1.
The c.1497delG nucleotide change is predicted to result in a translational frameshift (FS) changing Lys499 to Asn and to then encode an additional 11 ectopic amino acids prior to a premature stop codon (p.Lys499Asnfs∗13). This alteration results in elimination of the transporter’s final 82 amino acids, entailing a near complete deletion of the transporter’s cytoplasmic C terminus (Figure S2). Because the C terminus displays strong sequence conservation across many species (Figure S2) and encodes residues known to regulate surface trafficking,10 loss of this domain is likely to have profound functional consequences.
We investigated the functional impact of the c.1497delG variant via heterologous expression of cDNAs encoding mutant (FS) and wild-type (WT) CHT in transiently-transfected human embryonic kidney (HEK) 293 cells. Existing antibodies against CHT target the transporter’s C terminus,11 precluding their use in characterizing FS CHT. Thus, in order to detect WT and FS CHT, we added a hemagluttinin (HA) tag to the N termini of both transporters by using site-directed mutagenesis (Quick-Change kit, Stratagene). Cell-surface biotinylation experiments were performed as previously described.12 WT CHT migrated in transiently transfected HEK 293 cells as two broad bands of 48 kDa and 56 kDa; the 48 kDa band represents an immature, core-glycosylated species, and the 56 kDa band represents the mature, more highly glycosylated species that is enriched at the cell surface11 (Figure 2A). For equivalent amounts of transfected cDNA, reduced levels of both 48 kDa and 56 kDa products were observed, suggesting a reduced translation or stability of FS CHT. Doubling the amount of transfected transporter cDNAs produced a comparable increase in WT and FS CHT, maintaining their relative differences in protein level. Saturation analysis of [3H]-choline transport in FS-CHT-transfected cells demonstrated a significant reduction in transport activity relative to that in WT-CHT-transfected cells; this reduction was accounted for by a decrease in choline transport VMAX as opposed to a change in choline KM (Figure 2B). To determine whether the reduction in FS-CHT transport VMAX arises from a change in surface-CHT levels, we labeled cell-surface proteins with a membrane-impermeant biotinylating reagent and followed this with purification on Streptavidin beads, SDS-PAGE of total and surface extracts, and immunoblotting for the HA tag. As noted previously, two forms of WT and FS CHT were detected, and the mature, more substantially glycosylated form was enriched in cell-surface extracts (Figure 2C). Importantly, when the two transporter cDNAs were coexpressed (cDNA levels equal to 1× in Figure 2A), the combined levels of surface transporters of the two isoforms were reduced to ∼30% of that calculated from a simple addition of levels when the two cDNAs were transfected individually (Figure 2C; Student’s unpaired t test, p = 0.001, n = 5). These studies indicate that the FS CHT exerts a dominant-negative impact on total- and surface-protein levels, whether blotting for WT or FS CHT. Consistent with this finding, CHT activity in cells coexpressing WT and FS CHT constructs was found to be reduced significantly below that expected from a summation of transport activities from cells independently transfected with cDNA encoding either WT or FS CHT (Figure 2D). Notably, activity seen with dual transfection was even lower than observed when cells were transfected with FS-CHT-encoding cDNA, revealing that a more severe impact arises when cells produce CHTs with mismatched C termini.
Figure 2.
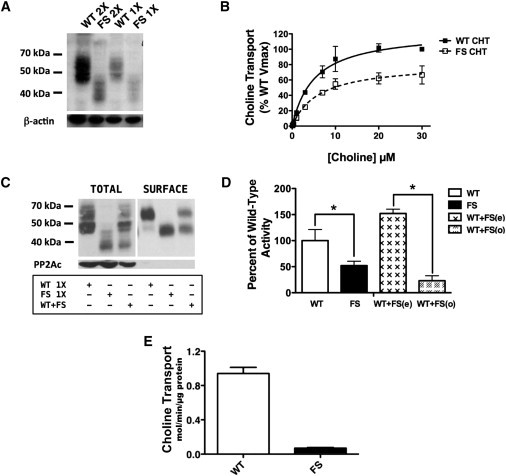
Immunoblotting and Cell-Surface Biotinylation of WT and FS CHT
(A) Levels of total WT and FS CHT after SDS-PAGE and immmunoblotting with HA antibody confirm FS-CHT truncation and reveal reduced expression of total FS CHT at both 2× (lanes 1 and 2) and 1× (lanes 3 and 4) protein. Stripping and blotting for β-actin reveal equal protein loaded across lanes. All experiments were conducted with the same total amount of transfected plasmid and were balanced with a pcDNA3 vector.
(B) Saturation analysis of HC-3-sensitive choline-transport activity in transfected HEK 293 cells demonstrates significantly reduced transport activity for FS CHT than for WT CHT. Transport deficits of FS-CHT-transfected wells are accounted for by a reduction in choline transport VMAX (33.4% reduction, p = 0.024, Student’s unpaired t test) as opposed to a change in choline KM (WT = 5.15 ± 1.12 μM, FS = 6.12 ± 1.72 μM, p = 0.66, Student’s unpaired t test). Specific choline uptake was determined by subtraction of the uptake obtained from transfected cells from that obtained from mock (vector) transfected cells acquired from assays conducted in parallel.
(C) Levels of total and surface WT and FS CHT are lower in cells transfected with both WT and FS CHT at 1× levels (as presented in A) than in cells separately transfected at 1× levels. All transfections were performed with identical amounts of total plasmid and were balanced by a pcDNA3 vector. Equivalent findings were obtained with cells transfected with the tagged WT and FS CHT constructs.
(D) Choline-transport activity is lower in FS CHT than in WT CHT (n = 5, ∗p = 0.035, Student’s unpaired t test). Observed transport activity of cells coexpressing WT and FS CHT (WT + FS(o)) falls significantly below that expected (WT + FS(e)) from a sum of the independent expression of these isoforms (n = 5,∗p = 0.005, Student’s unpaired t test).
(E) HC-3-sensitive choline-transport activity in whole-blood monocytes of dHMN-VII subjects (n = 4) and controls (n = 11) reveals a dramatic loss in dHMN-VII subjects (∗p = 0.72 × 10−6, Student’s one-tailed t test).
To investigate whether a similar reduction in CHT activity might be present in patient cells that natively express both mutant and WT SLC5A7 transcripts, we measured choline transport in whole-blood monocytes obtained from affected subjects and control individuals by using previously described methods.8 In these experiments, we readily detected HC-3-sensitive choline-transport activity in cells from control subjects (Figure 2E). In contrast, choline-transport activity in cells from individuals with the c.1497delG SLC5A7 variant was barely detectible, consistent with the minimal transport activity observed in cells transfected with cDNAs encoding both WT and FS CHT. Together, these findings provide strong evidence for a dominant-negative impact of the p.Lys499Asnfs∗13 alteration as opposed to effects expected from haploinsufficiency alone.
CHT is enriched in motor neurons and nerve terminals11 and is essential for NMJ signaling,13,14 providing a strong impetus for further evaluation of the structural and functional impact of the CHT variant. The NMJ is a specialized synapse designed to faithfully transmit electrical impulses from the presynaptic nerve terminal of the motor neuron to postsynaptic muscle cells via ACh. The genetically heterogeneous neuromuscular disorders known as the congenital myasthenic syndromes (CMSs) are well recognized to result from mutations in genes encoding presynaptic, postsynaptic, and synaptic NMJ proteins. Key characteristics of CMSs include fatiguing muscle weakness with a pathological decrement on repetitive muscle stimulation or abnormal jitter and blocking on single-fiber EMG. Onset of CMSs is typically congenital, and ophthalmoparesis, ptosis, and bulbar weakness are frequently seen. In contrast, onset of dHMN-VII is generally in the end of the first decade or in the middle of the second decade and occurs with a nonfatiguing progressive distal muscle wasting and weakness in association with vocal-cord paralysis due to involvement of the tenth cranial nerve (VCP; Table S4). The lack of a CMS phenotype in dHMN-VII subjects was unexpected and prompted us to repeat the electrophysiology studies in two affected subjects (V:19 [male, 56 years old] and VI:12 [male, 28 years old]) from the family (Figure 1). Repetitive stimulation showed no significant decrement, and the fact that single-fiber EMG showed an excess of jitter and blocking is suggestive of a disturbance of NMJ function (data not shown), but such changes could also be secondary to reinnervation. The changes were less marked in the younger of the two subjects (the disease in this subject progressed less). Approximately 5% of molecularly characterized CMS individuals are associated with defects in choline acetyltransferase (encoded by CHAT [MIM 118490]), the enzyme responsible for ACh synthesis, whereas the majority have defects in postsynaptic nicotinic ACh receptors (AChRs) rapsyn (encoded by RAPSN [MIM 601592]) or DOK7 (encoded by DOK7 [MIM 610285]).15–19
Although the phenotype associated with mutations in these genes differs significantly from that of dHMN-VII, it is noteworthy that vocal-cord paresis, frequently presenting as congenital stridor, is a feature of CMS particularly in association with DOK7 mutations and occasionally with mutations in CHAT.20,21 Possibly, this observation might be coincidental; however, the differences between dHMN-VII and CMS might relate to the unique contribution that CHT and the protein products of the CMS-associated genes make to cholinergic signaling in the context of the demands of different neuromuscular synapses. CHT is present (so far as is known) at all cholinergic synapses, but NMJs exhibit structural and functional variation across species, as well as between muscle-fiber types.22 Two forms of NMJs are known: (1) “en plaque,” where the muscle fiber has a single innervation and an NMJ diameter of 50 μm and (2) ”en grappe,” where the muscle fiber receives multiple synaptic contacts at NMJs of 10–16 μm in diameter. En grappe synapses innervate tonic fibers that are present on only a small number of mammalian muscle fibers and have been shown to release ACh by almost an order of magnitude longer than twitch terminals. En grappe NMJs are present on extraocular, stapedius, tensor tympani, laryngeal, and tongue muscle fibers. Evidence supports a role for CHT primarily in sustained, high-frequency ACh signaling23 given that reserve ACh stores are available for sustaining low-frequency signaling24 and because choline can be acquired through other mechanisms, such as the transporters that maintain choline phospholipid synthesis in all cells. Consequently, it is possible that the synapses affected in dHMN-VII are more dependent on CHT as a result of a requirement for sustained, high-frequency ACh release. Indeed, model-system studies with HC-3 have demonstrated a suppression of ACh release and cholinergic signaling only with sustained or higher-frequency stimulation.23 Alternatively, these synapses might be more sensitive to reduced CHT levels than are synapses with higher CHT levels. Structural and physiological differences exist among singly innervated fibers, where fast-twitch fibers display greater quantal release than do slow-twitch fibers, and thus the proportion of fibers within the affected motor systems might contribute sensitivity to the loss of CHT as well. Furthermore, unaffected neurons might also be protected by CTL1, a low-affinity CHT that has been reported to be present in motor neurons in rodents25 and to exhibit differential expression patterns across different NMJs.
Slc5a7-knockout mice display early postnatal paralysis, cyanosis, and lethality.13 Because a dominant-negative impact of the c.1497delG variant does not preclude the presence of a low amount of choline transport and because respiratory rates are much higher in mice than in humans, the lack of early-onset paralysis and respiratory dysfunction in our dHMN-VII-affected individuals might not be surprising. The deficits of our dHMN-VII subjects are more severe than they are in hemizygous Slc5a7 mice that are viable and do not exhibit an age-dependent motor neuropathy, although these animals do exhibit reduced CNS and autonomic levels of ACh and enhanced exercise-induced fatigue.26 Because NMJ morphology is altered in Slc5a7-knockout mice and shows similar alterations in Chat-knockout mice,27 the delayed nature of symptoms might also arise from differences in terminal plasticity that responds to reduced cholinergic signaling. This aspect might be illuminated by the assessment of NMJ morphology in the affected endplates of individuals with dHMN-VII and in studies of FS-Slc5a7 knockin transgenic models.
In summary, we provide genetic and functional evidence that dominantly acting SLC5A7 mutations underlie dHMN-VII, raising the possibility that CHT might have a role in idiopathic motor neuronopathies. Knowledge of a molecular lesion underlying dHMN-VII provides a genetic test for disease risk in affected families and might permit the development of rational pharmacotherapies. Furthermore, CHT or its regulation should be considered as a potential contributor to idiopathic motor neuronopathies.
Acknowledgments
The authors would like to thank the families described herein for participating in our study. The study was supported by the Medical Research Council and the Neurosciences Research Foundation, as well as by a National Institutes of Mental Health MERIT award to R.D.B. Special thanks also go to the family of J.M. for supporting this work.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Harding A.E. Hereditary spastic paraplegias. Semin. Neurol. 1993;13:333–336. doi: 10.1055/s-2008-1041143. [DOI] [PubMed] [Google Scholar]
- 2.Rossor A.M., Kalmar B., Greensmith L., Reilly M.M. The distal hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry. 2012;83:6–14. doi: 10.1136/jnnp-2011-300952. [DOI] [PubMed] [Google Scholar]
- 3.McEntagart M., Norton N., Williams H., Teare M.D., Dunstan M., Baker P., Houlden H., Reilly M., Wood N., Harper P.S. Localization of the gene for distal hereditary motor neuronopathy VII (dHMN-VII) to chromosome 2q14. Am. J. Hum. Genet. 2001;68:1270–1276. doi: 10.1086/320122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick K.J., McEntagart M., Alwan W., Reilly M., Crosby A.H. Refinement of the locus for distal hereditary motor neuronopathy VII (dHMN-VII) and exclusion of candidate genes. Genome. 2008;51:959–962. doi: 10.1139/G08-078. [DOI] [PubMed] [Google Scholar]
- 5.Pridmore C., Baraitser M., Brett E.M., Harding A.E. Distal spinal muscular atrophy with vocal cord paralysis. J. Med. Genet. 1992;29:197–199. doi: 10.1136/jmg.29.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Müller J.S., Mihaylova V., Abicht A., Lochmüller H. Congenital myasthenic syndromes: Spotlight on genetic defects of neuromuscular transmission. Expert Rev. Mol. Med. 2007;9:1–20. doi: 10.1017/S1462399407000427. [DOI] [PubMed] [Google Scholar]
- 7.Nakata K., Okuda T., Misawa H. Ultrastructural localization of high-affinity choline transporter in the rat neuromuscular junction: Enrichment on synaptic vesicles. Synapse. 2004;53:53–56. doi: 10.1002/syn.20029. [DOI] [PubMed] [Google Scholar]
- 8.Apparsundaram S., Ferguson S.M., George A.L., Jr., Blakely R.D. Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem. Biophys. Res. Commun. 2000;276:862–867. doi: 10.1006/bbrc.2000.3561. [DOI] [PubMed] [Google Scholar]
- 9.Bazalakova M.H., Blakely R.D. The high-affinity choline transporter: A critical protein for sustaining cholinergic signaling as revealed in studies of genetically altered mice. Handb Exp Pharmacol. 2006;(175):525–544. doi: 10.1007/3-540-29784-7_21. [DOI] [PubMed] [Google Scholar]
- 10.Ribeiro F.M., Black S.A., Cregan S.P., Prado V.F., Prado M.A., Rylett R.J., Ferguson S.S. Constitutive high-affinity choline transporter endocytosis is determined by a carboxyl-terminal tail dileucine motif. J. Neurochem. 2005;94:86–96. doi: 10.1111/j.1471-4159.2005.03171.x. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson S.M., Savchenko V., Apparsundaram S., Zwick M., Wright J., Heilman C.J., Yi H., Levey A.I., Blakely R.D. Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J. Neurosci. 2003;23:9697–9709. doi: 10.1523/JNEUROSCI.23-30-09697.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian Y., Galli A., Ramamoorthy S., Risso S., DeFelice L.J., Blakely R.D. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J. Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferguson S.M., Bazalakova M., Savchenko V., Tapia J.C., Wright J., Blakely R.D. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc. Natl. Acad. Sci. USA. 2004;101:8762–8767. doi: 10.1073/pnas.0401667101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnaswamy A., Cooper E. An activity-dependent retrograde signal induces the expression of the high-affinity choline transporter in cholinergic neurons. Neuron. 2009;61:272–286. doi: 10.1016/j.neuron.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Barišić N., Chaouch A., Müller J.S., Lochmüller H. Genetic heterogeneity and pathophysiological mechanisms in congenital myasthenic syndromes. Eur. J. Paediatr. Neurol. 2011;15:189–196. doi: 10.1016/j.ejpn.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Beeson D., Higuchi O., Palace J., Cossins J., Spearman H., Maxwell S., Newsom-Davis J., Burke G., Fawcett P., Motomura M. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 17.Ohno K., Quiram P.A., Milone M., Wang H.L., Harper M.C., Pruitt J.N., 2nd, Brengman J.M., Pao L., Fischbeck K.H., Crawford T.O. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor epsilon subunit gene: identification and functional characterization of six new mutations. Hum. Mol. Genet. 1997;6:753–766. doi: 10.1093/hmg/6.5.753. [DOI] [PubMed] [Google Scholar]
- 18.Ohno K., Engel A.G., Shen X.M., Selcen D., Brengman J., Harper C.M., Tsujino A., Milone M. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am. J. Hum. Genet. 2002;70:875–885. doi: 10.1086/339465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quiram P.A., Ohno K., Milone M., Patterson M.C., Pruitt N.J., Brengman J.M., Sine S.M., Engel A.G. Mutation causing congenital myasthenia reveals acetylcholine receptor beta/delta subunit interaction essential for assembly. J. Clin. Invest. 1999;104:1403–1410. doi: 10.1172/JCI8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinali M., Beeson D., Pitt M.C., Jungbluth H., Simonds A.K., Aloysius A., Cockerill H., Davis T., Palace J., Manzur A.Y. Congenital myasthenic syndromes in childhood: Diagnostic and management challenges. J. Neuroimmunol. 2008;201-202:6–12. doi: 10.1016/j.jneuroim.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 21.Jephson C.G., Mills N.A., Pitt M.C., Beeson D., Aloysius A., Muntoni F., Robb S.A., Bailey C.M. Congenital stridor with feeding difficulty as a presenting symptom of Dok7 congenital myasthenic syndrome. Int. J. Pediatr. Otorhinolaryngol. 2010;74:991–994. doi: 10.1016/j.ijporl.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 22.Hughes B.W., Kusner L.L., Kaminski H.J. Molecular architecture of the neuromuscular junction. Muscle Nerve. 2006;33:445–461. doi: 10.1002/mus.20440. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson S.M., Blakely R.D. The choline transporter resurfaces: New roles for synaptic vesicles? Mol. Interv. 2004;4:22–37. doi: 10.1124/mi.4.1.22. [DOI] [PubMed] [Google Scholar]
- 24.Rizzoli S.O., Betz W.J. Synaptic vesicle pools. Nat. Rev. Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- 25.O’Regan S., Traiffort E., Ruat M., Cha N., Compaore D., Meunier F.M. An electric lobe suppressor for a yeast choline transport mutation belongs to a new family of transporter-like proteins. Proc. Natl. Acad. Sci. USA. 2000;97:1835–1840. doi: 10.1073/pnas.030339697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bazalakova M.H., Wright J., Schneble E.J., McDonald M.P., Heilman C.J., Levey A.I., Blakely R.D. Deficits in acetylcholine homeostasis, receptors and behaviors in choline transporter heterozygous mice. Genes Brain Behav. 2007;6:411–424. doi: 10.1111/j.1601-183X.2006.00269.x. [DOI] [PubMed] [Google Scholar]
- 27.Brandon E.P., Lin W., D’Amour K.A., Pizzo D.P., Dominguez B., Sugiura Y., Thode S., Ko C.P., Thal L.J., Gage F.H., Lee K.F. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J. Neurosci. 2003;23:539–549. doi: 10.1523/JNEUROSCI.23-02-00539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.