

Background: The small heat shock proteins, αA-crystallin and αB-crystallin are considered to be two subunits of one single monolithic lens protein, α-crystallin.
Results: αA-Crystallin and αB-crystallin fractionate independent of each other and in two separate membrane compartments.
Conclusion: αA-Crystallin and αB-crystallin are two independent proteins in the lens.
Significance: These data provide functional insight into why αA-crystallin and αB-crystallin null mice have disparate phenotypes.
Keywords: Crystallin, Endoplasmic Reticulum (ER), Heat Shock Protein, Lens, Membrane, αA-Crystallin, αB-Crystallin, Lens, Rough ER, Smooth ER
Abstract
αA-Crystallin (αA) and αB-crystallin (αB), the two prominent members of the small heat shock family of proteins are considered to be two subunits of one multimeric protein, α-crystallin, within the ocular lens. Outside of the ocular lens, however, αA and αB are known to be two independent proteins, with mutually exclusive expression in many tissues. This dichotomous view is buoyed by the high expression of αA and αB in the lens and their co-fractionation from lens extracts as one multimeric entity, α-crystallin. To understand the biological function(s) of each of these two proteins, it is important to investigate the biological basis of this perceived dichotomy; in this report, we address the question whether αA and αB exist as independent proteins in the ocular lens. Discontinuous sucrose density gradient fractionation and immunoconfocal localization reveal that in early developing rat lens αA is a membrane-associated small heat shock protein similar to αB but with remarkable differences. Employing an established protocol, we demonstrate that αB predominantly sediments with rough endoplasmic reticulum, whereas αA fractionates with smooth membranes. These biochemical observations were corroborated with immunogold labeling and transmission electron microscopy. Importantly, in the rat heart also, which does not contain αA, αB fractionates with rough endoplasmic reticulum, suggesting that αA has no influence on the distribution of αB. These data demonstrate presence of αA and αB in two separate subcellular membrane compartments, pointing to their independent existence in the developing ocular lens.
Introduction
α-Crystallins are the most ubiquitous of all crystallins, the structural proteins of the ocular lens (1–4). Initially considered to be lens-specific, αB-crystallin (αB)2 was the first ubiquitous crystallin that was reported to be expressed outside of the lens (5); it was later joined by αA-crystallin (αA) (6, 7). αA and αB are two prominent members of the small heat shock family of proteins. They are expressed under various physiological and pathological conditions in various tissues and cells in culture (1, 2, 8). A notable feature of the extralenticular expression of αA and αB is their mutually exclusive presence in many tissues. For example, outside of the lens, highest expression of αB is seen in the heart, but there is no detectable αA in this tissue. Similarly, in the spleen where αA is expressed, there is no detectable αB (5, 7). These data strongly suggest that each of these two proteins exist and function independently of each other, yet based on an appreciable amount of physical and biochemical data, αA and αB are considered to be two subunits of one protein, α-crystallin, in the ocular lens. This conclusion is based primarily on two important observations: 1) the high expression of αA and αB in the lens and 2) demonstrated propensity of these two polypeptides for association and multimerization that leads to their chromatographic co-fractionation as a multimeric protein, α-crystallin. A large amount of work has gone into characterization of α-crystallin, and it is reported to contain 15–50 subunits of αA and αB in a 3:1 proportion with a molecular mass that ranges from 300,000 to 1,000,000 kDa (3, 4, 8).
αA and αB are encoded by two independent genes CRYAA located on chromosome 21 and CRYAB located on chromosome 11. Although they share significant similarity in the primary sequences of their proteins, their promoter sequences are different (2), suggesting independent regulation of their expression. In the rat, significant expression of αB is first seen in the developing heart (9). Indeed, during early development of the mouse lens, there are temporal differences in expression of these two genes; αA appears earlier than αB (10).
With the discovery of αB as an extralenticular protein (5), it became evident that these two polypeptides do not have to be part of a single protein (α-crystallin) and that they can exist as independent proteins in various non-lens tissues (7). Based on their differential insolubilization patterns with age (11) and their different physiological characteristics such as their phosphorylation patterns (12, 13), we have suggested that these proteins may function as independent proteins, even in the ocular lens (11). However, a clear demonstration of their independent existence in the ocular lens has thus far remained elusive, and they are still treated as two subunits of one protein, α-crystallin. Using biochemical fractionation and guided by the discovery that αB is a Golgi membrane-associated protein (14, 15), we demonstrate that αA and αB are associated with separate membranous compartments of the ocular lens cells, indicating that these two polypeptides have independent existence and, therefore, independent physiological functions in this tissue.
EXPERIMENTAL PROCEDURES
Animals and Tissues
Sprague-Dawley rats of various ages were purchased (Charles River Laboratories, Wilmington, MA) and acclimatized for 2 to 3 days with appropriate diet and water ad libitum according to the Department of Laboratory and Animal Medicine (UCLA) animal care and use protocol. Experiments were conducted according to the guidelines of Institutional Animal Research Committee (UCLA). The following lenses extracted from animals of different ages were used in this study: fetal day 18 (FD18), postnatal day 3 (P3), postnatal day 10 (P10), postnatal day 17 (P17), postnatal day 20 (P20), and post natal day 21 (P21).
Immunocytochemistry with Rat Lens Epithelia
P10 rat lens epithelial explants were cultured in minimal essential medium containing non-essential aminoacids, Earle's salts, 10% FBS, 1% l-glutamine, 1% penicillin, and streptomycin (Irvine Scientific, Santa Ana, CA) for 8 days, with medium changes every 48 h (14). The cells obtained from these cultures were seeded onto poly-l-lysine-coated (Sigma-Aldrich) microscope coverslips (Fisher Scientific). The cells were fixed with ice-cold methanol for 6 min at −20 °C and processed for immunocytochemistry and confocal microscopy with anti-αA (16) and anti-αB antibodies (15). GM130 (Golgi matrix protein marker) was localized with anti-mouse GM130: FITC (BD Transduction Laboratories) used at 1:50 dilution followed by nuclear staining with DAPI (4,6-diamidino-2-phenylindole) (Molecular Probes, Carlsbad, CA). Confocal images were acquired as z-stacks on a Leica TCS-SP multiphoton and confocal microscope using a 100× objective. The images were processed with Adobe Photoshop (version 6.0).
Colocalization Measurements in Confocal Images
For assessing the extent of co-localization between GM130 and αA and between GM130 and αB, the tiff images (0.5-μm-thin z sections) acquired with two photon confocal microscope (Leica TCS SP), were processed in Adobe Photoshop for presentation. For colocalization measurements, raw unprocessed images were opened in NIH ImageJ software (version 1.37c). The images were converted to an 8-bit grayscale, and the background was subtracted from the region of interest using the region of interest plug-ins. The “Colocalization Threshold” plug-in algorithm determines the threshold automatically and reduces background for each channel to eliminate the bias (17). This algorithm generates two coefficients (Pearson and Manders) per dual-channel image to compute the degree of colocalization (see supplemental Fig. S1). Differences between the colocalization coefficients were evaluated by Student's t test (**, p < 0.01).
Immunohistochemistry of the Rat Eye (Lens)
Postnatal day 10 (P10) rat eyes were dissected out and quickly immersed in PBS for 1 h followed by fixation in 4% paraformaldehyde overnight at 4 °C. The eyes were dehydrated, embedded in paraffin, sectioned (5 μm thin sections) on Leica RM2135 (Leica Microsystems) and processed for immunolabeling (18) with purified anti-αA (1:200 dilution) and anti-αB (1:200 dilution) using Immunopure Ultra-Sensitive ABC peroxidase staining kit (Pierce) and diaminobenzidine for detection (Ted Pella, Inc., Redding, CA). Glycerol gelatin (Sigma) was used for mounting, and images were acquired with a Zeiss confocal microscope using 4× and 40× objectives.
Fractionation of Golgi-enriched Membranes
Discontinuous sucrose gradients in 2.2-ml S55S tubes were used as described previously (14, 15). Twenty fractions (100-μl each) were collected from the bottom of the gradient by puncturing the tube with a 22-gauge needle. For this analysis, post-nuclear homogenates (1000 × g) were made either from whole lenses or from two anatomically dissected areas, lens epithelium + superficial cortex (LE+SC), and fiber mass (FM) (14). In some experiments, P10 lenses were exposed to brefeldin A (BFA) (1 μg/ml) (Fluka, Switzerland) in minimum essential medium at 37 °C for 90 min before homogenization and gradient analyses. One μl/lane of each fraction was electrophoresed on SDS-PAGE and immunoblotted with anti-αA. For a control run, purified human recombinant αA (50 μg) was analyzed similarly using 8 μl per lane of each gradient fraction (see Fig. 4).
FIGURE 4.
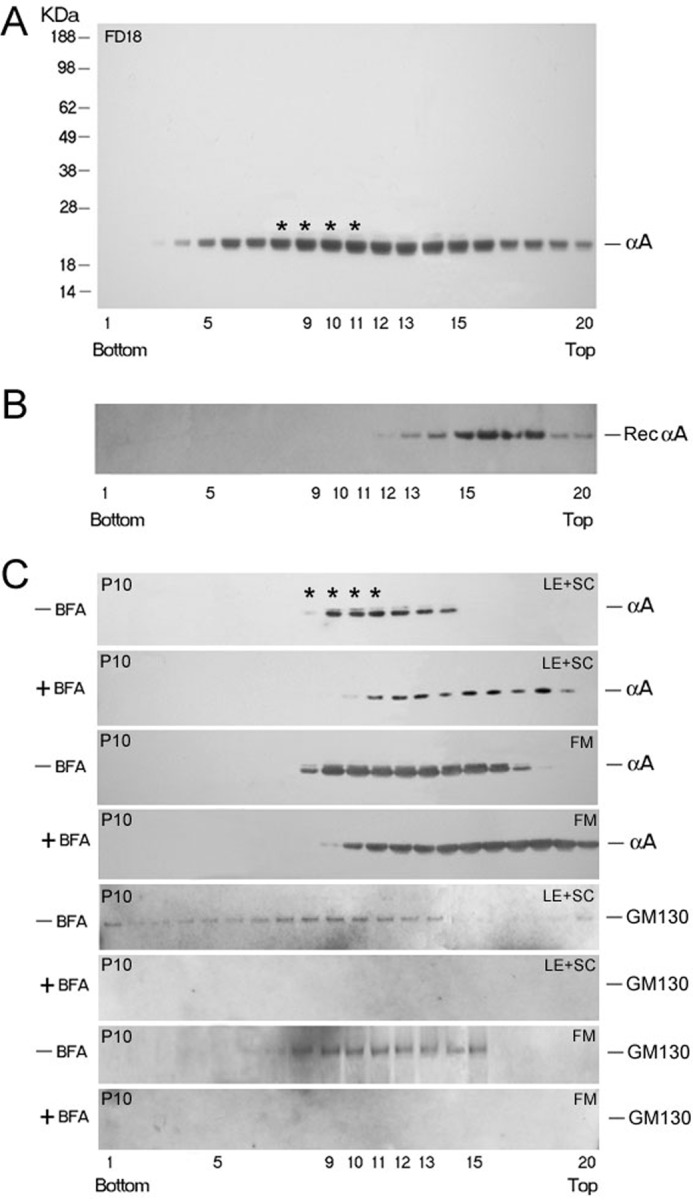
Discontinuous sucrose density gradient fractionation for analyses of Golgi-enriched membranes. A, distribution of αA (as detected by immunoblotting) in the gradient run with post-nuclear homogenate of whole FD18 rat lens (5.36 mg total protein). αA is distributed broadly, from the bottom (fraction 3) all through to the top of the gradient. Asterisks indicate the position of the Golgi enriched membrane fraction as determined by Golgi intrinsic membrane protein GM130 location (14, 15). The presence of αA in fractions 3–5, close to bottom (1.3 m sucrose) and in fractions 5–12 suggest that αA is associated with heterogeneous membrane components. B, purified recombinant αA (50 μg) was run in a separate gradient under similar conditions (Rec αA panel) as a control. C, sucrose gradient fractionation of post nuclear homogenates made from dissected lens epithelium + superficial cortex (LE+SC) and FM of P10 rat lenses. Lenses were incubated with +BFA for 90 min before fractionation. Note that αA associated with the Golgi in fractions 8–11 is susceptible to BFA treatment. The pattern shifts toward the top of the gradient (C, +BFA panels) because of the disorganization of the Golgi. Note that we could not detect any GM130 reactivity in the lens epithelium + superficial cortex (LE+SC) and FM in + BFA panels (bottom panel and third panel from bottom).
Fractionation of Homogenates for Enrichment of Various Membrane Compartments
An established procedure used previously for fractionation of free and bound polysomes was employed (19). The two isolated fractions have been previously exhaustively characterized by EM (19, 20). Postmitochondrial homogenates (7000 × g supernatants) were fractionated on discontinuous sucrose gradients as detailed below. A picture of the gradient tube showing separation of bound polysomes (BP) or rough endoplasmic reticulum (RER) and polysome-free smooth membranes (SM), and free polysomes from P10 rat heart is shown in Fig. 5A.
FIGURE 5.

Fractionation of smooth membranes and rough ER in FD18 rat lens and heart. A, picture (shown horizontally) of the fractionated gradient obtained with post mitochondrial supernatant of FD18 heart. The arrows indicate the positions of the bound polysomes (BP, also known as rough ER), the SM and free polysomes (FP). B, immunoblots of the gradient shown in A with anti-αA, anti-αB, anti-Ribophorin-1, anti-GM130, and anti-Golgin 58. Equal volumes (2.5 μl) from each fraction were used for immunoblotting. Note the absence of αA reactivity because there is no αA expressed in the heart (top panel). αB is seen in fractions 5–9 as is Ribophorin-1, which seems to associate with two discrete fractions of the bound polysomes (rough ER). It is also detected in the top half of the gradient in fractions 15 and 17). C, fractionation of FD18 lens post-mitochondrial supernatants. Note that αA (fractions 9–15) fractionates away from αB (fractions 6–8). Caveolin-1 (Cav-1) fractionates with SM in the same location where αA is seen. Note that Ribophorin-1 is mostly seen here with the RER. Transferrin, one more BP/RER marker, could not be detected in this gradient, although it is seen in P10 lens gradients (see Fig. 6). EXT, immune reactivities in aliquots of total cell extracts before fractionation. D, the distribution of total protein in FD18 lens and FD18 heart gradients. Note that the immune reactions seen for αA and αB do not correspond with this distribution pattern.
FD18 and P10 rat tissues (lens and heart) were homogenized in 0.35 m sucrose buffer containing 50 mm Tris-HCl, pH 7.6, 25 mm KC1, 10 mm MgCl2, and protease inhibitor mixture (Roche Diagnostics). The homogenate was centrifuged at 7000 × g for 10 min at 4 °C (Beckman Coulter Microfuge 22R) to pellet cell debris, nuclei, and mitochondria. 1.00 ml of post-mitochondrial supernatant was layered on top of 1.00 ml discontinuous sucrose gradient made up of 500 μl of 2.0 m sucrose (bottom) and 500 μl of 1.5 m sucrose (on top) in a 2.2-ml S55S centrifuge tube. The gradient was centrifuged at 70,000 × g for 16 h (Sorvall Discovery M150 S.E., Thermo Fisher Scientific, Inc.). Twenty (∼ 95-μl each) or 35 (∼50-μl each) fractions were collected from the bottom of the gradient as above for immunoblotting. The free polyribosomes pellet (19) at the bottom of the tube was washed free of sucrose and resuspended in homogenization buffer without sucrose (45 μl) before SDS-PAGE. The following total post-mitochondrial supernatant protein concentrations were analyzed: FD18 lens, 56 mg; FD18 heart, 65 mg; P10 lens, 720 mg; P10 heart, 300 mg.
Analyses of Multiple Gradient Fractions on a Single Immunoblot
With the prior knowledge that our antibodies react specifically with αB or αA and that no other bands are seen on these immunoblots (for example, see Fig. 4, top panel), we electrophoresed all 35 fractions on a single 15-lane SDS-PAGE gel (4% to 12% Bis-Tris NuPAGE gradient gels, Invitrogen) by multiple loadings, per lane, as follows. First, fractions 1–12 (load L1, see Fig. 7) were loaded, and electrophoresis was conducted for 12 min. The electrophoresis was stopped, the wells were rinsed with the running buffer, and the next 12 fractions 13–24 (load L2, see Fig. 7) were loaded, and electrophoresis was continued for another 9 min. The electrophoresis was again stopped; wells were rinsed and loaded for the third time with samples 25–35 (load L3, see Fig. 7), and electrophoresis was continued for an additional 40 min. Thus, all 35 fractions from a gradient could be analyzed on a single gel/immunoblot. The gel was processed for immunoblotting as detailed (14).
FIGURE 7.

αA and αB fractionate with smooth membranes and rough endoplasmic reticulum, respectively. Sucrose gradients similar to that shown in Fig. 6 were run with P10 heart and P10 lens post mitochondrial homogenates. Smaller volume fractions (35 fractions as opposed to 20 in Figs. 5 and 6) were collected from the bottom of the gradient. All 35 fractions were electrophoresed on a single SDS-PAGE gel and immunoblotted (see “Experimental Procedures”). We did this by multiple loadings into each well (thus each lane was loaded three times, 8–12 min apart); first, fractions 1–12, then 13–24, and finally 25–35 on the same gel. Four such gels were run (two from the heart and two from the lens) and immunoblotted. The numbers in each panel (1–35) represent the gradient fractions on each gel. These numbers are underneath the immunoreaction in each lane. Note that no reactions are seen in fractions 22–35 in all immunoblots. A, P10 lens immunoblots are shown (P10 lens αA and P10 lens αB). B, P10 heart immunoblots are shown (P10 heart αA and P10 heart αB). C, plots of the densitometer scans (arbitrary units) of the immunoblots obtained with anti-αA and anti-αB in the lens and in the heart shown in B. Dotted and dashed gray lines show total protein distribution in the heart and lens gradients. αB in the lens (panel P10 lens αB) and the heart (panel P10 heart αB) fractionates with rough ER. There is no αA in the heart (panel P10 heart αA), and therefore, there are no bands in this immunoblot. In the lens, αA fractionates with smooth membranes. X, blank lane; FP, free polysome pellet. L1, L2, and L3 refer to three loads (L1–L3, see “Experimental Procedures”).
Polyclonal antibodies against αA and αB were used at 1:5000 dilutions. Anti-αA is a polyclonal antibody raised against the N terminus 12 residues (2–13 of the rat αA) (16). Anti-αB is a polyclonal antibody raised against the C terminus, 13 residues (163–175) of the rat αB (14, 15). Anti-Transferrin, anti-Hsp70, anti-Ribophorin-1, anti-Ribophorin-1, and anti-Caveolin-1 (Santa Cruz Biotechnology, Santa Cruz, CA) were used at 1:2000 dilutions. Secondary antibody, goat anti-rabbit horseradish peroxidase (HRP) was used at 1:250,000 for αA or αB immunoblots and at 1:20,000 dilutions for all antibodies obtained from Santa Cruz Biotechnology. Anti-Golgin 58 (Abcam) and anti-GM130 (BD Transduction Laboratories) were used at 1:1000 dilutions.
Immunogold Labeling and Electron Microscopy
P10 rat lenses were fixed in phosphate-buffered saline (PBS, 0.1×, pH 7.4) containing 4% paraformaldehyde and 0.1% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) for 2 h at room temperature and then overnight at 4 °C. The fixed lenses were washed with filtered distilled water. The lenses were dehydrated by ascending alcohol gradient series followed by infiltration with ethanol : London Resin (LR) white series (2:1, 1:1, 1:2, and 1:4 ratio, 2-h each). After this, the samples were incubated with LR white resin for 2 h and then for 48 h with a fresh change of LR white resin on a rotating platform. The lenses were removed and transferred to gelatin capsules containing fresh LR white and allowed to polymerize for 24 h at 50–55 °C. Ultra-thin (60 nm) LR white lens sections were collected on nickel grids and preincubated with a drop of Tris-buffered saline containing 0.001% Triton X-100 containing 50 mm glycine and 0.1% NaBH4 for 20 min. The grids were rinsed with three changes of Tris-buffered saline containing 0.001% Triton X-100 and blocked with 1:20 dilution of normal goat serum (Pierce) in Tris-buffered saline containing 0.001% Triton X-100 for 30 min. The blocked grids were incubated with two different antibodies in one of the following combinations (the antibody incubations were done consecutively, each incubation period was for 24 h): 1) anti-αA and anti-αB, 2) anti-αA and anti-Ribophorin-1, and 3) anti-αB and anti-Ribophorin-1, with each at 1:200 dilutions. The grids were washed with Tris-buffered saline containing 0.001% Triton X-100 thoroughly and incubated with secondary antibodies tagged to either 12-nm or 18-nm gold particle (1:300 dilution) following a standard protocol (21).
RESULTS
Rat lens epithelial cells were cultured and used for immunofluorescence studies with anti-αA (red) and anti-GM130 (Golgi specific-membrane protein) (green) (Fig. 1). The GM130 and a fraction of αA co-localize in the perinuclear Golgi (Fig. 1, bottom panels, Merge, yellow); however, a lot of protein is in the cytoplasm (Fig. 1, compare red and yellow, bottom left panel). For the sake of comparison with αB, we repeated the previously published (14) localization with anti-αB. This is shown in supplemental Fig. S1. We used ImageJ software to quantify colocalization of GM130 with αA and GM130 with αB. The Pearson and Manders coefficients thus computed (see “Experimental Procedures”) indicate that there is better colocalization between αB and GM130 (supplemental Fig. S1) than between αA and GM130 (Fig. 1).
FIGURE 1.

Confocal images of αA localization in primary cultures of rat lens epithelial explants. The P10 rat lens epithelial explants in culture invariably contain nascent (differentiating) fiber cells (14). Perinuclear colocalization of αA (anti-αA, red, top panel) and GM130 (anti-mouse GM130; FITC, green, middle panel) is observed in lens epithelial cells (left column) as well as in the differentiating fiber cell (right column). The αA label (red) is predominantly outside of the Golgi (compare the colocalized yellow granules with the red stain in the bottom panel). Note the granular appearance of the colocalized proteins (bottom panel, yellow, Merge + DAPI) and as yet unrecognized presence of αA (red streaks), prominent in the nucleus in the fiber cell (right bottom panel). Nuclei are stained with DAPI (blue). Scale bar, 20 μm.
Lens epithelial explant cultures contain elongated cells (14), which may be differentiating lens fiber cells as indicated by the oval shape of the DAPI-stained nucleus and the perinuclear Golgi staining (with anti-GM130, Fig. 1, fiber cell, right panels). In these cells, the Golgi is typically lining the nucleus as indicated by GM130 immunofluorescence. The co-localization of αA (red) with GM130 (green) reveals a peculiar granular appearance in the perinuclear Golgi (Fig. 1, Merge + DAPI, bottom, right panel). We also note nuclear staining with anti-αA (Fig. 1, fiber cell panels).
We next wanted to compare immunolocalization of αA and αB in the native ocular lens. Before embarking on these analyses, we examined the temporal expression of αA and αB by immunoblotting (Fig. 2A). As previously reported (10, 22), the expression of αA is detected earlier than αB in the developing lens (Fig. 2A). These data also ascertain the specificity and lack of cross-reactivity of anti-αA and anti-αB used in this investigation. Fig. 2, B and C, shows immunolocalization of αA and αB in the native P10 lens. It is interesting to note that although anti-αA staining in the epithelium seems apical, it is discontinuous. On the other hand, with anti-αB, the staining in the central epithelium is robust (Fig. 2C) and clearly apical as established previously (14). Immunolocalization with anti-αA in the native lens was repeated using immunofluorescence (Fig. 3). Here, the central epithelium shows cytoplasmic presence of αA, without a clear polar pattern, whereas the proliferative zone shows enhanced labeling on the apical face of this epithelium (Fig. 3, middle right panel). In the equatorial region, the lens fiber mass shows streaks of αA stained Golgi along the elongated nuclei (Fig. 3, bottom panel). Note that there is very little immunoreactivity seen in the differentiating region, but as soon as differentiation sets in, we see αA staining streaks along the elongated nuclei. This pattern is similar to that seen with αB (14).
FIGURE 2.

Developmental expression and immunohistochemistry of αA and αB in the native ocular lens. A, immunoblot showing temporal expression pattern of αA and αB during lens development in the rat. Total protein extracts (0.2 μg/lane) from fetal day 18 (FD18) and postnatal days (P3, P10, P17, P21) were analyzed on two immunoblots. αA is expressed early in the FD18 rat lens, when there is no detectable αB. Human glioblastoma cell U373 MG total cell extract (20 μg), which only expresses αB, is shown in the last lane. Protein standards (kDa) are shown on the left. B and C, immunohistochemistry of αA and αB localization, respectively. Immunoperoxidase-diaminobenzidine-stained 4× image of the whole ocular lens is shown in the bottom panels and the central epithelium of this image is magnified (40×) and shown in the upper panels. Note that αA is apical in its location, which suggest its association with the apical Golgi, but there are discontinuities in its staining. The data shown in C confirm previously reported (14) colocalization of αB in the apical Golgi. Note definitive αB staining (C) in the apical epithelium in comparison with anti-αA staining in B (open arrowheads).
FIGURE 3.

Immunofluorescence of the native P10 lens with anti-αA. Different regions of the lens are shown. Central epithelium (CE) is shown in the top panel. Immunofluorescence (red) is seen in the cytoplasm of the lens epithelium without specific definition of immunofluorescence in the apical regions of the epithelium. The difference between immunoperoxidase staining (Fig. 2B) and immunofluorescence shown here may be because of the differential sensitivity of the two techniques, suggesting that there is αA in these cells that is not associated with the apical Golgi. In the proliferative zone (PZ, middle panel); however, the label is predominantly apical (open arrowheads). In the equatorial region (ER, bottom panel), streaks of αA are seen along the elongated nuclei (perinuclear location, thin arrows) in the differentiated fiber cells. The left column shows respective pre-immune controls. Nuclei are stained with DAPI (blue). Scale bar = 100 μm.
We have shown previously that αB is a Golgi membrane-associated protein in the developing lens (14). We, therefore, repeated analyses of lens post-nuclear homogenates on discontinuous sucrose density gradients (15) for fractionation of Golgi enriched membranes, as was done previously for αB (14). Postnuclear homogenates were made from whole FD18 and two anatomical domains of the P10 lenses, namely, lens epithelium plus superficial cortex, and the FM. The FD18 gradient shows that the protein is distributed all over, suggesting general association of αA with the membrane (Fig. 4A, top panel); this includes association with the Golgi membrane fraction(s) (asterisks, Fig. 4, FD18).
Analysis of the P10 lens postnuclear homogenates (Fig. 4C) show that there is no noticeable difference in the patterns obtained with the lens epithelium + superficial cortex (LE+SC) compared with FM. Based on the location of the Golgi marker GM130, which is seen mostly in the fractions 8–11 on this gradient (14, 15), and as indicated by the GM130 immunblots, part of αA on this gradient represents association with the Golgi membranes (indicated by asterisks, Fig. 4C, top panel). This is further ascertained by the use of BFA, which disorganizes the Golgi resulting in the disappearance of Golgi-associated protein from these fractions (for example, Fig. 4C, second panel from the top). The extent of loss of protein from these fractions upon BFA treatment identifies the Golgi-associated fractions qualitatively (compare +BFA versus −BFA panels in Fig. 4C). In LE + SC, 17.9% of the label is lost from Golgi fractions (8–11, Fig. 4C, top two panels). In the FM, this percentage goes higher up to 30.33%. However, in comparison with similar analyses done previously with αB (15), BFA has a much lesser impact on the pattern of αA distribution in this gradient (Fig. 4C, compare −BFA, FM to +BFA, FM), suggesting that more of this protein (αA) may be part of the non-Golgi membranes. This observation must also be considered in light of the fact that there is three times more αA than αB in the ocular lens. Based on these data, we conclude that although αA is associated with the Golgi, a large part of this protein is significantly associated with non-Golgi membranes. Additionally, numerous other side effects of BFA (23) treatment may have an impact on these interpretations, and it is possible that an appreciable amount of the protein, which these data suggest is associated with Golgi, may in fact be part of non-Golgi membrane compartments. We therefore investigated the possible existence of these two proteins in non-Golgi membrane compartments. We used a previously well characterized procedure involving discontinuous sucrose density gradient fractionation of membrane-bound polyribosomes or RER and SM (see “Experimental Procedures”) (19).
We first analyzed FD18 lens and FD18 heart. As expected, no GM130 or Golgin 58 (Golgi membrane markers) were detected, indicating absence of Golgi membranes in this gradient (Fig. 5, B and C, bottom two panels). In the extracts made from FD18 heart, which do not contain αA, αB fractionates with the BP or RER fraction (Fig. 5B). The RER is characterized by the presence of Ribophorin-1 (24, 25) in these fractions. αB is seen in fractions 5–9, overlapping with fractions 6 and 9 that contain Ribophorin-1 (Fig. 5B, FD18 heart). Although Ribophorin-1 is also seen in the top half of the gradient in fractions 15 and 17, αB is not found in these fractions. In the FD18 lens (Fig. 5C), αB is seen in fractions 6–8, which overlaps with fractions 5 and 7, which contain Ribophorin-1. These data suggest that αB fractionates with RER membranes as identified by the marker Ribophorin-1, it however does not explain why Ribophorin-1 is seen discontinuously only in specific fractions.
Analyses of FD18 lens extracts, which contain both αA as well as αB, show that these two proteins fractionate independently in two separate membrane fractions; αB fractionates with the BP (rough ER) membranes (with Ribophorin-1) (Fig. 5, B and C), and αA fractionates with the SM with Caveolin-1 (Fig. 5C) (26). We could not detect Transferrin, an RER-associated protein (27) in FD18 lens gradients (probably because of its low expression at this stage) but found it in RER fractions of the P10 lens gradients (Fig. 6), confirming the data obtained with the fetal lens (Fig. 5). Note that in this gradient (Fig. 6), αA fractionates with Hsp70 and Caveolin-1, whereas Flotillin-1, Ribophorin-1, and Transferrin fractionate with αB with RER fraction. We used Hsp70 as an additional marker for smooth membranes based on a recent report that suggests its role in protein trafficking and quality control in the ER (28), as was previously suggested for αA (29).
FIGURE 6.
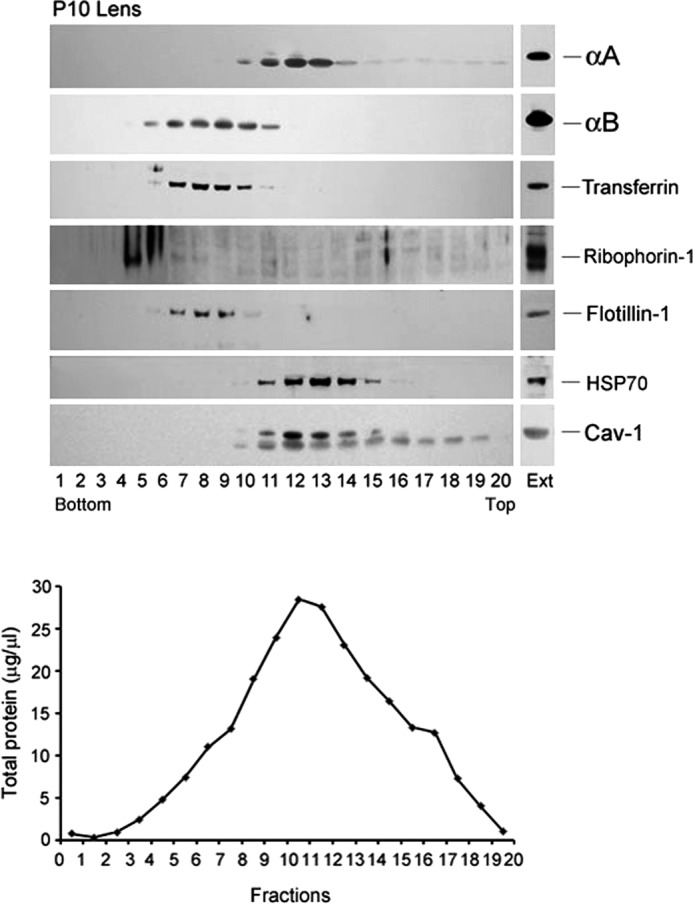
Fractionation of smooth membranes and rough ER from P10 rat lens. A similar gradient as in Fig. 5 was run. αA (top panel) fractionates with smooth membrane fractions 10–15 in which Caveolin-1 and HSP70 are detected (bottom two panels). αB is detected in fractions 5–11 (second panel) distinct from αA, in the rough ER, which is characterized by the presence of the marker Transferrin (third panel), Ribophorin-1 (in heavier polysomes, fourth panel), and Flotillin-1 (fifth panel). There is an overlap of αA and αB patterns in fractions 10 and 11. A light reaction for both αA as well as Caveolin-1 (Cav-1) is seen in the top fractions (15 onwards) in the gradient possibly because of the presence of high concentrations of αA in the ocular lens. EXT, immune reactivities in aliquots of total cell extracts before fractionation. The total protein in each fraction (μg/μl) is plotted in the lower panel. Note that there is no strict correspondence between immune reactions (particularly with αB) and the protein concentration profile.
There is an overlap in the presence of αA and αB between the two membrane fractions of the gradient (Fig. 6). An examination of the protein concentration data (Fig. 6, bottom panel) does not suggest close correspondence between the total protein distribution and αA and αB immunoreactivity. In the gradients shown in Figs. 5 and 6, the individual fraction volume was ∼100 μl (20 fractions in all). To avoid an overlap between the two membrane fractions, we reran the gradients with P10 lens and P10 heart extracts, collecting smaller volume fractions (35 in all per gradient) (Fig. 7, A and B). All fractions were electrophoresed on a single gel, allowing us an analysis of all of the 35 fractions on one immunoblot. This was made possible by multiple loadings into each lane of the gel (see “Experimental Procedures”). Fig. 7, A and B, shows the immunoblots, and Fig. 7C shows the density scans of these immunoblots. As indicated above, heart contains αB and no αA, whereas the lens contains both αA as well as αB. The data in Fig. 7, A and B, establish that αA and αB fractionate with two independent membrane domains.
We further examined the native lens with transmission electron microscopy in an effort to follow localization of αA, αB, and an RER marker such as Ribophorin-1 using gold immunocytochemistry (Fig. 8 and supplemental Fig. S2, A–D). Fig. 8 shows that Ribophorin-1 and αA do not label the same membrane domains, whereas Ribophorin-1 and αB label the RER membranes (with associated polyribosomes) in the lens fiber cells. Interestingly, αA and αB labeling reveals patterns similar to Ribophorin-1 and αA, suggesting that αA and αB associate with separate membranous compartments and hence, confirming the data obtained by gradient fractionation of the two membrane compartments (Figs. 5–7).
FIGURE 8.
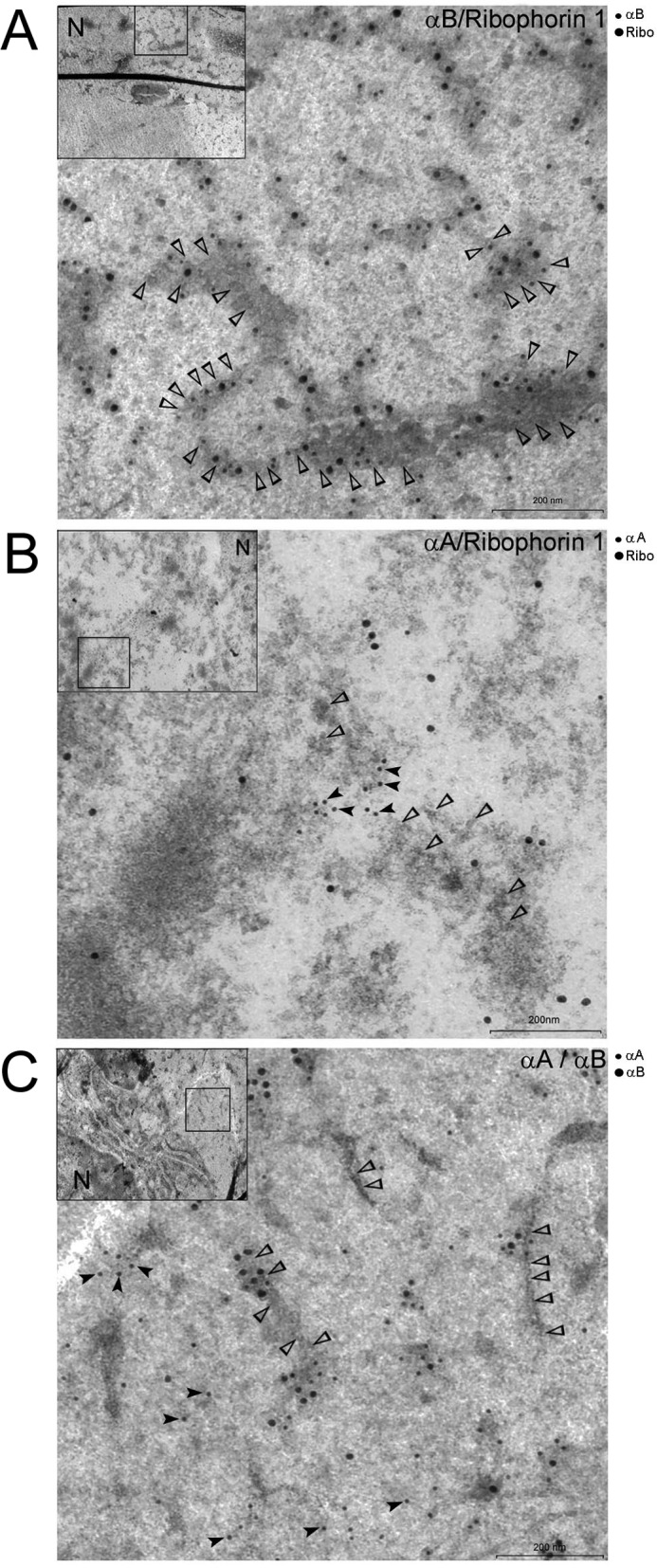
Localization of αB and Ribophorin-1 in native lens fiber cells. Shown above are three representative transmission electron microscopy images of the data obtained with immunogold labeling of αA, αB, and Ribophorin-1 (Ribo) in rat lens fiber cell ultrathin sections. Two antibodies were used for generating each picture as indicated in the top righthand corner of each micrograph. A, localization of αB (12-nm gold) and Ribophorin-1 (18-nm gold). Note that both the proteins are associated with membrane decorated with ribosomes (open arrowheads). B, micrograph showing labeling with anti-αA (12-nm gold, black arrowheads) and anti-Ribophorin-1 (18 nm gold). Note that there are very few 18-nm particles (Ribophorin-1) in the vicinity of 12-nm particles (αA); open arrowheads point to membrane-bound ribosomes. The two proteins do not seem to localize within the same membrane domains. C, micrograph showing labeling with anti-αA (12-nm gold) and anti-αB (18 nm). Open arrowheads point to membrane-bound ribosomes. This micrograph is similar to localization of αA and Ribophorin-1 shown in B. The two proteins do not share the same membrane domains. Low magnification images (insets) are shown in the top left corner of each micrograph. The square box in the inset shows the area magnified. Similar electron micrographs acquired from different regions of fiber cells are presented in supplemental Fig. S2, A–D. Scale bar, 200 nm. N, nucleus. Preimmune serum controls are presented in supplemental Fig. S2D.
DISCUSSION
This investigation addresses the dichotomy in the perceived perceptions of the status of two small heat shock proteins αA and αB, inside and outside of the ocular lens. The question, “ … are αA and αB two different proteins or two subunits of the same protein?” (2, 11), has not been addressed experimentally. In the ocular lens, αA and αB are considered as two subunits of one single protein, α-crystallin. α-Crystallin makes ∼30% of the total protein in the vertebrate lens, but concentrations of each of its subunits, αA and αB, outside of the lens are comparatively much smaller and catalytic. These two polypeptides share 58% sequence identity and a common conserved α-crystallin domain, characteristic of the small heat shock family of proteins (30).
We have previously examined the status of αB in various non-lens tissues and cells and found that this protein resides in the Golgi-enriched membrane fractions on a discontinuous sucrose density gradient (15, 31). It is part of the detergent resistant microdomains and is secreted out of the human retinal pigment epithelial cells in exosomes (31). Examination of the status of αB in the developing ocular lens in the rat showed that here also αB is associated with the Golgi membranes (14). These findings lead to the question whether αA, the other known subunit of α-crystallin, is also associated with the Golgi membranes. Historically, a fraction of α-crystallin (both αA and αB) has been reported to be membrane-associated in the normal adult transparent lens. These observations about the association of α-crystallins with the membranes were first made about four decades ago (32–35). We therefore examined αA status first in the lens epithelial explants in culture and then in the early developing lens.
Immunocytochemical localization of αA in the epithelial cell explants shows that although a large part of this protein is not seen in the Golgi, this polypeptide shows distinctly granular localization in the perinuclear Golgi in addition to its unmistakable presence in the nucleus (Fig. 1, fiber cell, right panel). The functional significance of the granular organization and its possible nuclear role remain to be understood.
Although the pattern of immunolocalization of αA in fiber cells looks very similar to the pattern obtained previously with αB (14), αA does not co-localize with the apical Golgi in the native lens epithelium as strictly as does αB (compare Fig. 2, B and C). This is further supported by colocalization analyses (supplemental Fig. S1). It is important to note here that lens epithelial cells are known to contain more αB than αA thus αA presence outside of the Golgi in these cells cannot be attributed to its higher concentration than αB.
The observation that a much smaller proportion of αA than αB is associated with the Golgi is supported by sucrose density gradient analyses of BFA-treated lenses. BFA disorganizes Golgi (36) and therefore, allows a rough assessment of the amount of protein that is associated with the Golgi membrane fractions when compared with untreated lenses (Fig. 4). Based on this analysis, a small but noticeable amount of this protein is found in the Golgi fractions (Fig. 4, fractions 8–10) as indicated by its disappearance from these fractions in +BFA gradients. However, BFA does have multiple effects on membranes that are not restricted to Golgi disorganization (23), which may further indicate that a significant amount of αA is not associated with the Golgi membranes (Fig. 4A).
As indicated above, the membrane association of αA has been known for the last four decades (35), yet it has remained unexplained because this protein has always been considered to be a soluble protein. In these early studies, only a fraction of αA was seen associated with the membranes. We believe this is because most of these studies were conducted with adult and/or mature lens preparations in which only a small population of metabolically active young fiber cells with intact αA, membrane association were represented (35). The current studies were done with young (fetal and postnatal D10) lenses that are predominantly composed of young differentiating fiber cells with intact αA-associated membranes.
In this investigation, we have presented data demonstrating that αA and αB proteins reside in separate membranous compartments of the ocular lens cells. Because lens contains high concentrations of proteins, it is generally difficult to process this tissue for anatomical immunolocalizations. Also, biochemical analyses are hampered by these high concentrations, which camouflage differences. We therefore had to use simpler well characterized techniques used previously for fractionation of bound and free polysomes (19, 20). The data presented in Figs. 5–7 suggest that αB sediments with RER, whereas αA stays with the SM fraction. It must however be noted that fractionations shown in the Figs. 5 and 6 did not show complete overlap in the location of αB and Ribophorin-1 (used as a marker for rough ER). This raises the question whether αB is associated with a specific subcellular membrane domain (37) of the RER. The answer to this question therefore remains ambiguous. At the same time, we know that αB is present in detergent-resistant membrane domains (31).
It must be noted that the experimental data presented in Figs. 5–7 were obtained using the whole FD18 and whole P10 lenses and therefore contain contributions both from the lens epithelium as well as the lens fiber mass. It is unlikely that αA and αB separation could have arisen artificially by homogenization and/or various manipulations as attested by the analyses of the post nuclear homogenate made from the fetal (FD18) and the P10 heart where there is no αA. In the heart extracts (Fig. 5, FD18 heart, and Fig. 7, P10 heart), αB fractionates to the same location as does αB in the FD18 and P10 lens extracts (rough ER, Figs. 5 and 6). These data, therefore, indicate that presence of αA in the lens homogenates has no influence on the fractionation of αB in this gradient (interestingly, there is 3× more αA than αB in the lens fiber cells) (2, 3, 8). These data thus clearly argue against existence of αA and αB as one protein in the developing lens. These observations were further corroborated by following immunolocalization of Ribophorin-1 and the two α-crystallins in the native lens fiber cells by immunogold labeling and transmission electron microscopy (Fig. 8 and supplemental Fig. S2, A–D). It is interesting to note that Ribophorin-1 and αB localize in the RER. Ribophorin-1 and αA do not show similar localization. Interestingly, αA and αB also do not seem to label the same compartment. In fact, αA and Ribophorin-1 and αA and αB localizations look very similar (Fig. 8, B and C, and supplemental Fig. S2, B and C), indicating that the two proteins (αA and αB) associate with different membrane domains or compartments.
The immunoblot density scans presented in Fig. 7 (bottom panel) show that αA distributes in two peak fractions. The first one and smaller of the two is in fractions 6–10 and the second in fractions 12–15. The first (fractions 6–10) overlaps with αB peak. Although it is possible that this overlap represents co-sedimentation of αA and αB, we believe this may represent αA in a separate minor compartment (possibly Golgi, although we did not detect Golgi markers in this gradient, see Fig. 5, B and C).
It must be recognized that both αA as well as αB have been considered as soluble proteins of the ocular lens, yet the data presented thus far in this investigation and elsewhere (14, 15, 31, 33, 35) provide definitive evidence that both these proteins are membrane-associated. Although the exact function of αA remains to be elucidated, its presence in the ER has been related to ER-associated degradation of denatured proteins; it may be involved in quality control of proteins destined for the cell surface as has been recently suggested for epithelial Na (+) channel expression in mouse cultured collecting duct cells (29). A similar function has recently been suggested for HSP70 (28). It is interesting to note that Hsp70 and αA have similar fractionation profiles on the sucrose gradient used for isolation of RER and SM (Fig. 6).
At this time, it is difficult to speculate about the granular nature of the co-localization of αA and GM130 in the perinuclear Golgi seen in Fig. 1. We believe that some of these as yet undefined non-crystallin functions (38) of αA may be the reason for early cataracts in αA null mice (39). The observations made in this investigation may also explain why mutations in αA and αB have disparate phenotypic consequences (38). αA knock-out leads to early cataracts in the developing eye (39), whereas the αB knock-out does not show such a phenotype (40). The appearance of early cataract in αA−/− mice and none in αB −/− mice suggests that the α-crystallin protein, if it did exist, either does not need a healthy αB for its function or that this protein is not composed of these two polypeptides. The data presented above does not support the perception of α-crystallin as a monolithic entity; instead, the data points to the independent existence of αA and αB in the ocular lens. It is important to point out that in these investigations, the focus has been only on complete αA and complete αB polypeptides and not truncated forms of these proteins (3).
The data presented here does not preclude the interaction of αA and αB to produce α-crystallin later in the life of the adult lens due to physiological changes in the maturing lens fiber cells. Terminal differentiation of the ocular lens fiber cells is attended by membrane changes, including the Golgi and ER breakdown (41–43) and a host of posttranslational modifications on these two proteins (3). However, it is clear from the data presented here that the two exist as independent proteins in the developing transparent lens and thus necessitate revision of the status of α-crystallins as a family of proteins in the ocular lens rather than one single monolithic entity.
Acknowledgment
We thank Jeff Cohen for excellent technical support.
This work was supported by NEI, National Institutes of Health grant to S. P. B. (1R01EY006044).

This article contains supplemental Figs. S1 and S2.
- αB
- αB-crystallin
- RER
- rough endoplasmic reticulum
- αA
- αA-crystallin
- FD18
- fetal day 18
- P3
- postnatal day 3
- FM
- fiber mass
- ER
- endoplasmic reticulum
- BP
- bound polysome
- SM
- smooth membrane.
REFERENCES
- 1. Andley U. P. (2007) Crystallins in the eye: Function and pathology. Prog. Retin. Eye Res. 26, 78–98 [DOI] [PubMed] [Google Scholar]
- 2. Bhat S. P. (2003) Crystallins, genes, and cataract. Prog. Drug. Res. 60, 205–262 [DOI] [PubMed] [Google Scholar]
- 3. Groenen P. J., Merck K. B., de Jong W. W., Bloemendal H. (1994) Structure and modifications of the junior chaperone α-crystallin. From lens transparency to molecular pathology. Eur. J. Biochem. 225, 1–19 [DOI] [PubMed] [Google Scholar]
- 4. Horwitz J. (2000) The function of α-crystallin in vision. Semin. Cell Dev. Biol. 11, 53–60 [DOI] [PubMed] [Google Scholar]
- 5. Bhat S. P., Nagineni C. N. (1989) αB subunit of lens-specific protein α-crystallin is present in other ocular and non-ocular tissues. Biochem. Biophys. Res. Commun. 158, 319–325 [DOI] [PubMed] [Google Scholar]
- 6. Kato K., Shinohara H., Kurobe N., Inaguma Y., Shimizu K., Ohshima K. (1991) Tissue distribution and developmental profiles of immunoreactive αB crystallin in the rat determined with a sensitive immunoassay system. Biochim. Biophys. Acta 1074, 201–208 [DOI] [PubMed] [Google Scholar]
- 7. Srinivasan A. N., Nagineni C. N., Bhat S. P. (1992) αA-crystallin is expressed in non-ocular tissues. J. Biol. Chem. 267, 23337–23341 [PubMed] [Google Scholar]
- 8. Horwitz J. (2003) α-Crystallin. Exp. Eye Res. 76, 145–153 [DOI] [PubMed] [Google Scholar]
- 9. Horwitz J. (1993) Proctor Lecture. The function of α-crystallin. Invest. Ophthalmol. Vis. Sci. 34, 10–22 [PubMed] [Google Scholar]
- 10. Robinson M. L., Overbeek P. A. (1996) Differential expression of αA- and αB-crystallin during murine ocular development. Invest. Ophthalmol. Vis. Sci. 37, 2276–2284 [PubMed] [Google Scholar]
- 11. Bhat S. P., Horwitz J., Srinivasan A., Ding L. (1991) αB-Crystallin exists as an independent protein in the heart and in the lens. Eur. J. Biochem. 202, 775–781 [DOI] [PubMed] [Google Scholar]
- 12. Chiesa R., McDermott M. J., Spector A. (1989) Differential synthesis and phosphorylation of the α-crystallin A and B chains during bovine lens fiber cell differentiation. Curr. Eye Res. 8, 151–158 [DOI] [PubMed] [Google Scholar]
- 13. Voorter C. E., de Haard-Hoekman W. A., Roersma E. S., Meyer H. E., Bloemendal H., de Jong W. W. (1989) The in vivo phosphorylation sites of bovine αB-crystallin. FEBS Lett. 259, 50–52 [DOI] [PubMed] [Google Scholar]
- 14. Gangalum R. K., Bhat S. P. (2009) αB-Crystallin: a Golgi-associated membrane protein in the developing ocular lens. Invest. Ophthalmol. Vis. Sci. 50, 3283–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gangalum R. K., Schibler M. J., Bhat S. P. (2004) Small heat shock protein αB-crystallin is part of cell cycle-dependent Golgi reorganization. J. Biol. Chem. 279, 43374–43377 [DOI] [PubMed] [Google Scholar]
- 16. Rao N. A., Saraswathy S., Wu G. S., Katselis G. S., Wawrousek E. F., Bhat S. (2008) Elevated retina-specific expression of the small heat shock protein, αA-crystallin, is associated with photoreceptor protection in experimental uveitis. Invest. Ophthalmol. Vis. Sci. 49, 1161–1171 [DOI] [PubMed] [Google Scholar]
- 17. Dunn K. W., Kamocka M. M., McDonald J. H. (2011) A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300, C723–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saint-Geniez M., Kurihara T., D'Amore P. A. (2009) Role of cell and matrix-bound VEGF isoforms in lens development. Invest. Ophthalmol. Vis. Sci. 50, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bloemendal H., Benedetti E. L., Bont W. S. (1974) Preparation and characterization of free and membrane-bound polysomes. Methods Enzymol. 30, 313–327 [DOI] [PubMed] [Google Scholar]
- 20. Benedetti E. L., Zweers A., Bloemendal H. (1968) Structural aspects of eye lens polyribosomes. Biochem. J. 108, 765–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Persson S., Havton L. A. (2009) Retrogradely transported fluorogold accumulates in lysosomes of neurons and is detectable ultrastructurally using post-embedding immuno-gold methods. J. Neurosci. Methods 184, 42–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brahma S. K., Sanyal S. (1987) Ontogeny of α-crystallin polypeptides during the lens development of a mutant mouse. Curr. Eye Res. 6, 1291–1297 [DOI] [PubMed] [Google Scholar]
- 23. Wagner M., Rajasekaran A. K., Hanzel D. K., Mayor S., Rodriguez-Boulan E. (1994) Brefeldin A causes structural and functional alterations of the trans-Golgi network of MDCK cells. J. Cell Sci. 107, 933–943 [DOI] [PubMed] [Google Scholar]
- 24. Kelleher D. J., Kreibich G., Gilmore R. (1992) Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kd protein. Cell 69, 55–65 [DOI] [PubMed] [Google Scholar]
- 25. Silberstein S., Kelleher D. J., Gilmore R. (1992) The 48-kDa subunit of the mammalian oligosaccharyltransferase complex is homologous to the essential yeast protein WBP1. J. Biol. Chem. 267, 23658–23663 [PubMed] [Google Scholar]
- 26. Robenek M. J., Severs N. J., Schlattmann K., Plenz G., Zimmer K. P., Troyer D., Robenek H. (2004) Lipids partition caveolin-1 from ER membranes into lipid droplets: updating the model of lipid droplet biogenesis. FASEB J. 18, 866–868 [DOI] [PubMed] [Google Scholar]
- 27. Takiguchi M., Mori M., Tatibana M. (1985) A simple and rapid procedure for high-yield isolation of essentially undegraded free and membrane-bound polysomes from rat liver. J. Biochem. 97, 1447–1459 [DOI] [PubMed] [Google Scholar]
- 28. Chanoux R. A., Robay A., Shubin C. B., Kebler C., Suaud L., Rubenstein R. C. (2012) Hsp70 promotes epithelial sodium channel functional expression by increasing its association with coat complex II and its exit from endoplasmic reticulum. J. Biol. Chem. 287, 19255–19265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kashlan O. B., Mueller G. M., Qamar M. Z., Poland P. A., Ahner A., Rubenstein R. C., Hughey R. P., Brodsky J. L., Kleyman T. R. (2007) Small heat shock protein alphaA-crystallin regulates epithelial sodium channel expression. J. Biol. Chem. 282, 28149–28156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caspers G. J., Leunissen J. A., de Jong W. W. (1995) The expanding small heat-shock protein family, and structure predictions of the conserved “α-crystallin domain.” J. Mol. Evol. 40, 238–248 [DOI] [PubMed] [Google Scholar]
- 31. Gangalum R. K., Atanasov I. C., Zhou Z. H., Bhat S. P. (2011) αB-Crystallin is found in detergent-resistant membrane microdomains and is secreted via exosomes from human retinal pigment epithelial cells. J. Biol. Chem. 286, 3261–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cenedella R. J., Chandrasekher G. (1993) High capacity binding of α crystallins to various bovine lens membrane preparations. Curr. Eye Res. 12, 1025–1038 [DOI] [PubMed] [Google Scholar]
- 33. Cobb B. A., Petrash J. M. (2000) Characterization of α-crystallin-plasma membrane binding. J. Biol. Chem. 275, 6664–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ifeanyi F., Takemoto L. (1990) Specificity of α crystallin binding to the lens membrane. Curr. Eye Res. 9, 259–265 [DOI] [PubMed] [Google Scholar]
- 35. Kibbelaar M. A., Bloemendal H. (1979) Fractionation of the water-soluble proteins from calf lens. Exp. Eye. Res. 29, 679–688 [DOI] [PubMed] [Google Scholar]
- 36. Fujiwara T., Oda K., Yokota S., Takatsuki A., Ikehara Y. (1988) Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 263, 18545–18552 [PubMed] [Google Scholar]
- 37. Suetsugu S., Toyooka K., Senju Y. (2010) Subcellular membrane curvature mediated by the BAR domain superfamily proteins. Semin. Cell Dev. Biol. 21, 340–349 [DOI] [PubMed] [Google Scholar]
- 38. Bhat S. P. (2004) Transparency and non-refractive functions of crystallins–a proposal. Exp. Eye Res. 79, 809–816 [DOI] [PubMed] [Google Scholar]
- 39. Brady J. P., Garland D., Duglas-Tabor Y., Robison W. G., Jr., Groome A., Wawrousek E. F. (1997) Targeted disruption of the mouse αA-crystallin gene induces cataract and cytoplasmic inclusion bodies containing the small heat shock protein αB-crystallin. Proc. Natl. Acad. Sci. U.S.A. 94, 884–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brady J. P., Garland D. L., Green D. E., Tamm E. R., Giblin F. J., Wawrousek E. F. (2001) αB-crystallin in lens development and muscle integrity: a gene knockout approach. Invest. Ophthalmol. Vis. Sci. 42, 2924–2934 [PubMed] [Google Scholar]
- 41. Bassnett S. (2002) Lens organelle degradation. Exp. Eye Res. 74, 1–6 [DOI] [PubMed] [Google Scholar]
- 42. Zelenka P. S. (1984) Lens lipids. Curr. Eye Res. 3, 1337–1359 [DOI] [PubMed] [Google Scholar]
- 43. Zigman S., Paxhia T., Marinetti G., Girsch S. (1984) Lipids of human lens fiber cell membranes. Curr. Eye Res. 3, 887–896 [DOI] [PubMed] [Google Scholar]