

Abstract
Protein phosphatase 2A (PP2A) is a multifunctional phosphatase that plays important roles in many cellular processes including regulation of cell cycle and apoptosis. Because PP2A is involved in so many diverse processes, it is highly regulated by both non-covalent and covalent mechanisms that are still being defined. In this study we have investigated the importance of leucine carboxyl methyltransferase-1 (LCMT-1) for PP2A methylation and cell function. We show that reduction of LCMT-1 protein levels by small hairpin RNAs causes up to a 70% reduction in PP2A methylation in HeLa cells, indicating that LCMT-1 is the major mammalian PP2A methyltransferase. In addition, LCMT-1 knockdown reduced the formation of PP2A heterotrimers containing the Bα regulatory subunit and, in a subset of the cells, induced apoptosis, characterized by caspase activation, nuclear condensation/fragmentation, and membrane blebbing. Knockdown of the PP2A Bα regulatory subunit induced a similar amount of apoptosis, suggesting that LCMT-1 induces apoptosis in part by disrupting the formation of PP2ABαAC heterotrimers. Treatment with a pancaspase inhibitor partially rescued cells from apoptosis induced by LCMT-1 or Bα knockdown. LCMT-1 knockdown cells and Bα knockdown cells were more sensitive to the spindle-targeting drug nocodazole, suggesting that LCMT-1 and Bα are important for spindle checkpoint. Treatment of LCMT-1 and Bα knockdown cells with thymidine dramatically reduced cell death, presumably by blocking progression through mitosis. Consistent with these results, homozygous gene trap knock-out of LCMT-1 in mice resulted in embryonic lethality. Collectively, our results indicate that LCMT-1 is important for normal progression through mitosis and cell survival and is essential for embryonic development in mice.
Protein phosphatase 2A (PP2A),2 a multifunctional serine/threonine phosphatase, has been implicated in the regulation of cell growth and proliferation, primarily at the G2/M transition, and in the development of human cancers (1). PP2A is composed of three functionally distinct subunits: A (structural), B-type (regulatory/targeting), and C (catalytic). Binding of different B-type subunits to the core PP2A heterodimer (A/C) alters its substrate specificity and targets PP2A to different substrates and subcellular locations. Although there are two genes each for the C and A subunits in mammalian cells, there are multiple families of B-type subunits ((B (B55/PR55), B′ (B56/PR61), B″ (PR72)) and a putative B‴ (striatin) family, each of which has multiple members (1). Thus, a variety of heterotrimeric PP2A holoenzymes exist that differ primarily in their regulatory/targeting B-type subunits. In addition, Polyomavirus middle tumor antigen and Polyomavirus and SV40 small tumor antigens act as viral B-type subunits, displacing certain cellular B-type subunits and binding to the PP2A core heterodimer, altering its activity (Refs. 2–9 and references therein).
Reversible methylation is one of the most specific cellular mechanisms known for regulating PP2A and, thus, may have promise as a mechanism-based therapeutic target. In mammalian cells PP2A is methylated on its catalytic subunit carboxyl-terminal leucine (Leu-309) α-carboxyl group by leucine carboxyl methyltransferase-1 (LCMT-1) (10–16) and is dem-ethylated by protein phosphatase methylesterase-1 (17–19). Methylation indirectly regulates PP2A function by altering the formation of PP2A holoenzymes, thus influencing the subcellular targeting and specificity of PP2A (8, 20–25). C subunit methylation has a differential effect on incorporation of different B-type subunits into PP2A holoenzymes. In Saccharomyces cerevisiae, where there is only one B subunit (Cdc55p) and one B′ subunit (Rts1p), methylation increases the formation of heterotrimers containing B subunit 20-fold while increasing B′ heterotrimer formation less than 2-fold (26). In mammalian cells, methylation is important for efficient assembly of BαAC heterotrimers (22, 24) but is not necessary for formation of PP2A complexes containing striatin family members or the viral B-type subunit, Polyomavirus middle tumor antigen (24). Based on experiments with a PP2A C subunit lacking leucine 309, methylation may differentially affect different B′ family members in mammalian cells as well (27).
Based on these data, loss of PP2A methylation would be predicted to have the greatest effect on the function of PP2A heterotrimers containing the B/B55 family of regulatory subunits (PP2ABAC). PP2ABAC trimers have been implicated in mitosis promoting factor (MPF/Cdk1-cyclin B1) regulation in Xenopus (28) and in the regulation of mitotic exit in yeast (29–31). Therefore, PP2A methylation may regulate entry into mitosis as well as mitotic progression and exit. Consistent with this possibility, the level of PP2A methylation has been reported to change in a cell cycle-dependent manner in rat and human cells (32, 33). PP2ABAC heterotrimers have also been shown to be essential in yeast for spindle checkpoint (34) and to regulate phosphorylation and degradation of human securin (35). In accord with this necessity for PP2ABAC heterotrimers, we and others have shown that PP2A methylation is required for spindle checkpoint in yeast as indicated by increased sensitivity of PP2A methyltransferase-disrupted cells to spindle-targeting drugs like nocodazole (23, 25). However, it is not known if PP2A methylation is important for regulation of mitosis in mammalian cells. In fact, nothing is known about the role of LCMT-1 or protein phosphatase methylesterase-1 in mammalian cell growth and proliferation, including whether LCMT-1 is the major PP2A methyltransferase in vivo. In this study we used lentiviral small hairpin RNA (shRNA) vectors to knock down LCMT-1 expression and investigate the importance of LCMT-1 in mammalian cells. We also tested whether LCMT-1 was necessary for embryonic development in mice. Collectively, our results indicate that LCMT-1 is the major PP2A methyltransferase in mammalian cells, that LCMT-1 is important for normal progression through mitosis and for cell survival, and that it is required for mouse embryonic development.
EXPERIMENTAL PROCEDURES
Antibodies
LCMT-1 was detected using an affinity-purified rabbit anti-LCMT-1 polyclonal antibody, RK3110, generated against a 17-amino acid peptide corresponding to residues 173–189 of human LCMT-1. PP2A Bα and C subunit were detected with mouse monoclonal antibodies from Upstate Bio-technology (clone 2G9) and BD Transduction Laboratories, respectively. Unmethylated PP2A C subunit was detected with a previously described (24) mouse monoclonal antibody (clone 4B7; available from Upstate Biotechnology, Stratagene and Santa Cruz Biotechnology). Methylated PP2A C subunit was detected with 2A10 mouse monoclonal antibody (Upstate Biotechnology, Inc.). HA-tagged proteins were immunoprecipitated and immunoblotted with the mouse monoclonal antibodies 12CA5 (available from Santa Cruz Biotechnology, Inc.) and 16B12 (Covance Research Products), respectively.
Cell Culture and the Creation of Stable Lines That Express LCMT-1- and Bα-directed shRNAs
HeLa cervical carcinoma cells were obtained from American Type Culture Collection. HCT116 colon cancer cells were obtained from Dr. Eva Schmeltz. Both lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. To create HeLa and HCT116 lines that stably express LCMT-1- or PP2A Bα-directed shRNAs, cells were infected with lentiviruses expressing shRNAs. These lentiviruses were generated using a three-plasmid-based lentivirus system (36) (available from The RNAi Consortium (TRC) at the Broad Institute). The targeted sequences in the shRNA lentiviruses we used were: L2, 5′-GCCATGTTCATAAACTACGAA-3′ (TRC ID TRCN0000035060); L3, 5′-CGTCGACATGATGGAGTT-GTA-3′ (TRC ID TRCN0000035061); Bα, 5′-GCAAGTGGC-AAGCGAAAGAAA-3′ (TRC ID TRCN0000002493). After infection with the appropriate lentivirus, cells were selected with puromycin (2 μg/ml) until a canary dish of uninfected parental cells was completely dead (usually 3 days). After incubating one additional day to allow the cells to recover from puromycin treatment, cells were then used for experiments, and aliquots of cells were frozen for later use. Cells were used within 10 days after selection/thawing.
Determination of the Level of PP2Ac Methylation
The steady-state level of PP2A catalytic subunit methylation was measured in lysates with a monoclonal antibody specific for unmethylated PP2A C subunit (4b7) using our published method (24), which is described in the legend to Fig. 1, B and C, in the current study. Quantitation of the 4b7 signal was performed with a Bio-Rad Fluor S-Max Chemilumimager and Bio-Rad Quantity One Software.
FIGURE 1. Analysis of LCMT-1 knockdown and PP2A methylation in LCMT-1 shRNA HeLa cells.
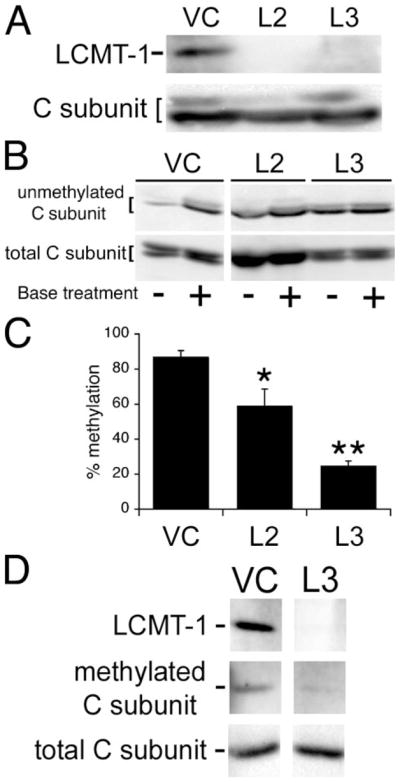
A, two different LCMT-1 shRNAs (L2 and L3) efficiently knockdown LCMT-1 protein levels in HeLa cells. VC cells and cells stably expressing the L2 and L3 LCMT-1 shRNAs were lysed, and lysates were probed for the steady-state levels of LCMT-1 and PP2A C subunit (C subunit; loading control) by immunoblotting. B, knockdown of LCMT-1 by expression of L2 or L3 shRNAs reduces PP2A C subunit methylation in HeLa cells. VC cells and LCMT-1 L2 and L3 shRNA cells were lysed, and the steady-state level of PP2A methylation in each cell line was determined using our previously published assay (24). Briefly, because base treatment demethylates the PP2A catalytic subunit, one aliquot of lysate from each line was treated for 5 min at 4 °C with 0.2 N NaOH to completely demethylate PP2A C subunit and was then neutralized (+lanes; 100% demethylated control), whereas another equal aliquot of lysate from each line was combined with preneutralized buffer (−lanes reflect endogenous methylation level). Then the untreated and base-treated aliquots were analyzed side by side on a 10% SDS-polyacrylamide gel followed by immunoblotting with a monoclonal antibody (4b7) specific for unmethylated C subunit. Demethylation of PP2A C subunit induced by L2 and L3 shRNA expression can be seen as an increase in the relative intensity of 4b7 signal in the minus versus plus lanes as compared with vector control cells. Also shown is an immunoblot of total PP2A C subunit showing that each pair of − and +lanes was loaded equally. C, the percent unmethylated PP2A catalytic subunit was determined by quantitatively comparing the amount of 4b7 signal in untreated samples (reflects level of endogenous unmethylated PP2A C subunit in each line) to that in the matched base-treated samples (100% demethylated controls) using a Bio-Rad Fluor-S Max Chemilumimager. Percent methylation was calculated by subtracting the percent of unmethylated PP2A from 100. The graph shows the averages and S.D. (error bars) of three independent experiments. Asterisks indicate significance versus vector control as assayed by t test (*, p = 0.0499; **, p = 0.0001). The C subunit can migrate as either singlets or doublets; whether double or single bands are seen can vary for the same sample from gel to gel. This pattern of migration in SDS-PAGE has been noted previously for endogenous PP2A C subunits (6, 8, 24, 32) and does not appear to be due to degradation. D, immunoblotting with the anti-methylated PP2A catalytic subunit antibody, 2A10, further strengthens the conclusion that LCMT-1 is the major PP2A methyltransferase in vivo. Lysates prepared from VC and L3 LCMT-1 knockdown cells were analyzed by immunoblotting with LCMT-1 antibody to demonstrate LCMT-1 knockdown and with 2A10 monoclonal antibody (methylated C subunit) to show L3 LCMT-1 knockdown causes a substantial reduction in PP2A catalytic subunit methylation. Also shown is an immunoblot of total PP2A C subunit showing that the lanes were loaded equally. Quantitation of the 2A10 signals from two separate experiments showed that there was greater than a 3-fold reduction in methylation in the L3 LCMT-1 shRNA knockdown cells.
Cell Transfections and Immunoprecipitations
The pCEP4 HA-tagged Bα construct was obtained from the laboratory of Dr. David M. Virshup (Department of Pediatrics and of Oncological Sciences, University of Utah). Vector control HeLa cells and HeLa cells that stably express shRNAs directed to LCMT-1 (L3) were plated at a density of 3.5 × 105 cells/60-mm dish and then transfected with pCEP4 HA-tagged Bα using FuGENE6 transfection reagent (Roche Diagnostics). At 72 h post-transfection, cells were lysed in an Nonidet P-40-containing lysis buffer (10% glycerol, 20 mM Tris, pH 8.0, 137 mM NaCl, 1% Nonidet P-40), and immunoprecipitation of N-terminally HA-tagged Bα was carried out with anti-HA tag monoclonal antibody, 12CA5, covalently cross-linked to protein A-Sepharose beads. To determine the amount of PP2A C subunit associated with each of the immunoprecipitated samples, the immunoprecipitates were analyzed by SDS-PAGE and immunoblotted with 16B12 anti-HA tag antibody and with anti-PP2A C subunit antibody, and then the relative amount of HA-tagged Bα and PP2A C subunit were determined by quantitation using a Bio-Rad Fluor S-Max Chemilumimager and Bio-Rad Quantity One Software.
Analysis of Cell Viability by Trypan Blue Exclusion
Vector control cells and cells that stably express shRNAs directed toward LCMT-1 (L2 or L3) or Bα were plated at a density of 2 × 104 cells/well in a 12-well dish, allowed to adhere overnight, washed to remove dead cells, and then incubated for 24 h at 37 °C. Cells were then harvested and checked for viability by trypan blue exclusion (0.4% trypan blue).
Analysis of DNA Condensation and Fragmentation Using DAPI Stain
Vector control HeLa cells and HeLa cells that stably express shRNAs directed toward LCMT-1 (L2 or L3) or Bα were plated at a density of 2 × 104 cells/well in a 12-well dish, allowed to adhere overnight, washed to remove dead cells, and then incubated for 24 h at 37 °C. Cells were then incubated with DAPI stain for 5 min at 37 °C. Cells were subsequently observed by fluorescence microscopy using an Olympus IX81 phase/fluorescence microscope and analyzed using Slidebook Microscope Analysis software (Intelligent Imaging, Inc.).
Caspase 3 in Cell Detection Assay
Vector control HeLa cells and HeLa cells that stably express shRNAs directed toward LCMT-1 (L2 or L3) or Bα were plated at a density of 1.5 × 105 cells/well in a 6-well dish, allowed to adhere overnight, washed to remove dead cells, and then incubated for 24 h at 37 °C. Cells were then harvested and gently pelleted, washed in media, and then incubated in media containing Red-DEVD-fmk caspase-3 activity indicator (DEVD-fmk conjugated to sulforhodamine; Calbiochem) and 10% fetal calf serum for 1 h in a 37 °C incubator per the manufacturer’s instructions. Cells were subsequently placed on a coverslip and observed by fluorescence microscopy using an Olympus IX81 phase/fluorescence microscope and analyzed using Slidebook Microscope Analysis software (Intelligent Imaging, Inc.).
z-DEVD-AFC Caspase 3 in Vitro Activity Assay
Vector control HeLa cells and HeLa cells that stably express shRNAs directed toward LCMT-1 (L2 or L3) or Bα were plated at a density of 1.5 ×105 cells/well in a 6-well dish, allowed to adhere overnight, washed to remove dead cells, and then incubated for 24 h at 37 °C. Cells were then washed 3 times with 1× phosphate-buffered saline and incubated in a Nonidet P-40-containing lysis buffer (10% glycerol, 20 mM Tris, pH 8.0, 137 mM NaCl, 1% Nonidet P-40) for 20 min on ice. The cells were then sonicated on ice for 5 × 2-s bursts. The supernatant was then centrifuged for 10 min at 13,000 μg. The protein concentration of the supernatant was assayed by the Lowry method (Bio-Rad). 25 μg of protein was incubated with z-DEVD-AFC (25 μM) (Calbiochem substrate IV) at 37 °C for 1 h. Caspase activity was measured using a Wallac Victor2 1420 Multilabel Counter fluorimeter (PerkinElmer Life Sciences) at 405-nm excitation and 535-nm emission wavelengths.
Effect of Caspase Inhibition on Cell Viability
Vector control HeLa cells and HeLa cells that stably express shRNAs directed toward LCMT-1 (L2 or L3) or Bα were plated at a density of 2 × 104 cells/well in duplicate 12-well dishes, allowed to adhere overnight, washed to remove dead cells, and then incubated with z-VAD-fmk pancaspase inhibitor (50 μM; BioMol) or with vehicle only for 24 h at 37 °C. Cells were then harvested and checked for viability by trypan blue exclusion (0.4% trypan blue), and the viability of each line ± caspase inhibitor was compared.
Phase Time-lapse Microscopy
Vector control HeLa cells and HeLa cells that stably express shRNAs directed toward LCMT-1 (L2 or L3) or Bα were plated at a density of 4 × 104 cells/well in a 6-well dish, allowed to adhere overnight, washed to remove dead cells, and then incubated at 37 °C in a temperature and atmosphere-controlled microscope stage incubator (M6; Zeiss Instruments) on an Olympus IX81 phase/fluorescence microscope equipped with a computer-driven motorized stage (ASI). Phase images were captured every 6 min for 24–36 h. Images were analyzed using Slidebook Microscope Analysis software (Intelligent Imaging, Inc.).
Nocodazole Treatment and Analysis of Cell Viability
Cells were plated at a density of 2 × 104 cells/well into duplicate 12-well dishes, allowed to adhere overnight, and washed to remove dead cells, and then one dish was incubated with nocodazole (200 ng/ml) and the other with vehicle control for 24 h at 37 °C. Cells were then harvested and assayed for viability by trypan blue exclusion (0.4% trypan blue) to determine any change in cell death for each line due to the presence of nocodazole.
Thymidine Block and Analysis of Cell Viability
Cells were plated at a density of 2 × 104 cells/well into triplicate wells in duplicate 12-well dishes, allowed to adhere overnight, washed to remove dead cells, and then incubated with thymidine (2 mM) or vehicle only for 24 h at 37 °C. Cells were then harvested and checked for viability by trypan blue exclusion (0.4% trypan blue) to determine any change in cell death for each line due to G1/S arrest induced by thymidine treatment.
Generation of LCMT-1 Knock-out Mice and Genotyping
LCMT 1+/− embryonic stem cells containing a gene trap insertion in the first intron of the LCMT1 locus were obtained from German Gene Trap Consortium, Neuherberg, Germany. These cells were expanded in culture and were injected into blastocysts from C57BL/6 donors using standard techniques by the Emory University School of Medicine Transgenic Mouse and Gene Targeting Core Facility. Resultant chimeric mice were bred to generate heterozygous F1 animals that were then intercrossed to determine whether homozygous knock-out of LCMT-1 was embryonic lethal.
RESULTS
Down-regulation of LCMT-1 Causes a Substantial Decrease in PP2Ac Methylation
To determine whether LCMT-1 was necessary for cell viability and normal cell cycle progression in mammalian cells, we decreased LCMT-1 expression in HeLa cells by stably expressing shRNAs to LCMT-1 via lentiviral vectors. Fig. 1A shows that two different LCMT-1 shRNAs, designated L2 and L3, were able to dramatically reduce LCMT-1 protein levels.
In yeast, loss of the LCMT-1 homolog, Ppm1p, results in loss of PP2A (Pph21p/Pph22p) methylation, indicating that Ppm1p is probably solely responsible for methylating PP2A under normal growth conditions (23, 25, 37). Although LCMT-1 has been shown to methylate PP2A in vitro (10–16), its contribution to PP2A methylation in vivo has not been quantitatively examined in mammalian cells. To determine the contribution of LCMT-1 to PP2A methylation in mammalian cells, we quantitated the steady-state levels of PP2Ac methylation in L2 and L3 LCMT-1 knockdown cells using our monoclonal antibody, 4b7, which is specific for unmethylated PP2A catalytic subunit (see “Experimental Procedures” and the legend for Fig. 1) (24). Fig. 1, B and C, show that the knockdown of LCMT-1 by L2 and L3 shRNAs caused a substantial (32 or 71%, respectively) reduction in PP2A catalytic subunit methylation in HeLa cells as compared with vector control (VC) cells. Interestingly, although LCMT-1 could not be detected easily in either the L2 or L3 cells (Fig. 1A), L3 was more efficient than L2 at reducing PP2A methylation (Fig. 1C). To further corroborate our results with the 4b7 anti-unmethylated PP2A catalytic subunit antibody, we performed a separate experiment in which we analyzed vector control and L3 LCMT-1 knockdown cells with another monoclonal antibody (2A10) that is specific for methylated PP2A catalytic subunit. The results in Fig. 1D show that 2A10 also detected a large reduction in methylated PP2A catalytic subunit in L3 LCMT-1 knockdown cells. Quantitation of results from two separate experiments showed that PP2A C subunit methylation was reduced >3-fold. Collectively, these results show that mammalian LCMT-1 is responsible for the majority of PP2A methylation, similar to the case for the S. cerevisiae LCMT-1 homolog, Ppm1p. They also demonstrate that the small amount of residual LCMT-1 protein in the L2 and L3 knockdown cells may be sufficient for methylating 25–60% of the PP2A in these cells.
Knockdown of LCMT-1 Greatly Reduces PP2A Bα Binding to C Subunit
In yeast, deletion of the gene encoding Ppm1p dramatically reduces the binding of PP2A B subunit to C subunit. We wanted to determine whether knockdown of LCMT-1 had similar effects on Bα binding to C subunit in mammalian cells. Because no antibody capable of immunoprecipitating endogenous Bα/C subunit complexes was available, we assayed the ability of transfected HA-tagged Bα (HA-Bα) to associate with endogenous C subunit in our vector control and L3 LCMT-1 shRNA HeLa cells. Fig. 2, A and B, show that methylation was greatly reduced in the L3 cells used for this experiment. Fig. 2, C and D, show that down-regulation of LCMT-1 by L3 shRNA greatly reduced C subunit association with HA-Bα. Thus, LCMT-1 is important for PP2ABαAC trimer formation in mammalian cells.
FIGURE 2. LCMT-1 knockdown reduces C subunit-Bα association.
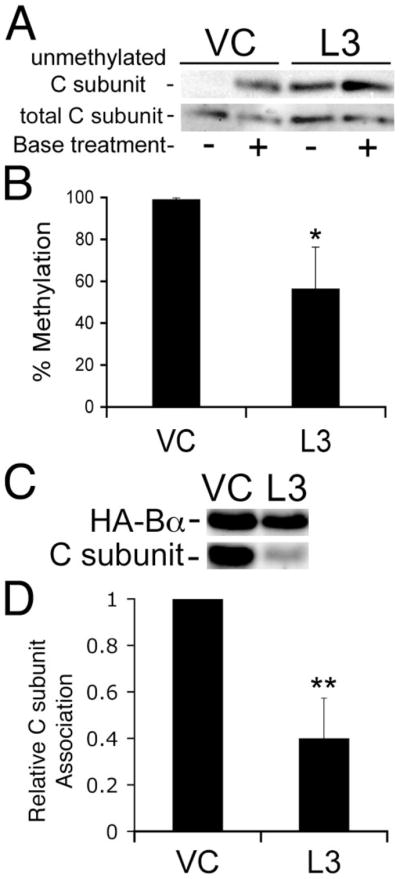
VC cells and cells stably expressing an shRNA directed against LCMT-1 (L3) were transfected with a construct expressing HA-tagged Bα (HA-Bα) or an empty vector. A, the steady-state level of PP2A methylation in VC and L3 lysates (5% of the input of each immunoprecipitate) was determined using our previously described assay using our 4b7 methylation-sensitive monoclonal antibody (see the legend for Fig. 1B). The reduction in methylation induced by expression of the L3 shRNA can be seen by the increased signal in the −lane. Also shown is an immunoblot of total PP2A C subunit showing that each pair of − and +lanes was loaded equally. B, percent methylation of the PP2A catalytic subunit was determined as described in the legend to Fig. 1C. The graph shows the averages and S.D. (error bars) of three independent experiments. The asterisk indicates significance versus vector control as assayed by t test (*, p = 0.02). C, HA-Bα immunoprecipitates of VC and L3 shRNA cells were probed for HA-tagged Bα (HA-Bα) and C subunit. The image shown is a representative immunoblot of three independent experiments. D, each band in panel C was quantitated using a Bio-Rad Fluor-S Max Chemilumimager. The relative amount of C subunit bound to HA-Bα (Relative C subunit association) was calculated as a measure of the efficiency of C subunit association with HA-Bα. The graph shown displays the average C subunit association with HA-Bα in the three experiments. Error bars show S.D. of the three independent experiments. Asterisks indicate significance versus vector control as assayed by t test (**, p = 0.0038).
Knockdown of LCMT-1 Induces Cell Death
In mammalian cells PP2A plays important roles in both cell cycle control and in apoptosis. We, therefore, analyzed the LCMT-1 knockdown cells and control cells to determine whether there was an effect of LCMT-1 knockdown on cell viability. After infection with L2 and L3 shRNA-expressing lentiviruses, we noticed that a portion of the HeLa cells knocked down for LCMT-1 was dying, whereas little to no death was seen in the cells infected with the control lentivirus. To determine the extent of death induced by LCMT-1 knockdown, we measured the percent of cells in each of these lines that died during a 24-h period in culture. Fig. 3A shows that both L2 and L3 shRNA knockdown HeLa cells had significantly more death (7–8%) during this period than the vector control cells (1%).
FIGURE 3. LCMT-1 or Bα knockdown causes cell death.
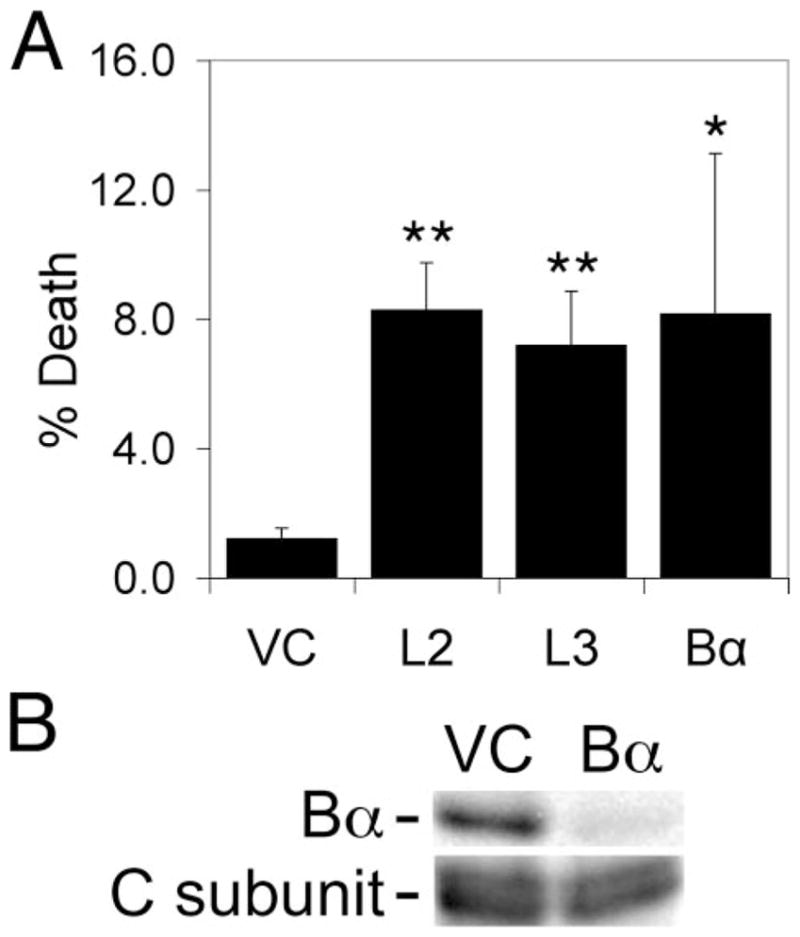
A, asynchronous VC cells, LCMT-1 shRNA knockdown cells expressing one of two different shRNAs to LCMT-1 (L2 or L3), and PP2A Bα regulatory subunit knockdown cells (Bα) were plated and allowed to adhere, washed to remove dead cells, and then harvested after 24 h and assayed for viability by trypan blue exclusion. Error bars show S.D. of five independent experiments. Asterisks indicate significance versus vector control as assayed by t test (L2**, p = 0.000005; L3**, p = 0.00004; Bα*, p = 0.014). B, PP2A Bα shRNA dramatically reduces Bα protein levels in HeLa cells. VC cells, cells stably expressing shRNAs directed against Bα were lysed, and lysates were probed for the steady-state levels of Bα and PP2A C subunit (loading control) by immunoblotting.
Because the stable formation of PP2A trimers containing the Bα regulatory subunit has been shown to be highly dependent on methylation of the PP2A C subunit (22, 24) and we showed above that Bα/C subunit complexes were reduced in LCMT-1 knockdown cells, it was possible that the cell death seen in L2 and L3 knockdown cells was due in part to impairment of PP2ABαAC trimer formation and function. We, therefore, tested whether knockdown of Bα would also induce death. Indeed, knockdown of Bα by a lentiviral shRNA vector, demonstrated in Fig. 3B, also caused an increase in cell death comparable with that induced by knocking down LCMT-1 (Fig. 3A). Although the increase in cell death in Bα knockdown cells was more variable than for L2 and L3, it was statistically significant (p < 0.05).
Knockdown of LCMT-1 Induces Caspase-dependent and Caspase-independent Cell Death
To begin to characterize the mechanism of death induced by knockdown of LCMT-1 or Bα, we analyzed our vector control, L2, L3, and Bα shRNA cells by phase time-lapse microscopy. In comparison to vector control cells, L2, L3, and Bα knockdown cells exhibited increased membrane blebbing (Fig. 4A and data not shown), a characteristic of caspase-mediated cell death (38). To further analyze this cell death, we next incubated each cell line with DAPI stain to detect condensed and fragmented DNA, which is also characteristic of apoptosis (39). Fig. 4B shows that LCMT-1 or Bα knockdown induced DNA condensation and fragmentation, supporting the idea that reduction of LCMT-1 induces apoptosis, perhaps in part by inhibition of Bα function.
FIGURE 4. Characterization of cell death induced by knockdown of LCMT-1 or Bα.

A, LCMT-1 knockdown causes membrane blebbing. Asynchronous LCMT-1 shRNA knockdown cells (L2) were observed for 24 –36 h by phase time-lapse microscopy. Six representative images of membrane blebbing in L2 knockdown cells are shown. B, knockdown of LCMT-1 or Bα causes DNA condensation and fragmentation in HeLa cells. Asynchronous unselected VC cells (data not shown), LCMT-1 shRNA knockdown cells (L2 or L3), and Bα knockdown cells infected with a multiplicity of infection of 3 were incubated with DAPI DNA stain to detect DNA condensation and fragmentation by immunofluorescence microscopy. Three representative images (top row, DAPI image; bottom row, differential interference contrast image) are shown for each cell line. C, LCMT-1 knockdown HeLa cells contain active caspases. Twenty-four hours after removal of dead cells by washing, asynchronous untreated VC cells, staurosporine-treated vector control cells (ST; positive control), LCMT-1 shRNA knockdown (L2 or L3), and Bα knockdown cells were harvested, incubated with Red-DEVD-fmk (Calbiochem) for 1 h at 37 °C, and assayed by immunofluorescence microscopy. Error bars show S.D. of results obtained from a minimum of three fields. Asterisks indicate significance versus vector control as assayed by t test (*, p =0.021; **, p =0.002). Similar results were obtained in two independent experiments. D, LCMT-1 knockdown HeLa cell lysates exhibit caspase activity. The same cells as used in panel C were harvested, lysed by sonication, and incubated with z-DEVD-AFC (Calbiochem) for 1 h at 37 °C and assayed for caspase activation using a fluorimeter. Caspase activity shown is in relative fluorescence units. Error bars show S.D. of results obtained triplicate wells. Asterisks indicate significance versus vector control as assayed by t test (ST**, p =0.0003; L2**, p =0.003; L3**, p =0.004). Similar results were obtained in three independent experiments, except that Bα had a statistically significant increase in caspase activity in one experiment. E, caspase inhibitor partially rescues cell death in LCMT-1 and Bα knockdown cells. The same cells as used in panel C were plated into duplicate plates, and dead cells were removed by washing. One plate of cells was treated with 50 μM z-VAD-fmk for 24 h, whereas the other was treated with vehicle only. Then the cells were harvested and assayed for trypan blue exclusion. A decrease in cell death in z-VAD-fmk-treated cells (black bars) as compared with untreated cells (vehicle control; white bars) can be seen. Error bars show S.D. of triplicate wells. Asterisks indicate significance versus vector control as assayed by t test (L2*, p =0.027; L3*, p =0.022; **, p =0.006). Similar results were obtained in two independent experiments.
Because the membrane blebbing observed in our knockdown cells is a characteristic of caspase activation (38), we wanted to determine whether LCMT-1 and Bα knockdown induce caspase activation. We first assayed caspase activation in live vector control, LCMT-1 knockdown, and Bα knockdown cells. Vector control cells treated with the pankinase inhibitor staurosporine were used as a positive control (40). The cell-permeable substrate we used for this assay, Red-DEVD-fmk, is a preferred substrate of caspases 3 and 7 but also can serve as a substrate for a number of other caspases (41). Fig. 4C shows that knockdown of LCMT-1 by either L2 or L3 shRNA caused an increase in caspase activity in vivo. Although knockdown of Bα consistently induced a small increase in caspase activation as detected by this assay, the increase was not statistically significant (Fig. 4C and data not shown). In a complementary approach, we assayed for caspase activity in lysates of our vector control and LCMT-1 and Bα knockdown cells using the caspase substrate z-DEVD-AFC. Again, staurosporine-treated vector control cells were used as a positive control. The results we obtained (Fig. 4D) are consistent with our results from the in vivo caspase assay. Knockdown of LCMT-1 by either L2 or L3 shRNA caused significant activation of caspases in three independent experiments. Knockdown of Bα on the other hand caused some caspase activation in all experiments, but only in one experiment was this increase significant (Fig. 4D and data not shown).
Although this result indicates that caspases are activated at least in the case of LCMT-1 knockdown cells, it does not show whether caspase activation is necessary for the observed apoptosis. To test whether the observed death requires caspase activation, we treated our LCMT-1 (L2 and L3) and Bα knockdown cells with the pancaspase inhibitor z-VAD-fmk (50 μM) to see if it would reduce cell death. Interestingly, we observed a reduction in cell death in all three knockdown cell lines (L2, L3, and Bα shRNA cells) as compared with vector control, but consistent with our caspase activation results, the reduction in cell death was always the least for the Bα knockdown cells (Fig. 4E and data not shown). These results suggest that down-regulation of LCMT-1 or Bα causes death by both caspase-dependent and caspase-independent pathways but that LCMT-1 knockdown causes more caspase-dependent cell death than Bα knockdown.
LCMT-1 or Bα Knockdown Causes Nocodazole Sensitivity
In our initial experiments using phase time-lapse microscopy, we found that a subset of LCMT-1 knockdown cells round up as if to enter mitosis, sometimes with a visible metaphase plate, and remain rounded for longer than expected for a normal mitosis, eventually undergoing apoptosis. Fig. 5 shows an example of such a cell that enters mitosis (metaphase plate is clearly visible in the cell at the 1.6h time point) and remains rounded until membrane blebbing begins ~14 h later. In yeast, we and others have shown that deletion of the gene PPM1, which encodes the yeast homolog of LCMT-1, or deletion of the gene CDC55, which encodes the yeast homolog of Bα, has no effect on cell viability under normal growth conditions unless the cells are sensitized by the microtubule-depolymerizing drug nocodazole, a phenotype characteristic of a spindle checkpoint defect (23, 25, 29, 34). To determine whether LCMT-1 and/or Bα knockdown results in a similar mitotic defect in mammalian cells as in yeast, we treated our LCMT-1 (L2 and L3) and Bα knockdown cells for 24 h with nocodazole (200 nM) and then assayed these cells to determine whether there were any changes in the amount of death as compared with untreated cells. The results in Fig. 6A show that although nocodazole treatment only caused a 1% increase in cell death in vector control cells, it induced a much greater increase in death in cells with reduced LCMT-1 levels (10–13%). Moreover, reduction of Bα levels also resulted in increased death in the presence of nocodazole (Fig. 6A).
FIGURE 5. Some LCMT-1 knockdown cells die after entering mitosis.

Asynchronous VC cells, LCMT-1 shRNA knockdown cells (L2 or L3), and Bα subunit knockdown cells were observed for 24 –36 h by phase time-lapse microscopy. A representative image of L3 knockdown cells at 0, 1.6, and 16 h is shown. The black arrow indicates the cell of interest through three panels. The metaphase plate (vertical white line) of this cell is visible in the middle panel (1.6-h time point).
FIGURE 6. LCMT-1 or Bα reduction causes nocodazole sensitivity in HeLa cells.

A, VC control HeLa cells, LCMT-1 shRNA knockdown cells (L2 or L3), and Bα shRNA cells were plated into duplicate plates, and dead cells were removed by washing. One plate of cells was treated with nocodazole for 24 h, whereas the other was treated with vehicle only. Then the cells were harvested and assayed for trypan blue exclusion. Shown is the net increase in cell death for each line resulting from nocodazole treatment (% death of each line in nocodazole minus the % death of the same line in vehicle control). Error bars show S.D. of three different experiments. Asterisks indicate significance versus vector control as assayed by t test (*, p = 0.022; L3**, p = 0.004; Bα**, p = 0.002). B, thymidine rescues cell death in LCMT-1 and Bα knockdown HeLa cells. The same cells as used in panel A were plated into duplicate plates, and dead cells were removed by washing. One plate of cells was treated with thymidine for 24 h, whereas the other was treated with vehicle only. Then the cells were harvested and assayed for trypan blue exclusion. Shown is the cell death in untreated (control; white bars) and thymidine-treated cells (black bars). Error bars show S.D. for triplicate wells. Asterisks indicate significance versus vector control as assayed by t test (L2**, p =0.002; L3**, p =0.003; Bα**, p = 0.002). Similar results were obtained two independent experiments.
Thymidine Treatment Rescues Cell Death Caused by LCMT-1 or Bα Knockdown
To determine whether the increased death caused by nocodazole was specifically caused by arresting cells in mitosis (and, thus, perhaps a spindle checkpoint defect) or was just due to cell cycle arrest, we also treated our vector control, LCMT-1, and Bα knockdown cells with thymidine, which has been used as a cell synchronization agent to arrest cells in G1/S. Strikingly, thymidine treatment caused a dramatic reduction in cell death in both LCMT-1 (L2 and L3) and Bα knockdown cells (Fig. 6), indicating that the increased cell death observed when cells are treated with nocodazole is specific to mitosis.
LCMT-1 Knockdown Also Causes Increased Death of HCT116 Colon Cancer Cells in Nocodazole
Increased sensitivity of HeLa LCMT-1 knockdown cells to nocodazole (Fig. 6A) suggested that LCMT-1 plays a role in spindle checkpoint and that drugs targeting LCMT-1 might have potential in combination chemotherapy with microtubule-targeting drugs. We wanted to determine next whether LCMT-1 knockdown would cause increased killing of another cancer cell type in nocodazole. The results presented in Fig. 7, A–C, show that L2 and L3 shRNAs also reduce LCMT-1 protein levels and PP2A methylation in HCT116 cells. Decreased methylation in the L2 and L3 cells was detected both by immunoblotting with 2A10 antibody specific for methylated PP2A catalytic subunit (Fig. 7A) and by using our methylation assay employing our monoclonal antibody specific for unmethylated PP2A catalytic subunit (Fig. 7, B and C). Importantly, Fig. 7D shows that HCT116 L2 and L3 LCMT-1 knockdown cells were more sensitive to nocodazole than were vector control cells. Similar to the results obtained for HeLa cells (Fig. 6A), an average increase in percent killing of 9% in nocodazole was seen with L2 and L3 shRNAs in three separate experiments, whereas vector control cells only showed an average increase in killing of 1.5% in the same experiments (Fig. 7D and data not shown).
FIGURE 7. LCMT-1 knockdown in HCT116 colon cancer cells also causes decreased PP2A C subunit methylation and increased nocodazole sensitivity.

A, two different LCMT-1 shRNAs (L2 and L3) efficiently knockdown LCMT-1 protein levels and reduce PP2A C subunit methylation in HCT116 cells. Lysates from VC cells and cells stably expressing the L2 and L3 LCMT-1 shRNAs were probed by immunoblotting for the steady-state levels of LCMT-1 and PP2A C subunit methylation (methylated PP2A C subunit; using an anti-methyl PP2A C subunit monoclonal antibody, 2A10). Total PP2A C subunit was also immunoblotted as a loading control. B, the steady-state level of PP2A methylation in VC, L2, and L3 lysates was also analyzed with our previously described assay using our 4b7 methylation-sensitive monoclonal antibody (see the legend for Fig. 1B). The untreated (−) and base-treated (+; 100% unmethylated control) aliquots were analyzed side by side on a 10% SDS-polyacrylamide gel followed by immunoblotting with 4b7, which is specific for unmethylated C subunit. Demethylation of PP2A C subunit induced by L2 and L3 shRNA expression can be seen as an increase in the relative intensity of 4b7 signal in the minus versus plus lanes of L2 and L3 as compared with the relative intensity of the minus and plus lanes in vector control. Also shown is an immunoblot of total PP2A C subunit showing that each pair of minus and plus lanes was loaded equally. C, quantitation of PP2A C subunit methylation reduction in HCT116 L2 and L3 LCMT-1 shRNA cells. The percent of unmethylated PP2A catalytic subunit in HCT116 cell lysates was determined by quantitatively comparing the amount of 4b7 signal in untreated samples (endogenous unmethylated PP2A C subunit) to that in the matched base-treated samples (100% demethylated controls) using a Bio-Rad Fluor-S Max Chemilumimager. Percent methylation was calculated by subtracting the percent of unmethylated PP2A from 100. The graph shows the averages of the results from two assays ± range. D, LCMT-1 knockdown in HCT116 cells causes increased sensitivity to nocodazole. HCT116 VC and L2 and L3 LCMT-1 shRNA knockdown cells were plated into duplicate plates, and dead cells were removed by washing. One plate of cells was treated with nocodazole for 24 h, whereas the other was treated with vehicle only. Then the cells were harvested and assayed for trypan blue exclusion. Shown is the net increase in cell death for each line resulting from nocodazole treatment (% death of each line in nocodazole minus % death of the same line in vehicle control). Percent death in the absence of nocodazole in this experiment was: VC, 1.74 ± 0.43; L2, 9.2 ± 1.5, p = 0.012; L3, 3.9 ± 0.7, p = 0.025). Error bars show S.D. for triplicate wells. Asterisks indicate significance versus vector control as assayed by t test (*, p = 0.028; **, p = 0.002). Similar results were obtained in three independent experiments, except in one case the p value for L3 was 0.06.
Homozygous Knock-out of LCMT-1 in Mice Causes Embryonic Lethality
Because our data indicated that LCMT-1 was necessary for normal progression through mitosis, we predicted that LCMT-1 knock-out would be embryonic lethal. To test this hypothesis, we generated an LCMT-1 knock-out mouse using LCMT-1+/− embryonic stem (ES) cells created by The German Gene Trap Consortium and tested whether live LCMT-1−/− pups could be obtained from crossing LCMT-1+/− mice with each other. Our results are shown in Fig. 8. Fig. 8A shows that our genotyping primer sets for wild-type LCMT-1 and for gene trap knock-out LCMT-1 both produce robust products using DNA from the original LCMT-1+/− ES cells or from progeny LCMT-1+/− or LCMT-1+/+ mice obtained from breeding. Fig. 8, B and C, show that LCMT-1 protein was reduced about 2-fold in the LCMT-1+/− ES cell line relative to control parental ES cells. Finally, Fig. 8D shows the average number of pups with each possible genotype obtained from eight LCMT-1+/− × LCMT-1+/− matings. If homozygous LCMT-1 loss had no effect on development, one would expect equal numbers of LCMT-1+/+ and LCMT-1−/− mice. However, although 19 LCMT-1+/+ mice and 26 LCMT-1+/− pups were born from these +/− × +/− crosses, no LCMT-1−/− pups were obtained from any of the crosses, indicating that loss of LCMT-1 is indeed embryonic lethal.
FIGURE 8. Homozygous knock-out of LCMT-1 in mice is embryonic lethal.
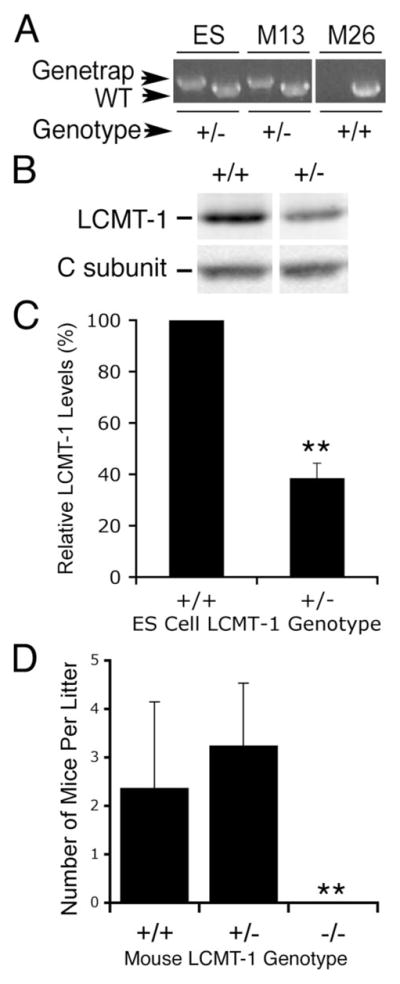
A, DNA was prepared from LCMT-1+/− mouse ES cells obtained from The German Gene Trap Consortium and from progeny mice (M13, M26) obtained from crossing of chimeric knock-out mice with C57BL/6 breeders. Each DNA was used in two PCR reactions, one using a primer pair specific for the wild-type LCMT-1 gene (WT; if positive, it indicates at least one LCMT-1 allele without gene trap insertion is present) and another using a primer pair specific for LCMT-1 having the gene trap insertion (Genetrap; forward primer matching the LCMT-1 intron and reverse primer matching the gene trap insert). Strong bands specific for the LCMT-1 Genetrap knock-out are seen in the LCMT-1+/− ES cells (control) and in the LCMT-1+/− mouse (M13), whereas the LCMT-1+/+ mouse (M26) clearly lacks the knock-out allele. B, LCMT-1 protein level is reduced in the LCMT-1+/− ES cells. Equal numbers of LCMT-1+/− ES cells (+/−) and control parental ES cells (+/+) were lysed and analyzed by SDS-PAGE and immunoblotting with LCMT-1 antibody. C, the results from one experiment from Fig. 7B performed in triplicate were quantitated, and the average and S.D. of the LCMT-1 levels in the LCMT-1+/− ES cells normalized to the control LCMT-1+/+ES cells are shown. Two separate experiments showed a reduction of >50%. D, no pups with homozygous gene trap knock-out of LCMT-1 were obtained from LCMT-1+/− × LCMT-1+/− mouse crosses. Eight crosses of LCMT-1+/− × LCMT-1+/− mice were performed, and pups were genotyped. Forty-five pups were obtained, 19 of which were LCMT-1+/− and 26 of which were LCMT-1−/−. No LCMT-1−/− mice were obtained. Shown are the average number of pups in each litter that were LCMT-1+/+, LCMT-1+/−, or LCMT-1−/−. Error bars indicate S.D. Asterisks indicate 99.8% confidence determined by t test that homozygous knock-out of LCMT-1 causes embryonic lethality (p = 0.002).
DISCUSSION
Carboxyl methylation of PP2A was discovered well over a decade ago, but its function remained obscure until the discovery that the addition and removal of the single methyl group on the PP2A carboxyl-terminal leucine could differentially regulate the assembly of certain PP2A heterotrimers (22–25). The cloning of LCMT-1 as a mammalian PP2A methyltransferase by De Baere et al. (16) has facilitated the investigation the contribution of this enzyme toward PP2A methylation and function. However, until the recent advent of small interfering RNA approaches for mammalian cells, much advancement in our understanding has come from experiments performed in yeast because of the ease of performing genetic analyses. In fact, before this study, nothing was known about the importance of LCMT-1 for normal cell cycle progression and cell survival. In this report we have utilized lentiviral shRNA and Genetrap knock-out approaches to investigate LCMT-1 function in mammalian cells. We show that a strong reduction in LCMT-1 not only results in a substantial reduction in PP2A methylation in mammalian cells but also causes a decrease in Bα subunit association with C subunit, induces apoptosis as evidenced by caspase activation, membrane blebbing, nuclear condensation and fragmentation, and cell death, and sensitizes HeLa and HCT116 cancer cells to the microtubule targeting drug nocodazole.
Although LCMT-1 was originally purified as a PP2A methyltransferase, only a small percentage of PP2A methyltransferase activity in the original cell extract was recovered (16), leaving open the question as to whether LCMT-1 is the major PP2A methyltransferase in mammalian cells. Our result showing that reduction of the steady-state level of PP2A C subunit methylation an average of 70% in HeLa cells and 63% in HCT116 cells by the LCMT-1 L3 shRNA clearly indicates that it is. Our observation that cells with ~10% of their normal amount of LCMT-1 protein have 30–70% of their normal amount of PP2A methylation suggests that either there is a second PP2A methyltransferase that is a minor contributor to PP2A methylation under normal growth conditions or cells have an excess of LCMT-1 for maintaining steady-state methylation levels of PP2A under cell culture conditions. The most obvious candidate for an additional PP2A methyltransferase would be the only known homolog of LCMT-1, LCMT-2. LCMT-2 was originally cloned by De Baere et al. (16), who showed that it could not methylate PP2A in vitro. Consistent with this in vitro mammalian result, a LCMT-2 homolog exists in yeast that does not seem to contribute detectably to PP2A methylation in vivo (23, 25, 37). These results suggest that LCMT-2 is not a PP2A methyltransferase. However, it still needs to be tested in vivo whether LCMT-2 contributes to PP2A methylation in mammalian cells. One possible explanation for the apparent excess of LCMT-1 is that it may be necessary for dynamic responses to cellular needs or for methylation of a currently unknown additional LCMT-1 substrate. Other possible LCMT-1 substrates include the PP2A-related phosphatases PP4 and PP6. Both of these phosphatases share ~60% amino acid identity with PP2A and have identical carboxyl-terminal amino acids, including a carboxyl-terminal leucine, the known site of carboxyl methylation for both PP2A and PP4 (42). Whether PP6 is methylated is not known. Further experiments will be necessary to determine whether LCMT-1 (or LCMT-2) methylates PP4 and/or PP6.
We have previously shown that Bα is the PP2A B-type regulatory subunit whose assembly into heterotrimers is most dependent on PP2A C subunit methylation (24). Our current coimmunoprecipitation results provide further support for this conclusion. LCMT-1 has been shown previously to associate with PP2A in vitro (22) and to methylate PP2A on its carboxyl-terminal leucine α carboxyl group (10–16). In this study we have demonstrated further that LCMT-1 down-regulation in mammalian cells reduces both PP2A C subunit methylation and the formation of PP2ABαAC heterotrimers and that down-regulation of these same heterotrimers by Bα knockdown can cause part of the phenotypes seen with LCMT-1 knockdown. Moreover, we have previously shown that loss of PP2ABAC heterotrimer formation results in selective loss of PP2A activity toward a PP2ABAC-specific substrate (8). Thus, it is possible that the cell death induced by LCMT-1 down-regulation may be due in part to dysregulation of Bα heterotrimer function. A similar amount of apoptosis resulted when protein levels of either Bα or LCMT-1 were greatly reduced. Moreover, the pancaspase inhibitor z-VAD-fmk could prevent a portion of the cell death induced by down-regulation of either LCMT-1 or Bα, indicating that in both cases caspase-dependent and caspase-independent death pathways contribute to cell death. This similarity in the cell death observed with either LCMT-1 or Bα reduction is again consistent with the possibility that LCMT-1 down-regulation induces death to some degree through Bα dysregulation. However, cell death induced by down-regulation of these two proteins does not appear to be identical. It was more difficult to detect caspase activation in vivo and in vitro with Bα knockdown cells, and the rescue by z-VAD-fmk was consistently lower for the Bα knockdown cells than for LCMT-1 knockdown cells. These differences suggest that LCMT-1 knockdown has other effects independent of Bα that contribute to cell death. In yeast, deletion of the gene that encodes the LCMT-1 homolog, Ppm1p, not only reduces the level of PP2ABAC heterotrimers 20-fold but also causes a smaller reduction (~2-fold) in PP2AB′AC heterotrimers (23, 25, 26). Therefore, one possible effect unique to LCMT-1 knockdown may be reduction of PP2AB′AC heterotrimers. Other possible targets of LCMT-1 that have yet to be tested also exist, such as PP2A heterotrimers, formed by other members (β, γ, δ, ε) of the B/B55 regulatory subunit family or by PP2A B″ regulatory subunits and PP4 and/or PP6 complexes. Although apoptosis-relevant targets of LCMT-1 other than Bα remain to be determined, our results clearly indicate that they exist.
The increased sensitivity of both LCMT-1 and Bα knockdown cells to the spindle-targeting drug, nocodazole, suggests that both of these proteins are important in mammalian cells for the spindle checkpoint, which ensures proper spindle formation and chromosomal attachment before progression from metaphase to anaphase. The fact that cell death was largely rescued by thymidine block-induced arrest in G1/S indicates that cell cycle arrest, which is induced in HeLa cells by either nocodazole or thymidine, is not the sensitizing factor. Instead, the effect is specific to mitosis. This death in nocodazole- and not in thymidine-treated cells is consistent with previous results from yeast showing that loss of the Bα homolog, Cdc55p, or of the LCMT-1 homolog, Ppm1p, causes increased sensitivity to nocodazole. However, it was not known with the increased complexity of mammalian cells whether this phenotype would be found in this system, where there are multiple members of each B-type regulatory subunit family. In mammalian cells the mechanistic details of the roles of B-type subunits in spindle checkpoint and mitotic exit are just beginning to emerge. Both the B family isoform, Bδ, and the B′ family isoform, B′γ, have recently been identified as negative regulators of sister chromatid separation (35, 43). Bδ was found to protect cohesins from degradation by stabilizing securin, a negative regulator of the cohesin protease, separase. B′γ was reported to protect cohesin degradation by binding to Shugoshin (hSgo1) and maintaining cohesins in a more stable, hypophosphorylated state. Our data suggest that Bα is also a negative regulator of the spindle checkpoint in mammalian cells. Whether it plays yet another distinct role or shares responsibility for dephosphorylation of cohesins or securin is not known. Overexpression data such as those presented in the Bδ/securin study do not rule out a similar role for Bα. Likewise, B′γ identification as an hSgo1 partner leaves open the possibility that other B-type subunits might also associate with Shugoshin proteins. Finally, a role for LCMT-1 in spindle checkpoint is consistent with a role for PP2A methylation in mitotic progression. Experiments are under way to explore this possibility further.
PP2A regulates apoptotic pathways both positively and negatively by targeting different substrates via its diverse regulatory subunits. Consequently, PP2A inhibitors and small interfering RNA down-regulation of PP2A C and A subunits have been reported to induce apoptosis or block apoptosis, depending on the system (44–50). In both Drosophila and mammalian cells, B56/B′ subunits have been reported to have an anti-apoptotic role (45, 46, 48). Some evidence has also been reported in mammalian cells for an anti-apoptotic role for B″ and B subunit families (48). In this latter study Strack et al. (48) presented results of four different assays analyzing the importance of B55/B family subunits for cell survival. In seeming contrast to our results, one of the assays that showed little effect was small interfering RNA knockdown of Bα. However, the efficiency of their Bα knockdown was not reported, making comparison to our results difficult. Moreover, in concert with our data, results from two of their other three assays supported an anti-apoptotic role for Bα. Our finding that Bα down-regulation induces a small amount of apoptosis that is dependent on progression through mitosis is also consistent with the ability of the adenovirus protein, E4orf4, to arrest cells in G2/M and induce apoptosis in cancer cells in a manner dependent on its ability to bind PP2A Bα (51–54). Our results indicate a pro-survival function for LCMT-1 as well. This function may be mediated to some extent through modulation of PP2ABαAC heterotrimer formation, but our data suggest that Bα -independent roles should also be considered.
The fact that knockdown of either Bα or LCMT-1 caused an enhancement of cell killing by nocodazole in HeLa cells raises the possibility that Bα heterotrimers and LCMT-1 may have potential as targets for combination chemotherapy with microtubule targeting drugs like taxanes. Importantly, both Bα and LCMT-1 knockdown HeLa cells were greatly protected from cell death by arrest in G1/S, presumably by blocking the progression of the knockdown cells into mitosis, where apoptosis was triggered due to a defective spindle checkpoint. One would expect that knockdown of these proteins would not have the same sensitizing effect to nocodazole in all cell types given that some cancer cells already have defects in checkpoints and cell cycle control. HCT116 cells also showed sensitization to nocodazole (Fig. 7). However, initial experiments with H1299 lung cancer cells gave a different result. Although these cells exhibit significant death when LCMT-1 is knocked down by either L2 or L3, this cell line is already more sensitive to nocodazole than HeLa or HCT116 cells and, not unexpectedly, does not show increased nocodazole sensitivity upon LCMT-1 knockdown by the L2 and L3 shRNAs (data not shown). Further experimentation will be necessary to investigate the potential of these proteins as novel drug targets.
Consistent with the observed effects of LCMT-1 reduction on cell viability, homozygous gene trap knock-out of LCMT-1 results in embryonic lethality. The expected ratio of LCMT-1+/+: LCMT-1+/−: LCMT-1−/− progeny mice from a cross of LCMT-1+/− mice is 1:2:1. We obtained a ratio of 1:1.4:0 (Fig. 8D) with sufficient numbers of progeny to clearly indicate that homozygous loss of LCMT-1 is lethal during development. This result could not arise simply from inviable gametes because both male and female LCMT-1+/− mice are fertile when bred to wild-type mice.3 Based on our results with cells, this lethality could result from defects in cell cycle or increased apoptosis, but it is also possible that additional signaling defects may also contribute. To our knowledge this is the first report that LCMT-1 is essential for development. In addition, the lower than expected numbers of LCMT-1+/− mice also raise the possibility that hemizygous loss of LCMT-1 may cause some embryonic lethality as well. Future experiments will be aimed at elucidating the nature and timing of developmental defects that lead to the death of embryos lacking this important methyltransferase enzyme.
Acknowledgments
We are grateful to Dr. William Hahn and The RNAi Consortium at the Broad Institute for lentiviral shRNA vectors and advice, to Dr. David M. Virshup for the HA-tagged Bα construct, to Dr. Eva Schmeltz for HCT116 cells, to the German Gene Trap Consortium for the LCMT-1+/− embryonic stem cells, to David Martin and colleagues in the Emory Transgenic facility for help with the LCMT-1 knock-out, to William Dalton for advice on caspase assays, and to Anita Corbett and Jennifer Jackson for critical reading of the manuscript.
Footnotes
This work was supported by NCI, National Institutes of Health Grant CA57327 (to D. C. P.) and Predoctoral Fellowship Award CA1236402 (to J. A. L.).
The abbreviations used are: PP2A, protein phosphatase 2A; LCMT-1, leucine carboxyl methyltransferase-1; shRNA, small hairpin RNA; Bα, PP2A B55α; C subunit, PP2A catalytic subunit; A subunit, PP2A structural subunit; HA, hemagglutinin; AFC, aminofluoromethylcoumarin; DAPI, 4′,6-diamidino-2-phenylindole; z-, benzyloxycarbonyl; fmk, fluoromethyl ketone; VC, vector control; ES, embryonic stem.
J. A. Lee and D. C. Pallas, unpublished information.
References
- 1.Janssens V, Goris J. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. Cell. 1990;60:167–176. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- 3.Yang SI, Lickteig RL, Estes R, Rundell K, Walter G, Mumby MC. Mol Cell Biol. 1991;11:1988–1995. doi: 10.1128/mcb.11.4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. Cell. 1993;75:887–897. doi: 10.1016/0092-8674(93)90533-v. [DOI] [PubMed] [Google Scholar]
- 5.Cayla X, Ballmer-Hofer K, Merlevede W, Goris J. Eur J Biochem. 1993;214:281–286. doi: 10.1111/j.1432-1033.1993.tb17922.x. [DOI] [PubMed] [Google Scholar]
- 6.Campbell KS, Auger KR, Hemmings BA, Roberts TM, Pallas DC. J Virol. 1995;69:3721–3728. doi: 10.1128/jvi.69.6.3721-3728.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glenn GM, Eckhart W. J Virol. 1995;69:3729–3736. doi: 10.1128/jvi.69.6.3729-3736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogris E, Gibson DM, Pallas DC. Oncogene. 1997;15:911–917. doi: 10.1038/sj.onc.1201259. [DOI] [PubMed] [Google Scholar]
- 9.Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Cancer Cell. 2004;5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- 10.Xie H, Clarke S. J Biol Chem. 1993;268:13364–13371. [PubMed] [Google Scholar]
- 11.Lee J, Stock J. J Biol Chem. 1993;268:19192–19195. [PubMed] [Google Scholar]
- 12.Xie H, Clarke S. J Biol Chem. 1994;269:1981–1984. [PubMed] [Google Scholar]
- 13.Favre B, Zolnierowicz S, Turowski P, Hemmings BA. J Biol Chem. 1994;269:16311–16317. [PubMed] [Google Scholar]
- 14.Li M, Damuni Z. Biochem Biophys Res Commun. 1994;202:1023–1030. doi: 10.1006/bbrc.1994.2031. [DOI] [PubMed] [Google Scholar]
- 15.Floer M, Stock J. Biochem Biophys Res Commun. 1994;198:372–379. doi: 10.1006/bbrc.1994.1052. [DOI] [PubMed] [Google Scholar]
- 16.De Baere I, Derua R, Janssens V, Van Hoof C, Waelkens E, Merlevede W, Goris J. Biochemistry. 1999;38:16539–16547. doi: 10.1021/bi991646a. [DOI] [PubMed] [Google Scholar]
- 17.Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC. J Biol Chem. 1999;274:14382–14391. doi: 10.1074/jbc.274.20.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie H, Clarke S. Biochem Biophys Res Commun. 1994;203:1710–1715. doi: 10.1006/bbrc.1994.2383. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Chen Y, Tolstykh T, Stock J. Proc Natl Acad Sci U S A. 1996;93:6043–6047. doi: 10.1073/pnas.93.12.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans DR, Hemmings BA. Mol Gen Genet. 2000;264:425–432. doi: 10.1007/s004380000302. [DOI] [PubMed] [Google Scholar]
- 21.Bryant JC, Westphal RS, Wadzinski BE. Biochem J. 1999;339:241–246. [PMC free article] [PubMed] [Google Scholar]
- 22.Tolstykh T, Lee J, Vafai S, Stock JB. EMBO J. 2000;19:5682–5691. doi: 10.1093/emboj/19.21.5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Tolstykh T, Lee J, Boyd K, Stock JB, Broach JR. EMBO J. 2000;19:5672–5681. doi: 10.1093/emboj/19.21.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu XX, Du X, Moreno CS, Green RE, Ogris E, Feng Q, Chou L, McQuoid MJ, Pallas DC. Mol Biol Cell. 2001;12:185–199. doi: 10.1091/mbc.12.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei H, Ashby DG, Moreno CS, Ogris E, Yeong FM, Corbett AH, Pallas DC. J Biol Chem. 2001;276:1570–1577. doi: 10.1074/jbc.M008694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gentry MS, Li Y, Wei H, Syed FF, Patel SH, Hallberg RL, Pallas DC. Eukaryot Cell. 2005;4:1029–1040. doi: 10.1128/EC.4.6.1029-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nunbhakdi-Craig V, Schuechner S, Sontag JM, Montgomery L, Pallas DC, Juno C, Mudrak I, Ogris E, Sontag E. J Neurochem. 2007;101:959–971. doi: 10.1111/j.1471-4159.2007.04503.x. [DOI] [PubMed] [Google Scholar]
- 28.Lee TH, Turck C, Kirschner MW. Mol Biol Cell. 1994;5:323–338. doi: 10.1091/mbc.5.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minshull J, Straight A, Rudner AD, Dernburg AF, Belmont A, Murray AW. Curr Biol. 1996;6:1609–1620. doi: 10.1016/s0960-9822(02)70784-7. [DOI] [PubMed] [Google Scholar]
- 30.Yellman CM, Burke DJ. Mol Biol Cell. 2006;17:658–666. doi: 10.1091/mbc.E05-04-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Ng TY. Mol Biol Cell. 2006;17:80–89. doi: 10.1091/mbc.E04-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turowski P, Fernandez A, Favre B, Lamb NJ, Hemmings BA. J Cell Biol. 1995;129:397–410. doi: 10.1083/jcb.129.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu T, Matsuzawa S, Mizuno Y, Kamibayashi C, Mumby MC, Andjelkovic N, Hemmings BA, Onoe K, Kikuchi K. Arch Biochem Biophys. 1997;339:210–217. doi: 10.1006/abbi.1996.9835. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Burke DJ. Mol Cell Biol. 1997;17:620–626. doi: 10.1128/mcb.17.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gil-Bernabe AM, Romero F, Limon-Mortes MC, Tortolero M. Mol Cell Biol. 2006;26:4017–4027. doi: 10.1128/MCB.01904-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 37.Kalhor HR, Luk K, Ramos A, Zobel-Thropp P, Clarke S. Arch Biochem Biophys. 2001;395:239–245. doi: 10.1006/abbi.2001.2558. [DOI] [PubMed] [Google Scholar]
- 38.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 39.Kerr JF, Wyllie AH, Currie AR. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Biochem Biophys Res Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 42.Honkanen RE, Golden T. Curr Med Chem. 2002;9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- 43.Kitajima TS, Sakuno T, Ishiguro K, Iemura S, Natsume T, Kawashima SA, Watanabe Y. Nature. 2006;441:46–52. doi: 10.1038/nature04663. [DOI] [PubMed] [Google Scholar]
- 44.Yan Y, Shay JW, Wright WE, Mumby MC. J Biol Chem. 1997;272:15220–15226. doi: 10.1074/jbc.272.24.15220. [DOI] [PubMed] [Google Scholar]
- 45.Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Proc Natl Acad Sci U S A. 2002;99:4221–4226. doi: 10.1073/pnas.072071699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Scuderi A, Letsou A, Virshup DM. Mol Cell Biol. 2002;22:3674–3684. doi: 10.1128/MCB.22.11.3674-3684.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chatfield K, Eastman A. Biochem Biophys Res Commun. 2004;323:1313–1320. doi: 10.1016/j.bbrc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 48.Strack S, Cribbs JT, Gomez L. J Biol Chem. 2004;279:47732–47739. doi: 10.1074/jbc.M408015200. [DOI] [PubMed] [Google Scholar]
- 49.Ray RM, Bhattacharya S, Johnson LR. J Biol Chem. 2005;280:31091–31100. doi: 10.1074/jbc.M503041200. [DOI] [PubMed] [Google Scholar]
- 50.Bonness K, Aragon IV, Rutland B, Ofori-Acquah S, Dean NM, Honkanen RE. Mol Cancer Ther. 2006;5:2727–2736. doi: 10.1158/1535-7163.MCT-06-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shtrichman R, Sharf R, Barr H, Dobner T, Kleinberger T. Proc Natl Acad Sci U S A. 1999;96:10080–10085. doi: 10.1073/pnas.96.18.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shtrichman R, Kleinberger T. J Virol. 1998;72:2975–2982. doi: 10.1128/jvi.72.4.2975-2982.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marcellus RC, Chan H, Paquette D, Thirlwell S, Boivin D, Branton PE. J Virol. 2000;74:7869–7877. doi: 10.1128/jvi.74.17.7869-7877.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shtrichman R, Sharf R, Kleinberger T. Oncogene. 2000;19:3757–3765. doi: 10.1038/sj.onc.1203705. [DOI] [PubMed] [Google Scholar]