

First Synthesis
The highly complex structure of syndecan-1 heparan sulfate glycopeptide was successfully assembled. The protective groups utilized and sequences for glycosyl linkage formation and protective group removal are critical for the success of synthesis. This is the first time this type of sulfated glycopeptides has been prepared, which lays the foundation to access other members of this important class of molecules.
Keywords: glycopeptides, glycosylation, synthetic design, sulfation, total synthesis
Heparan sulfate proteoglycans (HSPGs), which are composed of three parts: heparan sulfate (HS), a tetrasaccharide linker and the core protein, are ubiquitous components of mammalian cell surface and the extracellular matrix.[1] This family of molecules can serve as receptors for many adhesion molecules and growth factors, such as fibroblast growth factor, integrin, Wnt proteins and herpes simplex virus glycoprotein D.[2] HSPGs play important roles in a variety of biological processes including cancer development, angiogenesis, viral infection, and wound repair,[3] which render them interesting targets for the development of novel therapeutic interventions.[4] Although the biological activities of HSPGs have been largely attributed to the HS chains, there are increasing evidences that both the HS chain and the core protein can be important for directing the biological functions.[3a, 5] Naturally existing HSPGs are highly heterogeneous mixtures, where a single core protein contains diverse HS structures. This renders it extremely challenging to isolate HSPGs bearing a homogeneous HS chain, hindering the thorough understanding of the structure-function relationship. Chemical synthesis of HSPGs can reduce the structural heterogeneity and provide sufficient quantity for biological evaluations. Although much work has been done on synthesis of HS[6–7] and the tetrasaccharide linker,[8] no synthetic methods are available to date to prepare HSPGs. Herein, we report our studies in synthesis of HS glycopeptides containing homogenous HS glycans.
We are interested in human syndecan-1, an important HSPG on cell membrane.[4, 9] Our synthetic target is HS glycopeptide 1 from the extracellular domain of syndecan-1, which contains the binding sites for many receptors.[9d] The octasaccharide in 1 incorporates typical structural features of HSPGs, including the full tetrasaccharide linker, glucosamine (GlcN) α linked to both glucuronic acid (GlcA) and iduronic acid (IdoA), 2-O sulfation, 6-O sulfation and N-acetylation. Successful synthesis of this molecule will lay the groundwork for accessing other HS glycopeptides.
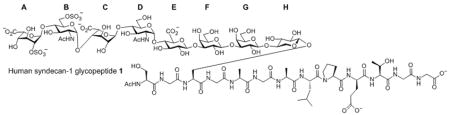
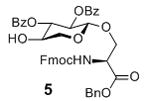
To prepare the octasaccharide of glycopeptide 1, our original strategy adapted a 2+5+1 glycosylation approach, where the linkages between units B (GlcN) and C (IdoA), and units G and H were to be formed by coupling disaccharide 2 with pentasaccharide 3 and pentasaccharide donor 4 with xyloside serine derivative 5 respectively. Although the α glycosyl linkage between GlcN and IdoA was formed stereospecifically using monosaccharide building blocks in our previous study,[10] the coupling between disaccharide donor 2 and pentasaccharide 3 failed to yield the desired heptasaccharide with donor decomposition products isolated as the major side products. Varying the protective groups on 2 and 3 did not improve the situation. The failure was attributed to the reduced glycosylation power of these building blocks towards unreactive acceptors compared to their monosaccharide counterparts. This was presumably due to their larger sizes and the electron withdrawing properties of additional glycan units, which can reduce their reactivities towards acceptors.[11] Besides the difficulty encountered in the 2+5 coupling, the 5+1 glycosylation of pentasaccharide donor 4 with the xyloside serine acceptor 5 was not productive either. These setbacks led us to consider a new 3+2+3 synthetic route, where the difficult linkages between B and C, G and H were to be formed earlier in the synthesis.
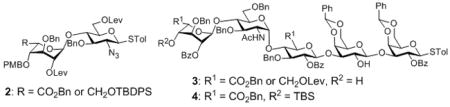
The “3 + 2 + 3” strategy called for the preparation of ABC trisaccharide 7, DE disaccharide 8 and FGH trisaccharide 9 (Figure 1). The synthesis of ABC trisaccharide 7 started from the coupling of donor 10 with acceptor 11 using the pre-activation protocol[12] with the promoter system of p-TolSCl/AgOTf.[13] This afforded α linked BC disaccharide 12 in 80% yield with complete stereoselectivity (Scheme 1a). All the pre-activation based glycosylation reactions were performed with a slight excess (1.1 eq) of glycosyl donor compared to the acceptor. Previously, when the p-methoxybenzyl (PMB) group was used to mask the 6-O position of glycosyl donors, the electron rich 6-OPMB moiety participated in the reaction forming an 1,6-anhydro sugar shutting down the desired glycosylation.[10] To avoid this side reaction, the PMB in 12 was exchanged with the tertbutyldiphenylsilyl group (TBDPS) followed by protective group adjustment generating disaccharide acceptor 13. Coupling of the idosyl thioglycoside 14 with disaccharide 13 produced ABC trisaccharide 7 (Scheme 1b).
Figure 1.
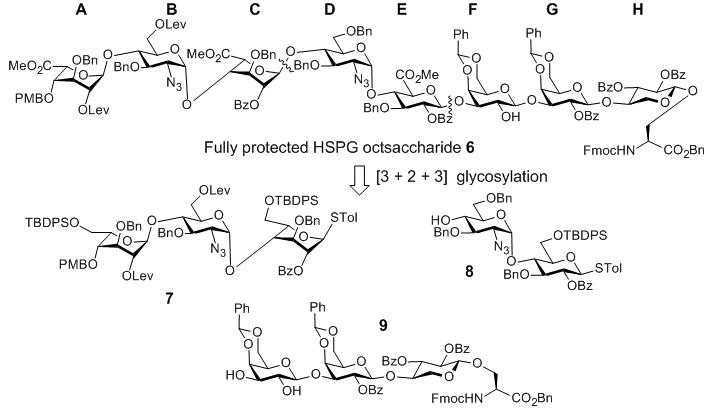
Retrosynthetic analysis of octasaccharide amino ester 6.
Scheme 1.
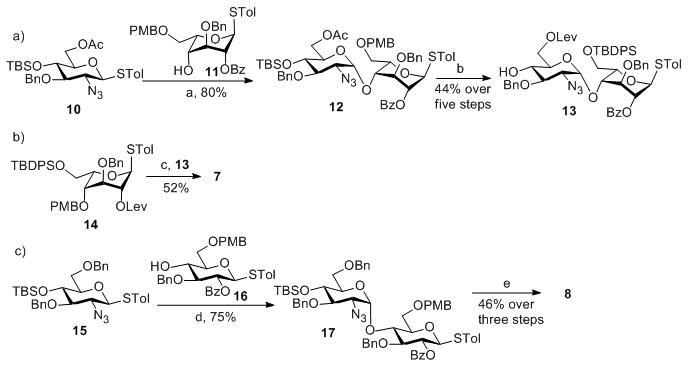
Reagents and conditions: a) AgOTf, p-TolSCl, MS 4Å, −78°C, then 11, TTBP, −78°C–0°C; b) (1) Mg(OMe)2, DCM, −20°C–0°C; (2) LevOH, EDC-HCl, DMAP, DCM; (3) DDQ, DCM/H2O; (4) HF/Pyridine; (5) TBDPSCl, imidazole, DCM; c) AgOTf, p-TolSCl, MS 4Å, −78°C, then 13, TTBP, −78°C–0°C; d) AgOTf, p-TolSCl, MS 4Å, −78°C, then 16, TTBP, −78°C–0°C; e) (1) DDQ, DCM/H2O; (2) HF/Pyridine; (3) TBDPSCl, imidazole, DCM.
The middle DE disaccharide 8 was formed in 75% yield by reaction of the glucosamine donor 15 with glucoside acceptor 16 under the pre-activation condition (Scheme 1c). Removal of the tertbutyldimethylsilyl (TBS) moiety with subsequent exchange of PMB with TBDPS produced disaccharide acceptor 8.
With the ABC and DE modules in hand, we turned our attention to the preparation of FGH trisaccharide acceptor 9. TBS protected xyloside donor 18 reacted with serine 19 followed by TBS removal leading to acceptor 5 (Scheme 2a). The formation of the challenging GH linkage was then tested using galactoside donor 20, which successfully glycosylated acceptor 5 in 81% yield (Scheme 2b). This suggested that the difficulty in pentasaccharide 4 glycosylation of 5 was indeed a result of the additional glycans on the pentasaccharide donor.
Scheme 2.
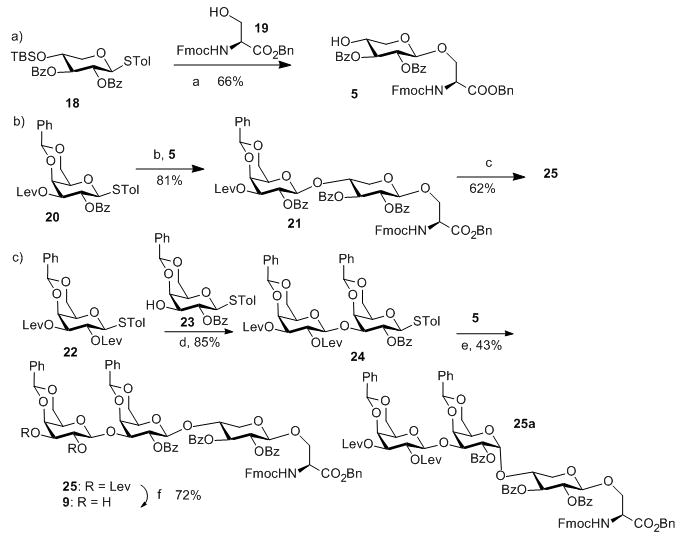
Reagents and conditions: a) (1) AgOTf, p-TolSCl, MS 4Å, −78°C, then 19, TTBP, −78°C–0°C; (2) Tf2O, THF, H2O; b) AgOTf, p-TolSCl, MS 4Å, −78°C, then 5, TTBP, −78°C–0°C; c) (1) AcOH, NH2NH2, DCM/MeOH; (2) 22, AgOTf, p-TolSCl, MS 4Å, TTBP, −78°C–0°C; d) AgOTf, p-TolSCl, MS 4Å, −78°C, then 23, TTBP, −78°C–0°C; e) AgOTf, p-TolSCl, MS 4Å, −78°C, then 5, TTBP, −78°C–0°C; f) AcOH, NH2NH2, DCM/MeOH.
To prepare trisaccharide 9, diLev protected galactoside donor 22 glycosylated acceptor 23 forming digalactoside 24 in 85% yield (Scheme 2c). 24 was then subjected to reaction with acceptor 5. The desired trisaccharide 25 was obtained in 43% yield in addition to the α anomer 25a (45%). We tested various glycosylation conditions (donor/acceptor ratio, solvent, temperature, etc). However, none of these trials improved the stereoselectivity. Considering the high stereoselectivity in formation of 21 and disaccharide donor 24 bearing 2-OBz moiety at the reducing end, which can function as a participating neighbouring group, the loss of stereoselectivity in reaction of 24 with 5 was intriguing and the exact reason is unknown. Several other groups have reported the loss of 1,2-trans selectivities using 2-O acyl protected galactosyl donors.[14] As an alternative, the Lev group in disaccharide 21 was removed and the resulting acceptor was glycosylated with galactoside 22 to generate trisaccharide 25 in 62% yield (Scheme 2b). The two Lev groups in trisaccharide 25 were removed by hydrazine acetate to afford FGH trisaccharide acceptor 9 (Scheme 2c).
Having prepared the three required modules, we investigated the union of these building blocks. The 3+2 coupling between trisaccharide donor 7 and disaccharide acceptor 8 proceeded smoothly producing pentasaccharide 26 in an excellent 93% yield. Subsequent “5 + 3” coupling between 26 and trisaccharide 9 gave octasaccharide 27 in 83% yield, thus completing the oligosaccharide backbone of the glycopeptide (Scheme 3). Octasaccharide 27 was treated with HF in pyridine to cleave the TBDPS protective groups and expose three primary hydroxyl groups, which were oxidized[15] followed by methyl ester formation[16] to afford octasaccharide amino ester 6 ready for peptide chain elongation (Scheme 3).
Scheme 3.
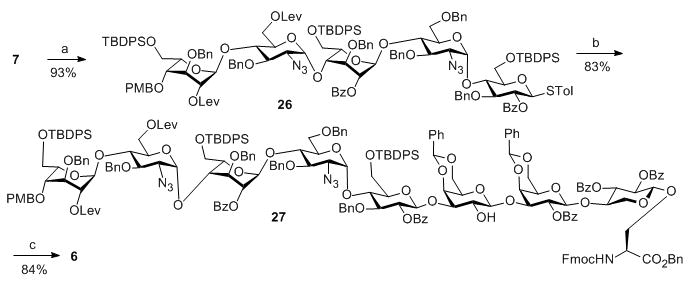
Reagents and conditions: a) AgOTf, p-TolSCl, MS 4Å, −78°C, then 8, TTBP, −78°C–0°C; b) AgOTf, p-TolSCl, MS 4Å, −78°C, then 9, TTBP, −78°C–0°C; c) (1) HF/pyridine; (2) TEMPO, BAIB, DCM/H2O/t-BuOH; (3) MeI, K2CO3, DMF.
To avoid the undesired reaction on the 2-OH of unit F in 6 during O-sulfation, we blocked this 2-OH with acetyl (Ac), followed by piperidine treatment exposing the amino group (Scheme 4a). The free amine 28 was then subject to coupling with dipeptide 29 to afford glycopeptide 30. Selective hydrogenation of 30 in the presence of NH4OAc[17] generated the free carboxylic acid 31 without affecting the benzyl ethers in the molecule.
Scheme 4.
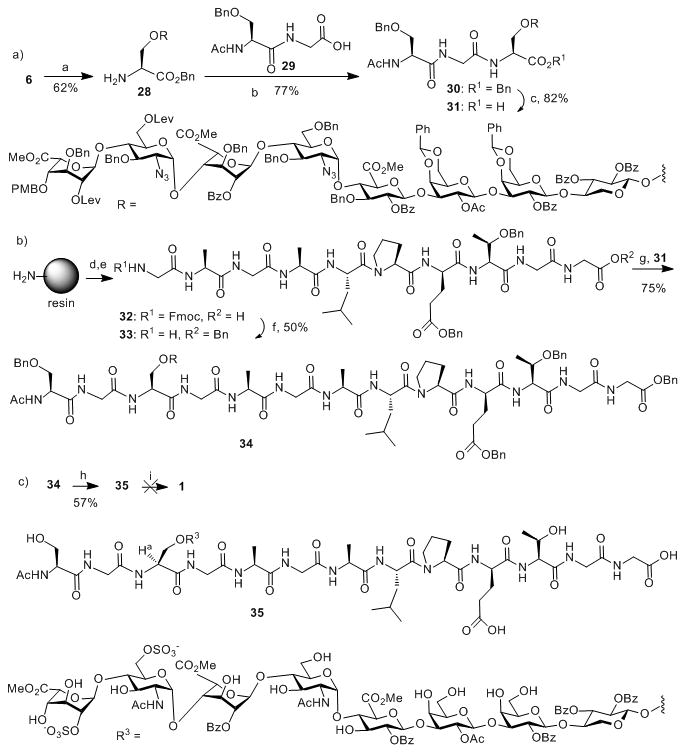
Reagents and conditions: a) (1) Ac2O, pyridine, 50 °C; (2) piperidine, DCM; b) HATU, DIPEA, DMF; c) Pd/C, H2, NH4OAc, DCM/MeOH; d) multiple rounds of automated solid phase peptide synthesis; e) TFA/H2O/Phenol/TIPS; f) (1) BnBr, DIPEA, DMF; (2) piperidine, DMF; g) HATU, DMF; h) (1) Zn, CuSO4 (sat.), Ac2O/THF/HOAc; (2) NH2NH2-H2O, HOAc, DCM/MeOH; (3) SO3-Et3N, DMF, 55 °C; (4) H2, Pd/C, DCM/MeOH; i) base catalyzed hydrolysis
Parallel to oligosaccharide synthesis, solid phase peptide synthesis was performed to prepare the C-terminal peptide 33 (Scheme 4b). As the final product will contain acid sensitive O-sulfates, the traditional strong acid promoted peptide deprotection needs to be avoided. Due to this consideration, the side chains of glutamic acid and threonine were protected as benzyl ester and benzyl ether respectively. The peptide was prepared on chloro-trityl resin and cleaved from solid phase by acid treatment (TFA/H2O/Phenol/TIPS) (Scheme 4b). The free carboxylic acid terminal was subsequently protected as benzyl ester and the N-terminal Fmoc was removed to generate the nonapeptide 33 with a free amino terminal. Coupling of octasaccharide carboxylic acid 31 with peptide 33 without any base additive[18] successfully produced glycopeptide 34 in 75% yield (Scheme 4b). It was important to omit the base during this step to avoid the β-elimination of the glycan chain from the peptide backbone.
To accomplish the deprotection of fully protected glycopeptide 34, the right reaction sequence is critical. Despite the fact that simultaneous hydrogenolysis of the benzyl ethers and the azido groups in HS oligosaccharide synthesis were reported before,[7i, 10, 19] treatment of 34 under catalytic hydrogenation condition led to a complex reaction mixture. To overcome this, we resorted to selective reduction of azide by zinc and copper sulfate with simultaneous acetylation of the newly liberated free amines (Scheme 4c).[20] The acetylated glycopeptides were then subjected to selective removal of the levulinoyl (Lev) groups by hydrazine acetate, followed by O-sulfation of the free hydroxyl groups and hydrogenation producing glycopeptide 35 (Scheme 4c). Although hydrolysis of the acetate and benzoate esters was the only step remaining to the final glycopeptides, it proved to be extremely challenging. We investigated a variety of reagents including LiOOH, NaOH and LiOH with pH values ranging from 8 to 11. Unfortunately, with pH around 8.5, Bz could not be removed while under higher pH, we observed significant degree of β-elimination of the glycan, which is a common problem in glycopeptide synthesis.[21]
The failure to deprotect 35 prompted us to redesign our deprotection sequence. Since β-elimination is presumably caused by deprotonation of the α-proton of the glycosylated serine (Ha marked in 35), we envision that if the serine carboxylic acid is kept in its free form, its deprotonation should raise the pKa value of the alpha-proton on the neighbouring carbon due to electrostatic repulsion, thus reducing the potential for β-elimination. Furthermore, Kihlberg and coworkers reported that partially deprotected glycopeptide was more stable against β-elimination compared to the fully protected form.[21] Thus, an alternative route was designed to first remove all the protecting groups on compound 30 except the three methyl esters, and to couple this partially deprotected glycopeptide to peptide 33. The assembled glycopeptide can be transformed to 1 via hydrogenation and mild base treatment since methyl esters are more sensitive to base hydrolysis. To test this, we converted 30 to its partially protected form 36 (azide reduction/acetylation, Lev removal, O-sulfation, hydrogenation and transesterification). For the transesterification step, pH was carefully maintained at 9.5 in order to minimize β-elimination while retaining the methyl esters by NaOMe treatment. All Ac and Bz groups were cleaved under this condition in 40 hours. With compound 36 in hand, we tested its coupling with peptide 33. It was shown that sulfates did not affect peptide coupling reactions.[18] Indeed, reaction of 36 with 33 produced glycopeptide 37 in 60% yield. Subsequent hydrogenation and methyl ester hydrolysis afforded the final glycopeptide 1 (Scheme 5).
In conclusion, we have established a strategy for the first synthesis of the highly complex structure of syndecan-1 HS glycopeptide. The protective groups utilized and the sequences for glycosyl linkage formation and protective group removal are critical for the successful synthesis. We believe the approach established and knowledge gained can be applied to the synthesis of other HS glycopeptides. Work in this regard as well as investigation of the exciting biological functions of the prepared HSPG bearing homogenous HS glycans is currently ongoing.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (CHE 1111550) and the National Institute of General Medical Sciences, NIH (R01GM072667).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author
References
- 1.Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Tumova S, Woods A, Couchman JR. Int J Biochem Cell Biol. 2000;32:269–288. doi: 10.1016/s1357-2725(99)00116-8. [DOI] [PubMed] [Google Scholar]
- 3.a) Choi Y, Chung H, Jung H, Couchman JR, Oh ES. Matrix Biol. 2011;30:93–99. doi: 10.1016/j.matbio.2010.10.006. [DOI] [PubMed] [Google Scholar]; b) Lambaerts K, Wilcox-Adelman SA, Zimmermann P. Curr Opin Cell Biol. 2009;21:662–669. doi: 10.1016/j.ceb.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Khotskaya YB, Dai Y, Ritchie JP, MacLeod V, Yang Y, Zinn K, Sanderson RD. J Biol Chem. 2009;284:26085–26095. doi: 10.1074/jbc.M109.018473. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bass MD, Morgan MR, Humphries MJ. Sci Signal. 2009;2:pe18. doi: 10.1126/scisignal.264pe18. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lin X. Development. 2004;131:6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 4.Yang Y, MacLeod V, Dai Y, Khotskaya-Sample Y, Shriver Z, Venkataraman G, Sasisekharan R, Naggi A, Torri G, Casu B, Vlodavsky I, Suva LJ, Epstein J, Yaccoby S, Shaughnessy JD, Barlogie B, Sanderson RD. Blood. 2007;110:2041–2048. doi: 10.1182/blood-2007-04-082495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) De Cat B, Muyldermans SY, Coomans C, Degeest G, Vanderschueren B, Creemers J, Biemar F, Peers B, David G. J Cell Biol. 2003;163:625–635. doi: 10.1083/jcb.200302152. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Iozzo RV. J Clin Invest. 2001;108:165–167. doi: 10.1172/JCI13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For representative reviews on HS synthesis, see: Dulaney SB, Huang X. Adv Carbohydr Chem Biochem. 2012;67:95–136. doi: 10.1016/B978-0-12-396527-1.00003-6.and references cited therein; Noti C, Seeberger PH. In: Chemistry and Biology of Heparin and Heparan Sulfate. Garg HG, Linhardt RJ, Hales CA, editors. Elsevier; Oxford: 2005. pp. 79–142.Petitou M, van Boeckel CAA. Angew Chem Int Ed. 2004;43:3118–3133. doi: 10.1002/anie.200300640.and references cited therein; Codee JDC, Overkleeft HS, van der Marel GA, van Boeckel CAA. Drug Discovery Today: Technol. 2004;1:317–326. doi: 10.1016/j.ddtec.2004.11.017.Poletti L, Lay L. Eur J Org Chem. 2003:2999–3024.Karst NA, Linhardt RJ. Curr Med Chem. 2003;10:1993–2031. doi: 10.2174/0929867033456891.
- 7.For representative publications on HS synthesis, see: Zulueta MML, Lin SY, Lin YT, Huang CJ, Wang CC, Ku CC, Shi Z, Chyan CL, Irene D, Lim LH, Tsai TI, Hu YP, Arco SD, Wong CH, Hung SC. J Am Chem Soc. 2012;134:8988–8995. doi: 10.1021/ja302640p.and references cited therein; Xu Y, Wang Z, Liu R, Bridges AS, Huang X, Liu J. Glycobiology. 2012;22:96–106. doi: 10.1093/glycob/cwr109.and references cited therein; Czechura P, Guedes N, Kopitzki S, Vazquez N, Martin-Lomas M, Reichardt NC. Chem Commun. 2011;47:2390–2392. doi: 10.1039/c0cc04686h.Tiruchinapally G, Yin Z, El-Dakdouki M, Wang Z, Huang X. Chem Eur J. 2011;17:10106–10112. doi: 10.1002/chem.201101108.Arungundram S, Al-Mafraji K, Asong J, Leach FE, Amster IJ, Venot A, Turnbull JE, Boons GJ. J Am Chem Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k.Baleux F, Loureiro-Morais L, Hersant Y, Clayette P, Arenzana-Seisdedos F, Bonnaffé D, Lortat-Jacob H. Nat Chem Biol. 2009;5:743–748. doi: 10.1038/nchembio.207.and references cited therein; Tatai J, Fügedi P. Tetrahedron. 2008;64:9865–9873.Chen J, Zhou Y, Chen C, Xu W, Yu B. Carbohydr Res. 2008;343:2853–2862. doi: 10.1016/j.carres.2008.06.011.Polat T, Wong CH. J Am Chem Soc. 2007;129:12795–12800. doi: 10.1021/ja073098r.Noti C, de Paz JL, Polito L, Seeberger PH. Chem Eur J. 2006;12:8664–8686. doi: 10.1002/chem.200601103.Codée JDC, Stubba B, Schiattarella M, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. J Am Chem Soc. 2005;127:3767–3773. doi: 10.1021/ja045613g.Fan RH, Achkar J, Hernandez-Torres JM, Wei A. Org Lett. 2005;7:5095–5098. doi: 10.1021/ol052130o.Poletti L, Fleischer M, Vogel C, Guerrini M, Torri G, Lay L. Eur J Org Chem. 2001:2727–2734.Petitou M, Herault JP, Bernat A, Driguez PA, Duchaussoy P, Lormeau JC, Herbert JM. Nature. 1999;398:417–422. doi: 10.1038/18877.Sinay P, Jacquinet JC, Petitou M, Duchaussoy P, Lederman I, Choay J, Torri G. Carbohydr Res. 1984;132:C5–C9. doi: 10.1016/0008-6215(87)80268-9.
- 8.For representative publications on tetrasaccharide linker synthesis, see: Huang TY, Zulueta MML, Hung SC. Org Lett. 2011;13:1506–1509. doi: 10.1021/ol200192d.Tamura J-i, Nakamura-Yamamoto T, Nishimura Y, Mizumoto S, Takahashi J, Sugahara K. Carbohydr Res. 2010;345:2115–2123. doi: 10.1016/j.carres.2010.06.019.Shimawaki K, Fujisawa Y, Sato F, Fujitani N, Kurogochi M, Hoshi H, Hinou H, Nishimura SI. Angew Chem, Int Ed. 2007;46:3074–3079. doi: 10.1002/anie.200604909.Thollas B, Jacquinet JC. Org Biomol Chem. 2004;2:434–442. doi: 10.1039/b314244b.Tamura J, Yamaguchi A, Tanaka J. Bioorg Med Chem Lett. 2002;12:1901–1903. doi: 10.1016/s0960-894x(02)00327-x.Allen JG, Fraser-Reid B. J Am Chem Soc. 1999;121:468–469.Yasukochi T, Fukase K, Suda Y, Takagaki K, Endo M, Kusumoto S. Bull Chem Soc Jpn. 1997;70:2719–2725.Neumann KW, Tamura J, Ogawa T. Glycoconjugate J. 1996;13:933–936. doi: 10.1007/BF01053188.Neumann KW, Tamura JI, Ogawa T. Bioorg Med Chem. 1995;3:1637–1650. doi: 10.1016/0968-0896(95)00151-4.Rio S, Beau JM, Jacquinet JC. Carbohydr Res. 1993;244:295–313. doi: 10.1016/0008-6215(83)85009-5.
- 9.a) Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IMS, Abildgaard N, Waage A, Børset M. Blood. 2000;95:388–392. [PubMed] [Google Scholar]; b) Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M. Nature Med. 1998;4:691–697. doi: 10.1038/nm0698-691. [DOI] [PubMed] [Google Scholar]; c) Kainulainen V, Wang H, Schick C, Bernfield M. J Biol Chem. 1998;273:11563–11569. doi: 10.1074/jbc.273.19.11563. [DOI] [PubMed] [Google Scholar]; d) Mali M, Jaakkola P, Arvilommi AM, Jalkanen M. J Biol Chem. 1990;265:6884–6889. [PubMed] [Google Scholar]
- 10.Wang Z, Xu Y, Yang B, Tiruchinapally G, Sun B, Liu R, Dulaney S, Liu J, Huang X. Chem Eur J. 2010;16:8365–8375. doi: 10.1002/chem.201000987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koeller KM, Wong CH. Chem Rev. 2000;100:4465–4493. doi: 10.1021/cr990297n.and references cited therein; Zeng Y, Wang Z, Whitfield D, Huang X. J Org Chem. 2008;73:7952–7962. doi: 10.1021/jo801462r.
- 12.a) Sun B, Srinivasan B, Huang X. Chem Eur J. 2008;14:7072–7081. doi: 10.1002/chem.200800757. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miermont A, Zeng Y, Jing Y, Ye XS, Huang X. J Org Chem. 2007;72:8958–8961. doi: 10.1021/jo701694k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang L, Wang Z, Li X, Ye XS, Huang X. Carbohydr Res. 2006;341:1669–1679. doi: 10.1016/j.carres.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang X, Huang L, Wang H, Ye XS. Angew Chem Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 14.a) Kato M, Hirai G, Sodeoka M. Chem Lett. 2011;40:877–879. [Google Scholar]; b) Jacquinet JC. Carbohydr Res. 2004;339:349–359. doi: 10.1016/j.carres.2003.10.012. [DOI] [PubMed] [Google Scholar]; c) Imamura A, Ando H, Korogi S, Tanabe G, Muraoka O, Ishida H, Kiso M. Tetrahedron Lett. 2003;44:6725–6728. [Google Scholar]; d) Chen L, Kong F. Tetrahedron Lett. 2003;44:3691–3695. [Google Scholar]
- 15.Van den Bos LJ, Codee JDC, Van dTJC, Boltje TJ, Van BJH, Overkleeft HS, Van der Marel GA. Org Lett. 2004;6:2165–2168. doi: 10.1021/ol049380+. [DOI] [PubMed] [Google Scholar]
- 16.a) Barbier M, Grand E, Kovensky J. Carbohydr Res. 2007;342:2635–2640. doi: 10.1016/j.carres.2007.08.019. [DOI] [PubMed] [Google Scholar]; b) Xie J. Eur J Org Chem. 2002:3411–3418. [Google Scholar]
- 17.Kim KS, Kim JH, Lee YJ, Lee YJ, Park J. J Am Chem Soc. 2001;123:8477–8481. doi: 10.1021/ja015842s. [DOI] [PubMed] [Google Scholar]
- 18.Kaczmarek P, Tocci GM, Keay SK, Adams KM, Zhang CO, Koch KR, Grkovic D, Guo L, Michejda CJ, Barchi JJ., Jr ACS Med Chem Lett. 2010;1:390–394. doi: 10.1021/ml100087a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JC, Lu XA, Kulkarni SS, Wen YS, Hung SC. J Am Chem Soc. 2004;126:476–477. doi: 10.1021/ja038244h. [DOI] [PubMed] [Google Scholar]
- 20.Vohra Y, Buskas T, Boons GJ. J Org Chem. 2009;74:6064–6071. doi: 10.1021/jo901135k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sjoelin P, Kihlberg J. J Org Chem. 2001;66:2957–2965. doi: 10.1021/jo001584q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.