

Cytochrome c oxidase (CcO) catalyzes the four electron reduction of dioxygen to form water in the terminal step of the electron transport chain.[1] Dioxygen binds to a unique heme-copper bimetallic active site, wherein the copper is ligated ~5 Å above the heme by three His residues.[2–3] One of these ligating His residues is covalently crosslinked to a nearby Tyr residue. The crosslinked Tyr is thought to participate in catalysis by providing the fourth electron needed to cleave the O-O bond in a net H• abstraction.[4] This hypothesis stems from the observation of an intermediate state (PM) in CcO that occurs after O-O bond cleavage, which is suggested to contain a tyrosyl radical based on chemical and spectral evidence.[5–8] The only observable enzymatic dioxygen intermediate before O-O bond rupture has been assigned as a ferric-superoxo species (A),[9–10] leading some to suggest this species is directly responsible for the net H• abstraction from the Tyr.[11–12] We and others favor an alternative mechanistic scenario, in which an unobserved peroxo intermediate functions as the active oxidant.[13–15] A putative peroxo moiety would take advantage of the His-Tyr crosslink and the copper ion as a pathway to access the fourth electron necessary for cleavage of its O-O bond. However, heme-peroxo-copper adducts are generally unreactive towards phenols, motivating efforts towards understanding factors required for O-O bond rupture by heme-copper sites. Recently, we reported preliminary evidence that the reaction of a heme-peroxo-copper adduct {[F8Fe]-O2-[CuAN]}+ (1, F8 = 5,10,15,20-tetrakis-(2,6-difluorophenyl)-porphyrinate, AN = bis(3-(dimethylamino)-propyl)-amine) with a coordinating base DCHIm (DCHIm = 1,5-dicyclohexylimidazole) results in formation of a discrete complex (2) that has enhanced reactivity towards phenols.[16] Herein we report the molecular and electronic structure of 2, which is an example of a heme-peroxo-copper complex in which the electronic state of the heme fragment is low-spin (LS).[17] Concomitant with the change in spin of the heme fragment from high-spin (HS) to LS upon conversion from 1 to 2, the Fe-O2-Cu core undergoes a change from µ-η2:η2 (“side-on”) in 1 to µ-1,2 (“end-on”) in 2 (Scheme 1). This novel bridging mode has not been observed previously in heme-copper model complexes, but it has been proposed in recent crystallographic studies on resting CcO.[18–20] However, comparison of the spectral features of resting CcO to those described herein for 2 reveal inconsistencies suggesting a reevaluation of the bridging mode of the peroxo group in resting CcO and providing insight into the electronic structure requirements for O-O bond cleavage.
Scheme 1.
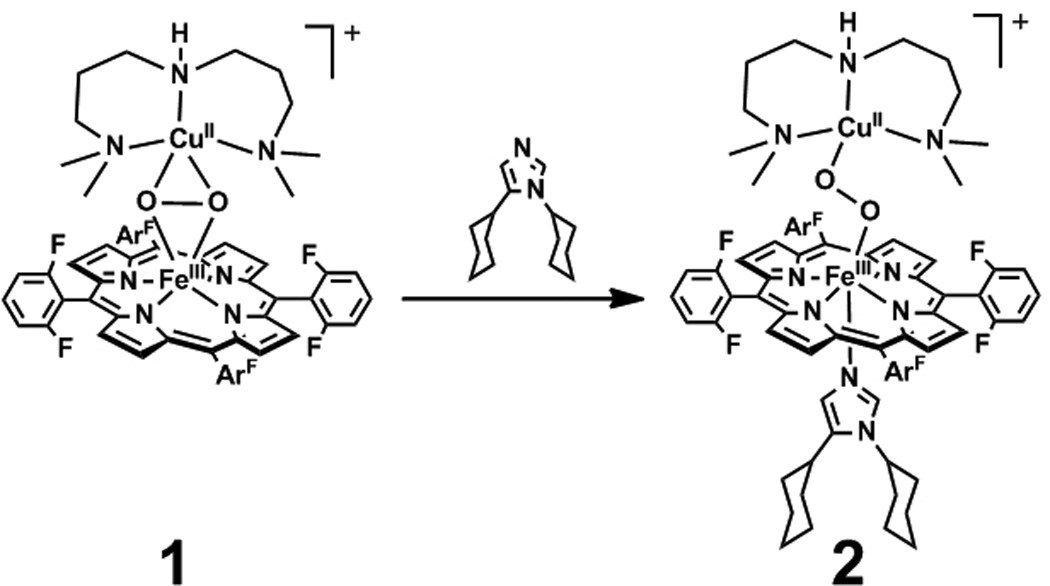
Reaction of 1 with DCHIm at cryogenic temperatures yields a discrete intermediate 2.
Low temperature reaction of a degassed THF solution of 1 with a molar equivalent of DCHIm generated 2 as indicated by changes in the optical spectrum (Figure 1). Specifically, a shift in the Soret from 418 nm (ε: 133.6 mM−1cm−1) to 421 nm (ε: 142.7 mM−1cm−1) was observed in addition to shift and collapse of the split Q-band of 1 (λmax, nm (ε, mM−1cm−1) = 538 (8.4), 561 (6.9)) to 537 nm (ε: 11.5 mM−1cm−1) in 2. While the spectrum of 2 is dominated by the heme spectral features, two low energy features were observed at 789 nm (ε: 1.5 mM−1cm−1) and 951 nm (ε: 0.9 mM−1cm−1). 2 is meta-stable at 193 K, decaying slowly to {[(F8)Fe]-O-[Cu(AN)]}+ with τ½ = 5 hr. 2 is EPR silent, consistent with the previously reported 2H-NMR data which indicated a diamagnetic ground state.[16]
Figure 1.

UV/Vis spectra of 1 (—) and 2 (—) at 193 K in THF overlaid with mid-energy region scaled by a factor of 10 and low-energy region scaled by a factor of 40.
Resonance Raman (rR) studies were preformed on 2 over a wide range of excitation energies. Excitation at 413 nm results in a spectrum dominated by heme vibrational features (Figure S1). Nonetheless, an isotope sensitive feature was observed at 575 cm−1, which downshifted to 552 cm−1 in the 18O2 isotopologue. This feature is identical to an isotope sensitive feature observed in independently prepared [(F8)Fe(O2)(DCHIm)], and thus is assigned on this basis as the νFe—O of a minor amount (< 20 %) of [(F8)Fe(O2)(DCHIm)] present in the sample (see Supporting Information).
In contrast to excitation at higher energies, excitation of 2 at low energy (775 nm) results in observation of five features, 796, 586, 475, 425, and 394 cm−1, three of which (796, 586, and 394 cm−1) shift to lower energy in the 18O2 isotopologue, to 754, 561, and 390 cm−1 (Figure 2). These features are not detected upon excitation of 1 or [(F8)Fe(O2)(DCHIm)]. The 586 cm−1 vibration is ~5× as intense as the other bands and is assigned as an Fe—O stretch on the basis of energy and isotopic shift (Δobs(calc) = 25 (26) cm−1). This energy compares well to the νFe—O of a variety of η1 (or “end-on”) heme-dioxygen adducts, such as a recent example of a heme-hydroperoxo adduct that displays a νFe—O at 570 cm−1.[21] The νFe—O band of a η2 (or “side-on”) heme-peroxo adduct would be at significantly lower energy, as observed in [(tmp)Fe(O2)]− (tmp = 5,10,15,20-tetrakis(2,4,6-trimethylphenyl) porphyrinate) at 470 cm−1.[21] The 796 cm−1 feature is consistent in energy and isotopic shift (Δ = 42 cm−1) with assignment as an intra-peroxide stretching (νO—O) mode. The energy of this νO—O feature in 2 is 40 cm−1 higher than that observed for 1.[22] The relatively low energy of νO—O in 1 was defined as resulting from backbonding of the filled Cu d orbitals into the strongly anti-bonding peroxo σ* orbital in the side-on structure.[16] Thus, the relatively high energy of the νO—O mode in 2 is indicative of end-on coordination of the peroxo group to Cu. This increase in the νO—O of the peroxo group upon going from side-on to end-on coordination to Cu is supported by DFT calculations (vide infra). The final isotope sensitive feature at 394 cm−1 has too small of an isotope shift (Δobs: 4 cm−1) to be a νCu—O mode (Δcalc: 12 cm−1), and also appears at too low an energy when compared to end-on Cu-peroxo dimers such as {[Cu(tmpa)]2(O2)}2+ (tmpa = tris(2-methylpyridyl)amine)) in which the νCu—O mode was observed at 561 cm−1.[23] The energy of the 394 cm−1 band is however consistent with its assignment as a trans-axial δN—Fe—O bend mode based on an analogous mode observed in [Fe(BLM)OH] (BLM = bleomycin) at 402 cm−1.[24] Taken together, the rR data indicate that the axial coordination of the imidazole base to 1 causes the peroxo bridge in 2 to adopt a µ-1,2 conformation. Further, the strong enhancement of the νFe—O and the lack of a detectable νCu—O feature indicate the assignment of the optical band at 789 nm as a peroxo → Fe charge transfer transition.
Figure 2.

Resonance Raman spectra of 2 prepared with natural abundance O2 (—) and 18O2 (- -) with 775 nm excitation at 77 K in THF. The spectrum presented is the composite of two separate acquisitions on different spots due to the range of the spectral window of the instrument at this excitation energy (separate ranges denoted by black bar), and the low energy region has been scaled by a factor of three to aid visualization.
To verify the coordination mode of the peroxo ligand in 2 and to obtain metrical parameters, Fe and Cu K-edge X-ray absorption spectroscopy (XAS) was performed (Figure 3, S2–S3, and Tables S1–S5). The XAS data confirm a six coordinate Fe(III) and a four coordinate Cu(II), which support a µ-1,2 peroxo bridge spanning the copper and heme derived from the rR data for 2. Specifically, the first shell of the Fe EXAFS was fit using one Fe-O/N contribution at 1.81 ± 0.02 Å and five Fe-N/O contributions at 2.02 ± 0.02 Å. An Fe•••Cu contribution was fit at 3.99 Å with its corresponding multiple-scattering (MS) Fe-O-Cu vector refined to 4.08 Å. However, this Fe•••Cu vector was not required in the fit due to overlap with porphyrin contributions. The Fe K-edge XAS of the independently prepared contaminant [(F8)Fe(O2)(DCHIm)] was also separately measured to assess its contribution to the XAS of 2; subtraction of < 20 % of the contaminant from the spectrum of 2 yielded no significant changes (see Supporting Information). The first-shell of the Cu EXAFS was fit with four Cu-N/O contributions at 1.98 ± 0.02 Å. Fits wherein the first shell was split to accommodate two scattering paths were not justified based on the resolution of the data (0.14 Å), but a split shell is consistent with the high σ2 value which reflects a distance distribution. The peak in the R = 3.3–4.3 Å range was fit with a Cu•••Fe single-scattering contribution at 4.01 Å with its corresponding MS Cu-O-Fe vector at 4.15 Å; however, this contribution is also not required by the data.
Figure 3.

EXAFS (inset) and non-phase-shift-corrected Fourier transform (- -), fit (—) of 2 in THF at 10 K. Top: Fe K-edge (data to k = 16 Å−1). Bottom: Cu K-edge (data to k = 12.8 Å−1). Phase shifts in the first shells are ~0.4 Å
Density functional theory (DFT) calculations were performed on 2. The calculations reproduce its spectrocopically deduced structure where the peroxo ligand bridges the Cu and Fe ions in a µ-1,2 fashion (Figure 4) and predict that the environment of the Fe is six coordinate pseudo-octahedral and the Cu is four coordinate D2d distorted square planar (τ = 47.4°, where τ = 0° for square planar and τ = 90° for tetrahedral). The peroxidic nature of the bridge is established computationally by a 1.40 Å O—O bond length, significantly shorter than the 1.46 Å O—O bond calculated for 1. This decrease in bond length correlates to the DFT calculated νO—O that increases from 821 cm−1 in 1 to 840 cm−1 in 2, reproducing the experimental νO—O trend (Table S8). The computed Fe—O bond length of 1.82 Å compares well to the EXAFS derived distance of 1.81 Å and the Cu—O bond at 1.90 Å compares reasonably to the EXAFS derived Cu—O/N scatterer envelope of 1.98 Å. The Cu—O bond length of 1.90 Å is somewhat shorter than the Cu—O bonds in 1 (2.00 and 2.09 Å) which reflects the change in going from five coordinate side-on in 1 to four coordinate end-on in 2. The Fe—O—O—Cu core dihedral angle is an acute 137.2° which leads to a relatively short Fe•••Cu distance of 4.01 Å. 2 is well described as a broken-symmetry (BS) singlet (ST = 0) that is calculated to be 2.2 kcal/mol lower in energy than the triplet, consistent with the diamagnetism observed in the 1H-NMR spectrum. In the singlet, the low-spin Fe(III) and the Cu(II) are anti-ferromagnetically coupled. The low-spin electronic configuration of the heme is a consequence of the axial ligation of imidazole, and leads to improved donation of the peroxo to iron,[25] contributing to the strong O—O bond.
Figure 4.

DFT optimized geometric structure of 2 using the spin-unrestricted BP86 functional on the BS (ST = 0) spin surface.
2 is a LS heme-peroxo-copper adduct in which the peroxo bridges the metals in a µ-1,2 fashion based on its spectral features and supported by DFT calculations. Crystallographic studies on “as-isolated” CcO[26] have suggested the presence of a bridging peroxo moiety also in a µ-1,2 geometry.[18–20] rR data on the resting enzyme show a feature at 755 cm−1 (647.1 nm excitation) which was attributed to an intra-peroxide stretch (νO—O) based on its disappearance upon exposure to cyanide.[27] Isotopically labeled (18O) resting CcO has not been reported, and a ligand-metal (νFe—O) mode is not present in the spectrum (400–1200 cm−1). In resting CcO, the heme a3 is HS (S = 5/2) based on: 1) the λmax of the heme Soret band at 424 nm, characteristic of a HS heme,[28] and 2) SQUID magnetization studies that demonstrated the heme a3—CuB pair are antiferromagnetically coupled to yield an overall S = 2 ground state.[29] Based on the spectral features of the bona fide µ-1,2 peroxo bridged structure described herein for 2, the proposed coordination mode of the peroxo in resting CcO is inconsistent with its spectroscopic observables. Specifically, the inconsistencies are: 1) a low intra-peroxide stretch is associated with backbonding from the copper, where the peroxo σ* orbital accepts charge from a Cu 3d orbital which weakens the O—O bond and is only possible for a peroxo bound side-on to a copper as demonstrated in 1 (Scheme 1), 2) end-on coordination of a peroxo to heme in 2 resulted in an intense νFe—O that is not observed in resting CcO, 3) the heme a3 found in resting CcO is HS despite the strong ligand field expected for a peroxo complex which would result in a LS heme electronic structure, as observed in 2 and in CcO with azide bound.[30] Taken together, the spectroscopy of resting CcO requires a weak or non-existent interaction of the peroxo with the ferric heme. If the spectral features of resting CcO are correctly interpreted, a more likely coordination mode for the peroxo to Cu is η2,[31] as in the recent crystallographic structure of a side-on nitrosyl bound to CuB in CcO.[32]
A weak interaction with the heme may also provide insight into the issue of why the peroxo in resting CcO does not reductively cleave to form PM, the intermediate in CcO formed after O-O cleavage. A weak interaction would limit overlap of the Fe d orbitals with the peroxo σ* orbital and disfavor the electron transfer required for cleavage.[33] The spin state change of the heme, which is associated with strong versus weak peroxo interaction with Fe, could also modulate reactivity. Indeed, the spin state change of HS 1 to LS 2 correlates to the activation of peroxo towards abstraction of the net H• from phenol, a prerequisite for reductive O—O bond cleavage. This spin state change could facilitate this reaction through thermodynamics (an S = 2 Fe(IV)=O heme product is disfavored by > 20 kcal/mol compared to an S = 1 Fe(IV)=O) and the kinetics associated with the spin state change. CcO would have additional constraints compared to 2 concerning spin, given the His-Tyr crosslink and proposed magnetic coupling between the Tyr radical and the Cu(II) ion in PM. Those factors that correlate the HS to LS electronic structure change to reactivity are being further investigated.
Experimental Section
UV/Vis samples were prepared by exposing a 4 mL of 0.36 mM equimolar solution of [(F8)FeII] and [CuI(AN)](BArF) in THF at −80 °C to dioxygen to generate 1. After the removal of the excess dioxygen, 1.0 equiv. (100 µL of 14.4 mM, 0.30 mg) of DCHIm was added to yield 2. rR samples were prepared in rubber septum sealed 5mm NMR tubes at 1mM concentration in an analogous manner. XAS samples were prepared at 5mM concentration in an analogous manner and loaded into 2 mm Delrin XAS cells with 38 µm Kapton windows. DFT calculations were performed using Gaussian03. [34], Molecular structures were optimized using the BP86 functional within the spin-unrestricted formalism. The basis sets employed on Fe, Cu and O2 were of triple-ζ quality with polarization (6–311g*). A double-ζ quality, split-valence basis was used on all other atoms (6–31g), and was augmented with polarization on the metal bound N atoms (6–31g*). An ultrafine integration grid was employed as was auto density fitting. Analytical frequency calculations were performed to ensure a stationary point had been reached and no imaginary frequency was found. Additional experimental details are available in the Supporting Information.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
These studies were supported by the NIH (DK031450 to EIS; GM60353 to KDK; RR001209 to KOH). MKE is supported by an NIH post-doctoral fellowship (GM085914). Computational resources were provided in part by the NSF through Teragrid (CHE080054N). Synchrotron resources were provided by the SSRL, the operation of which is supported by the DOE, Office of Basic Energy Science. The SSRL Structural Molecular Biology program is supported by the NIH (NCRR P41 RR001209) and the DOE Office of Biological and Environmental Research.
Contributor Information
Matthew T. Kieber-Emmons, Department of Chemistry, Stanford University, Stanford, CA 94305
Munzarin F. Qayyum, Department of Chemistry, Stanford University, Stanford, CA 94305
Yuqi Li, Department of Chemistry, The Johns Hopkins University, Baltimore, MD 21218.
Zakaria Halime, Department of Chemistry, The Johns Hopkins University, Baltimore, MD 21218.
Keith O. Hodgson, Department of Chemistry, Stanford University, Stanford, CA 94305 Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Stanford, CA 94309.
Britt Hedman, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Stanford, CA 94309.
Kenneth D. Karlin, Department of Chemistry, The Johns Hopkins University, Baltimore, MD 21218.
Edward I. Solomon, Department of Chemistry, Stanford University, Stanford, CA 94305; Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Stanford, CA 94309.
References
- 1.Ferguson-Miller S, Babcock GT. Chem. Rev. 1996;96:2889. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 3.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 4.Das TK, Pecoraro C, Tomson FL, Gennis RB, Rousseau DL. Biochemistry. 1998;37:14471. doi: 10.1021/bi981500w. [DOI] [PubMed] [Google Scholar]
- 5.Proshlyakov DA, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Appelman EH, Kitagawa T. J. Biol. Chem. 1994;269:29385. [PubMed] [Google Scholar]
- 6.Iwaki M, Breton J, Rich PR. Biochim. Biophys. Acta. 2002;1555:116. doi: 10.1016/s0005-2728(02)00265-7. [DOI] [PubMed] [Google Scholar]
- 7.Oganesyan VS, White GF, Field S, Marritt S, Gennis RB, Yap LL, Thomson AJ. J. Biol. Inorg. Chem. 2010;15:1255. doi: 10.1007/s00775-010-0683-5. [DOI] [PubMed] [Google Scholar]
- 8.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Science. 2000;290:1588. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 9.Varotsis C, Woodruff WH, Babcock GT. J. Biol. Chem. 1990;265:11131. [PubMed] [Google Scholar]
- 10.Han SW, Ching YC, Rousseau DL. Proc. Natl. Acad. Sci. U. S. A. 1990;87:2491. doi: 10.1073/pnas.87.7.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collman JP, Devaraj NK, Decreau RA, Yang Y, Yan YL, Ebina W, Eberspacher TA, Chidsey CED. Science. 2007;315:1565. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collman JP, Decreau RA, Sunderland CJ. Chem. Commun. 2006:3894. doi: 10.1039/b607277a. [DOI] [PubMed] [Google Scholar]
- 13.Blomberg MRA, Siegbahn PEM, Wikström M. Inorg. Chem. 2003;42:5231. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 14.Chufan EE, Puiu SC, Karlin KD. Acc. Chem. Res. 2007;40:563. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 15.Proshlyakov DA, Pressler MA, Babcock GT. Proc. Natl. Acad. Sci. U. S. A. 1998;95:8020. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halime Z, et al. Inorg. Chem. 2010;49:3629. doi: 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.A prior LS example was proposed in Collman JP, Herrmann PC, Boitrel B, Zhang X, Eberspacher TA, Fu L, Wang J, Rousseau DL, Williams ER. J. Am. Chem. Soc. 1994;116:9783.. However, this exhibits too small an isotope perturbation of νO-O (Δ = 18 cm−1). Another LS example was reported in Liu J, Naruta Y, Tani F. Angew. Chem. Int. Ed. 2005;44:1836. doi: 10.1002/anie.200462582., in which the ground state of the heme was inferred based on the porphyrin ν2 band.
- 18.Aoyama H, Muramoto K, Shinzawa-Itoh K, Hirata K, Yamashita E, Tsukihara T, Ogura T, Yoshikawa S. Proc. Natl. Acad. Sci. U. S. A. 2009;106:2165. doi: 10.1073/pnas.0806391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koepke J, Olkhova E, Angerer H, Müller H, Peng G, Michel H. Biochim. Biophys. Acta. 2009;1787:635. doi: 10.1016/j.bbabio.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Tiefenbrunn T, Liu W, Chen Y, Katritch V, Stout CD, Fee JA, Cherezov V. PLoS One. 2011;6:e22348. doi: 10.1371/journal.pone.0022348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J-G, Ohta T, Yamaguchi S, Ogura T, Sakamoto S, Maeda Y, Naruta Y. Angew. Chem., Int. Ed. Engl. 2009;48:9262. doi: 10.1002/anie.200904572. [DOI] [PubMed] [Google Scholar]
- 22.Chufan EE, Mondal B, Gandhi T, Kim E, Rubie ND, Moenne-Loccoz P, Karlin KD. Inorg. Chem. 2007;46:6382. doi: 10.1021/ic700363k. [DOI] [PubMed] [Google Scholar]
- 23.Baldwin MJ, Ross PK, Pate JE, Tyeklàr Z, Karlin KD, Solomon EI. J. Am. Chem. Soc. 1991;113:8671. [Google Scholar]
- 24.Liu LV, et al. Proc. Natl. Acad. Sci. U. S. A. 2010;107:22419. doi: 10.1073/pnas.1016323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.In a low-spin heme, the Fe dz2 is completely unoccupied. Thus, donation from the O22− π*σ into this orbital is more effective compared to a high-spin electronic configuration because of the lack of electron-electron repulsion in this bonding interaction.
- 26.The resting "as-isolated" form of the enzyme used for the crystallography is also called the "fast" fully oxidized form. See Moody AJ, Cooper CE, Gennis RB, Rumbley JN, Rich PR. Biochemistry. 1995;34:6838. doi: 10.1021/bi00020a030. for an overview of "fast" vs "slow".
- 27.Sakaguchi M, Shinzawa-Itoh K, Yoshikawa S, Ogura T. J. Bioenerg. Biomembr. 2010;42:241. doi: 10.1007/s10863-010-9282-y. [DOI] [PubMed] [Google Scholar]
- 28.Clore GM, Andrèasson LE, Karlsson B, Aasa R, Malmström BG. Biochemical Journal. 1980;185:139. doi: 10.1042/bj1850139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Day EP, Peterson J, Sendova MS, Schoonover J, Palmer G. Biochemistry. 1993;31:7855. doi: 10.1021/bi00082a003. [DOI] [PubMed] [Google Scholar]
- 30.Thomson AJ, Greenwood C, Gadsby PM, Peterson J, Eglinton DG, Hill BC, Nicholls P. J Inorg Biochem. 1985;23:187. doi: 10.1016/0162-0134(85)85025-x. [DOI] [PubMed] [Google Scholar]
- 31.The residual density between heme a3 and CuB was also modeled in an earlier study ( Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Science. 1998;280:1723. doi: 10.1126/science.280.5370.1723., footnote 9 therein) as a hydroperoxide bridge with the proton on the Fe bound oxygen atom, thus substantially weakening the peroxo-Fe interaction. However, it should be noted that typical Cu-OOH intraperoxide stretching frequencies are >820 cm−1.
- 32.Ohta K, Muramoto K, Shinzawa-Itoh K, Yamashita E, Yoshikawa S, Tsukihara T. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2010;66:251. doi: 10.1107/S1744309109055109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An alternative proposal has been presented in Kaila VRI, Oksanen E, Goldman A, Bloch DA, Verkhovsky MI, Sundholm D, Wikström M. Biochim. Biophys. Acta. 2011;1807:769. doi: 10.1016/j.bbabio.2010.12.016., in which the putative peroxo moiety in resting CcO is suggested as a superoxo moiety. However, such a species is inconsistent with the Raman spectral feature and would be LS.
- 34.Frisch MJ, et al. Wallingford, CT: Gaussian, Inc.; 2004. [Google Scholar]